

Abstract
Protein disulfide bond formation in Escherichia coli is catalyzed by the periplasmic protein DsbA. A cytoplasmic membrane protein DsbB maintains DsbA in the oxidized state by transferring electrons from DsbA to quinones in the respiratory chain. Here we show that DsbB activity can be reconstituted by co-expression of N- and C-terminal fragments of the protein, each containing one of its redox-active disulfide bonds. This system has allowed us (i) to demonstrate that the two DsbB redox centers interact directly through a disulfide bond formed between the two DsbB domains and (ii) to identify the specific cysteine residues involved in this covalent interaction. Moreover, we are able to capture an intermediate in the process of electron transfer from one redox center to the other. These results lead us to propose a model that describes how the cysteines cooperate in the early stages of oxidation of DsbA. DsbB appears to adopt a novel mechanism to oxidize DsbA, using its two pairs of cysteines in a coordinated reaction to accept electrons from the active cysteines in DsbA.
Keywords: disulfide bond formation/DsbB/electron transfer/Escherichia coli/oxidative protein folding
Introduction
Disulfide bonds are crucial for the folding, stability and function of many extracytoplasmic proteins (Gilbert, 1997). In Escherichia coli, the periplasmic protein DsbA acts as the primary catalyst of disulfide bond formation, utilizing its redox active site, Cys30–Pro31–His32–Cys33, which is located in a thioredoxin-like fold (Bardwell et al., 1991; Martin et al., 1993; Zapun et al., 1993). DsbA donates its Cys30–Cys33 disulfide to a pair of thiols on a target protein, thus leaving DsbA in the reduced form. The fact that DsbA is found exclusively in the oxidized form in normal cells (Kishigami et al., 1995) indicates that a mechanism exists for its reoxidation. This oxidation step is carried out by the cytoplasmic membrane protein DsbB which contains four cysteines essential for its function (Bardwell et al., 1993; Missiakas et al., 1993). DsbB restores disulfide bonds to DsbA by transferring electrons from the reduced DsbA to membrane-embedded quinones (Kobayashi et al., 1997; Bader et al., 1999, 2000). The reduced quinones are then oxidized by terminal oxidases aerobically using molecular oxygen or anaerobically using other electron acceptors such as nitrate or fumarate (Bader et al., 1999). Most of the structural disulfides in cells depend on this oxidative function of DsbB, as mutants lacking the protein are severely deficient in the oxidation of thiols of extracytoplasmic proteins. Many proteins that fail to form proper disulfide bonds are degraded rapidly in cells (Bardwell et al., 1991), indicating the importance of this pathway in the folding of exported proteins.
Escherichia coli also has a reductive pathway involved in disulfide bond formation. DsbC, a periplasmic protein with a thioredoxin-like fold and a CXXC motif (McCarthy et al., 2000), acts to reduce proteins with incorrectly paired cysteine residues. DsbC is maintained in the reduced active state (Rietsch et al., 1997) by the cytoplasmic membrane protein DsbD, which uses its six essential cysteines to transfer electrons from cytoplasmic thioredoxin onto DsbC (Stewart et al., 1999; Katzen and Beckwith, 2000). Electrons in this pathway ultimately derive from the small molecule NADPH which is required for the reduction of thioredoxin reductase.
DsbB spans the cytoplasmic membrane four times with both the N- and C-termini facing the cytoplasm. The activity of DsbB depends on its two pairs of conserved redox-active disulfide bonds, Cys41–Cys44 and Cys104– Cys130, located in the first and second periplasmic domains, respectively (Jander et al., 1994; Kobayashi and Ito, 1999; Figure 1A). The Arg48 residue plays an important role in the interaction of DsbB with quinones (Kadokura et al., 2000). That the Cys104–Cys130 pair is the direct donor of a disulfide to DsbA is suggested by the detection of a mixed-disulfide complex formed between Cys104 of DsbB and Cys30 of a DsbA variant lacking Cys33 (Guilhot et al., 1995; Kishigami and Ito, 1996). The DsbB Cys104–Cys130 pair is probably maintained in the oxidized state by the Cys41–Cys44 disulfide bond, since the oxidation of the Cys104–Cys130 pair requires Cys41 and Cys44, whereas the Cys41–Cys44 pair is still oxidized even in the absence of the Cys104–Cys130 pair (Kobayashi and Ito, 1999). Finally, the Cys41–Cys44 pair is kept oxidized by the function of the respiratory chain (Kobayashi and Ito, 1999). Further, Bader et al. (1999) showed that quinones can act as direct recipients of electrons from DsbB. Thus, electrons from DsbA are presumed to flow first to Cys104–Cys130 and from there to Cys41–Cys44, and then finally to quinones in the respiratory chain (Debarbieux and Beckwith, 1999).

Fig. 1. DsbB and its derivatives. (A) Sequence and topology of DsbB in the cytoplasmic membrane (Jander et al., 1994). DsbB was split into two polypeptides: α (from Met1 to Arg90) and β (from Pro85 to Arg176). For translation initiation, a methionine residue was placed at the N-terminus of β. Further, α and β were fused with triple FLAG (FLAG3) and His6-c-Myc sequences, respectively, for their detection. The essential cysteine residues of DsbB are marked with darker circle. In α, the non-essential cysteines (Cys8 and Cys49) were mutated to alanine and valine, respectively. (B) Structure of the operon fusion for the expression of α and β. Sequences encoding α and β were placed tandemly under the control of a lac promoter in a pSC101-derived low-copy plasmid, pHK544. A Shine–Dalgarno motif was put in front of each open reading frame.
While this model is consistent with existing data, many important questions still remain. For example, the mechanism of electron transfer between the two redox active sites within DsbB is unknown, as is the mechanism for transferring electrons from the first periplasmic domain to quinones in the respiratory chain.
Here we report a new approach to studying the process of electron transfer within the protein DsbB. In order to explore the communication between the two redox centers of DsbB, we have split the protein into two domains each containing two of the essential cysteines. These two domains when expressed separately in the same cell reconstitute active DsbB. Utilizing this new assay system, we characterized the process of electron transfer within DsbB, and found a novel mechanism employed by this enzyme to oxidize protein thiols. DsbB appears to coordinate action of all the four essential cysteines before releasing oxidized DsbA from the DsbA–DsbB complex, in contrast to previous models in which the disulfide in the second periplasmic domain is simply donated to cysteines in DsbA by a thiol–disulfide exchange reaction.
