

Abstract
A Sin3-interacting domain (SID) originally described in Mad proteins is necessary for both transcriptional repression and growth suppression by these transcription factors. We recently reported that a structurally and functionally related Mad1-like SID is also present in five Sp1-like repressor proteins (TIEG1, TIEG2, BTEB1, BTEB3 and BTEB4), demonstrating that SID–mSin3A interactions have a wider functional impact on transcriptional repression. SID–mSin3A interaction is necessary for the anti-proliferative function of Mad, TIEG and BTEB proteins. It remains to be established, however, whether the SID–mSin3A interaction is constitutive or regulated. Here, we describe that the Mad1-like SID domain of the Sp1-like repressor TIEG2 is inhibited by the epidermal growth factor (EGF)–Ras–MEK1–ERK2 signaling pathway, via phosphorylation of four serine/threonine sites adjacent to the SID. This phenomenon disrupts the SID–mSin3A interaction and thereby inhibits TIEG2’s repression activity. Thus, these results show for the first time that the repression of a SID-containing protein is regulated by signaling rather than functioning in a constitutive manner, extending our understanding of how the function of SID-containing repressors may be controlled.
Keywords: EGF/mSin3A/repression/SID/TIEG2
Introduction
A large number of studies have firmly established a key role for transcriptional repression in most cellular functions that regulate embryogenesis, maintain homeostasis and participate in mechanisms of disease, including cancer. An emerging concept in this field is that short peptide motifs are a common mechanism mediating repressor–corepressor interactions. For example, Hairy and Runt transcription factors function by recruiting the corepressor Groucho via the WRPW or VWRPY motifs (Chen and Courey, 2000). NCoR and SMRT interact with nuclear hormone receptors via the I/LXXI/VI motif, and ligand binding regulates this interaction (Minucci and Pelicci, 1999; Nagy et al., 1999). Also, a consensus motif, PVDLS/T, is required for the interaction of several repressor proteins with mCtBP2 (Turner and Crossley, 2001). Since repression by proteins containing these domains supports different types of cellular functions, the mechanisms of repressor–corepressor interactions are critical for the regulation of homeostasis. Therefore, significant efforts from many laboratories are focused on defining the repertoire of short motifs participating in repression. The results of these experiments will no doubt extend our understanding of gene silencing mechanisms and their impact on cell biology.
An important example of these repression mechanisms is the interaction between the corepressor mSin3A and a small motif called SID (Sin3-interacting domain) that contains the consensus φZZφφXφAAXXLE/D core sequence (Eilers et al., 1999; Brubaker et al., 2000). The SID was described originally in the Mad family of basic helix–loop–helix transcription factors and was also shown to be necessary for both their repressor and growth suppressive activities (Schreiber-Agus and DePinho, 1998). Interestingly, we have recently reported that five Sp1-like transcriptional repressors (TIEGs and BTEBs) also recruit the mSin3A–histone deacetylase complex through a conserved, α-helical motif with the core AA/VXXL sequence that is structurally and functionally similar to the Mad1 SID (Zhang et al., 2001). Other groups have also provided evidence for a wider participation of SID-like domains in transcriptional repression, such as the mammalian Pf1 (Yochum and Ayer, 2001) and the yeast Ume6 (Washburn and Esposito, 2001). Together, these studies have established that the SID represents a small structural motif that mediates interaction of various transcriptional repressors with the corepressor mSin3A. Furthermore, they reveal that SID–mSin3A interactions have been conserved throughout evolution, probably in part because they represent an efficient mechanism of repression selected to the organism’s advantage. Currently, however, whether repression by SID-containing proteins is constitutive or regulated remains to be established and, if regulated, the mechanisms underlying such a phenomenon need to be defined.
By analogy to nuclear receptors, we hypothesize that the repression activity of SID-containing proteins is likely to be regulated by differential association with the corepressors in various biological contexts. Potential mechanisms for this regulation include those that may act at the level of the SID domain itself, the corepressor mSin3A or modifications in the repressor that fall outside of the SID domain, affecting repressor–corepressor interactions. In contrast to nuclear receptors, the SID-containing proteins described thus far do not bind intracellular ligands. Therefore, other mechanisms, such as signaling-induced post-translational modification, at any of the levels mentioned above, are more likely to modulate their interactions with mSin3A. In this study, we have tested these hypotheses by studying the regulation of the SID-mediated transcriptional repression activity of the Sp1-like repressor TIEG2 by the epidermal growth factor (EGF)– Ras–MEK1–ERK2 signaling pathway. The results of these experiments show that the EGF–Ras–MEK1–ERK2 signaling pathway inhibits the repression activity of TIEG2 by mediating the phosphorylation of four sites immediately adjacent to the SID. This signaling-induced relief of the transcriptional repression of the TIEG2 SID results from a disruption of the binding between this domain and mSin3A. Therefore, TIEG2 is the first member of the TIEG/BTEB subgroup of Sp1-like transcriptional repressors shown to be regulated in this manner. Moreover, this is also the first evidence of a SID-containing protein that is regulated by signaling-induced phosphorylation of sites that exist outside of this motif. Thus, both concepts contribute to extend our understanding of the potential mechanisms that can regulate the function of these proteins.
Results
The repression activity of TIEG2 is regulated by EGF signaling
We examined the regulation of the transcriptional repression activity of TIEG2 by proliferative and anti-proliferative signaling pathways. NIH 3T3 cells, responsive to a wide variety of growth factors, were co-transfected with FLAG-tagged TIEG2 and BTE reporter plasmids and treated with either the anti-proliferative transforming growth factor-β (TGF-β) or the mitogenic EGF. Treatment with TGF-β, even at high doses, has no effect on the transcriptional repression activity of TIEG2 (Figure 1A, left panel). In contrast, treatment with EGF resulted in a dose-dependent relief of TIEG2 repression activity and reached saturation at a concentration of 25 ng/ml (Figure 1A, right panel). Similar effects were also observed following treatment of NIH 3T3 cells with other mitogenic factors such as hepatocyte growth factor and fibroblast growth factor, however to a much lesser degree (data not shown). In parallel experiments, Chinese hamster ovary (CHO) cells, which are EGF receptor deficient, were used for the reporter assay, along with a constitutively active form of the EGF receptor, vErbB. Figure 1B (left panel) shows that vErbB co-expression strongly inhibits the repression activity of TIEG2, similarly to EGF in NIH 3T3 cells. In addition, CHO cells were co-transfected with other members of the EGF pathway, such as Ras, MEK1 and ERK2. Figure 1B shows that constitutively active mutants of Ras and MEK1 (caRas and caMEK1) relieve TIEG2 repression, similarly to vErbB (Figure 1B, left panel). In contrast, cotransfection of CHO cells with dominant-negative Ras or ERK2 (dnRas and dnERK2), or treatment with the MEK1 inhibitior PD089059, antagonized the inhibitory effect of vErbB on TIEG2 repression activity (Figure 1B, right panel). Together, these data suggest that the proliferative EGF–Ras–MEK1–ERK2 signaling cascade is both sufficient and required for EGF-induced inhibition of TIEG2 repression activity.

