

Abstract
The vitamin D receptor (VDR) forms homo- or heterodimers on response elements composed of two hexameric half-sites separated by 3 bp of spacer DNA. We describe here the crystal structures at 2.7–2.8 Å resolution of the VDR DNA-binding region (DBD) in complex with response elements from three different promoters: osteopontin (SPP), canonical DR3 and osteocalcin (OC). These structures reveal the chemical basis for the increased affinity of VDR for the SPP response element, and for the poor stability of the VDR–OC complex, relative to the canonical DR3 response element. The homodimeric protein–protein interface is stabilized by van der Waals interactions and is predominantly non-polar. An extensive α-helix at the C-terminal end of the VDR DBD resembles that found in the thyroid hormone receptor (TR), and suggests a mechanism by which VDR and TR discriminate among response elements. Selective structure-based mutations in the asymmetric homodimeric interface result in a VDR DBD protein that is defective in homodimerization but now forms heterodimers with the 9-cis retinoic acid receptor (RXR) DBD.
Keywords: nuclear receptor/RXR/structure/TR/VDR
Introduction
The vitamin D receptor (VDR) (Baker et al., 1988) is a ligand-activated transcription factor that plays a central role in calcium homeostasis and has been implicated in regulating diverse biological functions, including cellular proliferation and differentiation (Abe et al., 1981; Bouillon et al., 1995; Feldman et al., 1997; DeLuca and Zierold, 1998). VDR belongs to the steroid and nuclear hormone receptor superfamily whose members include receptors for thyroid hormone (TR), all-trans retinoic acid (RAR), estrogen (ER), glucocorticoids (GR), 9-cis retinoic acid (RXR) and >150 others (Mangelsdorf and Evans, 1995; Mangelsdorf et al., 1995). Receptors bind as homo- or heterodimers to bipartite hormone response elements (HREs) via their DNA-binding domains (DBDs), which consist of a highly conserved 66 residue core made up of two zinc-nucleated modules that fold into a unified globular domain (Luisi et al., 1991; Khorasanizadeh and Rastinejad, 2001). An adjacent C-terminal extension (CTE) of the DBD imparts additional sequence or dimerization specificity. While some full-length receptors are able to form dimers in solution through their ligand-binding domains (LBDs), the DBDs themselves do not dimerize in the absence of their DNA target. However, in all of the cases reported to date, the DBDs and associated CTEs of the nuclear receptors generate the same pattern of DNA selectivity and dimerization as the full-length receptors from which they are derived (Mader et al., 1993; Perlmann et al., 1993; Towers et al., 1993; Zechel et al., 1994a,b).
Response elements typically consist of two hexameric half-sites whose consensus sequence, for the non-steroid receptors, is 5′-AGGTCA-3′. Diversity is achieved largely by varying the arrangement of the half-sites relative to one another—inverted, everted or direct repeats (DRs)—thereby restricting the dimeric species that can bind. Within the DR series of elements, nuclear receptors bind as heterodimers with RXR as the common partner. Further identity is imparted by varying the number of neutral base pairs separating the half-site repeats. This was formalized as the ‘1–5 rule’, which specifies the spacer required for high affinity binding of RXR–RXR (DR1), RXR–RAR (DR2), RXR–VDR (DR3), RXR–TR (DR4) and RXR–RAR (DR5) heterodimers (Umesono et al., 1991; Mangelsdorf and Evans, 1995). With the exception of the RAR–RXR–DR1 complexes, whose polarity is reversed (Kurokawa et al., 1994; Zechel et al., 1994b; Rastinejad et al., 2000), RXR occupies the upstream half-site in each element. VDR–VDR homodimers also bind to DR3 response elements.
The structures of dimeric nuclear receptor DBDs bound to DR4 (RXR–TR), DR2 (RevErb–RevErb) and DR1 (RXR–RXR and RAR–RXR) response elements have been determined and have provided key insights into the structural basis for response element spacer discrimination (Rastinejad et al., 1995, 2000; Zhao et al., 1998, 2000). Whereas the core DBDs all bind to the consensus half-site sequence in an identical manner, the geometry of the bipartite response element is read out by unique protein– protein dimerization contacts that match the spacing between the direct repeats. These contacts are frequently supported by simultaneous hydrogen bonds to the response element, thereby reinforcing the DNA dependence of the dimerization interaction. The non-conserved CTEs in all receptors examined to date adopt unique conformations. These conformations reflect the mechanism by which any sequence specificity outside of the hexameric half-site is imparted. Importantly, modeling has also shown the potential for the CTE of the downstream subunit to be employed as a molecular ‘bumper’ that makes severe steric clashes with the upstream subunit in the course of selecting against the wrong inter-half-site spacing.
VDR exhibits several properties that highlight its potential distinctiveness within the nuclear receptor superfamily. First among these is the mechanism of spacer discrimination. Unlike RevErb, whose CTE mediates recognition of sequences upstream of the DR2 hexameric half-sites, VDR does not show any preference for particular sequences outside of the half-site (Freedman et al., 1994). Moreover, modeling of RXR–TR DBDs on VDR-specific DR3 elements predicted that the CTE of TR would clash with the upstream RXR partner (Rastinejad et al., 1995). VDR may thus possess a unique CTE conformation and employ a novel mechanism of response element spacer discrimination. Secondly, as we demonstrate here, VDR DBD does not form heterodimers with RXR DBD on DR3 response elements; it forms only VDR homodimers. This is counter to the doctrine that the DBDs of the nuclear receptors recapitulate the DNA-binding specificity of their full-length counterparts. It is also puzzling because how would the VDR–RXR heterodimer form the intermolecular associations necessary for spacer discrimination if their DBDs do not associate? Finally, most naturally occurring VDR response elements do not contain consensus half-site sequences. These variations lead to differences in DNA-binding affinity that are likely to be important for the precise regulation of genes. The chemistry underlying such modulation of DNA affinity has yet to be revealed in this or any other hormone receptor system.
In order to understand the basis for DNA target specificity in VDR, we have solved the crystal structures of VDR DBD bound as a homodimer to a series of naturally occurring DR3 response elements. We used the homodimer for these studies because: (i) the homodimeric species exhibits ligand-dependent transcriptional activation in the presence of the co-activator proteins SRC-1 or TRAM-1 (Takeshita et al., 2000); (ii) the key VDR–DNA interactions, including the conformation and trajectory of the VDR CTE, are likely to be the same in the RXR–VDR heterodimer as they are in the homodimer because VDR DBD is common to both; (iii) the homodimer is experimentally accessible whereas the RXR–VDR DBD heterodimer to date could not be formed; and (iv) the structure of the homodimer is likely to be an important guide in designing constructs for future structural studies of the heterodimer. The structures we now report show the topology of assembly on DR3 response elements and the VDR surfaces that mediate dimeric association. The key interactions that lead to high and low affinity binding to non-consensus half-site sequences are also revealed. Surprisingly, the CTE of VDR resembles that of TR. From this, we outline a mechanism by which VDR discriminates among response element spacers. Finally, we propose a structural explanation for the remarkable stability of the VDR DBD homodimer and for the failure to observe RXR–VDR DBD heterodimers. We have exploited this understanding in the design of point mutants in VDR DBD that now allow the formation of RXR–VDR DBD heterodimers.
