

Abstract
The yeast Sir2 protein mediates chromatin silencing through an intrinsic NAD-dependent histone deacetylase activity. Sir2 is a conserved protein and was recently shown to regulate lifespan extension both in budding yeast and worms. Here, we show that SIRT1, the human Sir2 homolog, is recruited to the promyelocytic leukemia protein (PML) nuclear bodies of mammalian cells upon overexpression of either PML or oncogenic Ras (Ha-rasV12). SIRT1 binds and deacetylates p53, a component of PML nuclear bodies, and it can repress p53-mediated transactivation. Moreover, we show that SIRT1 and p53 co-localize in nuclear bodies upon PML upregulation. When overexpressed in primary mouse embryo fibroblasts (MEFs), SIRT1 antagonizes PML-induced acetylation of p53 and rescues PML-mediated premature cellular senescence. Taken together, our data establish the SIRT1 deacetylase as a novel negative regulator of p53 function capable of modulating cellular senescence.
Keywords: NAD-dependent deacetylase/p53/PML nuclear bodies/senescence/SIRT1
Introduction
It is becoming increasingly clear that the post-translational modifications of histones, within the context of chromatin, regulate gene expression. It has been proposed that these histone modifications constitute an epigenetic code, which can be interpreted in terms of chromatin structural changes leading to subsequent genome regulation (Strahl and Allis, 2000; Jenuwein and Allis, 2001). An example of such a signal is the acetylation of specific lysine residues in the conserved histone tails, which can be reversed by histone deacetylases (HDACs) (reviewed in Kouzarides, 1999). To date, three classes of HDACs have been discovered in eukaryotes, known as class I and II HDACs, and the silent information regulator 2 (Sir2) family (for reviews see Gray and Ekstrom, 2001; Khochbin et al., 2001).
Sir2 is an evolutionarily conserved protein, present in Archaea, Eubacteria through to humans (Brachmann et al., 1995; Frye, 1999, 2000). Originally discovered in budding yeast, ySir2 is a structural component of silent chromatin and is required for transcriptional silencing of the silent mating-type loci, telomeres and rDNA repeats (reviewed in Guarente, 1999; Gartenberg, 2000). Additionally, yeast Sir2 functions in double-stranded DNA repair, regulation of mitotic and meiotic cell cycle progression, suppression of rDNA homologous recombination, chromosome stability and modulation of lifespan (Brachmann et al., 1995; Cockell and Gasser, 1999; Guarente, 1999; Haber, 1999; Kaeberlein et al., 1999; Lin et al., 2000).
Recent studies have shown that ySir2 and its homologs possess NAD-dependent HDAC activity (Imai et al., 2000; Landry et al., 2000; Smith et al., 2000). Indeed, Sir2-like enzymes catalyze a unique reaction in which NAD cleavage is tightly coupled to histone deacetylation, thus distinguishing them from the previously characterized class I and II HDACs (Tanner et al., 2000; Tanny and Moazed, 2001). The hallmark of the Sir2 family is a conserved globular core domain of ∼250 amino acids, which harbors the catalytic activity and the three-dimensional structure of which has been elucidated (Finnin et al., 2001; Min et al., 2001). Mutations of conserved residues within this region lead to a loss of function in vitro and in vivo (Frye, 1999; Sherman et al., 1999; Tanny et al., 1999; Imai et al., 2000).
So far, little is known concerning the function of Sir2 homologs in organisms other than yeast. A recent study has shown that, like its yeast counterpart, Sir2 modulates lifespan in Caenorhabditis elegans, raising the exciting possibility that these enzymes link longevity, metabolism and chromatin silencing in many eukaryotic organisms (Guarente, 2000; Guarente and Kenyon, 2000; Tissenbaum and Guarente, 2001). As for the seven human homologs of Sir2, named SIRT1–7, no precise function has been described so far (Frye, 1999, 2000). However, a previous study showed that the core region of SIRT2 can function in transcriptional silencing when substituting that of ySir2 (Sherman et al., 1999). The cytoplasmic localization of certain members, as well as the conservation of the Sir2 family in prokaryotes, which on the whole do not possess histones, strongly suggests that other physiological substrates must exist (Afshar and Murnane, 1999; Perrod et al., 2001). Furthermore, a recent study shows that recombinant murine Sir2α deacetylates TAFI68, a subunit of TIF-Ib/SL1, and decreases RNA polymerase I transcription in vitro (Muth et al., 2001).
The nuclear bodies (NBs), often termed promyelocytic leukemia protein (PML) NBs, are discrete nuclear substructures and represent the natural accumulation sites of PML (reviewed in Matera, 1999; Seeler and Dejean, 1999; Zhong et al., 2000b). A cell typically contains 10–30 NBs per nucleus, although their number and size vary during the cell cycle (Everett et al., 1999; McNeil et al., 2000). NBs are dynamic structures, which are altered or disrupted in certain human diseases and in response to different cellular stresses. It is becoming increasingly apparent that NBs play a role in transcriptional regulation. Although many components of the NB have been identified to date, PML is the defining component and is essential for its proper formation (Ishov et al., 1999; Zhong et al., 2000a).
The PML protein was originally identified as part of the oncogenic fusion protein PML–retinoic acid receptor α (RARα) produced by the chromosomal translocation t(15;17) associated with acute promyelocytic leukemia (APL; reviewed in Melnick and Licht, 1999). Alternative splice variants exist, resulting in seven PML isoforms, named PML I–VII, which all contain an identical N-terminal region but differ in their C-terminal sequences (reviewed in Jensen et al., 2001). Although little is known to date concerning the specific roles of the PML variants, PML appears to have multiple functions: first, PML acts as a growth and tumor suppressor (Mu et al., 1994); secondly, PML is a mediator of multiple apoptotic signals (Quignon et al., 1998; Wang et al., 1998); and finally, PML acts both as a co-activator and co-repressor of various transcription factors, some of which localize to the NBs (Alcalay et al., 1998; Doucas et al., 1999; Fogal et al., 2000).
To date, several components of the NBs have been identified, and these include SUMO-1, Sp100, Sp140, CREB binding protein (CBP), BLM, Daxx, pRB, p53 and spectrin. Of particular interest is the tumor suppressor p53, which has been shown to physically interact with PML. p53 plays a key role in the control of cell growth, apoptosis and DNA repair, and is activated in response to many forms of stress (reviewed in Vogelstein et al., 2000; Ryan et al., 2001). The rapid induction of p53 is achieved through post-translational modifications leading to its stabilization and activation. In particular, acetylation of lysine residues in the C-terminus of p53 by P/CAF in vitro, and CBP/p300 in vitro and in vivo, has been reported (Sakaguchi et al., 1998; Liu et al., 1999; Ito et al., 2001). These acetyltransferases co-activate p53-mediated transcription in transient expression assays, and acetylation of p53 increases its DNA-binding activity in vitro (Avantaggiati et al., 1997; Gu and Roeder, 1997; Scolnick et al., 1997). More recently, acetylation has been linked to p53 stability and to the recruitment of co-activators (Barlev et al., 2001; Ito et al., 2001). In primary cells, p53 induces cellular senescence in response to oncogenic signals, and new evidence indicates that PML regulates this p53-dependent process (reviewed in Itahana et al., 2001; Pearson and Pelicci, 2001). Indeed, upon overexpression of a specific PML isoform, PML IV, p53 and CBP are recruited to the NBs, which favors the formation of a stable tricomplex with PML. It has been suggested that this results in increased p53 acetylation at lysine 382, enhancement of p53 activity and the induction of premature cellular senescence. Intriguingly, acetylation of lysine 382 occurs in serially passaged replicative senescent cells and is essential for optimal activation of p53 by PML (Pearson et al., 2000).
In this study, we investigated the function of SIRT1, the closest human homolog of ySir2. We show that SIRT1 is an active NAD-dependent HDAC that localizes to the NBs upon PML IV or oncogenic Ras overexpression and is able to physically interact with PML. SIRT1 binds p53 and promotes p53 deacetylation both in vitro and in vivo. Furthermore, SIRT1 represses p53-mediated transactivation in transient expression assays. Finally, SIRT1 co-localizes with p53 to the NBs upon PML IV upregulation, and antagonizes PML IV-induced cellular senescence, a process that is dependent on p53.
Results
SIRT1 is an active nuclear NAD-dependent HDAC
Of all human Sir2 proteins, SIRT1 shows the closest amino acid sequence homology to ySir2 (Frye, 1999). In order to investigate the function of SIRT1, we first set out to show that it possesses NAD-dependent HDAC activity, as reported for other members of the family. The full-length wild-type SIRT1 cDNA was cloned into a bacterial expression vector. In parallel, we constructed a point mutation in the SIRT1 cDNA coding sequence that converts a conserved histidine at position 363 to a tyrosine (hereafter designated SIRT1H363Y). The equivalent ySir2 mutant H364Y fails to support silencing in vivo and possesses impaired NAD-dependent HDAC activity in vitro (Tanny et al., 1999; Imai et al., 2000). Both wild-type and mutant SIRT1 proteins were expressed in Escherichia coli, purified and assayed for deacetylase activity. As shown in Figure 1A, the wild-type recombinant SIRT1 possesses strong NAD-dependent HDAC activity, whereas the activity of the SIRT1H363Y mutant is abrogated (compare columns 2 and 3). As reported previously for other Sir2 family members, the deacetylase activity associated with SIRT1 is not sensitive to trichostatin A (TSA), a potent inhibitor of class I and II HDACs (Figure 1B, compare columns 2 and 3). Recent reports, however, have demonstrated that the Sir2 family of enzymes catalyze a deacetylation reaction tightly coupled to NAD hydrolysis, resulting in the formation of nicotinamide and an acetyl-ADP-ribose product (Tanner et al., 2000; Sauve et al., 2001; Tanny and Moazed, 2001). We therefore hypothesized that the presence of excess nicotinamide in the deacetylase reaction would inhibit SIRT1’s activity. Indeed, as shown in Figure 1B, the addition of nicotinamide significantly compromises the NAD-dependent deacetylase activity of SIRT1.