Fig. 1. EGF signaling regulates TIEG2 repression activity. (A) NIH 3T3 cells were transiently transfected with FLAG-tagged TIEG2 along with pBTE reporter plasmids. Cells were treated with increasing amounts of TGF-β (0.1, 1 and 10 ng/ml) or EGF (5, 25 and 25 ng/ml) for 18 and 24 h, respectively. Note that TGF-β does not affect the repression activity of TIEG2, whereas EGF strongly antagonizes TIEG2-mediated repression. (B) CHO cells were transiently transfected with FLAG-tagged TIEG2 along with constitutively activate forms of the EGF receptor (vErbB), Ras (caRas) and MEK1 (caMEK1) or vErbB along with dominant-negative constructs of Ras (dnRas) and ERK2 (dnERK2), as indicated. vErbB-transfected cells were also treated with PD089059 (50 µM) prior to reporter assay. Note that vErbB, caRas and caMEK1 similarly relieve TIEG2-mediated repression. In addition, the inhibition of TIEG2 repression activity in vErbB-transfected cells is reversed by co-expression of dnERK2 and dnRas, as well as treatment with PD089059. Control western blots using cell extracts from transfected NIH 3T3 and CHO cells show that expression of TIEG2 is not changed by the various co-transfections and treatments.
Subsequently, we tested whether the antagonistic effect of EGF signaling changes the phosphorylation status of TIEG2 in vitro and in vivo. For the in vitro experiments, immunoprecipitated ERK2 was incubated with either GST alone or a GST fusion protein carrying full-length TIEG2 in the presence of [32P]ATP. Figure 2A illustrates that activated ERK2 (isolated from EGF-stimulated cells) phosphorylates TIEG2 in vitro (lane 4), but not GST alone (lane 2). However, ERK2 isolated from untreated cells does not phosphorylate TIEG2 (lane 3) or GST (lane 1). Control experiments in Figure 2B show that EGF treatment of NIH 3T3 cells causes ERK2 phosphorylation without changing its expression levels (lane 1 versus 2). Next, CHO cells were co-transfected with vErbB and FLAG-tagged TIEG2 followed by metabolic labeling with [32P]orthophosphate and immunoprecipitation with anti-FLAG antibodies. Figure 2C shows that co-transfection with vErbB results in increased phosphorylation of TIEG2 (lane 3 versus 4). Control experiments show that ERK2 is activated upon vErbB expression (lane 2 versus 4) and that cells transfected with vector alone do not exhibit TIEG2 phosphorylation (lanes 1 and 2). These results show that EGF signaling induces increased TIEG2 phosphorylation in vivo while in vitro assays demonstrate that TIEG2 is phosphorylatable by ERK2, suggesting that this kinase is a candidate to mediate the effects of this cascade. Because ERK2-mediated phosphorylation of transcription factors often changes their subcellular distribution (Cyert, 2001), we performed confocal laser scanning microscopy on CHO cells transfected with FLAG-tagged TIEG2 alone or in combination with caMEK1. Figure 2D demonstrates that FLAG-tagged TIEG2 is localized exclusively to the nucleus (red signal), which is consistent with previous reports (Cook et al., 1999), and that co-expression of caMEK1 does not alter its localization. Controls using an anti-phosphoERK2 antibody show that caMEK1 co-expression induces an increase in the signal of phosphorylated ERK2 in the cell nucleus (green). Overlaying the staining patterns demonstrates the nuclear localization of both proteins (yellow). Non-transfected cells or cells that were not incubated with the primary antibody did not exhibit immunostaining for FLAG-tagged TIEG2 (data not shown). Together, the results from the reporter, phosphorylation and localization experiments described above outline a mechanism of regulation of TIEG2 function whereby its repression activity is antagonized by EGF signaling-mediated phosphorylation via ERK2, which does not change the subcellular localization of this transcription factor.
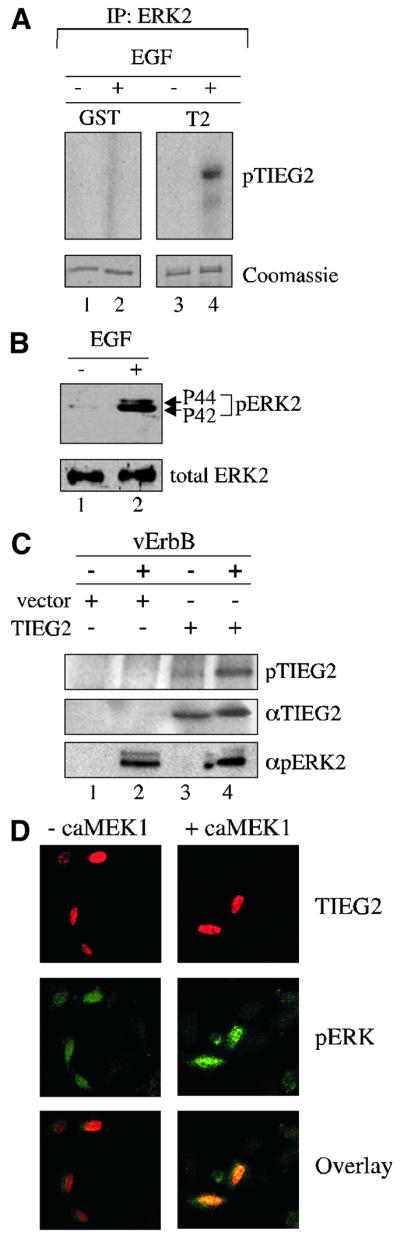
Fig. 2. EGF signaling induces phosphorylation of TIEG2 in vitro and in vivo. (A) Total ERK2 from either EGF-treated (25 ng/ml for 15 min) or untreated NIH 3T3 cell extracts was immunopurified and incubated with GST–TIEG2 or GST alone. Coomassie Blue gel analysis shows comparable amounts of GST proteins used in these assays. EGF-activated immunoprecipitated ERK2 strongly phosphorylates GST–TIEG2 (lane 4) in vitro, whereas ERK2 from untreated cells does not (lane 3). GST alone (lanes 1 and 2) is not phosphorylated. (B) Immunoprecipitated ERK2 used in (A) was subjected to western blot analysis using anti-phosphoERK2 (pERK2) and anti-total ERK2 antibodies. Note that EGF treatment leads to ERK2 phosphorylation without changing its expression. (C) CHO cells were co-transfected with FLAG-tagged full-length TIEG2 or the parental vector along with vErbB, followed by metabolic labeling with [32P]orthophosphate and immunoprecipitation with anti-FLAG antibodies. TIEG2 expression and vErbB-induced ERK2 activation were monitored by immunoblotting using anti-TIEG2 and anti-phosphoERK2 antibodies. [32P]orthophosphate incorporation was detected by audioradiography. Note that co-transfection with vErbB leads to increased phosphorylation of TIEG2 in vivo (lane 4 versus 3). Controls show that ERK2 is activated when vErbB is co-expressed (lanes 2 and 4). TIEG2 expression levels are unchanged by vErbB co-expression (lanes 3 and 4). (D) CHO cells were transfected with FLAG-tagged TIEG2 alone or together with caMEK1 and subjected to immunofluorescence analysis. Note that TIEG2 (red signal) was detected exclusively in the nucleus of FLAG-TIEG2-transfected cells. Note that co-expression of caMEK1 did not alter the subcellular localization of TIEG2. As a control, caMEK1 expression causes increased staining for the phosphorylated form of endogenous ERK2 (green). The overlay of the staining patterns demonstrates nuclear localization of both proteins (yellow).
