

Abstract
In mammals and Drosophila, apoptotic caspases are under positive control via the CED-4/Apaf-1/Dark adaptors and negative control via IAPs (inhibitor of apoptosis proteins). However, the in vivo genetic relationship between these opposing regulators is not known. In this study, we demonstrate that a dark mutation reverses catastrophic defects seen in Diap1 mutants and rescues cells specified for Diap1- regulated cell death in development and in response to genotoxic stress. We also find that dark function is required for hyperactivation of caspases which occurs in the absence of Diap1. Since the action of dark is epistatic to that of Diap1, these findings demonstrate that caspase-dependent cell death requires concurrent positive input through Apaf-1-like proteins together with disruption of IAP–caspase complexes.
Keywords: Apaf-1/apoptosis/Drosophila/IAP/PCD
Introduction
Apoptosis, or programmed cell death (PCD), is a cell deletion mechanism that is critical to metazoan survival. During development, PCD is essential for adjusting the number of cells within or between populations of cells, for the elimination of non-functional cells and for the sculpting of organs and tissues (reviewed in Jacobson et al., 1997; Vaux and Korsmeyer, 1999; Meier et al., 2000a). Physiologically, PCD is also pivotal in the maintenance of cell homeostasis and for host defense against viral pathogens. In addition, the misregulation of apoptosis is implicated in human diseases such as cancer and neurodegenerative-associated disorders (reviewed in Thompson 1995).
Studies initiated in the nematode, Caenorhabditis elegans, established that PCD is under genetic control and led to the identification of three essential components of the apoptotic pathway: ced-3, ced-4 and ced-9 (reviewed in Metzstein et al., 1998). Homologs of these ‘core’ apoptotic components are conserved throughout evolution along with many other activators, effectors and inhibitors of cell death. CED-3 represents the founding member of the proapoptotic cysteine proteases known as caspases (Yuan et al., 1993), whereas the antiapoptotic component, CED-9, shows structural and functional homology to the Drosophila and vertebrate Bcl-2 family of proteins (reviewed in Gross et al., 1999; Chen and Abrams, 2000). The third gene product, CED-4, bears homology to vertebrate Apaf-1 (reviewed in Hengartner, 1997) and Drosophila dark/Hac-1/Dapaf-1 (Kanuka et al., 1999; Rodriguez et al., 1999; Zhou et al., 1999) and is important in promoting apical caspase activation via the ‘apoptosome’ (Zou et al., 1999; Hengartner, 2000). The caspases, in turn, mediate PCD by cleaving selected intracellular proteins, including proteins of the nucleus, cytosol and cytoskeleton, thereby disabling important cellular processes and breaking down structural components of the cell (Nicholson and Thornberry, 1997; Salvesen and Dixit, 1997).
Molecular genetic studies of PCD in Drosophila led to the identification of three additional important cell death initiators known as reaper, grim and hid. The genes encoding these PCD initiators are linked within an ∼300 kb genomic interval referred to as the Reaper interval (reviewed in McCall and Steller, 1997; Rodriguez et al., 1998; Abrams, 1999). During embryonic development, most, if not all, cells are specified for PCD solely or combinatorially by reaper, grim and/or hid (White et al., 1994; Grether et al., 1995; Chen et al., 1996). These cell death initiators contain a short region of homology at the N-terminus, referred to as the RHG motif (Zhou et al., 1997), which is sufficient for binding to at least two of the Drosophila inhibitors of apoptosis proteins (IAPs): Diap1/Thread and Diap2 (Vucic et al., 1997, 1998; Kaiser et al., 1998; Lisi et al., 2000). In addition, the RHG of Reaper, Grim and Hid is capable of inducing apoptosis on its own (Vucic et al., 1998), so it has been proposed that disruption of caspase–IAP interactions through this domain may be of central importance in order for reaper, grim and hid to induce PCD (Meier and Evan, 1998; Goyal et al., 2000; Hay, 2000; Wu et al., 2001). In agreement with this model, Diap1 binds directly to at least two (i.e. drICE and Dronc) of the seven fly caspases and inhibits their cell killing activity (Wang et al., 1999; Meier et al., 2000b). Further more, caspase-induced killing in yeast can be blocked by co-expression of Diap1, and this effect can be reversed by co-expressing either Reaper, Grim or Hid (Wang et al., 1999). These interactions support a ‘liberation’ model in which the cell death activators control PCD by releasing caspases from IAP-mediated repression (Miller, 1999; Wang et al., 1999; Meier et al., 2000b). Consistent with this hypothesis, most, if not all, cells in Diap1 null animals die with signs of apoptotic DNA fragmentation early during embryogenesis (Wang et al., 1999; Goyal et al., 2000; Lisi et al., 2000). Because IAPs bind activated rather than the zymogenic forms of caspases, absence of these inhibitors in Diap1 null animals could release pre-existing enzymes from an otherwise repressed condition. Since zymogens exhibit intrinsic auto-activation properties and, under the right conditions, can induce cell death on their own (Salvesen and Dixit, 1997), caspase auto-activation followed by cascades of proteolysis could account for apoptosis in the absence of IAPs.
While it is clear that the balance of opposing regulatory forces determines the status of caspase activity in a given cell, the relative physiological contribution from positive regulators (Apaf-1/CED-4/Dark) and negative regulators (the IAP family) remains ill defined. In the worm, the mouse and the fly, mutations in positive regulators cause clear cell death-defective phenotypes, but this is not consistently true with respect to the negative regulators. Though absence of a single IAP gene promotes catastrophic dysregulation of caspases and apoptotic phenotypes in Drosophila (Wang et al., 1999; Lisi et al., 2000), surprisingly, neither of the two IAP homologs in C.elegans appear to function in cell death (Fraser et al., 1999). Murine IAPs can exert profound inhibitory effects in vitro and when overexpressed in cultured cells (Deveraux and Reed, 1999), but genetic knockouts of murine IAPs have only mild (or no) phenotypes, perhaps reflecting redundant functions among mammalian IAP family members (Holcik et al., 2000; Harlin et al., 2001). The Drosophila model is thus uniquely suited to assess the relationship between these positive and negative regulators of caspase action.
Loss of dark function is associated with cell death-defective phenotypes opposite to Diap1, and genetic evidence indicates that dark is also important for reaper, grim and hid death pathways. However, the in vivo genetic relationship between dark-dependent caspase activation and Diap1-dependent caspase inhibition is not known and, consequently, the possible connection between dark-associated functions and reaper, grim and hid remains unclear. In this study, we examine the apoptotic role of dark and determine its epistatic relationship to Diap1. Our results show that dark functions downstream or parallel to Diap1, arguing against the ‘liberation’ models, which postulate that IAPs alone are a pivotal determinant of whether a cell will die or live in Drosophila (Goyal et al., 2000; Meier et al., 2000b; Song et al., 2000). Instead, we show that important cell death-inducing cues must also converge upon dark in order for PCD to occur. These observations support a model of PCD whereby concurrent positive input, in the form of Dark-dependent caspase activity, occurs together with the disruption of IAP– caspase complexes to specify the apoptotic fate.