Fig. 1. SIRT1 is an active nuclear NAD-dependent HDAC. (A) Equivalent amounts of wild-type His-SIRT1 or mutant His-SIRT1H363Y were tested for HDAC activity in the presence (full column) or absence (open column) of 1 mM NAD+. Histone deacetylase activity is given as radioactivity (c.p.m.) of [3H]acetate released from an acetylated histone H4 peptide. Control assays containing only acetylated H4 peptide without any recombinant protein were performed. (B) NAD-dependent HDAC assay performed essentially as above in which GST–SIRT1 was tested for activity in the absence (2) or presence of either 2 µM TSA (3) or 5 mM nicotinamide (4). All reactions contain identical amounts of GST–SIRT1. (C) Asynchronous HeLa cells were analyzed for endogenous SIRT1 expression using an anti-SIRT1 antibody followed by incubation with an Alexa 488-conjugated secondary antibody (green). Nuclear DNA was stained with propidium iodide (red). Note the high level of propidium iodide staining in the nucleoli.
Having established that SIRT1 is an active enzyme, we next investigated the intracellular localization of SIRT1 in a human transformed cell line. For this purpose, we raised a polyclonal antibody against the C-terminal moiety of SIRT1. The antibody is specific for SIRT1 and does not cross-react with any other human SIRT (data not shown). Indirect immunofluorescence studies on HeLa cells, followed by confocal microscopy analysis, revealed a diffuse and relatively homogeneous nuclear SIRT1 distribution (Figure 1C, a and c). This is consistent with a previous study reporting a potential nuclear localization signal at the N-terminus of SIRT1 (Frye, 1999).
SIRT1 interacts with PML and is recruited to the NBs
Previously, PML has been reported to interact with chromatin regulators such as the acetyltransferase CBP, as well as with class I HDACs (Doucas et al., 1999; Khan et al., 2001; Wu et al., 2001). Together with the emerging role of the PML NB in transcriptional regulation, we wondered whether SIRT1 could interact with PML. We chose to study the PML IV isoform, which has previously been shown to interact with HDACs, as well as being implicated in the regulation of p53 activity and induction of premature senescence (Ferbeyre et al., 2000; Fogal et al., 2000; Pearson et al., 2000; Wu et al., 2001). In vitro, we observed a weak but specific interaction between glutathione S-transferase (GST)–SIRT1 and in vitro translated PML IV (data not shown). This suggests that post-translational modifications and/or associated factors mediating an indirect interaction may be required. Of particular relevance is the fact that PML is modified by SUMO-1, a small ubiquitin-related peptide, and this sumoylation event is necessary for the proper formation of PML NBs and the recruitment of NB-associated proteins, highlighting the importance of this modification in PML function (reviewed in Seeler and Dejean, 2001).
We next investigated whether SIRT1 and PML interact in vivo by performing co-immunoprecipitation experiments. HeLa cells were transfected with either an expression vector for Gal4PML IV or a control empty vector. Cell lysates were immunoprecipitated with anti-Gal4 antibody and the bound protein complexes were analyzed by western blotting using the anti-SIRT1 antibody. As shown in Figure 2A, endogenous SIRT1 interacts specifically with overexpressed PML IV (compare lanes 3 and 4). In a similar experiment, extracts from HeLa cells treated with arsenic trioxide (As2O3) were immunoprecipitated with either PGM-3, an antibody that recognizes an N-terminal epitope common to all PML isoforms, or an irrelevant antibody. Brief As2O3 treatments are known to enhance PML protein levels and sumoylation, and this subsequently leads to increased targeting of PML to the NBs and to enlarged NBs (data not shown; Zhu et al., 1997; Muller et al., 1998; Lallemand-Breitenbach et al., 2001). Figure 2B shows that endogenous PML and SIRT1 are able to interact specifically (compare lanes 2 and 3).



Fig. 2. SIRT1 interacts with PML and is recruited to the PML NBs. (A) Endogenous SIRT1 co-immunoprecipitates with PML IV. HeLa cells were transfected with either pcDNA3Gal4PML IV or empty pcDNA3Gal4, and the cell lysates were immunoprecipitated (IP) with anti-Gal4 antibody. The complexes were resolved by SDS–PAGE and analyzed by western blotting with anti-SIRT1 and anti-Gal4 antibodies as indicated. Endogenous SIRT1 and Gal4PML IV are indicated by arrows. (B) Endogenous SIRT1 and PML interact. HeLa cells were treated with 1 µM As2O3 for 4 h and whole-cell extracts (WCE) were immunoprecipitated with anti-PML or an irrelevant antibody. The complexes were resolved by SDS–PAGE and analyzed by western blotting with anti-SIRT1 antibody. Endogenous SIRT1 is indicated by an arrow. (C) Overexpression of PML IV in MEFs or WI38 cells results in the accumulation of both endogenous and GFP–SIRT1 in the NBs. MEFs were infected with PINCO-SIRT1 (panels 1–3) or PINCO-SIRT1 and pBABE-PML IV (panels 4–6). PML was detected with an anti-PML antibody followed by incubation with a Cy3-conjugated secondary antibody (red), while GFP–SIRT1 was revealed by the intrinsic green fluorescence of GFP. Merging of two colours results in yellow signal corresponding to co- localized proteins. MEFs were transfected with pcDNA3GFPPML IV (panels 7–9). WI38 cells were infected with pBABE-PML IV (panels 10–12) or pBABE (panels 13–15). Endogenous SIRT1 or mSir2α staining was revealed with anti-SIRT1 antibody followed by incubation with a Cy3- conjugated secondary antibody (red), while PML staining was monitored either by GFP fluorescence or with an anti-PML antibody followed by incubation with an Alexa 488-conjugated secondary antibody (green). (D) Expression of oncogenic Ras in MEFs recruits mSir2α to the NBs. MEFs were infected with pBABE-RasV12 (panels 1–3) or pBABE (panels 4–6). Endogenous PML and mSir2α stainings were performed as in (C).
The next issue we addressed was whether SIRT1 could be recruited to NBs by PML, as has been reported previously for other PML interactors (Ishov et al., 1999; Fogal et al., 2000; Boisvert et al., 2001). For this purpose, we constructed a PINCO-SIRT1 vector, which gives rise to retroviruses expressing a green fluorescent protein (GFP)-tagged SIRT1 protein. Primary mouse embryo fibroblasts (MEFs) were infected with SIRT1-expressing retroviruses and GFP fluorescence was monitored 4 days after infection. As expected, SIRT1 shows a uniform nuclear localization (Figure 2C, panel 2). However, no visible co-localization was observed between PML and SIRT1 (Figure 2C, panel 3). Next, MEFs were co-infected with PINCO-SIRT1 and pBABE-PML IV retroviruses. pBABE-PML IV is a retroviral expression vector for PML IV with a puromycin selectable marker. Following selection, cells were analyzed for GFP fluorescence and PML localization. Strikingly, when co-expressed with PML IV, SIRT1 shows a broad nuclear distribution with distinct speckles (Figure 2C, panel 5). These speckles correspond perfectly to the PML NBs, as demonstrated when both stainings are merged (Figure 2C, panel 6).
Next, we investigated the localization of endogenous SIRT1, or its murine counterpart mSIR2α, under similar conditions of PML IV upregulation. PML IV was overexpressed in both primary human diploid fibroblasts (WI38) and MEFs. Cells were then stained with anti-SIRT1 serum, which cross-reacts with endogenous mSIR2α (data not shown), and PML staining was monitored in parallel either by GFP fluorescence or with an anti-PML antibody. Figure 2C (panels 7 and 10) shows the typically enlarged NBs that form upon PML IV overexpression (Ferbeyre et al., 2000; Pearson et al., 2000). Observation of endogenous SIRT1 nuclear distribution, or respectively mSIR2α, together with the PML signal shows that both proteins co-localize at the enlarged NBs (Figure 2C, panels 9 and 12). The enrichment of SIRT1/mSIR2α in the NBs is significant, as it does not occur in mock-transfected/infected cells (Figure 2C, 13–15; data not shown). Staining with antibodies specific for other endogenous nuclear factors indicates that the vast majority are not recruited to the NBs upon PML IV overexpression (M.Faretta and S.Minucci, unpublished data), therefore demonstrating that PML IV does not cause the non-specific accumulation of endogenous nuclear proteins in the NBs. These results show that PML IV mediates partial relocalization of both endogenous and overexpressed SIRT1 to NBs. Interestingly, the residual SIRT1/mSIR2α staining observed in the nucleoplasm may suggest possible functions outside the NBs.
Expression of oncogenic Ras (Ha-rasV12) in primary fibroblasts is known to result in increased PML levels, concomitant with the enlargement and accumulation of NBs, similar to what occurs upon PML IV overexpression (Ferbeyre et al., 2000; Pearson et al., 2000). We therefore investigated endogenous mSIR2α and PML localization during activated Ras expression in MEFs. In infected pBABERasV12 cells, PML NBs show an increase in size and number compared with mock-infected cells, as shown previously (Figure 2D, panel 1 compared with 4). Strikingly, mSIR2α is seen to localize to some of these enlarged NBs (Figure 2D, panels 2 and 3), while no apparent co-localization of mSIR2α and PML is observed in mock-infected cells (panels 5 and 6). Importantly, these results demonstrate that oncogenic Ras, an upstream regulator of PML, is able to partially relocalize endogenous mSIR2α to the NBs.