Phosphorylation of TIEG2 occurs at ERK2 consensus sites adjacent to the Mad1-like SID
In vitro kinase assays using deletion mutants that carry either the N-terminal transcriptional regulatory domain (NTD) or the C-terminal DNA-binding domain (CTD) of TIEG2 were carried out to begin delineating the regions that are phosphorylated by ERK2. Figure 3A shows that recombinant ERK2 phosphorylates TIEG2 NTD (lane 3 versus 4) and to a lesser degree CTD (lane 5 versus 6). GST alone is not phosphorylated (lane 1 versus 2). In addition, activated MAP kinases JNK and p38 fail to phosphorylate the GST–NTD (data not shown). Next, we determined whether the N-terminus of TIEG2 is phosphorylated in vivo. For this purpose, CHO cells were transfected with TIEG2 NTD cloned as a GAL4 DNA-binding domain (DBD) fusion construct (GAL4 NTD) or GAL4 DBD alone, with or without caMEK1, followed by [32P]orthophosphate labeling and anti-GAL4 immunoprecipitations. Figure 3B shows that co-transfection with caMEK1 resulted in a significant increase of TIEG2 NTD phosphorylation (lane 3 versus 4). Similar results were obtained when GAL4 NTD was co-transfected with vErbB (data not shown). Expression of caMEK1 does not phosphorylate GAL4 DBD alone used as control (lane 1 versus 2). Subsequently, we focused on characterizing the effects of EGF–Ras–MEK1–ERK2 signaling on the N-terminal repression domain using GAL4-based reporter assays. GAL4 NTD or DBD alone was co-transfected into CHO cells along with a GAL4 luciferase reporter plasmid and vErbB, caRas or caMEK1. Figure 3C shows that GAL4 NTD represses transcription ∼6.6-fold (0.79 ± 0.1 GAL4 DBD versus 0.12 ± 0.04 GAL4 NTD). Co-transfection with vErbB, caRas or caMEK1 reduced GAL4 NTD-mediated repression activity to ∼1.3-fold (0.59 ± 0.1, 0.51 ± 0.09 and 0.51 ± 0.1, respectively). On the other hand, the MEK1 inhibitor PD089059 reverses vErbB inhibition of TIEG2 repression activity (0.20 ± 0.04). In contrast to the potent effect of the EGF–Ras– MEK1–ERK2 pathway on TIEG2 NTD repression activity, anisomycin, an activator of JNK and p38, only slightly affects the repression activity (0.29 ± 0.05). Control gel-shift assays using the GAL4 site show that vErbB and caMEK1 alter neither the expression nor the DNA-binding activity of these GAL4 constructs (data not shown). In addition, we performed gel-shift assays to determine whether the phosphorylation of the C-terminus affects TIEG2 DNA binding. Figure 3D shows that the rERK2-phosphorylated TIEG2 C-terminus retains its ability to bind a previously characterized TIEG2-binding site (lane 4 versus 5) (Cook et al., 1998). Taken together, these data suggest that TIEG2 repression activity is inhibited by the EGF–Ras–MEK1–ERK2 pathway through regulation of the N-terminal domain.

Fig. 3. EGF signaling inhibits TIEG2 N-terminal repression activity. (A) Equal amounts of GST fusion proteins carrying the N-terminal (GST–NTD) or the C-terminal (GST–CTD) domains of TIEG2 or GST alone were incubated with recombinant ERK2 (rERK2) (20 ng/µl) and [γ-32P]ATP as described in Materials and methods. Phosphorylation of the GST proteins was visualized by autoradiography. Note that rERK2 strongly phosphorylates GST–NTD (lane 3 versus 4) and to a lesser degree GST–CTD (lane 5 versus 6). GST alone is not phosphorylated (lane 1 versus 2). (B) CHO cells were transfected with TIEG2 NTD expressed as a GAL4 DBD fusion protein (GAL4 NTD) or GAL4 DBD alone (GAL4), with or without caMEK1. At 24 h post-transfection, cells were metabolically labeled with [32P]orthophosphate and subjected to anti-GAL4 immunoprecipitation. Phosphorylation of TIEG2 NTD was detected by autoradiography. Note that caMEK1 expression leads to increased phosphorylation of TIEG2 NTD (lane 3 versus 4). GAL4 DBD alone is not phosphorylated (lane 1 versus 2). To control for TIEG2 NTD expression, transfected CHO cells were labeled with [35S]methionine and analyzed by anti-GAL4 immunoprecipitation as described above. Note that TIEG2 NTD is expressed at comparable levels. (C) GAL4-based reporter assays were performed in CHO cells transfected with GAL4 DBD alone or GAL4 NTD along with a GAL4 luciferase reporter construct. Note that TIEG2 NTD repression (0.12 ± 0.04 versus 0.79 ± 0.1 GAL4 DBD alone) is strongly antagonized by co-expression of caMEK1 (0.51 ± 0.1), caRas (0.51 ± 0.09) or vErbB (0.59 ± 0.1). The MEK1 inhibitor PD089059 reverses the inhibition of TIEG2 NTD by vErbB (0.20 ± 0.04). Also note that anisomycin, a JNK and p38 activator, slightly reduces the NTD-mediated repression (0.29 ± 0.05). (D) GST–CTD and GST alone were subjected to rERK2 phosphorylation and used in a gel-shift assay with a probe containing a GC-rich binding site for TIEG2. The specific complex that forms between the GST–CTD and probe is indicated on the right (asterisk). Note that the DNA-binding activity of TIEG2 CTD is unaffected by rERK2 treatment (lane 4 versus 5). GST alone does not bind the GC probe (lanes 2 and 3); lane 1 shows the mobility of the GC-rich probe in the absence of binding proteins.