Results
Programmed cell death specified by reaper, grim and hid requires dark function
We examined the pattern of PCD within the embryonic central nervous system (CNS) midline glia of dark mutants (see Materials and methods for comments on dark alleles) using the marker strain P[1.0slit-lacZ] (Zhou et al., 1995). During stages 16–17 of embryogenesis, this marker labels three midline glia per segment in the CNS of wild-type embryos (Zhou et al., 1995; Dong and Jacobs, 1997; Figure 1A). However, there is a significant increase in the number of midline glia (∼8 cells/segment) present in darkCD4 mutants (Figure 1B). Thus, at least five supernumerary dark cells per segment (and sometimes many more) are not subject to a TUNEL-independent form of cell death nor are they delayed in their death program as reported for some cells in the Apaf-1 knockout mouse (Cecconi et al., 1998; Yoshida et al., 1998). Since 9–12 embryonic midline glia survive in Reaper interval [i.e. Df(3L)H99] mutants (Zhou et al., 1995; Dong and Jacobs, 1997), our data indicate that Dark is essential for most, if not all, cell death occurring in lineages labeled by this marker.
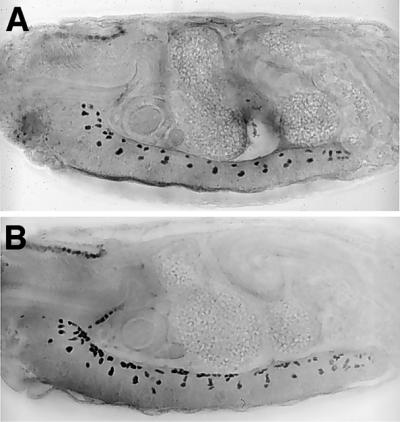
Fig. 1. Cells specified for apoptosis by genes in the reaper interval survive in the absence of dark function. Shown are anti-β-galactosidase staining of a stage 17 wild-type (A) and darkCD4 (B) mutant embryo bearing the P[1.0slit-lacZ] marker chromosome. Control yw embryos have approximately three P[1.0slit-lacZ]-expressing cells per segment at this point in development, whereas similarly staged darkCD4 mutants have ∼8 cells/segment.
dark is required for radiation-induced apoptosis mediated by reaper, grim and hid
We next asked whether dark might also play an essential role in damage-induced apoptosis mediated by reaper, grim and/or hid (White et al., 1994). We therefore examined the incidence of TUNEL-labeled cell death in wild-type (yw) and dark mutant embryos exposed to ionizing radiation, using previously described irradiation protocols that also induce the reaper gene (Nordstrom et al., 1996; Nordstrom and Abrams, 2000). As shown in Figure 2A, stage 9 wild-type embryos show widespread, damage-induced cell death throughout the embryo, whereas dark mutant embryos treated in parallel show very few and sparsely distributed apoptotic cells (Figure 2B and C). Thus, in addition to physiological PCD, dark plays a central role during pathological apoptosis engaged by reaper, grim and/or hid. Further more, consistent with the previously reported induction of a dark-enhancer trap reporter by UV irradiation (Zhou et al., 1999), we observed that expression of dark RNA itself is acutely responsive to ionizing radiation (Figure 2D).
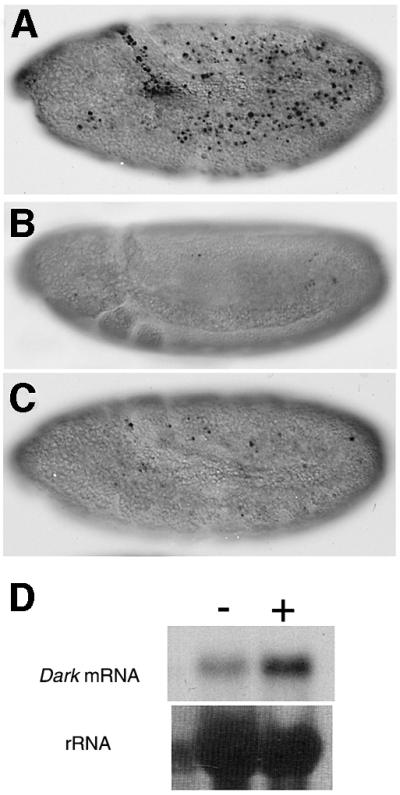
Fig. 2. dark is required for damage-induced apoptosis mediated by reaper, grim and/or hid. Nomarski micrographs of stage 9 wild-type (A) or dark (B and C) embryos labeled with TUNEL after X-ray treatment. (A) A wild-type embryo showing prominent cell death throughout. (B and C) In contrast, comparably staged, X-ray-treated dark embryos show little cell death after radiation treatment. Note that embryos bearing the Df(3L)H99 deletion (i.e. mutant for reaper, grim and hid) are similarly insensitive to X-ray-induced apoptosis (White et al., 1994; data not shown). (D) The top panel shows a northern blot of control (–) and X-ray-treated (+) wild-type embryos using a dark probe. Relative to control embryos, there is a dramatic increase of dark RNA in the X-ray-treated embryos. The lower panel depicts the same blot stained with methylene blue showing that similar amounts of total RNA were loaded in each lane.
dark is required for Diap-1-enhanced cell killing in the eye
Directed expression of reaper, grim and hid under the retina-specific GMR promoter induces widespread apoptotic death in the eye disc, producing a small/rough eye phenotype in adult flies (Grether et al., 1995; Chen et al., 1996; White et al., 1996). Mutations at dark and Diap1 modify these cell death phenotypes in opposite ways; loss of dark gene function suppresses cell killing by GMR-hid and GMR-grim transgenes (Rodriguez et al., 1999), and hypomorphic Diap1 alleles act as strong dominant enhancers of these same transgenes (Hay et al., 1995; Goyal et al., 2000; Lisi et al., 2000). To examine further the influence of dark and Diap1 upon apoptotic signaling and establish the order of gene action in this context, we undertook a series of epistasis studies. We tested whether dark alleles modify Diap1-mediated enhancement of Hid- or Grim-induced cell killing by introducing GMR-hid or GMR-grim tester strains into a genetic background heterozygous for Diap1 and homozygous for the darkCD4 mutation. While the thread5 (Diap15) loss-of-function mutation is still an effective dominant enhancer of GMR-grim- or GMR-hid-induced cell killing in flies heterozygous for dark (data not shown), Diap15 fails to modify cell killing of these same transgenes in flies homozygous for dark (Figure 3, for hid killing compare B and C, for grim killing compare E and F). These data show that a homozygous dark mutant background completely reverses the effect of heterozygozity for Diap1 when tested in the context of apoptotic signaling by grim and hid. Therefore, in classical genetic terms, dark is epistatic to Diap1 and suggests an order of gene action whereby dark functions either downstream or parallel to Diap1.