PML, SIRT1 and p53 co-localize in the NBs
Given the recent findings that mSIR2α can act on non-histone substrates, we speculated that one of the NB components could be a SIRT1 substrate (Muth et al., 2001). An obvious candidate was the tumor suppressor p53 whose function is regulated by post-translational modifications such as acetylation. Moreover, p53 is also recruited to the NBs by PML IV (Fogal et al., 2000). We therefore addressed whether PML, SIRT1 and p53 co-localized in the NBs. PML IV was expressed in U2OS cells and triple immunostaining was performed (Figure 3). As expected, PML IV and endogenous SIRT1 co-localize, as do PML IV and endogenous p53 (Figure 3, panels 6 and 7). Importantly, p53 and SIRT1 co-localize (Figure 3, panel 5) and the merge of the triple staining shows that they co-localize with PML in the NBs (panel 4). The triple co-localization is schematically represented in the graph in panel 8. Non-transfected U2OS cells show diffuse nuclear staining for endogenous p53 and SIRT1, and do not indicate any apparent co-localization between the two latter proteins and PML (panels 9–16). Thus, PML IV is able to recruit both endogenous SIRT1 and p53 simultaneously to the NBs.

Fig. 3. PML, SIRT1 and p53 co-localize in the NBs. U20S cells were either transfected with pcDNA3-PML IV (panels 1–7) or un-treated (panels 9–15). PML staining with mouse monoclonal PGM3 is shown in blue (AMCA), endogenous p53 staining with goat polyclonal anti-p53 in red (Cy3) and endogenous SIRT1 staining with rabbit anti-sirt1 in green (Alexa 488). Co-localization is shown in yellow (p53 and SIRT1), light blue (PML and SIRT1), violet (PML and p53) and white (triple co-localization). Panels 8 and 16 show the normalized intensity profiles along the lines showed in the insert respectively: p53 (red line), SIRT1 (green line) and PML (blue line). Blue peaks identify PML accumulation in the NBs. Peak distribution in panel 8 supports triple co-localization of p53, SIRT1 and PML.
SIRT1 binds p53 in vitro and in vivo
Next, we tested whether SIRT1 can bind p53 in vitro. We performed pull-down assays with either full-length GST–p53 or a series of GST–p53 fusions expressing only the N-terminal, middle or C-terminal regions of the protein, and in vitro translated SIRT1. We observed specific binding of SIRT1 to full-length p53 as well as to GST–p53(290–393), while weaker binding was seen with GST–p53(90–290) and no binding occurred with the N-terminal p53 fusion (Figure 4A). These results indicate that the C-terminal portion of p53, containing the acetylation sites reported so far, is sufficient to bind SIRT1.

Fig. 4. SIRT1 interacts with p53 in vitro and in vivo. (A) Pull-down experiment performed with in vitro translated (IVT) SIRT1 and GST–p53 (lane 3) or different GST–p53 fusions expressing different portions of the protein as indicated (lanes 4–6). Complexes were resolved by SDS–PAGE and visualized by autoradiography. IVT SIRT1 is indicated by an arrow. The exposure time of the input lane is half that of the binding reactions. (B and C) Whole-cell extracts (WCE) from untreated 293T cells or HCT116 UV-irradiated cells (20J/m2 for 5 or 20 h) were immunoprecipitated (IP) with either anti-SIRT1 or pre-immune sera, and the immunocomplexes analyzed by western blotting with anti-p53 (DO-1) antibody. Different exposures are shown in (C) in order to visualize p53 immunoprecipitated in the various samples. The asterisks indicate the Ig heavy chain.
Next, we investigated whether SIRT1 and p53 interact in vivo. Whole-cell extracts from 293T cells were immunoprecipitated with the anti-SIRT1 antibody or with the pre-immune serum. As shown in Figure 4B, p53 is specifically immunoprecipitated with the anti-SIRT1 antibody but not with the pre-immune serum (lanes 2 and 3). Interestingly, when similar experiments were performed with lysates from UV-irradiated HCT116 cells, the endogenous interaction between p53 and SIRT1 was increased compared with non-treated cells (Figure 4C, lanes 4–6). Similar results were obtained with UV-irradiated 293T cells (data not shown). In conclusion, these results show that SIRT1 interacts with p53 in vivo and that, importantly, this interaction is increased under conditions that activate p53.
SIRT1-mediated deacetylation of p53 occurs in vitro and in vivo
Having established that SIRT1 and p53 interact, a crucial issue to address was whether p53 could be a substrate for SIRT1. For this purpose we acetylated GST–p53 in vitro using recombinant p300 in the presence of [14C]acetylCoA. Following this acetylation step, active SIRT1 was added to half the reactions in the presence or absence of NAD+. All deacetylation reactions were performed in the presence of TSA. The samples were resolved on a gel and the acetylation status of p53 was monitored by autoradiography. As shown in Figure 5A (lane 2), GST–p53 is efficiently acetylated by recombinant p300. Comparison of lanes 3 and 4 shows a significant reduction of p53 acetylation signal only when SIRT1 is added in the presence of NAD+. The decrease in the acetylated signal is greater than is apparent in lane 4 if a correction is made for the slightly higher amount of GST–p53 used in that reaction, as shown by Ponceau staining in the bottom panel. Together, these results indicate that recombinant SIRT1 can deacetylate p53 in an NAD-dependent and TSA-insensitive manner. The p53 deacetylation reaction, however, is not complete (Figure 5A, lane 4). In vitro, p300 has been reported to acetylate lysines 373 and 382 (Sakaguchi et al., 1998; Liu et al., 1999). Therefore, SIRT1 might deacetylate one of these sites preferentially, explaining the residual acetylated p53 signal observed experimentally. We performed an in vitro deacetylation assay on GST–p53 as described above, followed by western blot analysis using a specific antibody that recognizes acetyl-lysine 382 of p53. Strikingly, SIRT1-mediated deacetylation of p53 results in no detectable signal with the anti-acetyl-lysine 382 antibody (Figure 5B, lane 4), implying that any residual acetylated p53 signal present in Figure 5A arises from acetyl-lysine 373. This suggests that SIRT1 preferentially recognizes lysine 382 and catalyzes a site-specific deacetylation reaction.

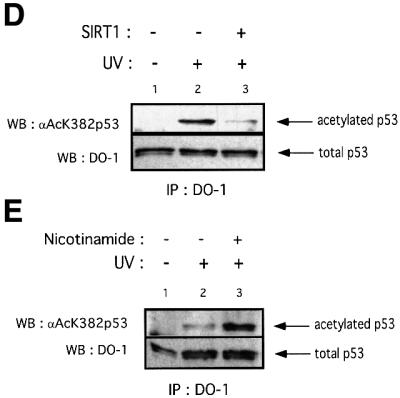

Fig. 5. SIRT1 can deacetylate p53 in vitro and in vivo. (A) Equivalent amounts of GST (lane 1) or GST–p53 (lanes 2–4) were acetylated in vitro by bacterially expressed p300 in the presence of [14C]acetylCoA. Recombinant SIRT1 was then added to the reactions in the presence (lane 4) or absence of 1 mM NAD+ (lane 3). Two micromolar TSA was added to all reactions. The samples were resolved by SDS–PAGE and acetylated p53 was visualized by autoradiography. Ponceau staining shows the total levels of GST–p53 used in the reactions. (B) An in vitro deacetylation reaction with GST–p53 and SIRT1 was performed as in (A), followed by western blot analysis with a specific antibody for acetylated lysine 382 (αAcK382p53). Acetylated GST–p53 is shown with an arrow. (C) 293T cells were co-transfected with combinations of the expression plasmids pcDNA3.1SIRT1, pcDNA3.1SIRT1H363Y and pcDNA3GAL4CBP(1099–1758) as indicated, or with empty vector controls (lane 1). Twenty-four hours post-transfection, whole-cell lysates were immunoprecipitated with DO-1 antibody and the complexes were analyzed by SDS–PAGE. The acetylation status of endogenous p53 was visualized using a specific antibody for acetylated lysine 382 (αAcK382p53). The bottom panel shows the total amount of p53 immunoprecipitated in each sample. (D) 293T cells were transfected with either pcDNA3.1SIRT1 (lane 3) or with empty vector control (lanes 1 and 2). Twenty-four hours post-transfection, cells were UV-irradiated (150 J/m2, lanes 2 and 3) and after 5 h, whole-cell extracts were prepared from control and UV-treated cells. Immunoprecipitations and western blotting were performed as in (C). (E) MCF7 cells were UV-irradiated (50 J/m2, lanes 2 and 3), and when indicated, nicotinamide was added to the media immediately following irradiation. Whole-cell extracts were prepared from control and UV-treated cells after 20 h. Immunoprecipitations and western blotting were performed as in (C). (F) SIRT1 overexpression in MEFs inhibits p53 acetylation. Immortalized MEFs were transfected with PINCO-SIRT1 and pcDNA3-p53. p53 overexpression was analyzed by DO-1 staining followed by Alexa 350-conjugated secondary antibody (blue, panel 1) while GFP fluorescence revealed GFP–SIRT1 staining (panel 3). Acetylated p53 was analyzed with anti-acetylated p53 antibody followed by Cy3-conjugated secondary antibody (red, panel 2). Mean fluorescence value of p53 and acetyl-p53 signals were measured in cells expressing low (gfpSIRT1–) and medium to high (gfpSIRT1+) GFPSIRT1 levels (panel 4; Student’s t-test, p = 0.002). The graph shows the normalized mean fluoresence intensities.