Sequence analysis shown in Figure 4A reveals the presence of four putative MAPK phosphorylation sites (T56, S94, S107 and S149) in the region immediately adjacent to the N-terminal SID domain of TIEG2 that conform to the ERK2 consensus target sequence of P-X-S/T-P (Jacobs et al., 1999). We created alanine substitution mutations of the consensus sites in the context of the GAL4 NTD construct. For the remainder of the paper, we will refer to these mutations as M1, M2, M3 and M4, corresponding to residues T56, S94, S107 and S149 mutated to alanine, respectively, and M5, corresponding to the mutant carrying alanine mutations of all four serine/threonine sites (Figure 4B). We then performed a GAL4 reporter assay in the absence or presence of caMEK1. Figure 4B shows that the repression activity of both the wild-type and mutant GAL4 NTD constructs is comparable in the absence of caMEK1 (lanes 2–7). Co-transfection with caMEK1 antagonizes the repression activity of wild-type NTD. The repression activity of the NTD mutants is also inhibited by caMEK1, although to a lesser degree than wild-type NTD. The M5 mutation leads to the most significant reduction of caMEK1-induced inhibition of the TIEG2 NTD repression (lane 7). Expression of caMEK1 does not modify the activity of the GAL4 DBD control (lane 1). To determine the effect of the alanine mutations on NTD phosphorylation, we performed in vivo phosphorylation studies combined with anti-GAL4 immunoprecipitation. Figure 4C shows that co-transfection with caMEK1 resulted in increased phosphorylation of wild-type NTD (lane 1 versus 2). The phosphorylation of TIEG2 NTD was slightly reduced by single mutations of the ERK2 sites (lanes 3, 4, 5 and 6). In contrast, the caMEK1-induced phosphorylation was abolished in the M5 mutant (lane 7). From these studies, we conclude that the four ERK2–MAPK consensus sites adjacent to the TIEG2 SID are targets of the EGF–Ras–MEK1–ERK2 pathway in vivo and that all four sites are needed for the antagonistic effect of this cascade on TIEG2 repression activity.

Fig. 4. ERK2 phosphorylation sites are adjacent to the TIEG2 SID. (A) A schematic representation of TIEG2 protein structure shows the three zinc finger motifs that bind DNA and repression domains SID, R2 and R3 within the NTD. Sequence analysis of the linker region found between SID and R2 reveals four putative MAP kinase phosphorylation sites (PLT56P, PQS94P, PVS107P and PSS149P). An additional site is present C-terminal to the zinc finger domains. (B) Alanine substitution mutants of the ERK2 sites were generated. GAL4 constructs encoding wild-type NTD (NTD WT), NTD with single point mutations (NTD M1–M4) or NTD with combined point mutations (NTD M5) were co-transfected into CHO cells along with the GAL4 reporter and caMEK1 as indicated. Note that co-transfection with caMEK1 antagonizes the repression activity of the wild-type NTD (lane 2) and to a lesser degree that of the GAL4 NTD mutants M1–M4 (lanes 3–6). The M5 mutant leads to the greatest reduction of MEK1-induced inhibition of the transcriptional repression activity. (C) As indicated, CHO cells were transfected with GAL4 NTD wild-type or mutant constructs along with caMEK1. Cells were then metabolically labeled with [32P]orthophosphate and anti-GAL4 immunoprecipitations were performed followed by audioradiography. Note that wild-type NTD phosphorylation is increased when caMEK1 is co-expressed (lanes 1 versus 2). CaMEK1-mediated phosphorylation of NTD M1–M4 (lanes 3, 4, 5 and 6, respectively) is reduced. NTD M5 abolishes caMEK1-induced phosphorylation (lane 7). Expression of the constructs was monitored by anti-TIEG2 western blots of cell lysates and shows that all constructs are expressed at comparable levels.
EGF-induced phosphorylation of TIEG2 disrupts its binding to mSin3A
Since the TIEG2 NTD is phosphorylated by ERK2 at sites adjacent to the SID, we hypothesized that the EGF–Ras–MEK1–ERK2 pathway may alter the activity of the SID rather than other repression domains. To test this hypothesis, we constructed various deletion mutants of the N-terminus and performed GAL4-based assays. Figure 5A shows a physical diagram of these deletion mutants (D1–D3). D1 lacks the SID but contains the four phosphorylation sites and the previously characterized repression domains R2 and R3. D2 contains only the SID. D3 contains the SID and adjacent ERK2 sites. Figure 5A shows that SID-containing constructs (D2 and D3; lanes 4 and 5, respectively) display a repression activity that is similar to that of the entire TIEG2 NTD (lane 2), whereas the SID minus D1 mutant (lane 3) displays a less potent repression activity. This result is identical to previously described data from our laboratory (Cook et al., 1999). Interestingly, caMEK1 antagonizes the repression activity of D3 (lane 5), whereas the activity of D1 was only slightly relieved (lane 3). In addition, D2 repression activity is unaffected by caMEK1. These data suggest that the repression activity associated with the SID is the main target of EGF–Ras–MEK1–ERK2 signaling. Thus, we investigated the role of these ERK2 sites in the MEK1-mediated inhibition of the SID repression activity by mutating them in the D3 construct (M1–M5 mutants). Figure 5B shows that, in the absence of caMEK1, all mutant GAL4 D3 constructs retain the repression activity as compared with wild-type D3 (lane 2 versus 3–7). The caMEK1-mediated inhibition of D3 repression is partially blocked by single point mutations of the phosphorylation sites (lanes 3–6), and mutation of all four phosphorylation sites nearly abolishes caMEK1-mediated inhibition of GAL4 D3 repression activity (lane 7), a result similar to what was observed in Figure 4B for the entire N-terminus. Together, these results indicate that although the ERK2 phosphorylation sites are necessary for the modulation of the SID repression activity, there is not a predominant site that single handedly is responsible for this phenomenon.

Fig. 5. EGF signaling targets SID repression activity. (A) Deletions of TIEG2 NTD expressing D1, D2 and D3 were cloned as GAL4 DBD fusion constructs and co-transfected into CHO cells in the absence or presence of caMEK1, along with the GAL4 luciferase reporter plasmid. Note that co-expression of caMEK1 strongly antagonizes the repression activity of NTD (lane 2) and D3 (lane 5), but not D2 (lane 4). Also note that D1 repression activity is slightly antagonized by caMEK1 co-transfection (lane 5). (B) Mutations M1–M5 were generated in the context of the GAL4 D3 construct, and GAL4-based reporter assays were performed. Note that caMEK1 co-expression inhibits the repression activity of wild-type D3 (lane 2) and that mutations M1–M4 partially block caMEK1 inhibition of SID repression activity (lanes 3–6). CaMEK1-mediated inhibition is almost abolished in D3 M5 (lane 7).