Results
Crystallization and structure determination
The structures of three human VDR DBD (residues 16–125)–DNA complexes (Figure 1) were solved and refined at 2.7–2.8 Å resolution. The protein was overexpressed in Escherichia coli and co-crystallized as a homodimer bound to three different DNA duplexes representing osteopontin (SPP), osteocalcin (OC) and consensus (DR3) response elements (Figure 1B). The structures were solved by molecular replacement, refined and independently verified with maps calculated from single wavelength anomalous dispersion (SAD) data collected at the zinc edge (Figure 3A). A portion of the final electron density map is shown in Figure 3B.

Fig. 1. Protein and DNA constructs used in the structure determination. (A) The human VDR DBD. Sequence numbers are for full-length hVDR and those in parentheses refer to the common hormone receptor DBD numbering scheme. Residues in italics are disordered in all of the structures. (B) The 18 bp DNA duplexes used in co-crystallization, shown 5′→3′ in the top strand. Half-sites are shown in boxes and are numbered by base pair. The DR3 sequence contains a direct repeat of two consensus half-sites. SPP is the mouse osteopontin VDRE and OC is the rat osteocalcin VDRE. Bases that differ from the consensus sequence are shaded gray and the structurally significant changes are highlighted in black. Estimates of relative binding of VDR DBD homodimers to each sequence are also shown.

Fig. 3. Experimental electron density and homodimeric assembly. (A) Unbiased experimental electron density from SAD phases. The map is contoured around the CTE of the upstream subunit of the VDR DBD–DR3 structure, which is shown as a Cα trace. (B) A portion of the 2Fo = Fc electron density map showing intersubunit dimerization contacts. (C) Stereo view of the dimerization interface in a van der Waals surface representation. (A) and (B) were made with Xtalview (McRee, 1999), and (C) was prepared with Ribbons.
Overall architecture
In each structure, the asymmetric unit consists of one VDR DBD homodimer–DNA complex. Figure 2 shows the overall topology of the VDR DBD–DR3 complex. Similar arrangements hold for the SPP and OC complexes. Each protein subunit fully engages its hexameric half-site and forms a head-to-tail dimer. The 3 bp spacer between the two half-sites dictates the relative displacement of the two protein subunits on the DNA: the VDR subunits are separated by a center–center distance of 9 bp and a relative azimuthal rotation of 45°.

Fig. 2. Overall architecture of the VDR DBD–DR3 complex. The red and blue Cα traces are the upstream and downstream subunits, respectively. Selected side chains are shown in green. Gray spheres are Zn atoms. The canonical half-site sequence is shown in gold, and the 5′-flanking base pairs and the spacer are shown in black. The structures of all three complexes presented here have the same overall architecture. The figure was made with Ribbons (Carson, 1991).
Although they were allowed to refine independently, the backbone atoms of the six VDR DBD polypeptides in the three structures are identical, with an average pairwise r.m.s. deviation of only 0.41 ± 0.082 Å. Alignment of the VDR core DBD with the DBD cores of TR, RXR and RAR within their respective protein–DNA complexes shows that they share a common structural motif. Such DBD alignments also result in the close superimposition of the DNA half-sites to which they are each bound, indicating that the manner in which each DBD sits on its half-site is nearly identical.
VDR assembly on DR3 elements
VDR binds preferentially and cooperatively to DR3- type response elements (Freedman and Towers, 1991; Umesono et al., 1991). The 3 bp spacer of the direct repeat response element separates the two hexameric half-sites and fixes their relative orientation. This in turn specifies the disposition of the two subunits of the DNA-bound homodimer such that specific surfaces from each protomer are juxtaposed. As seen in Figure 3C, the polypeptide dimerization contacts in the VDR homodimer involve the side chains of Pro61, Phe62 and His75 of the upstream protomer and residues Asn37, Glu92 and Phe93 of the downstream subunit. These six interfacial residues are invariant among the nine known VDRs from various species, and the combination of these six residues is unique among hormone receptors, thus underscoring the uniqueness of the homodimer interface.
The crystallographically observed role of the interfacial residues in VDR dimerization and cooperative assembly is supported by biochemical and mutagenesis studies. In particular, systematic mutagenesis of the CTE of VDR identified Phe93 as critical to DNA binding and in vivo reporter gene activation (Quack et al., 1998). Mutation of residues Asn37, Phe62, His75 and Phe93, the core of the hydrophobic dimer interface, abolished cooperative assembly on DR3 elements (Towers et al., 1993). Recently, it was shown that a chimera of the VDR core DBD with the TR hinge and LBD regions could not activate DR3 reporter genes (Miyamoto et al., 2001). However, inclusion of the hinge/CTE region of VDR restored transactivation from DR3 elements. This supports our observation that many of the important residues in dimerization on DR3 elements are C-terminal to the core DBD and without these residues cooperative assembly is abolished (Nakajima et al., 1994; Hsieh et al., 1999).
With the exception of one weak hydrogen bond between His75 and Asn37, the primary mechanism of association across the interfacial gap is via van der Waals contacts that produce a smooth, complementary interface (Figure 3C). The phenylalanine residue contributed to the interface from each protomer imparts a hydrophobic character to the complementary surfaces, and the removal of these residues from contact with solvent is likely to stabilize the homodimer strongly. The paucity of intersubunit hydrogen bonds, the hydrophobic character of the interface and the preponderance of van der Waals interactions distinguishes the VDR–VDR dimer interface from those of other hormone receptors such as RXR–TR, RXR–RXR and GR–GR, which involve many more cross-subunit stabilizing hydrogen bonds, or RAR–RXR, which employs DNA-buttressed polar side chains to stabilize the intersubunit interface.
Surprisingly, none of the VDR residues making dimerization interactions is supported by buttressing contacts with the DNA. Instead, within each protomer, the side chains of these residues are supported by van der Waals interactions with one another. For example, Phe93 positions Asn37 to make a hydrogen bond with His75 from the opposite protomer, and simultaneously supports the tertiary conformation of the first zinc module. On the upstream subunit, Pro61 restricts the conformation of Phe62. This results in the solvent exposure of the aromatic side chain, which favors interactions with the non-polar surface of the opposing subunit. These interactions are in marked contrast to those seen in the RevErb, RXR–TR and RAR–RXR complexes, where the residues participating in dimerization interactions were buttressed by simultaneous DNA contacts. Instead, the VDR homodimer resembles the homodimeric steroid receptors GR (Luisi et al., 1991) and ER (Schwabe et al., 1993), where DNA contacts stabilize the overall conformation of the receptor subunits but do not directly brace any of the interacting residues.
The fact that the VDR dimerization surfaces are tightly packed and lack direct DNA support is consistent with the likelihood that their tertiary conformation is insensitive to DNA binding. This would thus be a partial ‘pre-payment’ of the entropic cost of dimerization and, as we discuss later, may account for the relative stability of the VDR DBD homodimer compared with the RXR–VDR DBD heterodimer. However, even if the dimerization surfaces are pre-formed, they are still unlikely to support assembly of the VDR homodimer in the absence of DNA. The three VDR DBD dimer interfaces each bury only 340–380 Å2 of water-accessible surface, an area which is on a par with that buried by other nuclear hormone receptor dimer interfaces (290–480 Å2), but smaller than the >700 Å2 that normally constitutes a bona fide dimerization interface. Indeed, by gel filtration, non-DNA bound VDR DBD elutes as a monomer (Figure 7A, peak labeled ‘Free Protein’). Therefore, as is seen for other nuclear receptors, VDR DBD dimerization requires the allosteric action of properly spaced DNA half-sites to bring the DBDs into proper register before association can occur.