We next investigated whether SIRT1 mediates deacetylation of p53 in vivo. A portion of CBP containing the HAT domain was overexpressed in 293T cells either alone or in the presence of wild-type or mutant SIRT1. Endogenous p53 was immunoprecipitated with DO-1 antibody and the immunocomplexes were analyzed by western blotting using a specific antibody for acetylated lysine 382 of p53. As shown in Figure 5C (lane 2), overexpression of CBP HAT domain induces acetylation of endogenous p53 at lysine 382. This is consistent with a recent study which reports that p53 acetylation in vivo at this site is mediated by the p300/CBP acetyltransferases (Ito et al., 2001). Over expression of SIRT1 effectively reduces CBP-dependent p53 acetylation (lane 3). In contrast, overexpression of an inactive SIRT1 enzyme does not significantly affect p53 acetylation (lane 4, which should be corrected for the slightly lower amount of total p53 immunoprecipitated in that reaction, as shown). The bottom panel shows the amount of endogenous p53 immunoprecipitated in each sample. Both wild-type and mutant enzymes are expressed at similar levels, and SIRT1H363Y was demonstrated to bind p53 as efficiently as its wild-type counterpart (data not shown).
In order to investigate SIRT1-mediated p53 deacetylation within a physiological p53 activation pathway, 293T cells were transfected with SIRT1, UV irradiated, and the acetylation status of endogenous p53 monitored with a p53 antibody recognizing lysine 382. As shown in Figure 5D, overexpression of SIRT1 leads to a major reduction in p53 acetylation following UV treatment. However, the ultimate proof that SIRT1 regulates p53 acetylation levels in vivo necessitates inhibition of the endogenous enzyme followed by the analysis of p53 acetylation status. We therefore took advantage of the fact that nicotinamide compromised the activity of SIRT1 in vitro (see Figure 1B). MCF7 cells were treated with UV in the presence or absence of nicotinamide. The acetylation status of p53 was analyzed (Figure 5E). As expected, UV irradiation activated p53 and induced p53 acetylation (lane 2). However, cells grown in the presence of nicotinamide, following DNA damage, have greatly enhanced p53 acetylation levels (compare lanes 2 and 3). Treatment of non-UV-irradiated cells with nicotinamide does not result in any detectable p53 acetylation (data not shown), thus suggesting that the increase in p53 acetylation observed is caused by the inhibition of endogenous SIRT1. Similar results were obtained in HCT116 cells treated with etoposide, a chemical DNA-damaging agent (data not shown). Collectively, these results strongly support the notion that SIRT1 can mediate deacetylation of p53 at lysine 382 in vivo within the context of a physiological p53 activation pathway, and this regulation depends on its intact NAD-deacetylase activity.
Finally, we monitored SIRT1-mediated deacetylation of p53 in vivo by immunofluorescence. For this purpose, immortalized MEFs were transfected with p53- and GFP–SIRT1-expression plasmids. Cells were then stained with both anti-p53 and anti-acetylated p53 antibodies, while SIRT1 expression was followed by GFP. Both total and acetylated p53 signals show a uniform nuclear distribution (Figure 5F, panels 1 and 2). This indicates that overexpression of p53 promotes its acetylation. However, in a cell co-expressing GFP–SIRT1, the p53 acetylation signal is significantly decreased compared with a non-SIRT1-expressing cell (Figure 5F, compare panels 2 and 3). A small population of cells was analyzed and the mean fluorescence signals for total and acetylated p53 were quantified in both SIRT1-positive and -negative cells. The graph in Figure 5F (panel 4) shows that the level of acetylated p53 detected by immunofluorescence in SIRT1-expressing cells is >2-fold lower than in SIRT1-negative cells. This decrease of acetylated p53 is not caused by variations in the total p53 levels, as these are equivalent in both cell populations. These results demonstrate that SIRT1-induced deacetylation of p53 also occurs in primary fibroblasts.
SIRT1 represses p53-dependent transactivation
We next asked whether SIRT1 would control the transcriptional activation capacity of p53, given that it can deacetylate p53 both in vitro and in vivo. To study the effect of SIRT1 on p53 activity, we carried out transient transfection assays with a p53-responsive reporter, PGluc, which contains two consensus p53 response elements cloned upstream of the luciferase reporter (Ouchi et al., 1998). Assays were performed in a fibroblast cell line derived from MEFs with combinations of p53, SIRT1 and SIRT1H363Y expression vectors. Expression of p53 alone caused a >20-fold activation of the reporter gene (Figure 6, compare lanes 1 and 2). However, co-expression of p53 with SIRT1 results in a near complete loss of p53 transcriptional activity (lane 3). This inhibition of p53 transactivation is greatly alleviated when p53 is co-expressed with SIRT1H363Y (lane 4). Expression of either SIRT1 or SIRT1H363Y alone does not affect the basal level of transactivation of the reporter gene (lanes 5 and 6). These results show that SIRT1 is able to inhibit p53-dependent transcription in a manner dependent on its deacetylase domain.

Fig. 6. SIRT1 represses p53-mediated transactivation. (A) A luciferase reporter assay was performed in a fibroblast cell line derived from MEFs. Cells were transfected with 2 µg of PGluc (lanes 1–6) and 2.5 µg of pHKp53 (lanes 2–4), 1.5 µg of pcDNA3.1SIRT1 (lanes 3 and 5) or 1.5 µg of pcDNA3.1SIRT1H363Y (lanes 4 and 6). Relative luciferase units shown are the mean value of triplicates. (B) 293T cells were transfected with 0.1 µg of mdm2-luc (lanes 1–6) and 0.025 µg of pHKp53 (lanes 2–4), 0.5 µg of pcDNA3.1SIRT1 (lanes 3 and 5) or 0.5 µg of pcDNA3.1SIRT1H363Y (lanes 4 and 6). Relative luciferase units shown are the mean value of triplicates.
Next, we analyzed the ability of SIRT1 to repress p53-dependent transactivation on a natural genomic promoter, namely the p53-responsive mdm2 promoter. 293T cells were co-transfected with a reporter plasmid, mdm2-luc, which contains the mdm2 promoter cloned upstream of the luciferase gene, and combinations of p53, SIRT1 and SIRT1H363Y expression vectors (Figure 6B). Expression of p53 alone resulted in a 6-fold activation of the reporter plasmid, while co-expression of p53 with SIRT1 only activated the reporter gene ∼2-fold, thus a reduction of ∼60% (Figure 6B, compare lanes 1, 2 and 3). Co-expression of the catalytically inactive SIRT1 with p53 inhibited p53 transactivation by only ∼25% (lane 4). These results show that SIRT1 has the ability also to repress p53-dependent transcription in the context of a natural promoter.
SIRT1 rescues PML-induced cellular senescence
A connection between PML IV overexpression, recruitment of p53 to NBs, increased p53 acetylation and induction of premature cellular senescence has been suggested previously by Pearson et al. (2000). Given that SIRT1 can deacetylate p53, repress its transactivation capacity, and co-localizes with p53 and PML in the NBs, we asked whether SIRT1 can suppress PML IV-induced senescence. For this purpose, primary MEFs were infected with the relevant retroviruses expressing either GFP– SIRT1 or GFP–SIRT1H363Y in concert with PML IV. We have already shown that GFP–SIRT1 is recruited to the NBs under these conditions (see Figure 2C, panels 3–6). Infections with empty vectors were performed as controls. Cells were selected then counted over the subsequent 8 days. The growth curves are shown in Figure 7A. Expression of wild type SIRT1 or its mutant counterpart alone does not alter the growth pattern of the primary fibroblasts. As described previously, overexpression of PML IV in primary cells leads to immediate growth arrest, a hallmark of cellular senescence (Ferbeyre et al., 2000; Pearson et al., 2000). However, when SIRT1 is expressed along with PML IV, a near complete rescue of PML-induced growth arrest is seen. In contrast, this growth arrest was only partially relieved by co-expression of the catalytically inactive SIRT1H363Y. These results indicate that SIRT1 can antagonize PML IV-induced growth arrest.


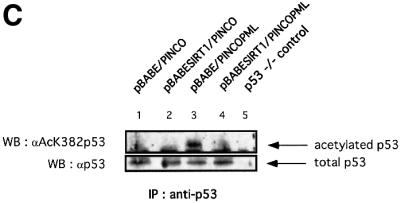
Fig. 7. SIRT1 antagonizes PML-induced cellular senescence. (A) Overexpression of SIRT1 in primary MEFs rescues PML IV-induced cell growth arrest. Wild-type MEFs were infected with the indicated retroviruses. Infected cells were selected, inoculated at 5 × 104 cells per well and counted for the subsequent 8 days. Day 0 is the first day after selection. The cell number at each time point shown on the growth curves represents the mean value of duplicate wells. (B) SIRT1 rescues a PML-induced senescent phenotype. WI38 cells were infected with the indicated retroviruses. Cells were selected, cultured for 8 days and then examined for GFP positivity, BrdU incorporation and acidic β-galactosidase activity. The BrdU-positive cells and β-galactosidase positive cells are shown as a percentage of GFP-positive cells. (C) SIRT1 inhibits PML-induced p53 acetylation. MEFs were infected with the indicated retroviruses. Following selection, cells were cultured for 4 days, lysates were prepared and p53 was immunoprecipitated with a rabbit polyclonal anti-p53 antibody. The acetylation status of endogenous p53 was visualized using a specific antibody for acetylated lysine 382 (αAcK382p53), while total p53 was revealed with an anti-p53 antibody (Ab-7). p53–/– cells are included as a negative control for the p53 immunoprecipitation.