Fig. 3. Diap1-mediated enhancement of hid- and grim-induced cell killing is reversed by loss of dark function. Light microscopy micrographs of (A) GMR-hid-1M/+, (B) GMR-hid-1M/+; Diap15/+, (C) darkCD4, GMR-hid-1M/darkCD4; Diap15/+, (D) GMR-grim-1/+, (E) GMR-grim-1/+; Diap15/+ and (F) darkCD4, GMR-grim-1/darkCD4; Diap15/+ fly eyes. (B) A fly wild-type for dark and heterozygous for the Diap1 loss-of-function allele, Diap15, shows enhancement of GMR-hid-1M cell killing. (C) In a fly homozygous for darkCD4, the Diap15 mutation fails to dominantly enhance hid-induced cell killing. (E) Diap15 enhances GMR-grim-1 retinal apoptosis in a fly wild-type for dark. (F) In contrast, Diap15 fails to enhance grim-induced cell killing in a darkCD4 mutant fly.
Degeneration of germline cells caused by dysregulation of Diap1 is reversed by dark
We also tested for possible genetic interactions between Diap1 and dark by examining whether germline phenotypes in females that carry two weak Diap1 alleles in trans to one another, thread6/thread8 (Diap18/6), were modified in a dark genetic background (see Materials and methods). As depicted in Table I, Diap16/8 mutant females are not absolutely sterile, but the vast majority (>90%) of these mutants fail to lay eggs when mated to yw males. However, when darkCD4; Diap16/8 females are scored similarly, egg laying is rescued in more than half of these animals (Table I). To assess directly the reversal of the Diap1 egg-laying defect by dark, we compared ovaries from Diap16/8 flies with ovaries from darkCD4; Diap16/8 mutants. Diap16/8 flies have severely stunted and irregular shaped ovaries that appear atrophied, while darkCD4; Diap16/8 females produce ovaries that are considerably larger and generally wild-type in appearance. Individual ovarioles dissected 4–5 days post-eclosion are shown in Figure 4. Whereas the vast majority of Diap16/8 late-stage egg chambers exhibit overtly degenerative phenotypes (Figure 4A), most darkCD4; Diap16/8 egg chambers are normal in morphology and number (Figure 4B). Thus, loss of dark function rescues functional ovaries from an otherwise degenerative fate caused by the misregulation of Diap1.
Table I. A dark mutation rescues defective egg laying caused by lesions at DIAP1.
| Female genotypea | Fail to lay eggs | nb |
|---|---|---|
| th6/th8 | 91% | 34 |
| darkCD4/+; th6/th8 | 69% | 54 |
| darkCD4/darkCD4; th6/th8 | 47% | 63 |
aSingle pair matings with yw males were set up using females of the genotype shown.
bIndividual vials with females were monitored for up to 14 days for eggs in food.
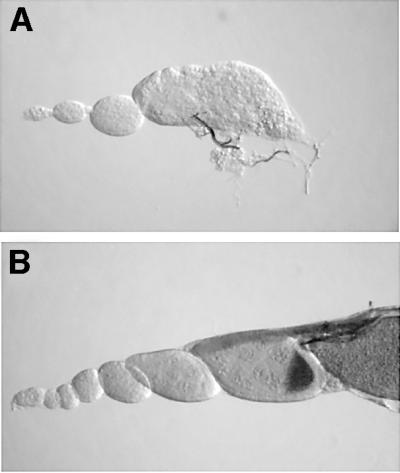
Fig. 4. Ovarian atrophy caused by Diap1 malfunction is reversed by dark. Nomarski color micrographs of Diap16/8 (A) and darkCD4/darkCD4; Diap16/8 (B) mutant ovarioles. (A) A Diap16/8 mutant ovariole that is poorly developed and atrophied. Note that the late egg chamber shows abnormal morphology and signs of degeneration. (B) By comparison, a double mutant ovariole shows improved distribution of egg chambers and advanced maturation of the oocyte.
dark blocks ectopic cell death in Diap1 mutant embryos
We next tested whether dark might similarly influence phenotypes associated with the complete absence of Diap1 function. For this purpose, we examined darkCD4; Diap15 double mutant embryos to determine whether dark might rescue defects associated with a null mutation for Diap1 (i.e. thread5/Diap15; see Materials and methods). Diap15 mutant animals progress normally through gastrulation but they arrest at the beginning of germ band extension (stage 7) and, within 90 min thereafter, adopt a characteristic morphology reflecting catastrophic events associated with widespread and synchronous apoptosis (Wang et al., 1999; this study). The nuclei of most, if not all, cells in Diap15 embryos exhibit apoptotic DNA fragmentation detected as extensive TUNEL labeling at a time, in wild-type embryos, that would otherwise correspond to extension of the germ band, stages 7–10 (Wang et al., 1999; see Figure 5B). However, in Diap1 mutant embryos that are also homozygous for dark, apoptotic cell death is remarkably attenuated (Figure 5C) and, quite strikingly, the developmental arrest characteristic of single Diap1 mutants does not occur (Figure 5, compare B and C). Instead, the vast majority of double mutants develop far beyond the embryonic stage at which single Diap1 mutants arrest. Whereas Diap15 mutants catastrophically arrest at stage 7, >90% of darkCD4; Diap15 embryos show clear signs of normal cell migration and differentiation such that morphological landmarks utilized (e.g. the appearance of the periodic bulges, germ band retraction, stomodeum) for purposes of staging through stage 10 are readily discernible (compare Figure 5A with C). Together, these observations establish that dark attenuates inappropriate apoptosis and reverses early morphogenetic arrest caused by the Diap1 null mutation. Thus, intact dark function is required for both ectopic apoptosis and the catastrophic arrest caused by the absence of Diap1. It is worth noting that, while most darkCD4; Diap15 embryos are spared from an early morphogenetic arrest, limited apoptosis does occur, and these animals are not spared from less severe abnormalities occurring at more advanced stages (compare Figure 5A with C with regard to TUNEL staining and embryo morphology). On rare occasions, we observed a novel class of darkCD4; Diap15 double mutants as shown in Figure 5D. These occurred at only 5% of the expected frequency and, while they fail to stain significantly for TUNEL, they also appear arrested at stage 7 like their single Diap1 mutant counterparts. Therefore, suppression of cell death per se is not always sufficient for morphogenetic ‘rescue’.