Fig. 7. Structure-based mutations and RXR–VDR DBD heterodimer formation. (A) A typical chromatogram showing isolation of dimeric DBD–DNA complexes on Superdex 75. (B) SDS–PAGE analysis of proteins in the complex peak. The lanes labeled ‘Load’ show the actual mixture of proteins applied to the column, and those labeled ‘Peak’ show the composition of the peak fraction of the protein–DNA complex peak. With full-length VDR–RXRΔAB–DR3 and RXR DBD–TR DBD–DR4, both proteins are recovered (lanes 2 and 7), indicating heterodimerization. With wild-type VDR and RXR DBD (lane 4), no RXR is observed. Since the protein:DNA ratio of the peak was determined to be 2:1, this indicates that only VDR homodimers are formed. In contrast, lane 5 shows that the mutant VDR and RXR DBDs form heterodimers since both proteins are recovered in the peak fraction. (C) Rationale behind structure-based mutations of VDR and RXR DBDs. Wild-type VDR and RXR DBDs are labeled V and R, mutant proteins are labeled V′ and R′, and half-site DNA is represented as an arrow. The favored dimeric species are boxed and the disfavored assembly pathway is indicated with an X.
C-terminal extension
The CTEs of nuclear receptors mediate receptor dimerization and contribute to response element binding affinity. In contrast to the core DBD, whose structure has been the same in all receptor DBDs solved to date, the structure of each CTE determined thus far has been unique. Moreover, each unique structure has been shown to be a key mediator of spacer discrimination. Since VDR is the only receptor known to date to bind to DR3 response elements, it was of considerable interest to visualize the structure of the VDR CTE and to compare it to that of TR and RevErb, which discriminate against non-DR4 or -DR2 response elements, respectively. Surprisingly, as shown in Figure 4, the CTE of VDR bears a striking resemblance to the CTE of TR (Rastinejad et al., 1995). Both CTEs exit the core DBD as short 3,10 helices, bend sharply four residues later and terminate in a long α-helix. We take this structural homology as evidence that VDR’s mechanism of spacer discrimination may be similar to that employed by TR. In contrast, the VDR CTE does not resemble the RevErb CTE (Zhao et al., 1998). This is consistent, however, with the observation that the RevErb CTE interacts with sequences upstream of the hexameric half-site and confers additional sequence specificity to the receptor. VDR has no such additional sequence specificity.

Fig. 4. The VDR CTE resembles that of TR and is present in solution. (A) Superimposition of VDR, TR and RevErb DBDs. Proteins were aligned using the recognition helix and half-site DNA. The residue at the end of the core region (Met89) is marked, as is the position of Asp97 of VDR and the position of RevErb that would correspond to residue 97. (B) Circular dichroism of free and complexed VDR DBD.
Interestingly, although the VDR and TR CTEs are structurally homologous, they make quantitatively different interactions with the DNA response element. The TR CTE makes 15 direct or water-mediated hydrogen bonds with the DNA, while the VDR CTE, which has a similar number of charged and basic residues, makes only two. Such differences cannot be explained solely by the lower resolution of the VDR structures. This suggests that the primary role of the VDR CTE is to mediate response element spacer discrimination, and not to provide additional DNA affinity.
The basis for response element spacer discrimination in VDR and TR
In order to visualize the potential unfavorable interactions that occur when VDR is bound to response elements with disfavored length spacers (DR1, DR2, DR4 and DR5), we used modeling to readjust the VDR DBD interface by changing the number of intervening base pairs in the DR3 response element. As seen in Figure 5A, for spacer lengths of <3 bp, the CTE helix of the downstream VDR molecule clashes with the backbone of the upstream partner. Therefore, unless the CTE helix is significantly rearranged or the DNA distorted, VDR cannot bind to these elements. In this regard, there is no evidence for such rearrangements: the orientation of the CTE relative to both the DNA and the core DBD is the same in all six independent copies of the protein described here. For DR4 and DR5 response elements, on the other hand, modeling predicts that the VDR subunits would be too far apart to make the direct contacts necessary to form a stable dimer interface, even after adjusting side chain rotamers to maximize potential interactions between the subunits (Figure 5B). Without the subsequent cooperative binding imparted by dimerization contacts, the affinity of VDR for the DNA would resemble the much weaker monomeric affinity of VDR for a single half-site. Spacer discrimination thus appears to be achieved by a combination of two mechanisms: (i) by restricting productive interactions to protomers assembled on the DNA only in the proper relative orientation, i.e. on a DR3 element; and (ii) by using the CTE terminal helix sterically to block assembly on response elements with shorter spacers. Note that because VDR DBD is also the downstream partner in the RXR–VDR–DR3 heterodimer, it is likely that the mechanism of spacer discrimination employed by this species resembles that seen in the homodimer. In particular, the CTE terminal helix is likely to restrict assembly on DR2 or shorter spacers. The precise nature of the productive interactions between RXR DBD and VDR DBD that produce cooperative interactions on DR3 targets but not on DR4 or longer, however, awaits the determination of the structure of this complex. Such experiments are now in progress (P.Shaffer and D.Gewirth, unpublished).
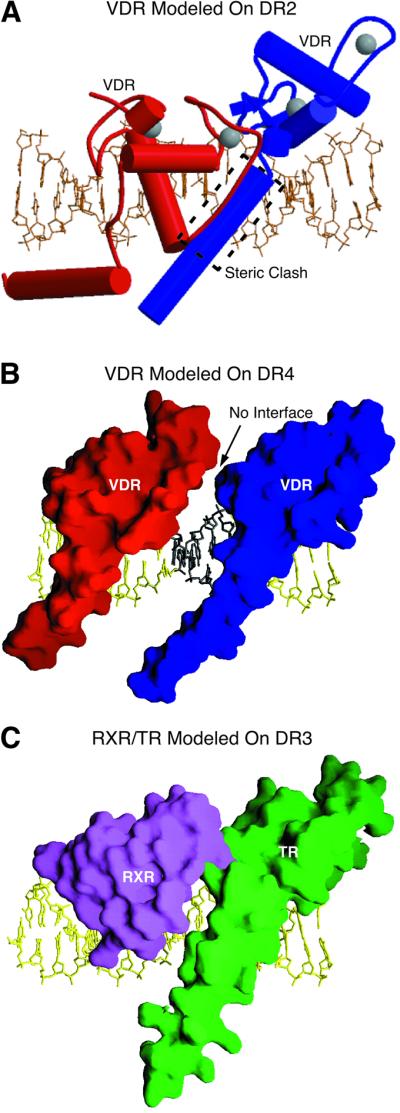
Fig. 5. Modeling studies based on VDR DBD structure. (A) Model of the VDR homodimer bound to a DR2 element. A likely steric clash is boxed. This figure was made with Molscript (Kraulis, 1991). (B) Model of the VDR homodimer bound to a DR4 element. Molecular surfaces of the proteins are shown. (C) Model of the RXR–TR heterodimer bound to DR3 DNA from the VDR homodimer structure. Proteins were placed by superimposing the backbone atoms of the core DBD and DNA half-site. (B) and (C) were prepared with GRASP (Nicholls et al., 1991).