In a similar series of experiments, WI38 cells were co-infected with either empty pBABE or pBABE-SIRT1 together with empty PINCO or PINCO-PML IV retroviruses. Cells were selected and then assayed for bromodeoxyuridine (BrdU) incorporation, which reflects the number of cells in S phase, and for acidic β-galactosidase (SA-β gal) activity, two markers commonly used in the analysis of senescent cells. Results are shown in Figure 7B. As observed for the growth curves, expression of SIRT1 alone does not significantly affect the percentage of cells in S phase or the percentage of SA-β gal-positive cells when compared with control infected cells. However, expression of PML IV results in a striking decrease of BrdU-positive cells and a huge increase in SA-β gal active cells, as reported previously (Ferbeyre et al., 2000; Pearson et al., 2000). Double-infected PML IV–SIRT1 cells exhibit a significant increase in the percentage of cells in S phase as compared with PML-infected cells, in addition to a marked decrease in the percentage of SA-β gal-positive cells. Taken together with the growth assay, these data indicate that overexpression of SIRT1 in primary cells can reverse PML IV-induced cellular senescence, and its intact deacetylase activity appears to be required for this process.
Given that PML IV-induced senescence is concomitant with increased p53 activation and acetylation at lysine 382, similar to what is observed during replicative senescence, we addressed the acetylation status of p53 in double-infected PML IV–SIRT1 cells. For this purpose, MEFs were co-infected with the relevant retroviruses, lysates were prepared from selected cells and p53 was immunoprecipitated. The acetylation status of p53 was assessed by western blotting using a specific antibody for acetylated lysine 382 and is shown in Figure 7C. No acetylation of endogenous p53 is observed in mock-infected and overexpressing SIRT1 cells (Figure 7C, lanes 1 and 2). As reported previously, PML IV expression in primary fibroblasts induces acetylation of endogenous p53 at lysine 382 (lane 3; Pearson et al., 2000). Strikingly, this acetylation signal is completely lost in double-infected PML IV–SIRT1 cells (lane 4). The total amount of endogenous p53 immunoprecipitated in each sample is equivalent and is shown in the lower panel. These results show that the antagonizing effect that SIRT1 exerts on PML-induced senescence is accompanied at the molecular level by a lack of p53 acetylation. This suggests that the growth-promoting activity of SIRT1 is mediated by p53 and is exerted, directly or indirectly, by the modulation of p53 acetylation levels.
Discussion
In this study, we have provided evidence for a function of SIRT1, the human Sir2 homolog. We show that the NAD-dependent deacetylase SIRT1 is recruited to the PML NBs together with its substrate p53. Furthermore, we demonstrate that SIRT1 can downregulate a p53-dependent process, namely the induction of cellular senescence, giving strength to a physiologically relevant role of SIRT1 in the p53 regulatory network.
Functional significance of SIRT1 and PML interaction
We report that SIRT1, a diffuse nuclear enzyme, is partially relocalized to the NBs upon PML IV upregulation or oncogenic Ras expression, an upstream element of the PML pathway. Previously, class II HDACs have been shown to accumulate in specific nuclear subdomains that are distinct from PML NBs (Downes et al., 2000). This is, therefore, the first demonstration that a deacetylase is recruited to the NB compartment. What function does SIRT1 exert there? The co-localization of SIRT1 and its substrate p53 within NBs suggests that the latter may function as nuclear organizing centers, which enable a given protein to have access to another, and/or provide a sufficiently high local concentration for NB-associated proteins to be active. However, other non-mutually exclusive hypotheses can be put forward. Indeed, the interaction of SIRT1 with PML may modulate PML function, or vice versa. Supporting this idea, PML has recently been shown to interact with multiple co-repressors and class I HDACs, and these interactions are required for the transcriptional repression function of PML (Khan et al., 2001; Wu et al., 2001). Therefore, the deacetylase activity of SIRT1 may be recruited by PML to contribute to PML-mediated transcriptional repression independently of the action of SIRT1 on p53.
SIRT1 adds to the growing list of co-activators, co-repressors and chromatin-associated proteins shown to be recruited to the NBs, thus giving strength to the emerging role of these nuclear speckles in the control of transcription. Furthermore, the previously reported organization of class II HDAC complexes into discrete subnuclear domains, together with the recruitment of multiple co-activators, co-repressors and chromatin regulators to the NB, indicate that the nucleus is highly compartmentalized and that the distinct subdomains must coordinate a variety of cellular processes (Hodges et al., 1998; Downes et al., 2000; Zhong et al., 2000b).
SIRT1: a novel negative regulator of p53 function
A major finding in this study is the demonstration that the acetylated form of the tumor suppressor p53 is a physiological substrate for SIRT1. Recently, acetylation of lysine residues in the C-terminus of p53 has been shown to occur in response to a wide variety of stresses and is invariably coupled to p53 activation (reviewed in Appella and Anderson, 2001; Ito et al., 2001). Moreover, p53 acetylation has been linked to p53 protein stability as well as optimal p53 transactivation in response to certain cellular stresses, and appears to be a tightly regulated event (Pearson et al., 2000; Ito et al., 2001). Indeed, two recent studies have reported p53 deacetylases: the first demonstrates that MDM2, a well established negative regulator of p53, can suppress p300/CBP-mediated p53 acetylation in vivo in a TSA-sensitive manner (Ito et al., 2001); the second report shows that a HDAC1-containing complex can bind and deacetylate p53, and that this interaction modulates p53-mediated cell growth arrest and apoptosis (Luo et al., 2000).
We now add an extra level of complexity to the regulation of p53 function by presenting evidence that SIRT1, an NAD-dependent deacetylase, is able to deacetylate p53 and modulate a p53-dependent process in vivo. Given the importance of acetylation in p53 function, it is not surprising to discover multiple enzymes capable of regulating this event. The most challenging issue now will be to understand the differential roles, if any, of the three p53 deacetylases and to investigate whether they perform redundant functions in vivo or, on the contrary, whether they are involved in specific p53 regulatory pathways. A relatively simple explanation for the existence of multiple p53 deacetylases would stem from the catalysis of acetyl-lysine-specific deacetylation reactions. Our results indicate that such a specificity may occur in the SIRT1-mediated deacetylation of p53 and would be directed against lysine 382. This has been proven further by an HPLC analysis in a recent report by Vaziri et al. (2001). Therefore, the NAD-dependent deacetylation of SIRT1 may work in conjunction with class I and II HDACs to finely coordinate the regulation of p53.
Recent studies demonstrated that p300/CBP-mediated co-activation of p53 gene expression occurs both through p53-targeted nucleosomal acetylation at the promoter level and through p53 acetylation per se, which itself enhances the association with co-activator complexes (Barlev et al., 2001; Espinosa and Emerson, 2001). Therefore, in addition to a direct deacetylation of p53 leading to inefficient co-activator binding, SIRT1 may mediate its repressive function through its recruitment to p53 target promoters, leading to transcriptionally silenced hypoacetylated nucleosomes. Supporting this hypothesis, SIRT1 has been found associated with the p21 promoter, a p53 target gene, in chromatin immunoprecipitation assays (N.Barlev and S.Berger, personal communication). However, future work will determine whether this is the case.
SIRT1 modulates PML-induced cellular senescence
This study strongly supports a role for SIRT1 in the regulation of premature cellular senescence, a proposed tumor-protection mechanism induced in response to certain cellular stresses (reviewed in Campisi, 2001). Our results indicate that SIRT1 antagonizes the induction of senescence, and this is concomitant with a lack of p53 acetylation. We propose a potential model for the induction of premature cellular senescence in response to PML IV upregulation (Figure 8). In this model, induction of PML promotes acetylation of p53 at lysine 382 by CBP and enhances its activity, thus resulting in p53 target gene expression and the onset of senescence. We propose that SIRT1 counteracts CBP-mediated acetylation, one possible explanation being the direct binding and deacetylation of p53, resulting in reduced p53 activity and target gene expression, allowing cells to continue growing. However, the exact mechanism of SIRT1’s action still remains to be elucidated and one cannot formally exclude at this stage indirect mechanisms acting on genes or as yet undiscovered substrates other than p53.

Fig. 8. Proposed model for the rescue of PML-induced premature senescence by SIRT1 overexpression. Upregulation of PML promotes acetylation of p53 by CBP and enhances its activity, leading to the expression of p53 target genes and the onset of senescence. We propose that SIRT1 expression antagonizes PML-induced acetylation of p53, thus modulating its activity and exerting a negative action on the induction of senescence.
In view of the recent findings that Sir2 modulates lifespan extension in yeast and worms, the discovery that a human Sir2 homolog is involved in cellular senescence is intriguing. Indeed, a loose link exists between organismal ageing and cellular senescence, namely cells taken from older organisms become senescent in culture after fewer serial passages than those taken from younger organisms (Campisi, 1996). Recently, this connection has been reinforced by the finding that p53, a key player in cellular senescence, is also involved in organismal ageing (Tyner et al., 2002). Therefore, both the Sir2 family and p53 appear to be implicated in ageing at the cellular level (senescence) and at the whole-organism level. An exciting possibility is the involvement of SIRT1/mSIR2α in organismal ageing in mammals. Intriguingly, a previous report demonstrating that TSA treatment of primary human cells induced a senescence-like state suggests that other class I or II HDACs may also be involved in this process (Ogryzko et al., 1996). However, the data presented here clearly indicate that the NAD-dependent deacetylase SIRT1 regulates premature cellular senescence induced by the tumor suppressors PML and p53.