Results
The strategy for expression of DsbB as complementary polypeptides
To study the process of electron transfer from one redox-active cysteine pair to another, we must be able to distinguish the different oxidation states of each partner. Furthermore, detection of mixed-disulfide intermediates between partners in such reactions is an important contribution to elucidating the mechanism of electron transfer (Guilhot et al., 1995; Kishigami and Ito, 1996; Frand and Kaiser, 1999). However, when it comes to studying the electron transfer reactions that take place within a protein, such as DsbB, the determination of the oxidation states or detection of disulfide-bonded intermediates becomes significantly more difficult. This laboratory has overcome this problem previously by splitting a membrane protein, DsbD, into separate polypeptide domains, each carrying a redox-active cysteine pair, and effectively reconstituting the thiol–disulfide exchange in vivo. This system enabled us to characterize the electron transfer reactions that occur between these polypeptides and between them and other proteins that are part of the reductive pathway (Katzen and Beckwith, 2000; unpublished data). We have proceeded to ask whether the same strategy might work to study electron transfer reactions carried out by the protein DsbB.
To separate each pair of cysteines of DsbB onto two different polypeptides, we have chosen to split the protein within the cytoplasmic loop (Figure 1A) for the following reasons. First, cuts within transmembrane segments were likely to be detrimental to the activity of the enzyme because they are important determinants of the topology of membrane protein (Popot and Engelman, 2000). Secondly, we thought we should avoid splitting in the segment between Ile45 and Arg48, since this region is important for the oxidation of DsbB by the electron transport chain (Kadokura et al., 2000; Kobayashi et al., 2001). Thirdly, a split after the third transmembrane segment would require the addition of a signal sequence to the C-terminal polypeptide to ensure the extracellular position of its N-terminus. In this case, any incomplete processing of the signal could make later analysis difficult. Finally, we reasoned that the cytoplasmic loop might not be important for the activity of the enzyme, since no specific function has been assigned to this region and its sequence is not conserved among DsbB homologs. We used slightly overlapping constructs, since the sequences flanking the transmembrane segments contribute to their correct membrane insertion. Further, we placed a triple FLAG tag in front of the N-terminal polypeptide and a His6- c-Myc tag at the end of the C-terminal polypeptide in order to allow separate detection of each polypeptide. We will call these polypeptides α and β, respectively. To express both α and β, the sequences encoding these fragments were placed tandemly under the control of a lac promoter on a pSC101-derived low copy plasmid (Figure 1B).
Co-expression of complementary polypeptides restores DsbB activity
To determine whether DsbB is still active when separated into two polypeptides, we expressed α and β in a ΔdsbB strain, and examined their ability to promote disulfide bond formation in the periplasmic protein β-lactamase. To determine the oxidation status of proteins including β-lactamase, we acid trap the protein in cell extracts and then alkylate any free cysteines with 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS). Because of the ∼0.5 kDa molecular weight of AMS, this modifi cation retards the mobility of the reduced form of proteins on gels, allowing ready distinction between the oxidized and the reduced forms of many proteins. Remarkably, the plasmid encoding the two separated DsbB domains completely restored disulfide bond formation to β-lactamase (Figure 2, lane 4) and maintained, albeit slightly less efficiently than full-length DsbB, 50–70% of DsbA in the oxidized form in a ΔdsbB strain (see Figure 3C, lane 8 and Figure 5A, lane 7). Plasmids coding for either α or β alone were unable to restore disulfide bond formation (Figure 2, lanes 3 and 5). However, the plasmid encoding both α and β did not allow disulfide bond formation in a ΔdsbA, ΔdsbB double mutant (data not shown), showing that the α and β are still functioning through the oxidation of DsbA. The α and β polypeptides expressed in a ΔdsbB strain also restored bacterial motility, a property which, in E.coli, depends on a functional disulfide bond formation pathway. However, when one of the complementary polypeptides was removed from the system, the cells lost motility (data not shown). Thus, these complementary polypeptides, α and β, assemble in the membrane into a functional unit that can act just like DsbB in the pathway of disulfide bond formation.
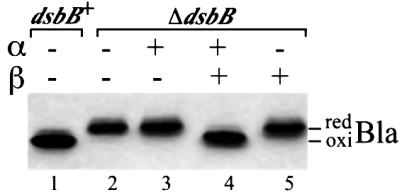
Fig. 2. Functionality of DsbB derivatives. The dsbB+ strain HK295 containing both pUC18 (carrying bla) and pHK544 (vector) was used in lane 1. All the other lanes use strain HK320 (ΔdsbB) containing both pUC18 and one of the following plasmids: pHK544 (lane 2), pHK555 for expression of α (lane 3), pHK553 for α and β (lane 4) and pHK556 for β (lane 5). Cells were grown at 37°C in M63 minimal Glc medium, and portions of exponentially growing culture were treated directly with trichloroacetic acid (TCA; final 5%) to precipitate whole-cell proteins. The TCA-treated samples were washed with acetone and subjected to alkylation with AMS. The proteins were separated with SDS–PAGE, and disulfide bond formation in a periplasmic protein, β-lactamase (Bla), was visualized with western blotting using anti-Bla antibody. Bla in lanes 1 and 2 provides the standard for the oxidized (oxi) and reduced (red) forms of this protein, respectively.

Fig. 3. In vivo redox state of α, β and DsbA. Strain HK320 (ΔdsbB) was transformed with one of the following plasmids: pHK544 (lanes 1–3), pHK555 (lanes 4–6), pHK553 (lanes 7–9) and pHK556 (lanes 10–12). Exponentially growing cells were harvested and were subjected to TCA precipitation, followed by treatment with (lanes 2, 5, 8 and 11) or without (lanes 1, 4, 7 and 10) AMS. The proteins in the latter samples served as oxidized standards. To generate the reduced standards, the above TCA precipitates were subjected to reduction with 100 mM DTT, a second TCA precipitation and modification with AMS (lanes 3, 6, 9 and 12). Proteins were separated with SDS–PAGE and visualized by western blotting with anti-FLAG (A), anti-Myc (B) and anti-DsbA (C) antibodies to detect α (tagged with FLAG3), β (tagged with His6-c-Myc) and DsbA, respectively. The positions of molecular mass standards are indicated on the left in kDa. Arrows and arrowheads indicate the α–β–DsbA mixed-disulfide complex and non-specific bands, respectively.

Fig. 5. Effect of reduction (A) and oxidation (B) in the periplasm on the formation of the mixed-disulfide ternary complex. (A) Strain HK320 (ΔdsbB) was transformed with pHK544 (lanes 1 and 6) or pHK533 (lanes 2–5 and 7–10). (B) The strain HK320 (lane 2) and the wild-type strain HK295 (lane 1) were transformed with pHK553. (A and B) Cells were grown in M63 minimal Glc medium, harvested at mid-log phase and subjected to TCA precipitation and modification with AMS. The indicated concentration of DTT was added to the culture 15 min before the harvest in (A). The samples were subjected to SDS–PAGE and western blotting with anti-FLAG in (A, lanes 1–5) and (B) and anti-DsbA in (A, lanes 6–10).