If the CTE helix acts as a steric block to assembly on incorrect response elements, we might expect that it is stable even in the absence of DNA. To address this question, we used circular dichroism (CD) to compare the spectrum of the free protein (residues 16–125) in solution with that of the DNA-bound protein (Figure 4B). Comparing the ellipticity of the two samples at 222 nm shows that there is no change in helical content of the protein upon DNA binding. This shows that the CTE helix of VDR is present even in the absence of the DNA target. As a control, similar experiments were conducted with VDR molecules that were truncated after residues 113 or 109. These variants have shorter terminal helices, and gave smaller absolute values of ellipticity at 222 nm (data not shown), which indicates that the terminal helix does indeed contribute to the overall ellipticity. Since the CTE helix in VDR makes almost no interactions with the DNA target and is stable in the absence of the DNA, the most likely role of the CTE helix in VDR is to prevent dimerization on incorrectly spaced response elements.
Previous modeling studies based on the RXR–TR–DR4 heterodimer structure suggested that the TR–DR4 complex could not accommodate dimeric binding to DR3 response elements due to steric interference between side chains of the TR CTE helix and the RXR backbone (Rastinejad et al., 1995). As shown here, however, VDR easily accommodates dimeric binding to DR3 response elements, and the structure of the TR half-complex is essentially identical to that of VDR (overall r.m.s. deviation of 0.79 Å). In order to explain this discrepancy, we modeled RXR and TR on the DR3 element by superimposing the RXR and TR proteins and their cognate half-sites on the VDR subunits and half-sites. Surprisingly, no steric interference between the RXR and TR atoms is seen (Figure 5C), implying that this is not the mechanism by which TR discriminates against DR3 elements. This conclusion is supported by experiments in which an excess of a DR3 response element was in fact able to compete with DR4 elements for RXR–TR binding (Kliewer et al., 1992b). Given the limitations of modeling studies, however, further insight into the mechanism of TR and VDR spacer discrimination must await determination of the structures of the appropriate non-cognate complexes.
The basis for differential half-site sequence affinity
The VDR DBD homodimer and the RXR–VDR heterodimer bind to naturally occurring vitamin D response elements (VDREs) with varying affinity. These differences are an important means of regulating levels of gene expression. At least 19 putative VDREs have been identified, and with few exceptions these are organized as DR3s that vary only in their precise half-site sequences (Toell et al., 2000). Gel shift, competition, and reporter gene activation studies have identified a hierarchy of affinities of VDR or VDR DBD for different response elements (Freedman and Towers, 1991; Nishikawa et al., 1993; Toell et al., 2000). The mouse SPP VDRE (see Figure 1B), one of the highest affinity elements known, supports both homo- and heterodimer binding. In contrast, the canonical DR3 element requires at least 10 times as much protein as the SPP VDRE in order to show a gel shift with VDR DBD homodimers, and is also a weaker response element for RXR–VDR heterodimers. Finally, the rat OC VDRE shows weak affinity for the RXR–VDR heterodimer and is not gel shifted at all by the VDR DBD homodimer.
In order to dissect the molecular basis for the SPP > DR3 > OC hierarchy of affinities, we solved the structures of the VDR DBD homodimer bound to each of these three VDREs. The protein–DNA interactions for each complex are depicted schematically in Figure 6. The key interactions between the VDR DBD protomers and the DNA half-sites are similar to those seen in previous structural determinations of hormone receptor– DNA complexes (Luisi et al., 1991; Schwabe et al., 1993; Gewirth and Sigler, 1995; Rastinejad et al., 1995, 2000; Zhao et al., 1998, 2000; Meinke and Sigler, 1999): four conserved residues in the recognition helix, Glu42, Lys45, Arg49 and Arg50, make sequence-specific base contacts in the major groove of the half-site. As expected, given their conserved core sequences, the core VDR DBD makes roughly the same number of base and backbone contacts as RXR, RAR, RevErb and TR do to their response elements.

Fig. 6. Protein–DNA contacts observed in the three response elements. (A) Base-specific contacts. The DNA is drawn underwound for clarity only. Hydrogen bonds are depicted as arrows, with the donor at the tail of the arrow. Dashed lines represent hydrogen bonds seen in only one of the two half-site complexes. If a side chain has more than one functional group, arrows contacting the same region on an oval arise from the same group. (B) Backbone contacts common to at least five of six half-sites. Dotted lines are interactions seen only in the upstream half-complex. (C) Details of the Glu42 and Arg50 hydrogen bonds in selected complexes showing key specifying interactions.
Response element affinity does not correlate to buried surface area, since the VDR homodimer–DNA interface buries ∼3100 ± 75 Å2 of water-accessible surface in each structure. The area of these interfaces is roughly equivalent to the 3180 and 3030 Å2 buried by the RXR–TR and RAR–RXR heterodimer DNA interfaces, respectively.
There are no significant differences in the overall geometry of each of the three response elements in our structures. Thus, the sequence-specific variation in affinity for different DR3-type response elements reflects the precise interactions between the VDR protomer and the bases of the half-site. The half-site of the high affinity SPP response element, GGTTCA, differs from the lower affinity AGGTCA consensus half-site at positions 1 and 3. Comparison of the VDR–SPP structure with that of the VDR–DR3 structure shows that the substitution of a T3:A3 base pair for a G3:C3 at the third position dictates the more favorable interaction in the SPP–VDR complex. In particular, the substitution of the larger purine (A3) for a pyrimidine (C3) results in a productive rearrangement of the side chain of Glu42 that allows it to buttress additional water-mediated hydrogen bonds to the DNA bases (Figure 6C). These extra hydrogen bonds increase the stability of the SPP complex.
In contrast, the key defect that results in dramatically reduced VDR DBD homodimer binding to the rat OC VDRE is found in the upstream, non-consensus, half-site (GGGTGA) at position 5. In all high affinity HREs, this position is a C:G base pair (top strand listed first) and the G of this pair accepts one or two strong hydrogen bonds from the guanidino nitrogens of Arg50 of the receptor. In the upstream OC half-site, however, the C5:G5 is replaced by a G5:C5. The C5 of this pair has no hydrogen bond acceptors and thus cannot form hydrogen bonds with Arg50 (Figure 6C). Importantly, although the Arg50 reorients to form a hydrogen bond to the phosphate backbone, difference distance matrix analysis shows that there is no significant reorientation of the protein backbone relative to the DNA backbone, compared with the DR3 and SPP complexes. This implies that it is only the loss of the specific base interaction at position 5 that causes the reduced VDR–OC affinity. While the loss of this interaction in the upstream OC half-site essentially destroys VDR homodimer binding, under physiological conditions it merely diminishes the overall affinity of the RXR–VDR heterodimer for the OC element. This is presumably because it is offset by the obligatory co-localization of the RXR and VDR DBDs imposed by the strong heterodimer interface formed between their LBDs. Such a DNA-independent dimer interface is not present in full-length VDR homodimers.