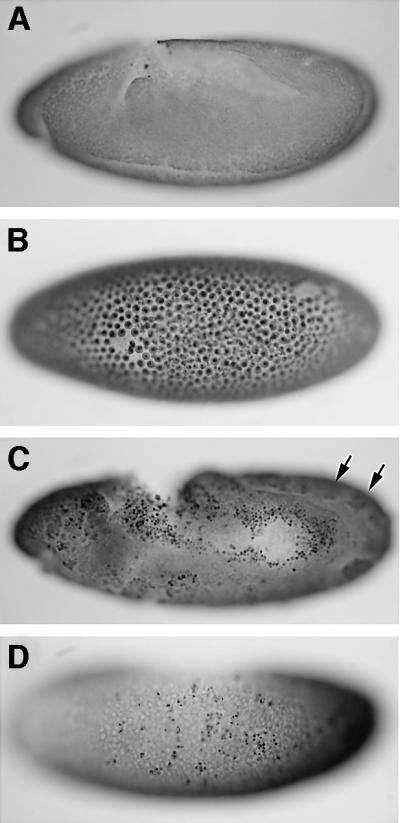
Fig. 5. Loss of dark reverses developmental arrest and prevents cell death in Diap1 embryos. Nomarski micrographs of 4- to 5-h-old embryos labeled by TUNEL. (A) Wild-type, (B) Diap15 and (C and D) darkCD4; Diap15 embryos. (A) At, or prior to, the extended germ band stage (stage 9), no programmed cell death occurs in wild-type embryos. (B) In contrast, Diap15 homozygous embryos of similar chronological age show widespread TUNEL labeling in virtually all cells and exhibit a characteristic ‘blastoderm’-like morphology. (C) By comparison, darkCD4; Diap15 mutant embryos progress considerably further in development (large arrows indicate the periodic bulges seen during stage 10) and are dramatically attenuated for TUNEL labeling. Note that even though this embryo shows germ band retraction, it has some inappropriate TUNEL labeling along with irregularities in embryonic development (i.e. an abnormal hole in the posterior midgut primordium). (D) A rare darkCD4; Diap15 mutant embryo which, though not rescued for the ‘blastoderm’-like morphology, shows TUNEL labeling in only a few cells [compare with (B)].
dark is required for excessive caspase activity observed in Diap1 embryos
We also examined epistasis between Diap1 and dark at the biochemical level by directly measuring caspase activity from stage 9–10 embryos using a fluorogenic DEVD substrate (Wang et al., 1999; see Materials and methods). Compared with wild-type (yw) strains, embryonic lysates from populations containing homozygous (stage 9–10) Diap15 mutants show highly elevated caspase activity. DEVDase activity in Diap15 lysates is elevated by levels ≥70-fold relative to lysates from control yw embryos (Figure 6). Enzymatic ‘hyperactivation’ in this context is thought to reflect the action of these proteases unrestrained by native inhibitors (Goyal, 2001). In contrast, lysates from darkCD4; Diap15 embryos show at least 80–90% lower DEVDase activity when compared with Diap15 lysates (Figure 6). Thus, a dark mutant background prevents ‘hyperactivation’ of caspase activity that otherwise occurs in the absence of Diap1 zygotic function. This finding indicates that liberation of caspases from negative regulation is not sufficient to achieve hyperactivation of caspases seen in Diap1 mutant embryos. To exclude the possibility that some portion of the activities measured in these assays might occur after the lysis step, we mixed Diap15 lysates with either wild-type or darkCD4 lysates and assayed caspase activity from these as well. We detected no synergistic effects indicative of post-lysis amplification of caspase activity in these mixed lysates (data not shown). Therefore, all measureable activity shown in Figure 6 reflects the authentic in vivo status in the embryo prior to lysis.

Fig. 6. Caspase ‘hyperactivation’ triggered by the removal of Diap1 requires dark function. Embryo protein extracts were analyzed for caspase activity by measuring Ac-DEVD-AFC release over time. Extracts derived from wild-type (yw), darkCD4 (dark), TM6/Diap15 (Diap1), and darkCD4; TM6/Diap15 (dark; Diap1) stocks were tested. Note that the inset shows yw and dark caspase activities, but at a smaller scale for ease of viewing. Each result was verified at least three times, and triplicate samples were recorded from each lysate. P < 0.005, two-tailed homoscedastic t-test.
It is worth noting here that lysates from yw and dark embryos were collected several hours before the onset of PCD (Abrams et al., 1993). These basal DEVDase measurements could therefore reflect a low constitutive flow of ‘leaky’ caspase activity occurring in the complete absence of apoptotic signals. Moreover, since activity in darkCD4 lysates is considerably lower (∼4-fold) than control yw lysates, positive input from Dark evidently contributes to ‘basal’ DEVDase levels (Figure 6).
Discussion
Disruption of caspase–Diap1 interactions by reaper/grim/hid alone is insufficient to induce PCD in vivo
Mutations in dark were shown previously to cause a variety of developmental defects, including an enlarged larval CNS (Rodriguez et al., 1999). We extend previous observations and establish that, in the absence of dark function, cells specified to die actually survive and also differentiate (Figure 1). The embryonic CNS midline glia is a well-studied cell lineage in the fly embryo (Zhou et al., 1995; Dong and Jacobs, 1997) and, though we only followed this particular lineage, it is likely that supernumerary cell types in other lineages also persist in dark mutants. Several important conclusions derive from our observations. First, the persistence of extra cells excludes trivial explanations for reduced TUNEL labeling in dark mutants (e.g. cell death in the absence of TUNEL labeling or redirected cell fates that prevent the specification of PCD) and reinforces models that favor a fundamental role for dark in embryonic PCD. Secondly, as previously shown in C.elegans (Metzstein et al., 1998) and in studies of Reaper interval Drosophila mutants (White et al., 1994), rescue from PCD can uncover a cryptic differentiation program. Thirdly, and most importantly, the same midline glial cells that fail to die in dark mutants also require the induced action of reaper, grim and hid (Zhou et al., 1995; Dong and Jacobs, 1997). While it could be argued that extra cells might arise from altered cell division or differentiation, we favor explanations relating to defective PCD since dark mutants are already known to exhibit reduced apoptosis in this tissue (Rodriguez et al., 1999) and Reaper interval mutants have a similar phenotype in midline glia but exhibit no defects in cell division (Zhou et al., 1995; Dong and Jacobs, 1997). Therefore, supernumerary embryonic cells present in darkCD4 mutants (Figure 1) probably survive because of lack of apoptosis instead of altered cell identity. Experi ments in Figure 1 also serve to highlight a requirement for dark function in normal PCD when reaper, grim and/or hid are expressed at physiological levels. It is noteworthy that Diap1 and at least two of the cell death initiators (i.e. reaper and hid) are expressed in pools of progenitor cells, which give rise to the embryonic midline glia and also play an important role in specifying the death of these cells (Zhou et al., 1995, 1997; Wing et al., 1998). Thus, although midline glia presumably receive death signals from reaper and hid leading to the disruption of Diap1–caspase interactions, these cells still fail to die and are even capable of differentiating normally in the absence of dark function. This inference supports conclusions from our epistasis studies (discussed below) indicating that a disruption of caspase–Diap1 interactions alone is insufficient for apoptosis, and suggests that dark functions as a co-effector of cell death signaling along with reaper, grim and/or hid (see model in Figure 7). These findings are consistent with our previous observations (Rodriguez et al., 1999) and the observed induction of dark expression upon UV (Zhou et al., 1999) or X-ray exposure (Figure 2D). As such, they validate models placing dark in the same pathway, downstream or parallel to these apoptotic activators. Finally, it is worth noting that the mammalian ortholog of dark, Apaf-1, functions downstream from the point at which mitochondrial factors including cytochrome c are released from mitochondria. If the same holds true for the fly protein, it follows that embryonic midline glia (and perhaps other cell types) can survive and differentiate beyond this mitochondrial ‘point of no return’.