Cysteine in α is required for the oxidation of β
The oxidized and reduced forms of α and β are readily distinguishable on gels after AMS alkylation (Figure 3A and B, lanes 7 and 9). This finding meant that we could determine the oxidation state of cysteines of α and β in cells grown under different conditions or carrying mutant versions of these components. In the presence of both α and β, β was completely oxidized (Figure 3B, lane 8). Removal of α from this system left β reduced (Figure 3B, lane 11), in agreement with the previous conclusion of Kobayashi and Ito (1999) that cysteines in the first periplasmic domain are required for the oxidation of cysteines in the second periplasmic domain. It should be noted, however, that α in this system was only partially oxidized (Figure 3A, lane 8), in contrast to its fully oxidized state when present in the full-length DsbB (Kobayashi and Ito, 1999). One possible explanation for this finding is that the oxidation of the two cysteines by the respiratory chain is less efficient due to the splitting of the protein. Recently, Xie et al. (2002) showed that a ubiquinone analog can bind to a part of the second periplasmic domain, suggesting that this region might also be involved in the interaction of DsbB with quinones, in addition to the region between Ile44 and Arg48 described above. Thus, it might be that the separation of the two regions in the split system caused less efficient oxidation of α by the respiratory function.
Our results indicate that the co-functioning of the split polypeptides in disulfide bond formation retains key properties of wild-type DsbB. First, introduction of cysteine to alanine mutations for each of the four essential cysteines of α and β rendered this system completely inactive (data not shown), indicating that these polypeptides use the same residues for catalysis as intact DsbB (Jander et al., 1994). Secondly, mutating either cysteine of α resulted in β remaining in the reduced state (Figure 4B, lanes 5 and 7). However, α remained partially oxidized with cysteine mutations in β (Figure 4A, lanes 9 and 11), providing further support for the conclusion that electrons flow from the cysteines in the second periplasmic domain to those of the first periplasmic domain (Kobayashi and Ito, 1999). Thirdly, we detected a β–DsbA mixed-disulfide complex under some conditions (see below), but not an α–DsbA complex, consistent with the previous findings that Cys104–Cys130 is the direct donor of a disulfide to the cysteines of DsbA. Finally, βC104A in cells expressing α and βC104A did not form a mixed-disulfide complex with DsbA even under the conditions that allow the accumulation of substantial amounts of mixed-disulfide complex between DsbA and DsbB (expression of DsbAC33A in a dsbA+-dsbB+ strain background; Guilhot et al., 1995; Kishigami and Ito, 1996) (data not shown). Introduction of a C41A, C44A or C130A mutation into the complementary polypeptides, however, did not prevent the formation of a mixed-disulfide bond between β and DsbAC33A under the same conditions (data not shown). Thus, Cys104 of β, as in the native DsbB, is the only cysteine that can interact directly with DsbA.

Fig. 4. Introduction of cysteine to alanine mutations into α and β. Strain HK320 (ΔdsbB) was transformed with one of the following plasmids: pHK544 (lanes 1 and 2), pHK553 (lanes 3 and 4), pHK559 for αC41A and β (lanes 5 and 6), pHK560 for αC44A and β (lanes 7 and 8), pHK561 for α and βC104A (lanes 9 and 10) and pHK562 for α and βC130A (lanes 11 and 12). Growing cells were harvested and were subjected to TCA precipitation and AMS alkylation (lanes 1, 3, 5, 7, 9 and 11). In lanes 2, 4, 6, 8, 10 and 12, the TCA precipitates were subjected to reduction with 100 mM DTT before modification with AMS. The samples were subjected to SDS–PAGE and western blotting with anti-FLAG (A) and anti-Myc (B) antibodies. red1 and red indicate the positions of singly (red1) and doubly alkylated (red) forms of proteins. Asterisks indicate the β–DsbA complex. Note that αC44A–β–DsbA migrates slightly faster than α–β–DsbA, reflecting differences in the number of free cysteine residues in each complex: αC44A–β–DsbA (one cysteine), α–β–DsbA (two cysteines).
α, β and DsbA form a disulfide-bonded ternary complex
Anti-FLAG antibody detected, in addition to the oxidized and reduced forms of α, a band of an apparent molecular mass of ∼45 kDa (Figure 3A, lane 8, indicated with an arrow). This latter band disappeared when samples were reduced with dithiothreitol (DTT) in the step before modification with AMS (Figure 3A, lane 9), suggesting that it represents a mixed disulfide. Anti-Myc and anti-DsbA antibodies also recognized the 45 kDa band (Figure 3, lane 8, see also Figure 5A, lane 7). We suggest that this mixed disulfide represents an α–β–DsbA ternary complex for the following reasons. First, the apparent molecular mass of this complex correlates well with the sum of the apparent molecular masses of the three postulated components (44 kDa). Secondly, the complex reacts especially with all three antibodies: anti-FLAG, anti-c-Myc and anti-DsbA. Thirdly, the band disappears when the reductant DTT is added (Figure 3, lane 9). Finally, its presence depends on simultaneous expression of α (Figure 3, lanes 8 and 11), β (Figure 3, lanes 5 and 8) and DsbA (data not shown). As with the previously discovered mixed disulfide between intact DsbB and DsbA, the amount of this apparent ternary complex was enhanced by the introduction of the C33A change into DsbA and eliminated by the introduction of the C30A change into DsbA (data not shown).
In addition to the ternary complex, we also detected, upon overexposure of the film, a minor band that was recognized by both anti-Myc and anti-DsbA antibodies but not by anti-FLAG antibody (data not shown, and Figure 4B, lane 3 and Figure 5A, lane 7). We conclude that this band represents a mixed disulfide formed between Cys104 of β and Cys30 of DsbA because: (i) this band forms only in the presence of both β and DsbA; (ii) it disappears upon DTT treatment of the sample; (iii) its apparent molecular mass agrees with the sum of those of β and DsbA; and (iv) mutating Cys30 of DsbA or Cys104 of β abolishes the formation of this complex (data not shown).
Characterization of the ternary complex
Characterization of mixed-disulfide complexes has contributed to our understanding of the mechanism of many enzymes that use cysteine thiols in their reactions. It seemed likely to us that the ternary complex of DsbA, β and α represented an intermediate in the pathway of electron transfer between these molecules. It is detected in cells where α and β are actively participating in the reoxidation of DsbA. Therefore, we proceeded to determine the exact pattern of disulfide bonding between these three polypeptides. Since a DsbA–DsbB mixed-disulfide intermediate (DsbA Cys30–DsbB Cys104) had been identified before and an analogous DsbA–β complex is described here, we presumed that the ternary complex must also contain an α–β mixed disulfide. Given the involvement of Cys104 of β in the complex with DsbA, it seemed likely that it was Cys130 of β that was disulfide bonded to a cysteine of α. To assess this proposed covalent interaction of Cys130 with a cysteine of α, we mutated each of the codons specifying the essential cysteines of β to alanine and determined the ability of the α and mutated β to form mixed-disulfide complexes.