RXR DBD and VDR DBD do not form heterodimers on DR3 response elements
While the VDR homodimer plays a role in DNA target recognition and activation (Cheskis and Freedman, 1994; Takeshita et al., 2000), the major activator of vitamin D-responsive genes is still the RXR–VDR heterodimer (Kliewer et al., 1992a). We tested the ability of RXR DBD and VDR DBD to form heterodimers on DR3 response elements using a gel filtration assay, which we employed because it allowed us to determine accurately both the protein composition and the stoichiometry of the dimeric protein–DNA complex. Such assays have been shown to correlate with other measures of protein–DNA binding, including electrophoretic mobility shift (Cheskis and Freedman, 1994; Chen et al., 1998; Juntunen et al., 1999). The protein composition of the peak containing the DNA–protein complex was established by SDS–PAGE, and the total amount of protein and DNA in the peak was measured by the Bradford method and by UV absorbance, respectively. When DR3-type response elements were used, the protein:DNA ratio in the complex peak was found to be 2.0 (± 0.1):1, as expected for homodimeric complexes. In all of the cases presented here, the protein composition across the peak containing the protein–DNA complex was uniform, indicating that the complexes contained a single species.
As seen in Figure 7B, lanes 3 and 4, VDR DBD does not form heterodimers with RXR DBD in the presence of DR3 targets. Only VDR DBD–DR3 homodimers were recovered. This phenomenon is independent of the length of the VDR CTE helix, of salt concentrations between 50 and 150 mM or a 10-fold molar excess of RXR DBD (not shown). As a control, RXRΔAB (an RXRα construct lacking the N-terminal AF-1 domain) and full-length VDR + 1,25 dihydroxyvitamin D3 were subjected to the same assay (lanes 1 and 2). In contrast to the results obtained with the isolated DBDs, but in agreement with prior published results (Yu et al., 1991; Kliewer et al., 1992a,b), full-length RXR and VDR formed heterodimers on DR3 elements. As a further control, RXR DBD and TR DBD were assayed for their ability to form heterodimers on a DR4 response element, to confirm that the assay recapitulates previously reported observations of DBD heterodimerization. Indeed, as seen in Figure 7B, lanes 6 and 7, only RXR–TR DBD–DR4 heterodimers were recovered. The failure to observe heterodimerization of VDR DBD with RXR DBD in the presence of the DR3 target thus places the VDR DBD at odds with the canonical view that the DBDs of steroid and nuclear receptors are sufficient for generating the same pattern of DNA selectivity and dimerization as full-length receptors (Mader et al., 1993; Perlmann et al., 1993; Zechel et al., 1994b). We note, however, that this observation does not contradict any published observations of RXR DBD–VDR DBD–DR3 heterodimers; such species have not been reported.
Formation of RXR DBD–VDR DBD heterodimeric complexes
Full-length heterodimers between RXR and VDR differentiate among correct and incorrect response elements, and there is no evidence for a DNA target recognition function outside of the DBDs and their CTEs. We therefore speculated that RXR DBD and VDR DBD fail to form heterodimers on DR3 response elements because of highly efficient competition from VDR DBD homodimers. This hypothesis predicts that if cooperativity between VDR DBD homodimers could be reduced, then the RXR–VDR DBD heterodimer would compete successfully with the VDR DBD homodimer for the DR3 target.
Guided by our VDR DBD homodimer structures, we made alanine substitution mutations for residues Pro61, Phe62 and His75 in the VDR DBD. We predicted that these mutations would destabilize the VDR DBD homodimeric interface without disrupting any potential RXR–VDR DBD heterodimer interface (see Figure 7C). This scheme exploits the fact that: (i) the direct repeat DR3 response element is polar and results in the asymmetric assembly of proteins on the two adjacent half-sites; (ii) heterodimers between RXR and VDR display a consistent and distinct polarity, with RXR binding to the upstream half-site and VDR to the downstream half-site (Perlmann et al., 1993; Quelo et al., 1994); and (iii) the VDR DBD residues making dimerization interactions do not directly stabilize the protein–DNA interface and can therefore be changed without altering the affinity of a VDR monomer for the DNA half-site. A Lys160Arg point mutant in the RXR DBD that may improve the affinity of this receptor for the DNA half-site (Rastinejad et al., 2000; Zhao et al., 2000) was also used in these experiments, although subsequent experiments with wild-type RXR DBDs have shown this mutation to be unnecessary (P.Shaffer and D.Gewirth, in preparation).
As seen in Figure 7B, lane 5, when stoichiometric amounts of the triple mutant of VDR DBD (Pro61Ala, Phe62Ala, His75Ala) and RXR DBD Lys160Arg were mixed with DR3 DNA and analyzed by the gel filtration assay, both proteins were observed in equal amounts in the fractions corresponding to the protein–DNA complex. This indicates the formation of the RXR–VDR DBD heterodimer. Inclusion of excess mutant VDR DBD or RXR DBD did not prevent heterodimer formation (not shown). The ability to mutate residues in VDR that change its dimerization behavior also demonstrates the accuracy and predictive power of these structures.
Discussion
Classically, the dimerization interfaces of the nuclear receptors are thought to have distinct but interdependent roles in receptor function. Partner selection is accomplished via a ligand-dependent association between the LBDs of the receptors. DNA target discrimination and binding, on the other hand, are mediated by a weaker DNA-dependent dimer interface between the two DBDs. In TR, the most extensively studied case, the relative stability of these dimerization interactions is consistent and reflects the transcriptionally active species: the preferred LBD partner is RXR, the preferred DBD partner is RXR, and the RXR–TR heterodimer preferentially activates transcription (Perlmann et al., 1996).
In contrast, the VDR DBD does not recapitulate the partner selection preference of the LBD. Thus, while the VDR LBD forms a stable, ligand-dependent heterodimer with the RXR LBD (Rochel et al., 2000), the preferred DBD partner, as we have shown here, is VDR, not RXR. Indeed, the VDR DBD homodimer has a compact, non-polar dimer interface, and the lack of any buttressing interactions between the dimer interface and the DNA highlights the likelihood that the interface is conformationally insensitive to the presence or absence of the DNA target. Compared with receptors that must fix or remodel side chain conformations upon DNA binding, a significant fraction of the entropic costs of homodimer formation has thus been pre-paid, and this may account for the unusual stability of the homodimer compared with the heterodimer. This analysis is supported by the demonstration here that mutation of the interfacial residues of the upstream partner allows preferential formation of the RXR–VDR DBD heterodimer.
The fact that the VDR DBD homodimer complex is energetically favored over the RXR–VDR DBD heterodimer would be puzzling in the context of a world in which only the RXR–VDR species is transcriptionally active. While the VDR homodimer may play a role in DNA target identification or transcriptional repression (Cheskis and Freedman, 1994, 1996), recently there has also been some evidence that the nuclear receptor co-activators SRC-1 and TRAP-1 may assist in the formation of stable ligand-dependent VDR homodimers on DR3 response elements, and that these complexes activate transcription in a ligand-dependent manner (Takeshita et al., 2000). If so, this provides a possible functional rationale for the unusual stability of the VDR DBD homodimer: the DBD–DBD interactions allow the formation of a transiently stable homodimer that is subsequently stabilized by co-activator binding. In this model, co-activator binding compensates for the lack of a stable homodimeric LBD dimer interface. The same study showed that co-activator proteins also form stable complexes with the RXR–VDR heterodimer, but in this case the heterodimer is already fully stabilized by the strong heterodimeric LBD interface and does not require the additional assistance of a strong DBD heterodimer interface.