Fig. 7. A ‘gas and brake’ model for the action of dark and Diap1 in cell death. Studies described here establish that dark functions downstream or parallel to the action of Diap1. We favor a parallel converging arrangement since, like Apaf-1, Dark acts positively upon apical procaspases while IAPs act negatively upon processed caspases. The model draws upon analogies to the ‘gas’ and ‘brake’ controlling forward movement (apoptosis) of a car. In order for PCD to occur, the gas (dark) must be engaged and the brake (Diap1) must be released in concert. Release of the brakes by reaper (rpr), grim or hid will only induce effective apoptosis (movement) if the gas (dark) is engaged concurrently by apoptotic stimuli.
Additional evidence that PCD requires coordinated dark-dependent caspase activation in conjuction with the release of IAP-mediated caspase inhibition comes from our tests of dark mutants under conditions of stress that elicit apoptotic responses by the embryonic cell death initiators. Like the Reaper interval mutants (White et al., 1994), we find that dark mutants also exhibit profound failures in cell death in response to ionizing radiation (Figure 2). Interestingly, the dark mutation itself does not interfere with the radiation induction of reaper mRNA (data not shown; Brodsky et al., 2000). We can therefore exclude the possibility that resistance to damage-induced apoptosis evident in irradiated dark mutants is caused by a disruption of upstream elements in the signaling pathway. Instead, the results raise the possibility that dark, like reaper (Brodsky et al., 2000), may function as an important effector of Drosophila p53- (Ollmann et al., 2000) mediated apoptosis. In addition, our findings reinforce an apoptotic requirement for dark (even when reaper is transcriptionally induced) and provide evidence for models where cell death signals do not converge on Diap1 alone (Figure 7). In future studies, it will be interesting to determine the range of damage signals that can engage dark activity, since this locus might also function in a broader range of damage signals beyond those provoked by radiation.
dark is epistatic to Diap1
In flies, Diap1 is thought to function as a rate-limiting brake on apoptotic cell death (reviewed in Hay 2000; Goyal, 2001). If, however, Diap1 were the most proximal rate-limiting regulator of apoptosis, then the presence or absence of dark function should have no influence on Diap1-dependent effects. If, on the other hand, dark and Diap1 exert interdependent functions, then the opposite outcome is predicted. The results from our studies provide consistent and compelling evidence favoring the latter scenario since, wherever tested, the influence of Diap1 upon apoptotic signaling is heavily dependent upon intact dark function. In the darkCD4 mutant, for instance, the Diap15 mutation fails to enhance grim- and hid-induced cell killing in the eye (Figure 3). These experiments examined Diap1 in the heterozygous condition, but the same outcome is also true when Diap1 is tested in the homozygous state. Early in embryonic development, Diap1 homozygotes exhibit catastrophic phenotypes including morphological arrest soon after gastrulation, extensive TUNEL-positive nuclei and hyperactivation of caspases (Wang et al., 1999). Each of these defects is profoundly dependent upon dark function since each is dramatically reversed by the dark mutation (Figures 5 and 6). In darkCD4; Diap15 double mutant embryos, we find no evidence for widespread apoptosis and, in fact, the large majority of these double mutants proceed through stages of embryogenesis well beyond the point at which single Diap1 mutants arrest. Thus, loss of dark not only suppresses apoptotic deaths that would otherwise occur, but definitively reverses a profound morphogenetic arrest that ensues in the absence of Diap1. Consequently, homozygosity at dark not only prevents the onset of apoptotic markers (i.e. DNA fragmentation and/or caspase activation), but actually preserves cells that are otherwise fated to die when normal checks upon caspases are removed. Rescue from inappropriate apoptosis in this instance is consistent with what we observed in the midline glia (Figure 1) but, rather than preserving cells fated for programmed death, rescue occurs even when signaling from IAP antagonists in the Reaper region is bypassed. We obtained a similar result, with similar implications, in the ovary, where ∼90% of heteroallelic Diap16/8 mutants show abnormal degeneration of early staged egg chambers and associated sterility (Figure 4; Table I). Again, loss of dark function not only suppresses the pathological effect, but actually reverses these degenerative defects to the extent that half of the darkCD4; Diap16/8 double mutant females produce normal egg chambers and are fertile (Figure 4; Table I). Thus, in both the ovary and the embryo, loss of dark rescues functional cells from apoptosis caused by misregulated Diap1.
Reversal of Diap1-dependent defects by dark is also evident when we directly assay caspase activity in early embryos. In contrast to the ‘hyperactivated’ caspase levels detected in Diap1 single mutant lysates, Diap15; darkCD4 lysates show levels of caspase activity that are suppressed >90% relative to Diap1 mutants (Figure 6). The unusually high caspase activity detected in Diap1 mutant embryos is thought to reflect the action of these proteolytic enzymes unimpeded by native inhibitors (Wang et al., 1999). However, studies here demonstrate that removing a negative regulator alone is not sufficient to achieve caspase hyperactivation, since dark is clearly required for this unrestrained activity. It is worth mentioning that the basal DEVDase activity in double mutants is still somewhat elevated compared with control embryos (Figure 6). This might indicate dark-independent caspase activity due to caspase auto-activation in the absence of Diap1 or, since it is possible that the darkCD4 allele used here is not a complete null, it could reflect hypomorphic dark function. Alternatively, since a role for Diap1 in cytokinesis has not been ruled out (Lisi et al., 2000), it is possible that this residual caspase activity ensues because of secondary developmental defects leading to cell death. Nevertheless, these enzymatic data extend our analyses to the biochemical level in a manner fully consistent with our phenotypic studies.