We have shown previously that the repression activity of TIEG2 SID requires interaction with mSin3A (Zhang et al., 2001). It is likely that the EGF–Ras–MEK1–ERK2 pathway regulates TIEG2 function by modulating the SID–mSin3A interaction. To test this hypothesis, we analyzed the effect of this pathway on the interaction of mSin3A with both full-length TIEG2 and the SID by immunoprecipitation assays combined with western blot analyses. Baseline experiments shown in Figure 6A illustrate the strong binding of endogenous mSin3A to TIEG2 (lane 1). In contrast, co-expression of either vErbB or caMEK1 markedly reduces the interaction of TIEG2 with mSin3A (lanes 2 and 3). Moreover, the vErbB-mediated disruption of the TIEG2–mSin3A interaction is reversed by treatment with the MEK1 inhibitor PD089059 (lane 4). We next studied whether the ERK2 sites adjacent to the SID are required for the interaction with mSin3A using the same deletion constructs as described in Figure 5A. Figure 6B illustrates that, in the absence of caMEK1, mSin3A co-immunoprecipitates with NTD, D2 and D3 (SID-containing constructs; lanes 2, 6 and 8, respectively) but not with D1 (SID minus construct; lane 4) or GAL4 DBD alone (lane 1). Co-expression of caMEK1, however, significantly reduces mSin3A binding by both the NTD (lane 2 versus 3) and D3 (lane 8 versus 9). On the other hand, mSin3A binding by D2, which has the SID but lacks the ERK2 sites, is unaffected (lane 6 versus 7). In addition, immunoprecipitations were performed using the GAL4 NTD and D3 constructs harboring the M5 mutations. Figure 6C shows that mutation of all ERK2 sites results in the loss of the ability of caMEK1 to regulate mSin3A binding (NTD M5, lane 3 versus 4; and D1 M5, 1 versus 2). These experiments reveal a mechanism by which the EGFR–Ras–MEK1–ERK2 signaling pathway antagonizes repression activity of TIEG2 by disrupting the SID–mSin3A interaction. This phenomenon involves the phosphorylation of four ERK2 sites located outside of, yet adjacent to, the SID.

Fig. 6. EGF signaling disrupts SID–mSin3A interaction. (A) CHO cells were transiently transfected with FLAG-tagged full-length TIEG2 along with vErbB or caMEK1 as indicated. Anti-FLAG immunoprecipitations were performed, and TIEG2 and mSin3A were detected by western blotting. Note that TIEG2 strongly binds endogenous mSin3A (lane 1) while vErbB or caMEK1 co-expression significantly reduces the binding of mSin3A by TIEG2 (lanes 2 and 3, respectively). The MEK1 inhibtor PD089059 reverses the vErbB-mediated reduction of mSin3A binding by TIEG2 (lane 4). Anti-TIEG2 western blot analysis shows comparable expression of TIEG2. (B) CHO cells were transfected with GAL4 NTD, NTD deletion mutants (see Figure 5) or GAL4 DBD alone. Anti-GAL4 immunoprecipitations were performed and mSin3A co-immunoprecipitation was examined by western blotting. Note that mSin3A co-immunoprecipitates with GAL4 NTD (lane 2) and deletion constructs D2 and D3 (lanes 6 and 8, respectively), but not with D1 (lane 4). CaMEK1 co-expression greatly reduces the binding of mSin3A to NTD (lane 3) and D3 (lane 9), but not to D2 (lane 7). Controls show that GAL4 DBD alone (lane 1) does not co-immunoprecipitate with mSin3A. (C) CHO cells were transfected with GAL4 NTD or D3 constructs carrying the M5 mutations. Note that mSin3A co-immunoprecipitation with NTD M5 (lanes 3 and 4) and D3 M5 (lanes 1 and 2) is not changed by co-expression of caMEK1. Western blot analysis shows that all GAL4 constructs exhibit comparable expression levels.
Discussion
The current study has investigated the potential mechanisms underlying the regulation of the transcriptional function of TIEG2, an Sp1-like protein containing a SID motif. These experiments were designed to test the hypothesis that repression by SID-containing proteins can be regulated at the level of SID–mSin3A interaction via post-translational modifications induced by distinct signaling pathways. In the case of TIEG2, we have previously reported that TGF-β up-regulates its mRNA expression while EGF does not exert any regulatory activity on the transcription of this gene (Cook et al., 1998). This TGF-β inducibility is consistent with the idea that TIEG proteins participate in expression pathways triggered by this growth factor, an idea supported by recently published data (Cook and Urrutia, 2000). In contrast, as shown in the current study, TGF-β does not modulate the transcriptional repression activity of TIEG2 (Figure 1A). It is noteworthy, however, that both EGF and the activated EGF receptor oncogene vErbB are efficient inhibitors of TIEG2 repression activity. Similarly, activated members of the EGF signaling pathway, such as Ras and MEK1, also inhibit this function (Figure 1B). Moreover, dominant-negative Ras and ERK2 as well as the MEK1 inhibitor PD089059 reverse the vErbB-mediated inhibition of TIEG2 repressor activity. These results are consistent with in vitro experiments showing that TIEG2 is phosphorylatable by active, immunoprecipitated ERK2, and in vivo assays showing that TIEG2 phosphorylation is induced by vErbB co-expression (Figure 2). Nuclear–cytoplasmic shuttling and regulation of the DNA-binding affinity of transcription factor are common modes of MAPK-controlled gene expression (Karin and Hunter, 1995; Black et al., 2001; Cyert, 2001; Yue and Mulder, 2001). Thus, we investigated whether the inhibition of TIEG2-mediated repression occurs via these mechanisms. However, the experiments in Figures 2D and 3D show that members of the EGF–Ras–MEK1–ERK2 pathway change neither the subcellular localization nor the DNA-binding activity of this protein. Together, the results discussed thus far implicate the EGF–Ras–MEK1–ERK2 pathway in the regulation of TIEG2, probably via the SID-mediated transcriptional repression activity.