All vitamin D-dependent genes are not transcribed at equal levels, and the modulation of transcriptional activity is likely to be due to many factors, including, possibly, whether transcription is stimulated by the VDR homodimer or the RXR–VDR heterodimer. Another factor, however, is the variation in the affinity of the receptor for the response element. There is significant deviation from the consensus half-site sequence in naturally occurring VDREs, and the results presented here show the stereochemical basis for this variation in response element affinity. Interestingly, the observed changes in affinity correlate with the gain or loss of hydrogen bonds between the protein and the bases of the DNA half-site. In contrast, an earlier study used a non-cognate steroid receptor–DNA complex to compare the structural basis for the near-absolute discrimination between steroid response element half-sites (AGAACA) and estrogen/nuclear receptor response element half-sites (AGGTCA) (Gewirth and Sigler, 1995). This study showed that discrimination was a function of the DNA target geometry, which led to the unfavorable incorporation of many more water molecules in the protein–DNA interface of the non-cognate complex. Together, this may point to a more general phenomenon whereby subtle variations in the energetics of DNA target affinity are modulated enthalpically via the gain or loss of hydrogen bonds, while stronger discrimination is achieved entropically, by the capture or liberation of solvent in the DNA–protein interface.
Materials and methods
Protein and DNA purification
The human VDR DBD (residues 16–125) was expressed in E.coli BL21/DE3 cells as inclusion bodies. Inclusion bodies were solubilized in 6 M guanidine-HCl buffer, renatured by dialysis and purified on SP Sepharose FastFlow (pH 7.4), Source 15S (pH 6.9) and Superdex 75 (100 mM NaCl) (all Pharmacia). Protein concentration was determined by amino acid analysis and Bio-Rad assay. Homogeneity was assessed by SDS–PAGE.
RXR DBD and TR DBD were overexpressed in E.coli as GST fusions and purified as described previously (Rastinejad et al., 1995).
Synthetic oligonucleotides were purchased from the Keck Oligo nucleotide Synthesis Facility at Yale University and were detritylated and purified on a reversed-phase column (Rainin Dynamax-300 Å PureDNA). Concentrated, purified strands were annealed by heating to 95°C and slowly cooling to room temperature.
Crystallization and data collection
Samples for co-crystallization contained DNA and protein concentrations of 0.33 and 0.66 mM, respectively, in 5 mM Tris pH 7.6, 50 mM NaCl and 2 mM dithiothreitol (DTT). Crystals were grown by the hanging drop vapor diffusion method at 18°C by the addition of 2 µl of the complex to an equal volume of reservoir solution (4–6% polyethylene glycol 4000, 50 mM MES pH 5.6, 5 mM MgCl2, 10% glycerol and 10 mM DTT). Diffraction quality crystals (typical dimensions 0.15 × 0.15 × 0.8 mm) in the space group P43212 grew in 1–3 weeks. The addition of glycerol and magnesium led to a dramatic enlargement of the crystal volume.
Crystals were equilibrated gradually into a reservoir solution which also contained 20% 2-methyl-2,4-pentanediol before being flash cooled in liquid nitrogen. Diffraction data were collected at –180°C on beamlines 14BMC or 19BM at the Advanced Photon Source using CCD detectors. Data were indexed and reduced using HKL2000 (Otwinowski and Minor, 1997). Diffraction from crystals of VDR DBD–DNA complexes extended beyond 2.5 Å in the direction parallel to the long unit cell axis, but was substantially weaker in the perpendicular directions, reflecting the underlying 4:1 anisotropy of the unit cell dimensions. This necessitated the use of crystals of large volume, as well as high brilliance synchrotron X-ray sources.
Data collection and refinement statistics are summarized in Table I. SAD data, keeping the Bijovet pairs separate, were also collected on the zinc edge (1.2828 Å) for all three structures and used to calculate unbiased maps that independently confirmed the results derived from the molecular replacement models.
Table I. Summary of data collection and refinement statistics for VDR–DNA complexes.
| Diffraction data/Complex | SPP | DR3 | OC | |||
|---|---|---|---|---|---|---|
| Source | APS-14BMC | APS-19BM | APS-19BM | APS-19BM | APS-19BM | APS-19BM |
| Space group | P43212 | P43212 | P43212 | |||
| a, c (Å) | 62.14, 241.75 | 61.81, 242.34 | 61.15, 241.80 | |||
| Wavelength (Å) | 1.0000 | 1.2828 | 1.0332 | 1.2828 | 1.0332 | 1.2828 |
| Resolutiona (Å) | 50–2.70 | 50–3.1 | 50–2.80 | 50–3.0 | 50–2.70 | 50–3.0 |
| Last shell (Å) | 2.80–2.70 | 3.21–3.10 | 2.90–2.80 | 3.11–3.00 | 2.80–2.70 | 3.11–3.00 |
| Unique reflections | 13 744 | 16 105 | 12 384 | 17 291 | 13 127 | 17 551 |
| Completeness (%) | 98.2 (99.9) | 99.8 (98.9) | 99.5 (99.0) | 96.9 (86.0) | 96.7 (77.6) | 99.7 (99.0) |
| Average I/σI | 22.9 (2.3) | 18.9 (3.0) | 27.2 (2.3) | 27.1 (2.1) | 34.4 (2.9) | 25.8 (3.9) |
| Redundancy | 5.9 | 8.3 | 14.1 | 12.6 | 12.5 | 5.3 |
| Rmergeb (%) | 8.1 (76) | 11.1 (27) | 10.0 (39) | 9.9 (48) | 7.1 (32) | 6.3 (36) |
| Phasing powerc | 1.31 | 1.74 | 1.50 | |||
| FOM (after DM)d |
|
0.18 (0.97) |
|
0.25 (0.85) |
0.23 (0.92) | |
| Crystallographic refinement | ||||||
| Resolution range (Å) | 50–2.70 | 50–2.80 | 50–2.70 | |||
| Reflections (F >2σF) | 12 490 (10 433) | 11 200 (9350) | 11 999 (10 321) | |||
| Non-solvent atoms | 2100 | 2204 | 2237 | |||
| Solvent atoms | 35 | 31 | 34 | |||
| Protein model (upstream/downstream) | 22–110/22–106 | 22–114/22–120 | 21–114/21–121 | |||
| Side chains truncated to alanine | 4/5 | 6/14 | 6/12 | |||
| R.m.s. deviation from ideality | ||||||
| Bond lengths (Å) | 0.0146 | 0.0154 | 0.0157 | |||
| Bond angles (°) | 1.66 | 1.69 | 1.78 | |||
| R-value (F >2σF)e (%) | 22.6 (20.9) | 21.4 (19.6) | 22.3 (20.6) | |||
| Rfree (F >2σF) | 28.9 (27.0) | 27.2 (25.4) | 27.5 (25.8) | |||
aThe resolution limit was defined as I/σI ≥2.0.
bRmerge = ∑hkl∑i|Ii(hlk) – <I(hkl)>|/∑hkl∑II(hkl).
cPhasing power = <[|FH|/E]>, where E is the residual lack of closure.
dFigure of merit = <|∑P(α)eiα/∑P(α)|>, where α is the phase and P(α) is the phase probability distribution.
eR = ∑|Fo – Fc|/∑Fo: 10% of reflections were used to calculate Rfree.