As expected, the cysteine to alanine mutation at residue 104 eliminated the α–β–DsbA complex (Figure 4, lanes 9 and 10). This mutation, however, enhanced the formation of the α–β complex. This finding indicates that Cys130 can form a disulfide bond with one of the two cysteines in α. In contrast, the mutation at Cys130 resulted in the complete elimination of the α–β and α–β–DsbA complexes (Figure 4, lanes 11 and 12). These results reveal the distinct functions of the two cysteine residues, Cys104 and Cys130, in the pathway of disulfide bond formation: Cys104 is devoted to the direct interaction with Cys30 of DsbA, whereas Cys130 is the active cysteine that can participate in the formation of the mixed-disulfide complex with a cysteine in the first periplasmic domain of DsbB.
To identify the cysteine residue in the first periplasmic domain that directly interacts with Cys130, we mutated essential cysteines of α and examined their effects on the formation of the mixed disulfide between α and β. The C44A mutant still retained its ability to form α–β and, albeit in an apparently reduced amount, the α–β–DsbA complexes (Figure 4, lanes 7 and 8), indicating that Cys41, the sole cysteine on αC44A, can form a disulfide bond with Cys130 of β. In marked contrast, the cysteine to alanine mutation at residue 41 completely eliminated the formation of a disulfide between α and β (Figure 4, lanes 5 and 6), indicating that this residue is required for the interdomain mixed-disulfide bond formation. Altogether, these results indicate that Cys41 is the active cysteine involved in the direct interaction with Cys130.
Formation of α–β–DsbA ternary complex is enhanced by low levels of DTT
To investigate further the possibility that the ternary complex detected in cells expressing active α and β is an intermediate in the pathway to oxidation of DsbA, we devised two tests. We reasoned that if we generated conditions that resulted in an increased ratio of reduced to oxidized DsbA, thus requiring DsbB to act more frequently as an oxidant of DsbA, any intermediates in the pathway might be present in higher concentration in the cells. To increase the level of reduced DsbA, we treated growing cultures with low concentrations of the reductant DTT for 15 min and assessed its effect on the ability of cells to form the ternary complex. In the absence of DTT, ∼30% of DsbA was in its reduced form (Figure 5A, lane 7). In the presence of low concentrations of DTT, >90% of DsbA was reduced (Figure 5A, lanes 8–10). Simul taneously, we observed the enhanced accumulation of the α–β–DsbA complex (Figure 5A, lanes 2–5 and 7–10), consistent with our proposal that this complex is an intermediate in the reaction. In addition to the α–β–DsbA complex, we observed enhanced accumulation of the α–β and β–DsbA complexes upon treatment with low concentrations of DTT, indicating that either (i) these complexes are also intermediates in the reaction or (ii) these complexes arose as a result of the resolution of pre-formed α–β–DsbA with the attack by DTT thiol.
Since causing DsbA to be present mostly in the reduced state enhanced the formation of ternary complex, we further tested our model by creating conditions where DsbA was predominantly in the oxidized state. Under these conditions, there would be less call for the α and β polypeptides to participate in the oxidation of DsbA. We did this by determining the levels of ternary complex in cells in which wild-type DsbB was also being expressed, thus providing an additional oxidant for DsbA. The presence of dsbB+ on the chromosome of such a strain greatly reduced the amount of the ternary complex (Figure 5B, lanes 1 and 2), providing further evidence that the ternary complex was formed in the process of oxidation of DsbA.
Discussion
In this study, we have explored the process of electron transfer within DsbB and between DsbB and DsbA, by splitting DsbB into two polypeptides (α and β) each containing only two cysteine residues. These separate domains when expressed in the same cell are able to reconstitute DsbB activity efficiently. This split system has allowed us to elucidate details of the mechanism of DsbB action, not readily studied when DsbB is in its intact state. Our results have led us to propose a novel mechanism employed by this enzyme to oxidize protein thiols.
Using the split DsbB system, we have detected a ternary complex that has the properties of an intermediate in the transfer of electrons between DsbB and DsbA. This complex consists of the three polypeptides, α, β and DsbA, linked together by two disulfide bonds. These bonds exist between Cys30 of DsbA and Cys104 of β, and between Cys130 of β and Cys41 of α.
Our studies, and particularly the finding of the ternary complex, led us to propose the following picture of electron transfer in the oxidation of reduced DsbA by fully oxidized DsbB. The process begins with the nucleophilic attack by the deprotonated thiol (Cys30) of reduced DsbA on the Cys104–Cys130 disulfide of DsbB (Figure 6A, stage I), leading to formation of the mixed-disulfide complex between Cys104 of DsbB and Cys30 of DsbA. This reaction results in the transfer of one electron from DsbA to DsbB, rendering the Cys130 residue reduced (Figure 6A, stage II). Then, the thiol of the reduced Cys130 attacks the disulfide in the first periplasmic domain (Figure 6A, stage III) to form an interdomain mixed-disulfide complex between Cys130 in the second periplasmic domain and Cys41 in the first periplasmic domain of DsbB (Figure 6A, stage IV). This reaction transfers one electron from the second periplasmic domain to the first periplasmic domain and renders Cys44 reduced. This interdomain electron transfer reaction occurs prior to the movement of the second electron from DsbA to DsbB (Figure 6A, stage III), generating a ternary complex in which the first and second periplasmic domains of DsbB together with DsbA are connected by two disulfide bonds (Figure 6A, stage IV). Finally, the attack by Cys33 of DsbA on the disulfide bond between Cys30 of DsbA and Cys104 of DsbB resolves the Cys30–Cys104 disulfide bond or quinones drive the reaction, in either case yielding oxidized DsbA. It is tempting for us to speculate that the reduced Cys44 in the ternary complex (Figure 6A, stage IV) further attacks a quinone molecule to transfer one electron from Cys44 to quinone, which then triggers the release of oxidized DsbA from the DsbA–DsbB complex (not shown in Figure 6A). This model is consistent with the findings that the DsbB R48H mutant, which showed reduced ability to interact with quinone, accumulated DsbA–DsbB complex in vivo (Kadokura et al., 2000). We have not obtained evidence here that sheds light on the final steps in this process in which DsbB becomes reoxidized by transfer of electrons into the respiratory chain.