Interestingly, since Apaf-1/Dark adaptor proteins act upon procaspases (Zou et al., 1999) while IAPs preferentially inhibit processed caspases (Goyal, 2001), our results further suggest that pre-existing levels of processed caspases in most cells are probably not high enough to achieve an apoptotic threshold. Instead, a positive cell death stimulus from dark is required for the unusually high levels of caspase activation seen in Diap1–/– embryos. It is also evident that Dark-dependent ‘basal’ levels of DEVDase are detected in these assays (Figure 6) several hours before the onset of embryonic PCD (Abrams et al., 1993), and therefore authentic effector caspase (e.g. DrICE, Dcp-1) activity occurs even in the absence of overt apoptotic signals. These data indicate that constitutive levels of active effector caspases, not derived from autoproteolysis but instead promoted by Apaf-1-like adaptor proteins, may exist in many and perhaps all viable cells.
Taken together, a strict Diap-1–caspase ‘liberation’ model does not explain sufficiently the evidence described. We show that the action of dark is epistatic to that of Diap1, demonstrating an order of gene action whereby Dark functions either downstream or parallel to Diap1. Therefore, simple derepression of caspases via an IAP inhibitory bridge does not account adequately for epistasis between Diap1 and dark. Put another way, our results support the notion that apoptotic cell death in vivo results from the simultaneous activation of caspases by dark and the derepression of caspases by reaper, grim and/or hid. Accordingly, our findings are inconsistent with models that presume that Diap1 is the sole effector of reaper, grim and hid and that cells are ‘pre-loaded’ with sufficient levels of IAP-inhibited processed caspases to achieve cell killing. Instead, we favor a ‘gas and brake’ model whereby positive input from Apaf-1/Dark adaptors, together with removal of IAP inhibition, drives caspase activation to levels that exceed a threshold necessary for apoptosis (Figure 7).
Materials and methods
Genetics of dark alleles
All crosses and experiments were carried out at 25°C using the previously described darkCD4 mutation (Rodriguez et al., 1999) derived from a nearby P-element referred to as P1041 or l(2)k11502 (http://www. flybase.net/). The l(2)k11502 strain contains a P-element mapping ∼1.3 kb upstream of the dark translation initiation site. This P-element (referred to as dapaf1K1 in Kanuka et al., 1999) was thought to be a lethal allele of the dark locus (Zhou et al., 1999). However, we find that this insertion, referred to here as darkP1041, is at best a weak hypomorphic dark allele (Rodriguez et al., 1999). For instance, darkP1041 shows only slight cell death defects in the embryo and barely detectable defects in adults as compared with darkCD4 animals (Kanuka et al., 1999; Rodriguez et al., 1999; Zhou et al., 1999). Secondly, while darkCD4 blocks most if not all cells induced by X-ray treatment, darkP1041 is indistinguishable from wild-type (data not shown). Thirdly, in situ hybridization analysis indicates that darkCD4 causes a severe reduction in dark mRNA distribution throughout the embryo (data not shown), whereas darkP1041 reportedly only disrupts expression in the procephalic head region (Zhou et al., 1999). We should note that in studies with darkCD4 mutants, we find no evidence for extra photoreceptor cells in the pupal eye (data not shown). Also note that the yw strain is used as control here and throughout our studies since it is the parental genotypic backgound for the production of darkCD4.
Diap-1 and dark epistatic tests in the retina
For the GMR-grim cell killing experiments, females of the genotype yw; darkCD4, P[GMR-grim]-1/darkCD4; Diap15/+ were examined for suppression of induced grim killing in the eye as compared with yw; P[GMR-grim]-1/+; Diap15/+ females. For GMR-hid killing experiments, females of the genotype, yw; darkCD4, P[GMR-hid]-1M/darkCD4; Diap15/+ were compared with yw; P[GMR-hid]-1M/+; Diap15/+ females. We were unable to construct the appropriate strains to perform similar experiments with GMR-reaper transgenes. All crosses were carried in parallel at 25°C. Micrographs were taken using a Coolpix Nikon digital camera.
Mutant embryo collections and TUNEL staining
Previously, it was demonstrated that maximal TUNEL cell death of Diap15 null embryos occurs during late stage 9 (Wang et al., 1999). Therefore, stage 9–10 embryos were collected and apoptosis assayed using TUNEL labeling. Briefly, embryos were collected for 40 min from adults of the genotype yw; darkCD4/darkCD4; TM6/Diap15, which previously had been pre-cleared twice, and the embryos were aged for ∼4 h. Diap1 mutant embryos were collected from adults of the genotype yw; TM6/Diap15 in parallel as described above. TUNEL labeling is essentially as in Rodriguez et al. (1999). In addition, the 3,3′-diaminobenzidine–peroxide signal was enhanced with nickel chloride and the embryos were pre-cleared using methyl salicylate and viewed using a Zeiss microscope (Patel, 1994).
Caspase activity assays from embryos
For the caspase activity experiments, embryos were collected from either (i) yw, (ii) yw; TM6/Diap15 or (iii) yw; darkCD4; TM6/Diap15 fly strains. Pools of ∼50–100 dechorionated embryos were transferred to an Eppendorf tube with a brush and suspended in 100 µl of buffer A (20 mM HEPES–KOH pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM sodium EDTA, 1 mM sodium EGTA, 1 mM dithiothreitol and 0.1 mM phenylmethylsulfonyl fluoride). Embryos were crushed with a disposable Kontes pestle in a 1.5 ml Eppendorf tube for ∼1 min. Cellular debris and lipids were separated from the soluble fraction by a 15 min spin at 15 000 g at 4°C, and the protein concentrations were measured and normalized by addition of buffer A. A 5 µg aliquot of protein extract was incubated with 50 µM AcDEVD-AFC (Calbiochem) substrate in a final volume of 20 µl in a 384 microtiter plate. Fluorescence was monitored over time with excitation at 360 nm and emission at 465 nm in a SpectraFluor Plus plate reader (Tecan).