Finally, two groups have recently published similar findings in which they show that SIRT1 can bind and deacetylate p53 in vivo, and furthermore, that SIRT1 modulates p53-dependent apoptosis in response to oxidative stress and DNA damage (Luo et al., 2001; Vaziri et al., 2001). Taken together with our results, it appears that SIRT1 is implicated in multiple stress-activated regulatory pathways that relay to p53.
Materials and methods
Recombinant DNA
Full-length SIRT1 cDNA (DDBJ/EMBL/GenBank accession No. NM-012238) was cloned by PCR from the human testis Marathon cDNA library. SIRT1H363Y mutant was generated by site-directed mutagenesis. Both wild-type and mutant cDNAs were cloned into pcr4TOPO (Invitrogen), then subcloned into pGEX2TK (Pharmacia) for bacterial GST fusion expression, pet30a (Novagen) for expression of His-tagged proteins, pcDNA3.1B (Invitrogen) for myc-tagged mammalian expression, PINCO (Grignani et al., 1998) and pBABE (Morgenstern and Land, 1990) for retroviral mediated gene transfer. A SIRT1 HindIII–NotI fragment from SIRT1pcr4TOPO was subcloned into pET28 (Novagen) for expression of His6-SIRT1(506–747) fusion protein. PINCO-PML IV, pBABE-PML IV, pBABERasV12 (Pearson et al., 2000), pcDNA3 GAL4CBP(1099–1758) (Martinez-Balbas et al., 1998), pcDNA3 GFPPML IV, pcDNA3GAL4PML IV and pcDNA3PML IV (Clontech) have been described elsewhere. pGEX2TK plasmids containing the p53 truncations, pHK-p53 and pcDNA3p53 for mammalian expression were constructed by PCR. All constructs were verified by DNA sequencing.
NAD-dependent HDAC assay
HDAC assays were carried out essentially as described (Brehm et al., 1998) in 100 µl of NAD–HDAC buffer containing 50 mM Tris–HCl pH 8.8, 4 mM MgCl2, 0.2 mM dithiothreitol (DTT) with 150 000 c.p.m. of a tritium-labeled acetylated H4 peptide. One millimolar NAD+ (Sigma) was added to reactions when indicated. For inhibition experiments, 2 µM TSA (Wako Bioproducts) or 5 mM nicotinamide (Sigma) was added to reactions. Assays were performed in triplicate.
Protein expression and in vitro binding assays
GST fusion proteins were expressed in E.coli strain XA90 using the pGEX (Pharmacia) vector system. Purification of GST fusions from crude bacterial lysates was performed as described previously (Bannister and Kouzarides, 1996). His-tagged SIRT1 fusions were expressed in E.coli strain BLR using the pET (Novagen) vector system. Bacteria were lysed with BPER buffer (Pierce) and the protein was purified from the lysate on an Ni-NTA–agarose column (Qiagen) according to the manufacturer’s instructions. In vitro transcription/translation was carried out using the TNT system (Promega) and binding assays were performed as described previously (Fuks et al., 2000).
Cell culture, transfections and retroviral infections
HeLa, 293T, HCT116, MCF7, U2OS and the spontaneously immortalized cells derived from MEFs were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% FBS, penicillin, streptomycin and glutamine (all Gibco-BRL). WI38 cells were maintained in DMEM with 10% North American serum, penicillin, streptomycin and glutamine (all Gibco-BRL). Cells were grown at 37°C in an atmosphere containing 5% CO2. Primary MEFs were maintained in DMEM with 10% North American serum, non-essential amino acids, 50 µM β-mercaptoethanol, penicillin, streptomycin and glutamine (all Gibco-BRL). Cells were grown at 37°C in an atmosphere containing 9% CO2. Transfections were performed with the standard calcium phosphate method, except for the immunofluorescence studies, where MEFs were transfected with 2 µg of total DNA using Fugene reagent (Boehringer Mannheim, Germany). Infections of primary fibroblasts were performed by retroviral mediated gene transfer using Phoenix packaging cells as described previously (Serrano et al., 1997). The PINCO vector expresses a GFP visible within 24 h, allowing determination of infection efficiencies. Thirty-six hours following infection, cells were selected with 3 µg/ml puromycin. Day 0 was considered 3 days after initiation of chemical selection when all non-infected cells were dead.
Senescence analysis
In the growth curve experiments, cells were plated at 50 000 cells per well in duplicate and counted on successive days. β-galactosidase analysis was carried out essentially as described (Dimri et al., 1995). To visualize BrdU incorporation, infected cells were plated on coverslips, incubated for 1 h in the presence of 10 µM BrdU, fixed, and nuclei incorporating BrdU were visualized by immunostaining using a pure monoclonal anti-BrdU antibody and a FITC-conjugated secondary antibody (both from Becton Dickinson, USA).
Immunoprecipitation experiments
HeLa and 293T cells in culture dishes (15 cm diameter) were transfected with 40 µg of expression vectors. 293T, MCF7 and HCT116 cells were UV irradiated with 150, 50 and 20 J/m2, respectively. When indicated, 1 µM As2O3 and 5 mM nicotinamide (all Sigma) were added to the culture media. Cells were washed in ice-cold PBS and lysed in RIPA buffer (50 mM Tris pH 8, 150 mM NaCl, 5 mM EDTA, 1% NP-40, 0.1% SDS, 0.5% DOC plus protease inhibitors) at 4°C for 30 min. Lysates were cleared by centrifugation (input samples were taken at this stage), protein levels were quantified and equal amounts of lysate were incubated with 3 µg of anti-Gal4 mouse monoclonal antibody (RK5C1; Santa Cruz), anti-p53 mouse monoclonal antibody (DO-1; Santa Cruz), anti-SIRT1 rabbit polyclonal antibody or pre-immune serum, or anti-PML mouse monoclonal antibody (PGM-3; Santa Cruz) for 2 h. Fifty microliters of a slurry of protein A/G–Sepharose beads (Pharmacia) were added and incubation continued for a further 2 h with rotation at 4°C. Precipitates were washed five times in RIPA and resuspended in loading buffer for SDS–PAGE analysis. Immunoprecipitation experiments with primary fibroblasts were carried out 8 days after infection essentially as described above. Briefly, cells were washed twice in ice-cold PBS before being lysed in 50 mM Tris–HCl pH 7.8, 150 mM NaCl, 2 mM EDTA, 2 mM DTT, 1% Triton X-100 plus protease inhibitors, and snap frozen. Equal amounts of lysate were immunoprecipitated with a rabbit polyclonal anti-p53 antibody (393fl; Santa Cruz).
Antibodies and western blot analysis
Western blotting was performed according to standard procedures. The following primary antibodies were used: His-SIRT1(506–747) fusion protein was used as an antigen to produce a specific rabbit polyclonal anti-SIRT1 antibody; mouse monoclonal anti-Gal4 (RK5C1; Santa Cruz); mouse monoclonal anti-p53 (DO-1; Santa Cruz); sheep polyclonal anti-p53 (Ab-7; Oncogene); rabbit polyclonal anti-acetylK382p53 (kindly provided by E.Appella). Primary antibodies were revealed by HRP-conjugated secondary antibodies (Abcam), followed by enhanced chemiluminesence (Amersham).
Immunofluoresence analysis
Cells were cultured directly on glass coverslips and then fixed with 4% paraformaldehyde. After washing with PBS, cells were permeabilized with 0.1% Triton X-100 in PBS for 10 min, blocked and incubated with primary antibodies. Primary antibodies used were mouse monoclonal anti-hPML (PGM-3; Santa Cruz), mouse monoclonal anti-p53 (DO-1; Santa Cruz), rabbit polyclonal anti-acetylated p53 (Ab-1; Oncogene), rabbit polyclonal anti-SIRT1 and mouse polyclonal anti-murine PML (Contegno et al., 2002). Cells were then washed three times in PBS and incubated with goat anti-mouse and/or anti-rabbit Cy3-conjugated (Amersham), Alexa 488- or Alexa 350-conjugated (Molecular Probes) secondary antibodies. Simultaneous detection of PML, p53 and SIRT1 in U2OS cells was obtained by using mouse monoclonal anti-hPML (PGM-3; Santa Cruz), rabbit polyclonal anti-SIRT1 antibody and goat polyclonal anti-p53 (clone 393; Santa Cruz). AMCA-conjugated donkey anti-mouse (Jackson Immunoresearch), Cy3-conjugated donkey anti-goat (Amersham) and Alexa 488-conjugated donkey anti-rabbit (Molecular Probes) antibodies were employed for the detection step. Coverslips were mounted in a 90% glycerol solution containing diazabicyclo-(2.2.2)octane antifade (Sigma). Coverslips were examined under an AX-70 Provis (Olympus) fluorescence microscope and images collected by a Hamamatsu c5985 b/w camera. Image analysis (line profiling and mean fluorescence calculations) was performed using NIH Image software (W.Rasband, NIH).
Gene reporter assays
Immortalized MEFs or 293T cells were transfected at 40–60% confluency with a total of 10 µg of DNA. The mdm2-luc plasmid was a kind gift from Dr Xin Lu. Cells were washed 24 h after transfection and incubated for an additional 24 h before harvesting. Luciferase assays were performed in triplicate using the luciferase reporter assay system (Promega) according to the manufacturer’s instructions.