Deletion mutagenesis and reporter assays reveal that the repression activity of the SID is the main target of EGF signaling inhibition of TIEG2 (Figure 5). This occurs through phosphorylation of four ERK2 sites adjacent to the SID, and mutations of all these sites largely relieve MEK1-induced inhibition of SID repression activity (Figures 4 and 5). Furthermore, a construct containing the SID alone, without ERK2 sites, is sufficient to repress transcription, although this activity is unresponsive to MEK1, suggesting that sites outside of this domain have a regulatory function on SID–mSin3A interactions (Figure 5). Thus, we examined whether EGF signaling inhibits TIEG2 by this mechanism. Indeed, Figure 6A shows that the EGF signaling cascade reduces the binding of TIEG2 to endogenous mSin3A. In addition, similarly to the repression activity, the SID–mSin3A interaction is disrupted by overexpressing caMEK1. This effect appears to be dependent on the presence of the adjacent ERK2 sites, since MEK1 loses its effect when the four consensus sites are mutated or deleted (Figure 6B and C). However, deletion of the SID leads to only a moderate decrease in the TIEG2 N-terminus repression activity, due to the presence of other repression domains. In contrast, caMEK1 overexpression causes a much greater relief of repression. This suggests that the EGF–Ras–MEK1– ERK2 pathway may have a second function in modulating TIEG2 repression activity, in addition to disrupting mSin3A interaction. We anticipate that the discovery of other corepressors that work outside the SID, together with experiments similar to the ones described here, will contribute further to the understanding of TIEG2 function. Taken together, the results of reporter and binding assays demonstrate that EGF signaling, through phosphorylation of ERK2 sites adjacent to the TIEG2 SID, antagonizes TIEG2 repression activity by disrupting SID–mSin3A interaction. This regulatory mechanism may involve the secondary structure of the SID in such a way that the mSin3A interaction surface of this domain becomes shielded upon phosphorylation. Alternatively, the SID may remain exposed to mSin3A but adopt a different tertiary structure after phosphorylation of the ERK2 sites. Interestingly, recent NMR studies have shown that, in the case of Mad1, the SID and the PAH2 domain of mSin3A are able to induce extensive folding of each other, undergoing highly hydrophobic interactions (Brubaker et al., 2000). This suggests that the proper exposure of the interacting surfaces is required for binding. Thus, we anticipate that additional data, using stringent biophysical methods to monitor the folding state of the SIDs in response to post-translational modifications of adjacent regions, will differentiate between these mechanisms.
Thus far, four different types of protein have been shown to contain functional SID motifs. Although these motifs are structurally similar, two different types of SID have been described based on affinity. The SID present in members of the Mad family binds mSin3A with higher affinities than the SID from Sp1-like proteins (J.-S.Zhang and R.Urrutia, unpublished data). Two recently characterized proteins, Pf1 and Ume6, also appear to belong to this second group (Washburn and Esposito, 2001; Yochum and Ayer, 2001). Based on these data, it is likely that competition for mSin3A serves as a mechanism for regulating the function of these proteins. For instance, high affinity SIDs (e.g. Mad1) may be able to displace low affinity domains (TIEG2) under certain physiological conditions, though evidence supporting this idea is lacking. On the other hand, an alternative form of regulation could occur by the mechanism shown in this study, namely differential phosphorylation of SID-containing proteins that changes the affinity for mSin3A binding. According to this mechanism, phosphorylation by distinct signaling cascades selectively may activate, inactivate or not have any effect on the function of different SID-containing proteins. Phosphorylation has been shown as a mechanism for regulating the repression activity of some members of the Mad/Max/Myc network of transcription factors, though this occurs at the level of DNA binding (Berberich and Cole, 1992; Bousset et al., 1993). For example, the C-terminus of Mad1 contains motifs that are targets of casein kinase II (CKII) phosphorylation, which mediates its DNA binding and repression activities (Barrera-Hernandez et al., 2000). However, it has not been reported whether the phosphorylation status leads to differential binding of corepressors. Interestingly, the reverse effect, namely the regulation of corepressors by phosphorylation, has been reported recently, though not for mSin3A. For example, SMRT phosphorylation by MAPKs is associated with reduced affinity for its target transcription factors (Hong and Privalsky, 2000), whereas CKII phosphorylation of SMRT leads to stabilization of the SMRT–nuclear hormone receptor complex (Zhou et al., 2001). Similarly, CKII phosphorylation of the Groucho/TLE corepressor is involved in a mechanism that promotes the transcriptional repression of Hes1–Groucho/TLE protein complex (Nuthall et al., 2002). However, we found no evidence of mSin3A phosphorylation in response to EGF–Ras–MEK1–ERK2 signaling under the conditions where this pathway phosphorylates TIEG2 and disrupts SID–mSin3A interactions (data not shown), suggesting that different corepressor pathways are regulated differentially. Thus, the data in this report, together with the fact that several SID-containing proteins exhibit putative phosphorylation sites within the N-terminal domain (like TIEG2) as revealed by computer-assisted mapping (NetPhos2, http://www.cbs.dtu.dk/services/NetPhos/; data not shown), suggest that similar mechanisms should be explored for other SID-containing proteins.
In summary, the disruption of the SID–mSin3A interaction, through phosphorylation of adjacent ERK2 sites, reveals the existence of a mechanism by which SID-containing proteins may be regulated by signaling rather than functioning in a constitutive manner. These results demonstrate that, in addition to DNA binding, nuclear localization and modifications in the corepressor themselves, disruption of SID–mSin3A interactions by modification of the SID-containing repressor may be an important mechanism of regulation for these proteins.
Materials and methods
Plasmid construction
Standard molecular biology techniques were used to clone TIEG2 and its various mutants into the pCMV-Tag2 (Stratagene, La Jolla, CA), pGEX (Amersham Pharmacia, Piscataway, NJ) and pM (Clontech, Palo Alto, CA) vectors for expression as FLAG-tagged, GST fusion or GAL4 DBD fusion proteins, respectively. GST fusion constructs include full-length TIEG2 (GST–TIEG2; amino acids 2–512), the C-terminal domain (GST– CTD; amino acids 379–495) and the N-terminal domain (GST–NTD; amino acids 2–371). GAL4 DBD constructs include NTD (amino acids 2–371) and deletion constructs D1 (amino acids 42–371), D2 (amino acids 2–53) and D3 (amino acids 2–150). The GAL4, pBTE and RSV-Renilla reporters have been described previously (Kaczynski et al., 2001; Zhang et al., 2001). Single point mutations of the ERK2 phosphorylation consensus sites adjacent to the SID of TIEG2 were generated using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer’s suggestions. The pFC-MEK1 vector (Clontech), vErbB and RasV12 were used as constitutive active mutants of MEK1, the EGF receptor and Ras, respectively. The dominant-negative constructs of Ras (dnRas) and ERK2 (dnERK2) contained mutations N17 and K52R, respectively. All constructs were verified by sequencing.