Fertility assays and ovary dissections
Since the molecular nature of the thread8 (Diap18) mutation is still undefined, we wanted to safeguard against the possibility that it might represent a mutation at a locus other than Diap1. Therefore, we tested the Diap18 strain for non-complementation against a deletion that removes the Diap1 gene, the Diap15 mutation, and also tested for dominant suppression or enhancement of GMR-hid-1M and P[GMR-reaper]-M (GMR-reaper) cell killing phenotypes in the eye. While we find that Diap18 is lethal in trans to the Diap1 deficiency and Diap15, we fail to see any modification of cell killing of GMR-hid-1M and GMR-reaper (data not shown), suggesting that it might represent a very weak Diap1 mutation. The Diap16 (also known as th6B) allele has a C412Y missense mutation within the RING domain of Diap1 and is believed to represent a type 1 gain-of-function mutation because it enhances induced cell killing by grim and reaper but not hid (Lisi et al., 2000). Adult females of the appropriate genotype were obtained from crossing Diap16/TM6C to Diap18/TM6C flies. Diap16/Diap18 transheterozygous flies were selected based on the absence of the dominant balancer. Flies of the genotype CyO/darkCD4; TM6/Diap16 were mated to CyO/darkCD4; TM6/Diap18 in order to obtain dark homozygous Diap1 transheterozygous flies. Single females of the appropriate genotype were mated to yw males and monitored for egg laying over the course of 1–2 weeks. Ovary dissections were carried out in phosphate-buffered saline and stained for several hours in FeNap staining buffer (Ashburner, 1989).
Immunohistochemistry
Immunohistochemistry was performed as described in Patel (1994). β-galactosidase was detected using a mouse monoclonal antibody (Promega) followed by detection with anti-horseradish peroxidase-conjugated secondary antibodies using the Vectastain Elite Kit (Vector Labs) (Zhou et al., 1995).
Acknowledgments
Acknowledgements
We thank J.Nambu for providing us with the P[1.0slit-lacZ] strain, J.Kennison for the Diap1 mutant strains and sharing unpublished observations, and X.Wang for valuable advice on caspase activity assays and also making comments on the manuscript. P.C. was supported by a grant from the Leukemia and Lymphoma Society. This work was supported by grants from the NIH/NIA (R01 AG12466) and the American Cancer Society to J.M.A.
References
- Abrams J.M. (1999) An emerging blueprint for apoptosis in Drosophila. Trends Cell Biol., 9, 435–440. [DOI] [PubMed] [Google Scholar]
- Abrams J.M., White,K., Fessler,L. and Steller,H. (1993) Programmed cell death during Drosophila embryogenesis. Development, 117, 29–44. [DOI] [PubMed] [Google Scholar]
- Ashburner M. (1989) Drosophila: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Brodsky M.H., Nordstrom,W., Tsang,G., Kwan,E., Rubin,G.M. and Abrams,J.M. (2000) Drosophila p53 binds a damage response element at the reaper locus. Cell, 101, 103–113. [DOI] [PubMed] [Google Scholar]
- Cecconi F., Alvarezbolado,G., Meyer,B.I., Roth,K.A. and Gruss,P. (1998) Apaf1 (Ced-4 homolog) regulates programmed cell death in mammalian development. Cell, 94, 727–737. [DOI] [PubMed] [Google Scholar]
- Chen P. and Abrams,J.M. (2000) Drosophila apoptosis and Bcl-2 genes: outliers fly in. J. Cell Biol., 148, 625–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P., Nordstrom,W., Gish,B. and Abrams,J.M. (1996) Grim, a novel cell death gene in Drosophila. Genes Dev., 10, 1773–1782. [DOI] [PubMed] [Google Scholar]
- Deveraux Q.L. and Reed,T.C. (1999) IAP family proteins—suppressors of apoptosis. Genes Dev., 13, 239–252. [DOI] [PubMed] [Google Scholar]
- Dong R. and Jacobs,J.R. (1997) Origin and differentiation of supernumerary midline glia in Drosophila embryos deficient for apoptosis. Dev. Biol., 190, 165–177. [DOI] [PubMed] [Google Scholar]
- Fraser A.G., James,C., Evan,G.I. and Hengartner,M.O. (1999) Caenorhabditis elegans inhibitor of apoptosis protein (IAP) homologue BIR-1 plays a conserved role in cytokinesis. Curr. Biol., 9, 292–301. [DOI] [PubMed] [Google Scholar]
- Goyal L. (2001) Cell death inhibition: keeping caspases in check. Cell, 104, 805–808. [DOI] [PubMed] [Google Scholar]
- Goyal L., McCall,K., Agapite,J., Hartwieg,E. and Steller,H. (2000) Induction of apoptosis by Drosophila reaper, hid and grim through inhibition of IAP function. EMBO J., 19, 589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grether M.E., Abrams,J.M., Agapite,J., White,K. and Steller,H. (1995) The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev., 9, 1694–1708. [DOI] [PubMed] [Google Scholar]
- Gross A., McDonnell,J.M. and Korsmeyer,S.J. (1999) BCL-2 family members and the mitochondria in apoptosis. Genes Dev., 13, 1899–1911. [DOI] [PubMed] [Google Scholar]
- Harlin H., Reffey,S.B., Duckett,C.S., Lindsten,T. and Thompson,C.B. (2001) Characterization of XIAP-deficient mice. Mol. Cell. Biol., 21, 3604–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay B.A. (2000) Understanding IAP function and regulation: a view from Drosophila. Cell Death Differ., 7, 1045–1056. [DOI] [PubMed] [Google Scholar]
- Hay B.A., Wassarman,D.A. and Rubin,G.M. (1995) Drosophila homologs of baculovirus inhibitor of apoptosis proteins function to block cell death. Cell, 83, 1253–1262. [DOI] [PubMed] [Google Scholar]
- Hengartner M.O. (1997) Apoptosis—ced-4 is a stranger no more. Nature, 388, 714–715. [DOI] [PubMed] [Google Scholar]
- Hengartner M.O. (2000) The biochemistry of apoptosis. Nature, 407, 770–776. [DOI] [PubMed] [Google Scholar]
- Holcik M., Thompson,C.S., Yaraghi,Z., Lefebvre,C.A. MacKenzie,A.E. and Korneluk,R.G. (2000) The hippocampal neurons of neuronal apoptosis inhibitory protein 1 (NAIP1)-deleted mice display increased vulnerability to kainic acid-induced injury. Proc. Natl Acad. Sci. USA, 97, 2286–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson M.D., Weil,M. and Raff,M.C. (1997) Programmed cell death in animal development. Cell, 88, 347–354. [DOI] [PubMed] [Google Scholar]
- Kaiser W.J., Vucic,D. and Miller,L.K. (1998) The Drosophila inhibitor of apoptosis D-IAP1 suppresses cell death induced by the caspase drICE. FEBS Lett., 440, 243–248. [DOI] [PubMed] [Google Scholar]