In vitro p53 deacetylation assay
Equal amounts of GST and GST–p53 (2 µg) were first acetylated in vitro using a His-p300(1071–1715) fusion protein (20 000 c.p.m. activity on histones) and [14C]acetylCoA (NEN) in IPH buffer as described previously by Bannister and Kouzarides (1996). Samples were then washed, equilibrated in 100 µl of NAD–HDAC buffer and recombinant active GST–SIRT1 (20 000 c.p.m. deacetylase activity on H4 peptide) was added to the reactions in the presence or absence of 1 mM NAD+. Two micromolar TSA was added to all samples. Reactions were incubated at 37°C for 90 min before being resolved by SDS–PAGE and blotted on nitrocellulose membrane. Autoradiography was performed at –70°C overnight.
Acknowledgments
Acknowledgements
We thank E.Appella for providing the anti-acetylK382p53 antibody and X.Lu for the mdm2-luc plasmid. We would like to thank Andy Bannister for suggestions and critical reading of the manuscript. E.L. was funded by a grant from Cancer Research UK, U.M.B. by an HFSP grant (RG-196/98). S.M. and P.G.P. were funded by grants from the Italian Association for Cancer Research, the Italian Foundation for Cancer Research, the Italian Ministry of Health and the European Community.
References
- Afshar G. and Murnane,J.P. (1999) Characterization of a human gene with sequence homology to Saccharomyces cerevisiae SIR2. Gene, 234, 161–168. [DOI] [PubMed] [Google Scholar]
- Alcalay M., Tomassoni,L., Colombo,E., Stoldt,S., Grignani,F., Fagioli,M., Szekely,L., Helin,K. and Pelicci,P.G. (1998) The promyelocytic leukemia gene product (PML) forms stable complexes with the retinoblastoma protein. Mol. Cell. Biol., 18, 1084–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appella E. and Anderson,C.W. (2001) Post-translational modifications and activation of p53 by genotoxic stresses. Eur. J. Biochem., 268, 2764–2772. [DOI] [PubMed] [Google Scholar]
- Avantaggiati M.L., Ogryzko,V., Gardner,K., Giordano,A., Levine,A.S. and Kelly,K. (1997) Recruitment of p300/CBP in p53-dependent signal pathways. Cell, 89, 1175–1184. [DOI] [PubMed] [Google Scholar]
- Bannister A.J. and Kouzarides,T. (1996) The CBP co-activator is a histone acetyltransferase. Nature, 384, 641–643. [DOI] [PubMed] [Google Scholar]
- Barlev N.A., Liu,L., Chehab,N.H., Mansfield,K., Harris,K.G., Halazonetis,T.D. and Berger,S.L. (2001) Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell, 8, 1243–1254. [DOI] [PubMed] [Google Scholar]
- Boisvert F.M., Kruhlak,M.J., Box,A.K., Hendzel,M.J. and Bazett-Jones,D.P. (2001) The transcription coactivator CBP is a dynamic component of the promyelocytic leukemia nuclear body. J. Cell Biol., 152, 1099–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann C.B., Sherman,J.M., Devine,S.E., Cameron,E.E., Pillus,L. and Boeke,J.D. (1995) The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev., 9, 2888–2902. [DOI] [PubMed] [Google Scholar]
- Brehm A., Miska,E.A., McCance,D.J., Reid,J.L., Bannister,A.J. and Kouzarides,T. (1998) Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature, 391, 597–601. [DOI] [PubMed] [Google Scholar]
- Campisi J. (1996) Replicative senescence: an old lives’ tale? Cell, 84, 497–500. [DOI] [PubMed] [Google Scholar]
- Campisi J. (2001) Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol., 11, S27–S31. [DOI] [PubMed] [Google Scholar]
- Cockell M.M. and Gasser,S.M. (1999) The nucleolus: nucleolar space for RENT. Curr. Biol., 9, R575–R576. [DOI] [PubMed] [Google Scholar]
- Contegno F., Cioce,M., Pelicci,P.G. and Minucci,S. (2002) Targeting protein inactivation through an oligomerization chain reaction. Proc. Natl Acad. Sci. USA, 99, 1865–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri G.P. et al. (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl Acad. Sci. USA, 92, 9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucas V., Tini,M., Egan,D.A. and Evans,R.M. (1999) Modulation of CREB binding protein function by the promyelocytic (PML) oncoprotein suggests a role for nuclear bodies in hormone signaling. Proc. Natl Acad. Sci. USA, 96, 2627–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downes M., Ordentlich,P., Kao,H.Y., Alvarez,J.G. and Evans,R.M. (2000) Identification of a nuclear domain with deacetylase activity. Proc. Natl Acad. Sci. USA, 97, 10330–10335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa J.M. and Emerson,B.M. (2001) Transcriptional regulation by p53 through intrinsic DNA/chromatin binding and site-directed cofactor recruitment. Mol. Cell, 8, 57–69. [DOI] [PubMed] [Google Scholar]
- Everett R.D., Lomonte,P., Sternsdorf,T., van Driel,R. and Orr,A. (1999) Cell cycle regulation of PML modification and ND10 composition. J. Cell Sci., 112, 4581–4588. [DOI] [PubMed] [Google Scholar]
- Ferbeyre G., de Stanchina,E., Querido,E., Baptiste,N., Prives,C. and Lowe,S.W. (2000) PML is induced by oncogenic ras and promotes premature senescence. Genes Dev., 14, 2015–2027. [PMC free article] [PubMed] [Google Scholar]
- Finnin M.S., Donigian,J.R. and Pavletich,N.P. (2001) Structure of the histone deacetylase SIRT2. Nature Struct. Biol., 8, 621–625. [DOI] [PubMed] [Google Scholar]
- Fogal V. et al. (2000) Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J., 19, 6185–6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye R.A. (1999) Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem. Biophys. Res. Commun., 260, 273–279. [DOI] [PubMed] [Google Scholar]
- Frye R.A. (2000) Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun., 273, 793–798. [DOI] [PubMed] [Google Scholar]
- Fuks F., Burgers,W.A., Brehm,A., Hughes-Davies,L. and Kouzarides,T. (2000) DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nature Genet., 24, 88–91. [DOI] [PubMed] [Google Scholar]
- Gartenberg M.R. (2000) The Sir proteins of Saccharomyces cerevisiae: mediators of transcriptional silencing and much more. Curr. Opin. Microbiol., 3, 132–137. [DOI] [PubMed] [Google Scholar]
- Gray S.G. and Ekstrom,T.J. (2001) The human histone deacetylase family. Exp. Cell Res., 262, 75–83. [DOI] [PubMed] [Google Scholar]
- Grignani F., Kinsella,T., Mencarelli,A., Valtieri,M., Riganelli,D., Lanfrancone,L., Peschle,C., Nolan,G.P. and Pelicci,P.G. (1998) High-efficiency gene transfer and selection of human hematopoietic progenitor cells with a hybrid EBV/retroviral vector expressing the green fluorescence protein. Cancer Res., 58, 14–19. [PubMed] [Google Scholar]
- Gu W. and Roeder,R.G. (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell, 90, 595–606. [DOI] [PubMed] [Google Scholar]
- Guarente L. (1999) Diverse and dynamic functions of the Sir silencing complex. Nature Genet., 23, 281–285. [DOI] [PubMed] [Google Scholar]
- Guarente L. (2000) Sir2 links chromatin silencing, metabolism, and aging. Genes Dev., 14, 1021–1026. [PubMed] [Google Scholar]
- Guarente L. and Kenyon,C. (2000) Genetic pathways that regulate ageing in model organisms. Nature, 408, 255–262. [DOI] [PubMed] [Google Scholar]
- Haber J.E. (1999) Sir-Ku-itous routes to make ends meet. Cell, 97, 829–832. [DOI] [PubMed] [Google Scholar]
- Hodges M., Tissot,C., Howe,K., Grimwade,D. and Freemont,P.S. (1998) Structure, organization, and dynamics of promyelocytic leukemia protein nuclear bodies. Am. J. Hum. Genet., 63, 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S., Armstrong,C.M., Kaeberlein,M. and Guarente,L. (2000) Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature, 403, 795–800. [DOI] [PubMed] [Google Scholar]
- Itahana K., Dimri,G. and Campisi,J. (2001) Regulation of cellular senescence by p53. Eur. J. Biochem., 268, 2784–2791. [DOI] [PubMed] [Google Scholar]
- Ishov A.M., Sotnikov,A.G., Negorev,D., Vladimirova,O.V., Neff,N., Kamitani,T., Yeh,E.T., Strauss,J.F.,III and Maul,G.G. (1999) PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J. Cell Biol., 147, 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito A., Lai,C.H., Zhao,X., Saito,S., Hamilton,M.H., Appella,E. and Yao,T.P. (2001) p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J., 20, 1331–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K., Shiels,C. and Freemont,P.S. (2001) PML protein isoforms and the RBCC/TRIM motif. Oncogene, 20, 7223–7233. [DOI] [PubMed] [Google Scholar]
- Jenuwein T. and Allis,C.D. (2001) Translating the histone code. Science, 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M., McVey,M. and Guarente,L. (1999) The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev., 13, 2570–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M.M. et al. (2001) Role of PML and PML-RARα in Mad-mediated transcriptional repression. Mol. Cell, 7, 1233–1243. [DOI] [PubMed] [Google Scholar]
- Khochbin S., Verdel,A., Lemercier,C. and Seigneurin-Berny,D. (2001) Functional significance of histone deacetylase diversity. Curr. Opin. Genet. Dev., 11, 162–166. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. (1999) Histone acetylases and deacetylases in cell proliferation. Curr. Opin. Genet. Dev., 9, 40–48. [DOI] [PubMed] [Google Scholar]