Structure determination and refinement
The three structures were solved and refined using CNS (Brünger et al., 1998). Initial phases for the VDR DBD–DR3 complex were obtained by molecular replacement, with the search model consisting of the TR portion of the refined RXR–TR DBD–DR4 complex, PDB code 2NLL. Only the core DBD region bound to its half-site DNA was used, and all non-conserved residues (50%) were truncated to alanines.
Simulated annealing omit maps revealed density for the additional 6 bp, missing side chains and residues C-terminal to the core DBD. The extended model was built using O (Jones et al., 1991). Manual rebuilding was followed by simulated annealing refinement using a maximum likelihood target, overall anisotropic B-factor, and bulk solvent correction. In later rounds of refinement, restrained individual B-factors were added and the resolution extended to 2.8 Å. The SPP and OC VDR–DNA complexes were solved and refined in a similar manner, except that the starting model was the refined VDR–DR3 homodimer complex and the initial refinement used rigid body rotation. The extent of these models and the refinement statistics are given in Table I.
Stereochemistry was assessed using Procheck (Laskowski et al., 1993), and clashes were identified using contact dots (Word et al., 2000). Graphics presented here used Ribbons (Carson, 1991), GRASP (Nicholls et al., 1991), XtalView (McRee, 1999) and Molscript (Kraulis, 1991).
Circular dichroism
CD measurements were made at room temperature using an Aviv Model 202 spectrometer. Spectra were obtained in 1 nm steps with 5 s averaging. Samples were made in and blanked against phosphate-buffered saline. The spectrum for unbound protein contained 10 µM VDR DBD and the spectrum for the homodimeric complex contained 10 µM protein and 5 µM DR3–DNA to form 5 µM of homodimeric complex. The spectrum of the complex was corrected for DNA background.
Site-directed mutagenesis
Oligonucleotide-directed point mutations in VDR and RXR were constructed with the QuikChange Site-Directed Mutagenesis Kit (Stratagene). Point mutations were confirmed by sequencing.
Gel filtration assay
DNA (10 nmol) was mixed with VDR and RXR DBDs (final concentration 10 µM DNA) and applied to a 1.6 × 70 cm Superdex 75 column (Pharmacia) equilibrated with 25 mM Tris pH 7.6, 50 mM NaCl, 2 mM DTT flowing at 1 ml/min. Individual fractions (2 ml) from the peak containing the protein–DNA complex were precipitated quantitatively with 4 vols of acetone at –20°C for 30 min, pelleted, air dried and resuspended in 1/20th of their original volume. SDS–PAGE was carried out using 20% PhastGels (Pharmacia) and proteins visualized by Coomassie Blue staining. The DR3 DNA duplex used in the VDR assays had a top strand sequence of 5′-CGACAGGTCACGAAGGTCAC-3′. The experiments with full-length VDR and RXR were conducted in the same manner except that the column used was Superdex 200.
Acknowledgments
Acknowledgements
We thank Karen Soldano for preparative assistance early on in this project, and thank the staff at APS-14BMC and APS-19BM, especially Drs Norma Duke and Andrzej Joachimiak, for access and assistance. Coordinates and structure factors have been deposited with the PDB (1KB2, 1KB4, 1KB6).
References
- Abe E., Miyaura,C., Sakagami,H., Takeda,M., Konno,K., Yamazaki,T., Yoshiki,S. and Suda,T. (1981) Differentiation of mouse myeloid leukemia cells induced by 1 α,25-dihydroxyvitamin D3. Proc. Natl Acad. Sci. USA, 78, 4990–4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker A.R., McDonnell,D.P., Hughes,M., Crisp,T.M., Mangelsdorf, D.J., Haussler,M.R., Pike,J.W., Shine,J. and O’Malley,B.W. (1988) Cloning and expression of full-length cDNA encoding human vitamin D receptor. Proc. Natl Acad. Sci. USA, 85, 3294–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouillon R., Okamura,W.H. and Norman,A.W. (1995) Structure–function relationships in the vitamin D endocrine system. Endocr. Rev., 16, 200–257. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr., D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Carson M. (1991) Ribbons 2.0. J. Appl. Crystallogr., 24, 958–961. [Google Scholar]
- Chen Z., Iyer,J., Bourguet,W., Held,P., Mioskowski,C., Lebeau,L., Noy,N., Chambon,P. and Gronemeyer,H. (1998) Ligand- and DNA-induced dissociation of RXR tetramers. J. Mol. Biol., 275, 55–65. [DOI] [PubMed] [Google Scholar]
- Cheskis B. and Freedman,L.P. (1994) Ligand modulates the conversion of DNA-bound vitamin D3 receptor (VDR) homodimers into VDR–retinoid X receptor heterodimers. Mol. Cell. Biol., 14, 3329–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheskis B. and Freedman,L.P. (1996) Modulation of nuclear receptor interactions by ligands: kinetic analysis using surface plasmon resonance. Biochemistry, 35, 3309–3318. [DOI] [PubMed] [Google Scholar]
- DeLuca H.F. and Zierold,C. (1998) Mechanisms and functions of vitamin D. Nutr. Rev., 56, S4–S10; discussion S75. [DOI] [PubMed] [Google Scholar]
- Feldman D., Glorieux,F.H. and Pike,J.W. (eds) (1997) Vitamin D. Academic Press, San Diego, CA.