Fig. 6. Proposed mechanisms for the early steps in the reoxidation of DsbA by DsbB. A model based on results presented herein (A) and a model proposed by us and others previously (B). Both models share the initial steps (stages I and II). See Discussion for details.
An alternative model was suggested previously for the steps involved in the interaction between DsbB and DsbA (Figure 6B) (Kishigami and Ito, 1996; Debarbieux and Beckwith, 1999). According to this proposal, Cys33 of DsbA attacks the disulfide bond between Cys30 of DsbA and Cys104 of DsbB to release oxidized DsbA from the complex (Figure 6B, stage III′). This reaction transfers a second electron from DsbA to DsbB, leading to the reduction of both cysteines in the second periplasmic domain of DsbB (Figure 6B, stage IV′).
Several of our findings are consistent with the model we present here (Figure 6A) and not with the previously proposed model (Figure 6B). The existence of an intermediate ternary complex between α, β and DsbA cannot be explained by the alternative model. This latter model predicts an intermediate stage in which oxidized DsbA along with reduced β is present (Figure 6B, stage IV′), followed by stages in the reoxidation of β by α. That the ternary complex is an intermediate in the pathway is supported by our studies on the effects of increasing or lowering the level of reduced DsbA. Increasing the amount of this substrate by adding low concentrations of DTT to cultures enhanced the accumulation of the ternary complex; while decreasing the amount of substrate by co-expressing wild-type DsbB dramatically decreased the formation of the ternary complex. These two findings are predicted by the model in which the ternary complex is an intermediate formed in the process of oxidation of DsbA by α and β. DTT is a strong reductant that also acts to resolve pre-formed mixed-disulfide complexes. The fact that the formation of the ternary complex was enhanced with DTT, even with its ability as a reductant, suggests that this complex was formed by a forward reaction driven by the supply of oxidizing equivalents from the respiratory chain.
Furthermore, in cells expressing wild-type active α and β, we did not detect any reduced β, an intermediate predicted by the alternative model (Figure 6B, stage IV′). Consistent with this latter finding, we do not detect any partially reduced full-length DsbB when this enzyme is active. The only species observed are the fully oxidized DsbB and the DsbA–DsbB complex (Kadokura et al., 2000). These results suggest that either (i) electrons from the reduced cysteines in the second periplasmic domain are so quickly transferred to the first periplasmic domain and then onto quinones that the enzyme does not accumulate the putative intermediate, partially reduced DsbB or (ii) this feature of the alternative model was incorrect.
More specific details of our model are provided by several lines of evidence presented here. First, that a cysteine in the second periplasmic domain can interact directly with a cysteine in the first periplasmic domain is supported by the capture of mixed-disulfide complexes of α and β. Secondly, we did not observe any α or β complexes other than the following: α–β–DsbA, α–β and β–DsbA. The results indicate that these interactions are stable and specific. Thirdly, Cys41 and Cys130 are the active cysteines involved in the covalent interaction between the two periplasmic domains. The evidence for this conclusion includes the findings that the introduction of C41A and C130A mutations completely abolished the complexes while the C44A or C130A substitution mutations did not disrupt the covalent interaction between α and β.
Our systematic analysis of the function of the four essential cysteines of DsbB revealed that Cys130 is devoted to covalent interaction with the first periplasmic domain, while Cys104 is devoted to interaction with Cys30 of DsbA. In contrast, it was implied previously that Cys104, which directly interacts with DsbA, might also be involved in the interaction with the Cys41–Cys44 disulfide (Kobayashi and Ito, 1999). Mechanically, the separation of reactivities as shown here may be important because it allows the reduced Cys130 in the binary complex to attack the Cys41–Cys44 bond even before the resolution of the disulfide bond that connects Cys104 of DsbB and Cys30 of DsbA. Otherwise, DsbA has to be resolved from Cys104 of DsbB in order for the Cys104 to attack Cys41–Cys44.
Ordinary, thiol–disulfide exchange reactions are carried out between two thiols and one disulfide. We describe here a novel mechanism that involves the coordinated action of four cysteines of DsbB in the early stages of oxidation of DsbA. Why, then, does the oxidation of two thiols of DsbA require this coordination? DsbA is highly oxidizing in vivo and in vitro, i.e. the reduced form of this protein is energetically favored (Bardwell et al., 1991; Zapun et al., 1993; Grauschopf et al., 1995). After the formation of the DsbA Cys30–DsbB Cys104 disulfide, there would be a strong tendency favoring the reverse reaction, with Cys130 attacking the disulfide bond, thus restoring reduced DsbA. This is favored because of the strong tendency of DsbA to remain reduced. However, DsbB may be arranged structurally so that after the freeing of Cys130, this amino acid is positioned to react more readily with the Cys41–Cys44 disulfide bond. The product of this reaction, a Cys130–Cys41 disulfide bond within DsbB, will prevent Cys130 from resolving the DsbA Cys30–DsbB Cys104 disulfide. This latter reaction would have regenerated the unwanted reduced form of DsbA. Thus, the coordinate action of the cysteines of DsbB might be required because of the need to oxidize highly oxidizing DsbA.
Theoretically, if the redox potential of the cysteine pair of the second periplasmic domain is higher than that of DsbA, and if that of the first periplasmic domain is even higher than that of the second periplasmic domain, the non-coordinated model (Figure 6B) would appear reasonable. However, we think it unlikely for the following reasons. First, DsbA has the most oxidizing cysteine pair ever characterized. Secondly, DsbB mutants in which the two amino acids between Cys41 and Cys44 were replaced by those of the CXXC motifs from a variety of other oxidoreductases still retained DsbB activity (Kobayashi et al., 2001).
The novel mechanism invoked here to explain the oxidation of protein thiols for DsbB may not be restricted to bacteria. In the endoplasmic reticulum of the yeast Saccharomyces cerevisiae, a membrane-associated protein, Ero1p, catalyzes the oxidation of a luminal protein, Pdi1p, which, in turn, donates its disulfide to a target protein (Frand and Kaiser, 1999). Like DsbB, Ero1p contains two redox-active disulfides that are required for the activity of this enzyme (Frand and Kaiser, 2000). In addition, its substrate, Pdi1p, is highly oxidizing like DsbA. Thus, Ero1p might adopt a coordinated mechanism similar to that used by DsbB for the oxidation of Pdi1p.
Our report describes the successful reconstitution of the activity of a membrane protein by expressing two membrane-embedded domains of the protein. Several other cases have been reported where membrane protein function has been restored by expressing separately domains of such proteins that ordinarily are joined in a single polypeptide chain (Bibi and Kaback, 1990; Berkower et al., 1996; Wilkinson et al., 1997). The splitting system described here and for the previously studied DsbD (Katzen and Beckwith, 2000) has proven to be particularly powerful for the study of electron transfer between domains of a protein. In this study, we split DsbB at the cytoplasmic loop that connects the second and the third transmembrane segments. We did not have to use any additional signal to preserve the topology of the split version of the membrane protein. This is in contrast to the DsbD studies where we split this membrane protein into two periplasmic domains and one membrane-embedded domain. In that case, it was necessary to add a signal peptide to maintain the topology of the C-terminus. This work shows that the separately expressed membrane-embedded domains of a protein provide a powerful tool for the detailed analysis of enzyme mechanisms in vivo.