- Kanuka H., Sawamoto,K., Inohara,N., Matsuno,K., Okano,H. and Miura,M. (1999) Control of the cell death pathway by Dapaf-1, a Drosophila Apaf-1/CED-4-related caspase activator. Mol. Cell, 4, 757–769. [DOI] [PubMed] [Google Scholar]
- Lisi S., Mazzon,I. and White,K. (2000) Diverse domains of THREAD/DIAP1 are required to inhibit apoptosis induced by REAPER and HID in Drosophila. Genetics, 154, 669–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall K. and Steller,H. (1997) Facing death in the fly: genetic analysis of apoptosis in Drosophila.Trends Genet., 13, 222–226. [DOI] [PubMed] [Google Scholar]
- Meier P. and Evan,G. (1998) Dying like flies. Cell, 95, 295–298. [DOI] [PubMed] [Google Scholar]
- Meier P., Finch,A. and Evan,G. (2000a) Apoptosis in development. Nature, 407, 796–801. [DOI] [PubMed] [Google Scholar]
- Meier P., Silke,J., Leevers,S.J. and Evan,G.I. (2000b) The Drosophila caspase DRONC is regulated by DIAP1. EMBO J., 19, 598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzstein M.M., Stanfield,G.M. and Horvitz,H.R. (1998) Genetics of programmed cell death in C.elegans—past, present and future. Trends Genet., 14, 410–416. [DOI] [PubMed] [Google Scholar]
- Miller L.K. (1999) An exegesis of IAPs: salvation and surprises from BIR motifs. Trends Cell Biol., 9, 323–328. [DOI] [PubMed] [Google Scholar]
- Nicholson D.W. and Thornberry,N.A. (1997) Caspases—killer proteases. Trends Biochem. Sci., 22, 299–306. [DOI] [PubMed] [Google Scholar]
- Nordstrom W. and Abrams,J.M. (2000) Guardian ancestry: fly p53 and damage-inducible apoptosis. Cell Death Differ., 7, 1035–1038. [DOI] [PubMed] [Google Scholar]
- Nordstrom W., Chen,P., Steller,H. and Abrams,J.M. (1996) Activation of the reaper gene during ectopic cell killing in Drosophila. Dev. Biol., 180, 213–226. [DOI] [PubMed] [Google Scholar]
- Ollmann M. et al. (2000) Drosophila p53 is a structural and functional homolog of the tumor suppressor p53. Cell, 101, 91–101. [DOI] [PubMed] [Google Scholar]
- Patel N. (1994) Imaging neuronal subsets and other cell types in wholemount Drosophila embryos and larvae. In Goldstein,L.S.B. and Fyrberg,E.A. (eds), Drosophila melanogaster: Practical Uses in Cell and Molecule Biology. Academic Press, San Diego, CA, pp. 446–485.
- Rodriguez A., Chen,P. and Abrams,J.M. (1998) Molecular prophets of death in the fly. Am. J. Hum. Genet., 62, 514–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A., Oliver,H., Zou,H., Chen,P., Wang,X.D. and Abrams,J.M. (1999) Dark is a Drosophila homologue of Apaf-1/CED-4 and functions in an evolutionarily conserved death pathway. Nature Cell Biol., 1, 272–279. [DOI] [PubMed] [Google Scholar]
- Salvesen G.S. and Dixit,V.M. (1997) Caspases—intracellular signaling by proteolysis. Cell, 91, 443–446. [DOI] [PubMed] [Google Scholar]
- Song Z., Guan,B., Bergman,A., Nicholson,D.W., Thornberry,N.A., Peterson,E.P. and Steller,H. (2000) Biochemical and genetic interactions between Drosophila caspases and the proapoptotic genes rpr, hid, and grim. Mol. Cell. Biol., 20, 2907–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson C.B. (1995) Apoptosis in the pathogenesis and treatment of disease. Science, 267, 1456–1462. [DOI] [PubMed] [Google Scholar]
- Vaux D.L. and Korsmeyer,S.J. (1999) Cell death in development. Cell, 96, 245–254. [DOI] [PubMed] [Google Scholar]
- Vucic D., Kaiser,W.J., Harvey,A.J. and Miller,L.K. (1997) Inhibition of reaper-induced apoptosis by interaction with inhibitor of apoptosis proteins (IAPs). Proc. Natl Acad. Sci. USA, 94, 10183–10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucic D., Kaiser,W.J. and Miller,L.K. (1998) Inhibitor of apoptosis proteins physically interact with and block apoptosis induced by Drosophila proteins Hid and Grim. Mol. Cell. Biol., 18, 3300–3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.L., Hawkins,C.J., Yoo,S.J., Muller,H.A.J. and Hay,B.A. (1999) The Drosophila caspase inhibitor DIAP1 is essential for cell survival and is negatively regulated by HID. Cell, 98, 453–463. [DOI] [PubMed] [Google Scholar]
- White K., Grether,M., Abrams,J.M., Young,L., Farrell,K. and Steller,H. (1994) Genetic control of programmed cell death in Drosophila. Science, 264, 677–683. [DOI] [PubMed] [Google Scholar]
- White K., Tahaoglu,E. and Steller,H. (1996) Cell killing by the Drosophila gene reaper. Science, 271, 805–807. [DOI] [PubMed] [Google Scholar]
- Wing J.P., Zhou,L., Schwartz,L.M. and Nambu,J.R. (1998) Distinct cell killing properties of the Drosophila reaper, head involution defective, and grim genes. Cell Death Differ., 5, 930–939. [DOI] [PubMed] [Google Scholar]
- Wu J.W., Cocina,A.E., Chai,J., Hay,B.A. and Shi,Y. (2001) Structural analysis of a functional DIAP1 fragment bound to grim and hid peptides. Mol. Cell, 8, 95–104. [DOI] [PubMed] [Google Scholar]
- Yoshida H., Kong,Y.Y., Yoshida,R., Elia,A.J., Hakem,A., Hakem,R., Penninger,J.M. and Mak,T.W. (1998) Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell, 94, 739–750. [DOI] [PubMed] [Google Scholar]
- Yuan J., Shaham,S., Ledoux,S., Ellis,H.M. and Horvitz,H.R. (1993) The C.elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1B-converting enzyme. Cell, 75, 641–652. [DOI] [PubMed] [Google Scholar]
- Zhou L., Hashimi,H., Schwartz,L.M. and Nambu,J.R. (1995) Pro grammed cell death in the Drosophila central nervous system midline. Curr. Biol., 5, 784–790. [DOI] [PubMed] [Google Scholar]
- Zhou L., Schnitzler,A., Agapite,J., Schwartz,L.M., Steller,H. and Nambu,J.R. (1997) Cooperative functions of the reaper and head involution defective genes in the programmed cell death of Drosophila central nervous system midline cells. Proc. Natl Acad. Sci. USA, 94, 5131–5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L., Song,Z.W., Tittel,J. and Steller,H. (1999) HAC-1, a Drosophila homolog of APAF-1 and CED-4 functions in developmental and radiation-induced apoptosis. Mol. Cell, 4, 745–755. [DOI] [PubMed] [Google Scholar]
- Zou H., Yuchen,L., Xuesong,L. and Wang,X. (1999) An APAF-1/cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem., 274, 11549–11556. [DOI] [PubMed] [Google Scholar]