- Freedman L.P. and Towers,T.L. (1991) DNA binding properties of the vitamin D3 receptor zinc finger region. Mol. Endocrinol., 5, 1815–1826. [DOI] [PubMed] [Google Scholar]
- Freedman L.P., Arce,V. and Perez Fernandez,R. (1994) DNA sequences that act as high affinity targets for the vitamin D3 receptor in the absence of the retinoid X receptor. Mol. Endocrinol., 8, 265–273. [DOI] [PubMed] [Google Scholar]
- Gewirth D.T. and Sigler,P.B. (1995) The basis for half-site specificity explored through a non-cognate steroid receptor–DNA complex. Nature Struct. Biol., 2, 386–394. [DOI] [PubMed] [Google Scholar]
- Hsieh J.C. et al. (1999) Characterization of unique DNA-binding and transcriptional-activation functions in the carboxyl-terminal extension of the zinc finger region in the human vitamin D receptor. Biochemistry, 38, 16347–16358. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Juntunen K., Rochel,N., Moras,D. and Vihko,P. (1999) Large-scale expression and purification of the human vitamin D receptor and its ligand-binding domain for structural studies. Biochem. J., 344, 297–303. [PMC free article] [PubMed] [Google Scholar]
- Khorasanizadeh S. and Rastinejad,F. (2001) Nuclear-receptor inter actions on DNA-response elements. Trends Biochem. Sci., 26, 384–390. [DOI] [PubMed] [Google Scholar]
- Kliewer S.A., Umesono,K., Mangelsdorf,D.J. and Evans,R.M. (1992a) Retinoid X receptor interacts with nuclear receptors in retinoic acid, thyroid hormone and vitamin D3 signalling. Nature, 355, 446–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer S.A., Umesono,K., Noonan,D.J., Heyman,R.A. and Evans,R.M. (1992b) Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature, 358, 771–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis P.J. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Kurokawa R., DiRenzo,J., Boehm,M., Sugarman,J., Gloss,B., Rosenfeld,M.G., Heyman,R.A. and Glass,C.K. (1994) Regulation of retinoid signalling by receptor polarity and allosteric control of ligand binding. Nature, 371, 528–531. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., Moss,D.S. and Thornton,J.M. (1993) Main-chain bond lengths and bond angles in protein structures. J. Mol. Biol., 231, 1049–1067. [DOI] [PubMed] [Google Scholar]
- Luisi B.F., Xu,W.X., Otwinowski,Z., Freedman,L.P., Yamamoto,K.R. and Sigler,P.B. (1991) Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature, 352, 497–505. [DOI] [PubMed] [Google Scholar]
- Mader S., Chen,J.Y., Chen,Z., White,J., Chambon,P. and Gronemeyer,H. (1993) The patterns of binding of RAR, RXR and TR homo- and heterodimers to direct repeats are dictated by the binding specificites of the DNA binding domains. EMBO J., 12, 5029–5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangelsdorf D.J. and Evans,R.M. (1995) The RXR heterodimers and orphan receptors. Cell, 83, 841–850. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf D.J. et al. (1995) The nuclear receptor superfamily: the second decade. Cell, 83, 835–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRee D.E. (1999) XtalView/Xfit—a versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol., 125, 156–165. [DOI] [PubMed] [Google Scholar]
- Meinke G. and Sigler,P.B. (1999) DNA-binding mechanism of the monomeric orphan nuclear receptor NGFI-B. Nature Struct. Biol., 6, 471–477. [DOI] [PubMed] [Google Scholar]
- Miyamoto T. et al. (2001) The role of hinge domain in heterodimerization and specific DNA recognition by nuclear receptors. Mol. Cell. Endocrinol., 181, 229–238. [DOI] [PubMed] [Google Scholar]
- Nakajima S., Hsieh,J.C., MacDonald,P.N., Galligan,M.A., Haussler,C.A., Whitfield,G.K. and Haussler,M.R. (1994) The C-terminal region of the vitamin D receptor is essential to form a complex with a receptor auxiliary factor required for high affinity binding to the vitamin D-responsive element. Mol. Endocrinol., 8, 159–172. [DOI] [PubMed] [Google Scholar]
- Nicholls A., Sharp,K.A. and Honig,B. (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins, 11, 281–296. [DOI] [PubMed] [Google Scholar]
- Nishikawa J., Matsumoto,M., Sakoda,K., Kitaura,M., Imagawa,M. and Nishihara,T. (1993) Vitamin D receptor zinc finger region binds to a direct repeat as a dimer and discriminates the spacing number between each half-site. J. Biol. Chem., 268, 19739–19743. [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Perlmann T., Rangarajan,P.N., Umesono,K. and Evans,R.M. (1993) Determinants for selective RAR and TR recognition of direct repeat HREs. Genes Dev., 7, 1411–1422. [DOI] [PubMed] [Google Scholar]
- Perlmann T., Umesono,K., Rangarajan,P.N., Forman,B.M. and Evans,R.M. (1996) Two distinct dimerization interfaces differentially modulate target gene specificity of nuclear hormone receptors. Mol. Endocrinol., 10, 958–966. [DOI] [PubMed] [Google Scholar]
- Quack M., Szafranski,K., Rouvinen,J. and Carlberg,C. (1998) The role of the T-box for the function of the vitamin D receptor on different types of response elements. Nucleic Acids Res., 26, 5372–5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quelo I., Kahlen,J.P., Rascle,A., Jurdic,P. and Carlberg,C. (1994) Identification and characterization of a vitamin D3 response element of chicken carbonic anhydrase-II. DNA Cell Biol., 13, 1181–1187. [DOI] [PubMed] [Google Scholar]
- Rastinejad F., Perlmann,T., Evans,R.M. and Sigler,P.B. (1995) Structural determinants of nuclear receptor assembly on DNA direct repeats. Nature, 375, 203–211. [DOI] [PubMed] [Google Scholar]
- Rastinejad F., Wagner,T., Zhao,Q. and Khorasanizadeh,S. (2000) Structure of the RXR–RAR DNA-binding complex on the retinoic acid response element DR1. EMBO J., 19, 1045–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochel N., Wurtz,J.M., Mitschler,A., Klaholz,B. and Moras,D. (2000) The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol. Cell, 5, 173–179. [DOI] [PubMed] [Google Scholar]
- Schwabe J.W., Chapman,L., Finch,J.T. and Rhodes,D. (1993) The crystal structure of the estrogen receptor DNA-binding domain bound to DNA: how receptors discriminate between their response elements. Cell, 75, 567–578. [DOI] [PubMed] [Google Scholar]
- Takeshita A., Ozawa,Y. and Chin,W.W. (2000) Nuclear receptor coactivators facilitate vitamin D receptor homodimer action on direct repeat hormone response elements. Endocrinology, 141, 1281–1284. [DOI] [PubMed] [Google Scholar]
- Toell A., Polly,P. and Carlberg,C. (2000) All natural DR3-type vitamin D response elements show a similar functionality in vitro. Biochem. J., 352, 301–309. [PMC free article] [PubMed] [Google Scholar]
- Towers T.L., Luisi,B.F., Asianov,A. and Freedman,L.P. (1993) DNA target selectivity by the vitamin D3 receptor: mechanism of dimer binding to an asymmetric repeat element. Proc. Natl Acad. Sci. USA, 90, 6310–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umesono K., Murakami,K.K., Thompson,C.C. and Evans,R.M. (1991) Direct repeats as selective response elements for the thyroid hormone, retinoic acid and vitamin D3 receptors. Cell, 65, 1255–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Word J.M., Bateman,R.C.,Jr, Presley,B.K., Lovell,S.C. and Richardson,D.C. (2000) Exploring steric constraints on protein mutations using MAGE/PROBE. Protein Sci., 9, 2251–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu V.C. et al. (1991) RXRβ: a coregulator that enhances binding of retinoic acid, thyroid hormone and vitamin D receptors to their cognate response elements. Cell, 67, 1251–1266. [DOI] [PubMed] [Google Scholar]
- Zechel C., Shen,X.Q., Chambon,P. and Gronemeyer,H. (1994a) Dimerization interfaces formed between the DNA binding domains determine the cooperative binding of RXR/RAR and RXR/TR heterodimers to DR5 and DR4 elements. EMBO J., 13, 1414–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechel C., Shen,X.Q., Chen,J.Y., Chen,Z.P., Chambon,P. and Gronemeyer,H. (1994b) The dimerization interfaces formed between the DNA binding domains of RXR, RAR and TR determine the binding specificity and polarity of the full-length receptors to direct repeats. EMBO J., 13, 1425–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q., Khorasanizadeh,S., Miyoshi,Y., Lazar,M.A. and Rastinejad,F. (1998) Structural elements of an orphan nuclear receptor–DNA complex. Mol. Cell, 1, 849–861. [DOI] [PubMed] [Google Scholar]
- Zhao Q., Chasse,S.A., Devarakonda,S., Sierk,M.L., Ahvazi,B. and Rastinejad,F. (2000) Structural basis of RXR–DNA interactions. J. Mol. Biol., 296, 509–520. [DOI] [PubMed] [Google Scholar]