Materials and methods
Strains and plasmids, and primers used in this work are listed in Tables I and II, respectively.
Table I. Strains and plasmids used in this work.
| Strain or plasmida | Relevant genotypes and/or characteristics |
|---|---|
| Strains | |
| HK295 | MC1000 Δara714 leu+ |
| HK320 | HK295 ΔdsbB |
| JP313b | MC4100 Δara714 |
| MC1000c | araD139 Δ(araABC-leu)7697 galU galK Δ(lac)X74 rpsL thi |
| Plasmidsd | |
| pAM238e | lac promoter, pSC101-based, Spcr |
| pBAD18f | pBR-based, ampicillinr, rrnBT1T2 terminator |
| pHK517 | pAM238 with DsbB-His6-c-Myc |
| pHK521 | pLAC-YC with α |
| pHK522 | pAM238 with β-His6-c-Myc |
| pHK537 | pAM238 with FLAG3-DsbB-His6-c-Myc |
| pHK538 | pLAC-YC with FLAG3-α |
| pHK543 | pAM238 with FLAG3-α and β-His6-c-Myc |
| pHK544 | pAM238 with rrnBT1T2 terminator |
| pHK552 | pAM238 with FLAG3-DsbB |
| pHK553 | pHK544 with FLAG3-α and β-His6-c-Myc |
| pHK555 | pHK544 with FLAG3-α |
| pHK556 | pHK544 with β-His6-c-Myc |
| pHK559 | pHK544 with FLAG3-αC41A and β-His6-c-Myc |
| pHK560 | pHK544 with FLAG3-αC44A and β-His6-c-Myc |
| pHK561 | pHK544 with FLAG3-α and βC104A-His6-c-Myc |
| pHK562 | pHK544 with FLAG3-α and βC130A-His6-c-Myc |
| pLAC-YCg | lacUV5 promoter, pACYC184-based, Camr |
| pWM76h | pQE70 with DsbB-His6 |
aUnless otherwise indicated, the source of the strain is the present work.
cLaboratory collection.
dAll the DsbB derivatives contain C9A and C49V mutations.
Table II. Primers used in this work.
| Name | Sequence (5′ to 3′) |
|---|---|
| DSBH9 | AAAGGTACCAAAGAGGAGAAATTAAGCATGCTGCGATTTTTGAAC |
| DSBH11 | CTTCTAGACTACAGCAGATCTTCTTCAGAAATCAGTTTCTGTTCGTGATGGTGATGGTGATGAGATCTGCGACC |
| DSBH15 | CTTCTAGACTAGCGACCGAACAGATCACGTTTTTTCGCTTT |
| DSBH22 | AAAGGTACCAAAGAGGAGAAATTAAGCATGCCGAAAACTCCGCTGCGTTATGTAGCGATGG |
| DSBH23 | ACTTCTAGACTAACGCAGCGGAGTTTTCGGGGCGATCGCGCCAATCAG |
| DSBH31 | AAGGATCCCTGCGATTTTTGAACCAAGCTTCACAAGGC |
| DSBH32 | AAGGATCCCTTGTCATCGTCATCCTTGTAATCGATGTCATGATCTTTATAATCACCGTCATGGTCTTTGTAGTCCATGCTTAATTTCTCCTCTTTGGTACC |
| DSBH42 | AATGAATTGGTTTAAACTGCGCACTCTATGCATATTGCAGGGAAATGATTATTCCGGGGATCCGTCGACC |
| DSBH43 | AAATATAGCGGCAGGAAAAAAGCGCTCCCGCAGGAGCGCCGAATGGATTAGTGTAGGCTGGAGCTGCTTC |
| DSBH46 | GTGATGTTACTGAAACCTGCGGTGCTCTGTATTTATGAA |
| DSBH47 | TTCATAAATACAGAGCACCGCAGGTTTCAGTAACATCAC |
| DSBH48 | CTGAAACCTTGCGTGCTCGCCATTTATGAACGCGTCGCG |
| DSBH49 | CGCGACGCGTTCATAAATGGCGAGCACGCAAGGTTTCAG |
| DSBH50 | CCTTCGCCGTTTGCCACCGCGGATTTTATGGTTCGTTTC |
| DSBH51 | GAAACGAACCATAAAATCCGCGGTGGCAAACGGCGAAGG |
| DSBH56 | AAAGAATTCTATGGTACCAAAGAGGAGAAATTAAGCATGGAC |
| DSBH57 | AAAGAGCTCAAAATCTAGACTAACGCAGCGGAGTTTTCGGGGC |
| DSBH58 | TTTTCTGCAGGCTGTTTTGGCGGATGAGAGAAGATTTTC |
| DSBH59 | AAAAAAGCTTGAGTTTGTAGAAACGCAAAAAGGCCATCC |
Strains
All the strains are derivatives of HK295 which was constructed by P1 transduction of the Δara714 leu+ region of JP313 into MC1000. To delete the chromosomal dsbB gene of HK295, we employed the method described by Datsenko and Wanner (2000). The primers used were DSBH42 and DSBH43. We noticed that the dsbB::kan5 allele ordinarily used as dsbB null (Bardwell et al., 1993; Kadokura et al., 2000; Kobayashi et al., 2001) is inappropriate for the current splitting system because even β alone can partially complement the disulfide defect of the strain. Later, we sequenced dsbB::kan5 to find a mini-Tn10-kan insertion after Gln95 of DsbB, leaving the α portion entirely intact. Thus, the partial complementation could have occurred as a consequence of co-expression of the N-terminal fragment of DsbB from the chromosome and β from the plasmid. Importantly, however, other than that described above, we did not observe any difference in phenotype between the dsbB::kan5 strain and the ΔdsbB strain.