Cell culture and reporter assays
CHO and NIH 3T3 cells were obtained from the American Type Culture Collection (ATCC, Rockville, MD) and cultured in Ham’s F12 medium and Dulbecco’s modified minimal essential medium, respectively, supplemented with 5% fetal bovine serum, 5% normal calf serum, 100 U/ml streptomycin and 100 U/ml penicillin (Gibco/Life Technologies, Rockville, MD). CHO or NIH 3T3 cells were transiently transfected using LipofectAMINE™ (Gibco/Life Technologies), as described previously (Kaczynski et al., 2001; Zhang et al., 2001). Briefly, cells were cultured in 24-well tissue culture plates and co-transfected with FLAG-tagged TIEG2 or various GAL4 deletion mutants of the N-terminus and appropriate reporter plasmids. As indicated, cells were co-transfected with vErbB, caRas, dnRas, caMEK1 or dnERK2 or treated with TGF-β (0.1–10 ng/ml), EGF (5–50 ng/ml), PD089059 (50 µM) or anisomycin (5 µg/ml). The parental pCMV-Tag2 vector or GAL4 DBD alone was used as a control for basal transcriptional activity. As a control for transfection efficiency, all conditions included co-transfection with the RSV-Renilla luciferase control plasmid. At 24 h after transfection, luciferase assays were performed with a Turner 20/20 luminometer and the Dual-Luciferase-Reporter Assay System in accordance with the manufacturer’s suggestions (Promega). In GAL4 assays, relative luciferase activity is expressed as 5 × GAL4 luciferase values normalized to Renilla luciferase values ± SD. For BTE reporter assays, the relative luciferase activity ± SD is the ratio of Renilla-normalized pBTE6 to pBTE0 values. All reporter studies were performed in triplicate in at least three independent experiments with similar results.
Phosphorylation assays
The GST fusion proteins used in this assay were generated as described previously (Kaczynski et al., 2001; Zhang et al., 2001). Briefly, expression of GST–TIEG2, GST–NTD or GST–CTD was induced in BL21 cells (Stratagene) by addition of 2 mM isopropyl-β-d-thiogalactopyranoside (Sigma, St Louis, MO). Fusion proteins were purified using glutathione–Sepharose 4B affinity chromatography according to the manufacturer’s suggestions (Amersham Pharmacia). To perform in vitro kinase assays, fusion proteins were incubated with recombinant ERK2 (20 ng/µl; Stratagene) for 30 min at 30°C in a buffer containing 25 mM HEPES pH 7.5, 10 mM magnesium acetate, 50 µM cold ATP and 2 µCi of [γ-32P]ATP. Endogenous ERK2 was immunoprecipitated using anti-total ERK2 antibodies (Santa Cruz, Santa Cruz, CA) from NIH 3T3 cells treated with serum-free medium or medium containing 25 ng/ml EGF (Austral Biologicals, San Ramon, CA) for 15 min, followed by lysis in a buffer containing 20 mM Tris–HCl, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4 and 1 µg/ml leupeptin. Immuno precipitated ERK2 was incubated with GST fusion proteins in a buffer containing 25 mM Tris–HCl pH 7.5, 5 mM β-glycerophosphate, 2 mM dithiothreitol (DTT), 0.1 mM Na3VO4 and 10 mM MgCl2, with 2 µl of 10 µM (cold) ATP, 1.0 µg of GST–TIEG2 and 4 µCi of [γ-32P]ATP for 30 min. The kinase reactions were terminated by addition of SDS–PAGE loading dye. For in vivo phosphorylation studies, CHO cells were transfected with FLAG-tagged TIEG2, various GAL4 constructs or parental vectors along with constitutively active members of the EGF signaling pathway, as indicated. At 24 h after transfection, cells were incubated in serum- and phosphate-free medium for 6–10 h, followed by metabolic labeling with [32P]orthophosphate for 4 h. Immunopre cipitations using anti-GAL4 or anti-FLAG agarose-conjugated antibodies were performed as described below. All samples were resolved by SDS–PAGE, and phosphorylation was detected by autoradiography.
Immunoprecipitation and western blotting
NIH 3T3 cells or CHO cells were treated with various signal transducing agents or transfected with expression vectors as indicated. At 24 h after transfection, cells were harvested and cell extracts were obtained as described previously (Kaczynski et al., 2001; Zhang et al., 2001). Protein concentrations were measured using the BCA method (Pierce, Rockford, IL). For immunoprecipitation, IgG-pre-cleared cell extracts were incubated at 4°C for 3 h with agarose-conjugated anti-GAL4 DBD (Santa Cruz) or anti-FLAG M2 (Sigma) antibodies, and resultant complexes were collected by centrifugation. Where indicated, cells were metabolically labeled with [35S]methionine prior to immunoprecipitation. Western blot analysis was performed on immunoprecipitates or cell extracts using PDVF membranes as described previously (Kaczynski et al., 2001; Zhang et al., 2001). Membranes were probed with the following antibodies: anti-mSin3A (Santa Cruz), anti-TIEG2 (Transduction Laboratories, Lexington, KY), anti-GAL4 DBD (Santa Cruz), anti-ERK2 (Santa Cruz), anti-phosphoERK2 (Santa Cruz) and peroxidase-conjugated secondary antibodies (Sigma). Lumilight (Roche/Boehringer Mannheim) was used for visualization of the immunoreactive proteins as specified by the manufacturer.
Confocal microscopy
CHO cells, grown on chambered coverslips, were transfected with FLAG-tagged TIEG2 with or without caMEK1 using LipofectAMINE™ (Gibco/Life Technologies). At 24 h post-transfection, cells were washed, fixed and blocked as previously described (Gebelein and Urrutia, 2001). Samples were probed with anti-TIEG2 antibodies (Transduction Laboratories) and anti-phosphoERK2 antibodies (Santa Cruz). Phos phorylated ERK2 was detected with a secondary antibody coupled to Alexa 588 (green) and TIEG2 was visualized by a secondary antibody coupled to Alexa 468 (red). Coverslips were mounted on glass slides and cells were observed with a Zeiss LSM-510 confocal laser scanning microscope (Carl Zeiss, Inc., Oberkochen, Germany) using a 63× oil immersion objective with a numerical aperture of 1.25.
Gel-shift assay
Gel-shift assays were performed essentially as described previously (Gebelein and Urrutia, 2001; Kaczynski et al., 2001). Briefly, a double-stranded 5′-ATTCGATCGGGGCGGGGCGAGC-3′ probe was end-labeled with [γ-32P]ATP and incubated with GST and GST–CTD proteins. Samples were resolved on a 4% non-denaturating polyacrylamide gel and analyzed by autoradiography. Where indicated, GST–CTD or GST alone was subjected to treatment with recombinant ERK2 and non-isotope ATP, as described above for in vitro kinase assays, prior to gel-shift analysis.
Acknowledgments
Acknowledgements
We thank Dr M.L.Privalsky (University of California) for the vErbB construct, Dr J.S.Gutkind (National Institutes of Health) for the caRas, dnRas and dnERK2 constructs, Dr M.Wagner for his assistance in performing laser scanning microscopy and Dr Brian Gebelein for critically reviewing the manuscript. R.U. is funded by the National Institute of Health Grants DK52913 and DK56620, the Mayo Cancer Center and the Lustgarten Foundation. V.E. is supported by the Deutsche Krebshilfe.