- Lallemand-Breitenbach V. et al. (2001) Role of promyelocytic leukemia (PML) sumolation in nuclear body formation, 11S proteasome recruitment, and As2O3-induced PML or PML/retinoic acid receptor α degradation. J. Exp. Med., 193, 1361–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry J., Sutton,A., Tafrov,S.T., Heller,R.C., Stebbins,J., Pillus,L. and Sternglanz,R. (2000) The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc. Natl Acad. Sci. USA, 97, 5807–5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.J., Defossez,P.A. and Guarente,L. (2000) Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science, 289, 2126–2128. [DOI] [PubMed] [Google Scholar]
- Liu L., Scolnick,D.M., Trievel,R.C., Zhang,H.B., Marmorstein,R., Halazonetis,T.D. and Berger,S.L. (1999) p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell. Biol., 19, 1202–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J., Su,F., Chen,D., Shiloh,A. and Gu,W. (2000) Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature, 408, 377–381. [DOI] [PubMed] [Google Scholar]
- Luo J., Nikolaev,A.Y., Imai,S., Chen,D., Su,F., Shiloh,A., Guarente,L. and Gu,W. (2001) Negative control of p53 by Sir2α promotes cell survival under stress. Cell, 107, 137–148. [DOI] [PubMed] [Google Scholar]
- Martinez-Balbas M.A., Bannister,A.J., Martin,K., Haus-Seuffert,P., Meisterernst,M. and Kouzarides,T. (1998) The acetyltransferase activity of CBP stimulates transcription. EMBO J., 17, 2886–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matera A.G. (1999) Nuclear bodies: multifaceted subdomains of the interchromatin space. Trends Cell Biol., 9, 302–309. [DOI] [PubMed] [Google Scholar]
- McNeil S., Javed,A., Harrington,K.S., Lian,J.B., Stein,J.L., van Wijnen,A.J. and Stein,G.S. (2000) Leukemia-associated AML1/ETO (8;21) chromosomal translocation protein increases the cellular representation of PML bodies. J. Cell Biochem., 79, 103–112. [PubMed] [Google Scholar]
- Melnick A. and Licht,J.D. (1999) Deconstructing a disease: RARα, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood, 93, 3167–3215. [PubMed] [Google Scholar]
- Min J., Landry,J., Sternglanz,R. and Xu,R.M. (2001) Crystal structure of a SIR2 homolog–NAD complex. Cell, 105, 269–279. [DOI] [PubMed] [Google Scholar]
- Morgenstern J.P. and Land,H. (1990) Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res., 18, 3587–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu Z.M., Chin,K.V., Liu,J.H., Lozano,G. and Chang,K.S. (1994) PML, a growth suppressor disrupted in acute promyelocytic leukemia. Mol. Cell. Biol., 14, 6858–6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S., Matunis,M.J. and Dejean,A. (1998) Conjugation with the ubiquitin-related modifier SUMO-1 regulates the partitioning of PML within the nucleus. EMBO J., 17, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muth V., Nadaud,S., Grummt,I. and Voit,R. (2001) Acetylation of TAF(I)68, a subunit of TIF-IB/SL1, activates RNA polymerase I transcription. EMBO J., 20, 1353–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogryzko V.V., Hirai,T.H., Russanova,V.R., Barbie,D.A. and Howard,B.H. (1996) Human fibroblast commitment to a senescence-like state in response to histone deacetylase inhibitors is cell cycle dependent. Mol. Cell. Biol., 16, 5210–5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouchi T., Monteiro,A.N., August,A., Aaronson,S.A. and Hanafusa,H. (1998) BRCA1 regulates p53-dependent gene expression. Proc. Natl Acad. Sci. USA, 95, 2302–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson M. and Pelicci,P.G. (2001) PML interaction with p53 and its role in apoptosis and replicative senescence. Oncogene, 20, 7250–7256. [DOI] [PubMed] [Google Scholar]
- Pearson M. et al. (2000) PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature, 406, 207–210. [DOI] [PubMed] [Google Scholar]
- Perrod S., Cockell,M.M., Laroche,T., Renauld,H., Ducrest,A.L., Bonnard,C. and Gasser,S.M. (2001) A cytosolic NAD-dependent deacetylase, Hst2p, can modulate nucleolar and telomeric silencing in yeast. EMBO J., 20, 197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quignon F., De Bels,F., Koken,M., Feunteun,J., Ameisen,J.C. and de The,H. (1998) PML induces a novel caspase-independent death process. Nature Genet., 20, 259–265. [DOI] [PubMed] [Google Scholar]
- Ryan K.M., Phillips,A.C. and Vousden,K.H. (2001) Regulation and function of the p53 tumor suppressor protein. Curr. Opin. Cell Biol., 13, 332–337. [DOI] [PubMed] [Google Scholar]
- Sakaguchi K., Herrera,J.E., Saito,S., Miki,T., Bustin,M., Vassilev,A., Anderson,C.W. and Appella,E. (1998) DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev., 12, 2831–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve A.A., Celic,I., Avalos,J., Deng,H., Boeke,J.D. and Schramm,V.L. (2001) Chemistry of gene silencing: the mechanism of NAD+-dependent deacetylation reactions. Biochemistry, 40, 15456–15463. [DOI] [PubMed] [Google Scholar]
- Scolnick D.M., Chehab,N.H., Stavridi,E.S., Lien,M.C., Caruso,L., Moran,E., Berger,S.L. and Halazonetis,T.D. (1997) CREB-binding protein and p300/CBP-associated factor are transcriptional coactivators of the p53 tumor suppressor protein. Cancer Res., 57, 3693–3696. [PubMed] [Google Scholar]
- Seeler J.S. and Dejean,A. (1999) The PML nuclear bodies: actors or extras? Curr. Opin. Genet. Dev., 9, 362–367. [DOI] [PubMed] [Google Scholar]
- Seeler J.S. and Dejean,A. (2001) SUMO: of branched proteins and nuclear bodies. Oncogene, 20, 7243–7249. [DOI] [PubMed] [Google Scholar]
- Serrano M., Lin,A.W., McCurrach,M.E., Beach,D. and Lowe,S.W. (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell, 88, 593–602. [DOI] [PubMed] [Google Scholar]
- Sherman J.M., Stone,E.M., Freeman-Cook,L.L., Brachmann,C.B., Boeke,J.D. and Pillus,L. (1999) The conserved core of a human SIR2 homologue functions in yeast silencing. Mol. Biol. Cell, 10, 3045–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.S. et al. (2000) A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc. Natl Acad. Sci. USA, 97, 6658–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl B.D. and Allis,C.D. (2000) The language of covalent histone modifications. Nature, 403, 41–45. [DOI] [PubMed] [Google Scholar]
- Tanner K.G., Landry,J., Sternglanz,R. and Denu,J.M. (2000) Silent information regulator 2 family of NAD-dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose. Proc. Natl Acad. Sci. USA, 97, 14178–14182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanny J.C. and Moazed,D. (2001) Coupling of histone deacetylation to NAD breakdown by the yeast silencing protein Sir2: evidence for acetyl transfer from substrate to an NAD breakdown product. Proc. Natl Acad. Sci. USA, 98, 415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanny J.C., Dowd,G.J., Huang,J., Hilz,H. and Moazed,D. (1999) An enzymatic activity in the yeast Sir2 protein that is essential for gene silencing. Cell, 99, 735–745. [DOI] [PubMed] [Google Scholar]
- Tissenbaum H.A. and Guarente,L. (2001) Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature, 410, 227–230. [DOI] [PubMed] [Google Scholar]
- Tyner S.D. et al. (2002) p53 mutant mice that display early ageing-associated phenotypes. Nature, 415, 45–53. [DOI] [PubMed] [Google Scholar]
- Vaziri H., Dessain,S.K., Ng Eaton,E., Imai,S.I., Frye,R.A., Pandita,T.K., Guarente,L. and Weinberg,R.A. (2001) hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell, 107, 149–159. [DOI] [PubMed] [Google Scholar]
- Vogelstein B., Lane,D. and Levine,A.J. (2000) Surfing the p53 network. Nature, 408, 307–310. [DOI] [PubMed] [Google Scholar]
- Wang Z.G., Ruggero,D., Ronchetti,S., Zhong,S., Gaboli,M., Rivi,R. and Pandolfi,P.P. (1998) PML is essential for multiple apoptotic pathways. Nature Genet., 20, 266–272. [DOI] [PubMed] [Google Scholar]
- Wu W.S., Vallian,S., Seto,E., Yang,W.M., Edmondson,D., Roth,S. and Chang,K.S. (2001) The growth suppressor PML represses transcription by functionally and physically interacting with histone deacetylases. Mol. Cell. Biol., 21, 2259–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S., Muller,S., Ronchetti,S., Freemont,P.S., Dejean,A. and Pandolfi,P.P. (2000a) Role of SUMO-1-modified PML in nuclear body formation. Blood, 95, 2748–2752. [PubMed] [Google Scholar]
- Zhong S., Salomoni,P. and Pandolfi,P.P. (2000b) The transcriptional role of PML and the nuclear body. Nature Cell Biol., 2, E85–E90. [DOI] [PubMed] [Google Scholar]
- Zhu J., Koken,M.H., Quignon,F., Chelbi-Alix,M.K., Degos,L., Wang,Z.Y., Chen,Z. and de The,H. (1997) Arsenic-induced PML targeting onto nuclear bodies: implications for the treatment of acute promyelocytic leukemia. Proc. Natl Acad. Sci. USA, 94, 3978–3983. [DOI] [PMC free article] [PubMed] [Google Scholar]