Plasmids
The parental plasmid pHK517 (encoding DsbB-His6-c-Myc under lac promoter control) was constructed by amplifying a fragment from pWM76 (Bader et al., 1999) using primers DSBH9 and DSBH11, and cloning it in between the KpnI and XbaI sites of pAM238 (Spcr, pSC101 ori). All the DsbB derivatives described in this work lack all the non-essential cysteines of wild-type DsbB due to the C8A and C49V mutations derived from pWM76.
pHK521 (encoding DsbB α) was constructed by amplifying the α region from pHK517 using primers DSBH9 and DSBH23, digesting the product with KpnI and XbaI, and cloning it into a pACYC184-based lacUV5 promoter vector, pLAC-YC. To fuse a FLAG3 epitope to the N-terminus of α, pHK521 was used as a template for PCR amplification with DSBH31 and DSBH32. The product was digested at BamHI sites in the primers and self-ligated, yielding pHK538 encoding FLAG3-α. To construct pHK522 (encoding β-His6-c-Myc under lac promoter control), a β-His6-c-Myc region was amplified from pHK517 with primers DSBH11 and DSBH22, and cloned into the KpnI and XbaI sites of pAM238. The same cloning strategy as for pHK538 was used to fuse the FLAG3 epitope to DsbB-His6-c-Myc to construct pHK537 encoding FLAG3-DsbB-His6-c-Myc. Using this double-tagged DsbB as a calibration standard, we determined that the expression level of α is ∼10-fold higher than that of β in a strain co-transformed with pHK538 and pHK522, probably due to the differences in the copy number and the promoter employed between the two constructs. To avoid such imbalance in the expression level of α and β, we constructed pHK543 (encoding both FLAG3-α and β-His6-c-Myc under its lac promoter) by amplifying the FLAG3-α region from pHK538 using primers DSBH56 and DSBH57, digesting the product with EcoRI and SacI, and cloning it into pHK522 between the lac promoter and the β-His6-c-Myc region. The operon fusion plasmid pHK543 expressed similar amounts of FLAG3-α and β-His6-c-Myc.
To compare the expression levels of these DsbB derivatives with that of wild-type DsbB from the chromosome, a FLAG3-DsbB region was amplified from pHK537 using primers DSBH15 and DSBH56, and cloned into the KpnI and XbaI sites of pAM238, yielding pHK552. Usage of an antibody recognizing the C-terminal 14 amino acid sequence of DsbB (Kadokura et al., 2000) allowed us to compare the expression level of FLAG3-DsbB from pHK552 with that of wild-type DsbB from chromosome. Subsequent utilization of FLAG3-DsbB and FLAG3-DsbB-His6-c-Myc as calibration standards permitted us to determine that the expression levels of FLAG3-α and β-His6-c-Myc from pHK543 were ∼10-fold lower that of DsbB from chromosome.
Plasmid pAM238 and its derivative pHK543 do not have a specific signal after the cloning site that acts to terminate transcription from the lac promoter. Since introduction of a strong terminator often stabilizes the transcript and enhances the production of the gene product, we reasoned that we might be able to elevate the expression level of FLAG3-α and β-His6-c-Myc by inserting a strong terminator, rrnBT1T2, downstream of the coding sequences for these polypeptides. Thus the rrnBT1T2 terminator was amplified from pBAD18 with primers DSBH58 and DSBH59, digested with PstI and HindIII, and cloned into pHK543, yielding pHK553. FLAG3-α and β-His6-c-Myc were expressed from pHK553 at levels about one-third as high as that of wild-type DsbB from the chromosome and complemented the disulfide defect of a ΔdsbB strain well. The empty vector for pHK553, pHK544, was constructed by inserting the same rrnBT1T2 terminator into the PstI and HindIII sites of pAM238.
To construct pHK555 (encoding FLAG3-α), pHK553 was digested with XbaI to remove β-His6-c-Myc, and then self-ligated. pHK556 (β-His6-c-Myc) was constructed in a similar way to pHK555 using KpnI to remove FLAG3-α from pHK553.
When indicated, cysteine to alanine substitution mutations were introduced into the constructs using the QuickChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). The mutagenic primers used were DSBH46 and DSBH47 (for C41A), DSBH48 and DSBH49 (C44A), DSBH50 and DSBH51 (C104A), and DSBH52 and DSBH53 (C130A). All the constructions were verified by DNA sequencing.
In the Results and Discussion, we call the FLAG3-α and β-His6-c-Myc polypeptides simply α and β, respectively.
Media and growth conditions
Cells were grown aerobically at 37°C with shaking in NZ medium or M63 minimal glucose medium supplemented with the appropriate antibiotics. All the DsbB variants analyzed in this work were placed under the lac promoter–operator of a low-copy plasmid (pSC101 ori) and expressed in strains lacking lacI in the M63 minimal glucose medium. Presumably, their expression levels were determined through the balance of catabolite repression (by the presence of glucose in medium) and derepression (caused by the absence of the lacI gene in these strains).
Redox state analysis
To analyze the formation of mixed disulfides and to determine the in vivo redox states of proteins, the free cysteine residues of proteins were acid trapped and alkylated with the high molecular mass reagent AMS (Molecular Probes, Eugene, OR) as described (Kadokura et al., 2000). Alkylated samples were separated by SDS–PAGE, and stained with respective antibodies, essentially as indicated previously. Antibodies used were anti-Myc (A-14) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), anti-FLAG (M2) (Sigma, St Louis, MO), anti-β-lactamase (5 Prime 3 Prime, Inc., Boulder, CO) and anti-DsbA (Bardwell et al., 1993).
Acknowledgments
Acknowledgements
We thank Barry L.Wanner for plasmids, and Julia Romano for advice on protein tagging. We are grateful to the lab members, especially Hongping Tian, Daniel Ritz, Federico Katzen and Ron Ortenberg for helpful discussions throughout the work. This work was supported by NIH grant (GM41883) to J.B. J.B. is an American Cancer Society Research Professor.