References
- Barrera-Hernandez G., Cultraro,C.M., Pianetti,S. and Segal,S. (2000) Mad1 function is regulated through elements within the carboxy terminus. Mol. Cell. Biol., 20, 4253–4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berberich S.J. and Cole,M.D. (1992) Casein kinase II inhibits the DNA-binding activity of Max homodimers but not Myc/Max heterodimers. Genes Dev., 6, 166–176. [DOI] [PubMed] [Google Scholar]
- Black A.R., Black,J.D. and Azizkhan-Clifford,J. (2001) Sp1 and kruppel-like factor family of transcription factors in cell growth regulation and cancer. J. Cell. Physiol., 188, 143–160. [DOI] [PubMed] [Google Scholar]
- Bousset K., Henriksson,M., Luscher-Firzlaff,J.M., Litchfield,D.W. and Luscher,B. (1993) Identification of casein kinase II phosphorylation sites in Max: effects on DNA-binding kinetics of Max homo- and Myc/Max heterodimers. Oncogene, 8, 3211–3220. [PubMed] [Google Scholar]
- Brubaker K., Cowley,S.M., Huang,K., Loo,L., Yochum,G.S., Ayer,D.E., Eisenman,R.N. and Radhakrishnan,I. (2000) Solution structure of the interacting domains of the Mad–Sin3 complex: implications for recruitment of a chromatin-modifying complex. Cell, 103, 655–665. [DOI] [PubMed] [Google Scholar]
- Chen G. and Courey,A.J. (2000) Groucho/TLE family proteins and transcriptional repression. Gene, 249, 1–16. [DOI] [PubMed] [Google Scholar]
- Cook T. and Urrutia,R. (2000) TIEG proteins join the Smads as TGF-β-regulated transcription factors that control pancreatic cell growth. Am. J. Physiol. Gastroenterol. Liver Physiol., 278, G513–G521. [DOI] [PubMed] [Google Scholar]
- Cook T., Gebelein,B., Mesa,K., Mladek,A. and Urrutia,R. (1998) Molecular cloning and characterization of TIEG2 reveals a new subfamily of transforming growth factor-β-inducible Sp1-like zinc finger-encoding genes involved in the regulation of cell growth. J. Biol. Chem., 273, 25929–25936. [DOI] [PubMed] [Google Scholar]
- Cook T., Gebelein,B., Belal,M., Mesa,K. and Urrutia,R. (1999) Three conserved transcriptional repressor domains are a defining feature of the TIEG subfamily of Sp1-like zinc finger proteins. J. Biol. Chem., 274, 29500–29504. [DOI] [PubMed] [Google Scholar]
- Cyert M.S. (2001) Regulation of nuclear localization during signaling. J. Biol. Chem., 276, 20805–20808. [DOI] [PubMed] [Google Scholar]
- Eilers A.L., Billin,A.N., Liu,J. and Ayer,D.E. (1999) A 13-amino acid amphipathic α-helix is required for the functional interaction between the transcriptional repressor Mad1 and mSin3A. J. Biol. Chem., 274, 32750–6. [DOI] [PubMed] [Google Scholar]
- Gebelein B. and Urrutia,R. (2001) Sequence-specific transcriptional repression by a full length Kruppel-associated box-zinc finger protein. Mol. Cell. Biol., 21, 928–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S.H. and Privalsky,M.L. (2000) The SMRT corepressor is regulated by a MEK-1 kinase pathway: inhibition of corepressor function is associated with SMRT phosphorylation and nuclear export. Mol. Cell. Biol., 20, 6612–6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs D., Glossip,D., Xing,H., Muslin,A. and Kornfeld,K. (1999) Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK2 MAP kinase. Genes Dev., 13, 163–175. [PMC free article] [PubMed] [Google Scholar]
- Kaczynski J., Zhang,J.-S., Ellenrieder,V., Conley,A., Duenes,T., Kester, H., van der Burg,B. and Urrutia,R. (2001) The Sp1-like protein BTEB3 inhibits transcription via the BTE box by interacting with mSin3A and HDAC-1 co-repressors and competing with Sp1. J. Biol. Chem., 276, 36749–36756. [DOI] [PubMed] [Google Scholar]
- Karin M. and Hunter,T. (1995) Transcriptional control by protein phosphorylation: signal transmission from the cell surface to the nucleus. Curr. Biol., 5, 747–757. [DOI] [PubMed] [Google Scholar]
- Minucci S. and Pelicci,P.G. (1999) Retinoid receptors in health and disease: co-regulators and the chromatin connection. Semin. Cell Dev. Biol., 10, 215–225. [DOI] [PubMed] [Google Scholar]
- Nagy L. et al. (1999) Mechanism of corepressor binding and release from nuclear hormone receptors. Genes Dev., 13, 3209–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuthall H.N., Husain,J., McLarren,K.W. and Stifani,S. (2002) Role for Hes1-induced phosphorylation in Groucho-mediated transcriptional repression. Mol. Cell. Biol., 22, 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber-Agus N. and DePinho,R. (1998) Repression by the Mad(Mx1)–Sin3 complex. BioEssays, 20, 808–818. [DOI] [PubMed] [Google Scholar]
- Turner J. and Crossley,M. (2001) The CtBP family: enigmatic and enzymatic transcriptional co-repressors. BioEssays, 23, 683–690. [DOI] [PubMed] [Google Scholar]
- Washburn B.K. and Esposito,R.E. (2001) Identification of the Sin3-binding site in ume6 defines a two-step process for conversion of Ume6 from a transcriptional repressor to an activator in yeast. Mol. Cell. Biol., 21, 2057–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yochum G.S. and Ayer,D.E. (2001) Pf1, a novel PHD zinc finger protein that links the TLE corepressor to the mSin3A–histone deacetylase complex. Mol. Cell. Biol., 21, 4110–4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue J. and Mulder,K.M. (2001) Transforming growth factor-β signal transduction in epithelial cells. Pharmacol. Ther., 91, 1–34. [DOI] [PubMed] [Google Scholar]
- Zhang J.-S., Moncrieffe,M.C., Kaczynski,J., Ellenrieder,V., Prendergast,F.G. and Urrutia,R. (2001) A conserved α-helical motif mediates the interaction of Sp1-like transcriptional repressors with the corepressor mSin3A. Mol. Cell. Biol., 21, 5041–5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Gross,W., Hong,S.H. and Privalsky,M.L. (2001) The SMRT corepressor is a target of phosphorylation by protein kinase CK2 (casein kinase II). Mol. Cell. Biochem., 220, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]