References
- Bader M., Muse,W., Ballou,D.P., Gassner,C. and Bardwell,J.C. (1999) Oxidative protein folding is driven by the electron transport system. Cell, 98, 217–227. [DOI] [PubMed] [Google Scholar]
- Bader M.W., Xie,T., Yu,C.A. and Bardwell,J.C. (2000) Disulfide bonds are generated by quinone reduction. J. Biol. Chem., 275, 26082–26088. [DOI] [PubMed] [Google Scholar]
- Bardwell J.C., McGovern,K. and Beckwith,J. (1991) Identification of a protein required for disulfide bond formation in vivo. Cell, 67, 581–589. [DOI] [PubMed] [Google Scholar]
- Bardwell J.C., Lee,J.O., Jander,G., Martin,N., Belin,D. and Beckwith,J. (1993) A pathway for disulfide bond formation in vivo. Proc. Natl Acad. Sci. USA, 90, 1038–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkower C., Taglicht,D. and Michaelis,S. (1996) Functional and physical interactions between partial molecules of STE6, a yeast ATP-binding cassette protein. J. Biol. Chem., 271, 22983–22989. [DOI] [PubMed] [Google Scholar]
- Bibi E. and Kaback,H.R. (1990) In vivo expression of the lacY gene in two segments leads to functional lac permease. Proc. Natl Acad. Sci. USA, 87, 4325–4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko K.A. and Wanner,B.L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl Acad. Sci. USA, 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debarbieux L. and Beckwith,J. (1999) Electron avenue: pathways of disulfide bond formation and isomerization. Cell, 99, 117–119. [DOI] [PubMed] [Google Scholar]
- Frand A.R. and Kaiser,C.A. (1999) Ero1p oxidizes protein disulfide isomerase in a pathway for disulfide bond formation in the endoplasmic reticulum. Mol. Cell, 4, 469–477. [DOI] [PubMed] [Google Scholar]
- Frand A.R. and Kaiser,C.A. (2000) Two pairs of conserved cysteines are required for the oxidative activity of Ero1p in protein disulfide bond formation in the endoplasmic reticulum. Mol. Biol. Cell, 11, 2833–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil D. and Bouche,J.P. (1991) ColE1-type vectors with fully repressible replication. Gene, 105, 17–22. [DOI] [PubMed] [Google Scholar]
- Gilbert H.F. (1997) Protein disulfide isomerase and assisted protein folding. J. Biol. Chem., 272, 29399–29402. [DOI] [PubMed] [Google Scholar]
- Grauschopf U., Winther,J.R., Korber,P., Zander,T., Dallinger,P. and Bardwell,J.C. (1995) Why is DsbA such an oxidizing disulfide catalyst? Cell, 83, 947–955. [DOI] [PubMed] [Google Scholar]
- Guilhot C., Jander,G., Martin,N.L. and Beckwith,J. (1995) Evidence that the pathway of disulfide bond formation in Escherichia coli involves interactions between the cysteines of DsbB and DsbA. Proc. Natl Acad. Sci. USA, 92, 9895–9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman L.M., Belin,D., Carson,M.J. and Beckwith,J. (1995) Tight regulation, modulation and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol., 177, 4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jander G., Martin,N.L. and Beckwith,J. (1994) Two cysteines in each periplasmic domain of the membrane protein DsbB are required for its function in protein disulfide bond formation. EMBO J., 13, 5121–5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadokura H., Bader,M., Tian,H., Bardwell,J.C.A. and Beckwith,J. (2000) Roles of a conserved arginine residue of DsbB in linking protein disulfide-bond-formation pathway to the respiratory chain of Escherichia coli. Proc. Natl Acad. Sci. USA, 97, 10884–10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzen F. and Beckwith,J. (2000) Transmembrane electron transfer by the membrane protein DsbD occurs via a disulfide cascade. Cell, 103, 769–779. [DOI] [PubMed] [Google Scholar]
- Kishigami S. and Ito,K. (1996) Roles of cysteine residues of DsbB in its activity to reoxidize DsbA, the protein disulphide bond catalyst of Escherichia coli. Genes Cells, 1, 201–208. [DOI] [PubMed] [Google Scholar]
- Kishigami S., Akiyama,Y. and Ito,K. (1995) Redox states of DsbA in the periplasm of Escherichia coli. FEBS Lett., 364, 55–58. [DOI] [PubMed] [Google Scholar]
- Kobayashi T. and Ito,K. (1999) Respiratory chain strongly oxidizes the CXXC motif of DsbB in the Escherichia coli disulfide bond formation pathway. EMBO J., 18, 1192–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T., Kishigami,S., Sone,M., Inokuchi,H., Mogi,T. and Ito,K. (1997) Respiratory chain is required to maintain oxidized states of the DsbA–DsbB disulfide bond formation system in aerobically growing Escherichia coli cells. Proc. Natl Acad. Sci. USA, 94, 11857–11862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T., Takahashi,Y. and Ito,K. (2001) Identification of a segment of DsbB essential for its respiration-coupled oxidation. Mol. Microbiol., 39, 158–165. [DOI] [PubMed] [Google Scholar]
- Martin J.L., Bardwell,J.C. and Kuriyan,J. (1993) Crystal structure of the DsbA protein required for disulphide bond formation in vivo. Nature, 365, 464–468. [DOI] [PubMed] [Google Scholar]
- McCarthy A.A., Haebel,P.W., Torronen,A., Rybin,V., Baker,E.N. and Metcalf,P. (2000) Crystal structure of the protein disulfide bond isomerase, DsbC, from Escherichia coli. Nature Struct. Biol., 7, 196–199. [DOI] [PubMed] [Google Scholar]
- Missiakas D., Georgopoulos,C. and Raina,S. (1993) Identification and characterization of the Escherichia coli gene dsbB, whose product is involved in the formation of disulfide bonds in vivo. Proc. Natl Acad. Sci. USA, 90, 7084–7088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogliano J., Lynch,A.S., Belin,D., Lin,E.C. and Beckwith,J. (1997) Regulation of Escherichia coli cell envelope proteins involved in protein folding and degradation by the Cpx two-component system. Genes Dev., 11, 1169–1182. [DOI] [PubMed] [Google Scholar]
- Popot J.L. and Engelman,D.M. (2000) Helical membrane protein folding, stability and evolution. Annu. Rev. Biochem., 69, 881–922. [DOI] [PubMed] [Google Scholar]
- Rietsch A., Bessette,P., Georgiou,G. and Beckwith,J. (1997) Reduction of the periplasmic disulfide bond isomerase, DsbC, occurs by passage of electrons from cytoplasmic thioredoxin. J. Bacteriol., 179, 6602–6608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz D., Lim,J., Reynolds,C.M., Poole,L.B. and Beckwith,J. (2001) Conversion of a peroxiredoxin into a disulfide reductase by a triplet repeat expansion. Science, 294, 158–160. [DOI] [PubMed] [Google Scholar]
- Stewart E.J., Katzen,F. and Beckwith,J. (1999) Six conserved cysteines of the membrane protein DsbD are required for the transfer of electrons from the cytoplasm to the periplasm of Escherichia coli. EMBO J., 18, 5963–5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson B.M., Esnault,Y., Craven,R.A., Skiba,F., Fieschi,J., K’Epes,F. and Stirling,C.J. (1997) Molecular architecture of the ER translocase probed by chemical crosslinking of Sss1p to complementary fragments of Sec61p. EMBO J., 16, 4549–4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T., Yu,L., Bader,M.W., Bardwell,J.C.A. and Yu,C.A. (2002) Identification of the ubiquinone-binding domain in the disulfide catalyst disulfide bond protein B. J. Biol. Chem., 277, 1649–1652. [DOI] [PubMed] [Google Scholar]
- Zapun A., Bardwell,J.C. and Creighton,T.E. (1993) The reactive and destabilizing disulfide bond of DsbA, a protein required for protein disulfide bond formation in vivo. Biochemistry, 32, 5083–5092. [DOI] [PubMed] [Google Scholar]