

Abstract
The vesicular stomatitis virus (VSV) matrix protein (M) interacts with cellular membranes, self-associates and plays a major role in virus assembly and budding. We present the crystallographic structure, determined at 1.96 Å resolution, of a soluble thermolysin resistant core of VSV M. The fold is a new fold shared by the other vesiculovirus matrix proteins. The structure accounts for the loss of stability of M temperature-sensitive mutants deficient in budding, and reveals a flexible loop protruding from the globular core that is important for self-assembly. Membrane floatation shows that, together with the M lysine-rich N-terminal peptide, a second domain of the protein is involved in membrane binding. Indeed, the structure reveals a hydrophobic surface located close to the hydrophobic loop and surrounded by conserved basic residues that may constitute this domain. Lastly, comparison of the negative-stranded virus matrix proteins with retrovirus Gag proteins suggests that the flexible link between their major membrane binding domain and the rest of the structure is a common feature shared by these proteins involved in budding and virus assembly.
Keywords: budding/matrix protein/membrane interaction/structure/vesicular stomatitis virus
Introduction
Enveloped viruses acquire their membranes by budding from host cell membranes. One viral protein, the matrix protein, plays a major role in budding of negative-stranded viruses, including orthomyxoviruses (Gomez-Puertas et al., 2000), filoviruses (Timmins et al., 2001) or rhabdoviruses (Flamand, 1970; Mebatsion et al., 1999). Vesicular stomatitis virus (VSV) has long served as a model for studies of rhabdovirus budding. VSV is an enveloped virus with a single-stranded non-segmented RNA genome of negative polarity coding for five proteins; its genome is associated with the nucleoprotein (N), the large protein (L) and the phosphoprotein (P) to form the nucleocapsid. Together, L and P proteins form the RNA-dependent RNA polymerase involved in the transcription of the genome in viral mRNAs, and in its replication. The matrix protein (M) is involved in later steps of the infectious cycle. It condenses the nucleocapsid into a tightly coiled helical structure (Newcomb and Brown, 1981; Newcomb et al., 1982) and, probably as a consequence, inhibits the transcription of the viral genome (Clinton et al., 1978; Martinet et al., 1979). This tightly coiled structure seems to be formed near cellular membranes where M is bound (Odenwald et al., 1986; Flood and Lyles, 1999), and gets enveloped during budding by a lipid bilayer containing the viral glycoprotein. Strong evidence that M plays a major role in VSV budding comes from studies of VSV temperature-sensitive (ts) mutants affected in budding that were shown to be mutated in the M gene and were complemented by wild-type plasmid-derived M (Flamand, 1970; Lyles et al., 1996).
M is a small 229 amino acid protein (26.6 kDa) com prising a positively charged N-terminal part (there are eight lysine residues in the first 20 residues). The function of M is not limited to viral assembly and budding. M expression causes the rounding of cells, a phenotype characteristic of VSV infection (Blondel et al., 1990; Melki et al., 1994). M also enhances the availability of the cellular translation machinery by inhibiting the export of RNAs from the nucleus (Her et al., 1997; Petersen et al., 2000; von Kobbe et al., 2000). In Xenopus oocytes, this inhibition is triggered by the interaction of M with nuclear pores (Petersen et al., 2000; von Kobbe et al., 2000) and may be related to M inhibition of host cell transcription (Black and Lyles, 1992; Black et al., 1994).
Two biochemical properties of VSV M have been studied: auto-association and membrane binding. VSV M self-associates into large multimers at physiological NaCl concentration (McCreedy et al., 1990), a process initiated by trypsin-sensitive nucleation sites. Increasing the salt concentration reverses this self-association (Gaudin et al., 1995). M self-association could be important for nucleocapsid compaction and may constitute a driving force for budding. VSV M cell membrane association is also necessary for budding (Lenard, 1996). This interaction was proven to be extremely stable in vivo as it is not destabilized by harsh treatments such as 2 M KCl or pH 11 (Bergmann and Fusco, 1988; Chong and Rose, 1993, 1994). The regions of the protein involved in membrane association are not completely defined: hydrophobic photolabelling using [125I]TID has identified the N-terminal region of the protein (Lenard and Vanderoef, 1990), but M mutants deleted from the N-terminal domain still interact with cellular membranes, suggesting that another domain of the protein is involved (Chong and Rose, 1994; Ye et al., 1994).
Due to its tendency to self-associate, M could not be crystallized. In an attempt to isolate a fragment of VSV M for crystallization, we had shown previously that trypsin-cleaved M [Mt, residues 44–229 (Ogden et al., 1986)] is recruited in M aggregates, but does not aggregate on its own, and thermolysin-cleaved M (Mth) neither self-associates nor is recruited in aggregates (Figure 1A; Gaudier et al., 2001). Here we report the three-dimensional structure of the Mth of VSV Indiana (Orsay strain), which is the first structure of a rhabdovirus protein to be determined. The Mth fold is a new fold. The structure shows that M of VSV ts mutants deficient in budding is destabilized with respect to the wild type, which accounts for their phenotype. The structure also enables us to identify the regions of M involved in its interaction with membranes. Comparison to the structures and domain organization of other negative-stranded virus matrix proteins and of retrovirus Gag proteins suggests that these viruses employ a similar strategy for the membrane targeting part of the budding process.

Fig. 1. (A) Schematic of VSV M sequence. Mt and Mth are trypsin and thermolysin digestion products of M, respectively; Mth is the species whose structure was determined crystallographically. (B) Ribbon diagram of VSV M. Rainbow colouring, from blue to red, describes the N-terminal to C-terminal direction of the polypeptide chain. The interruption of the chain in the large five-stranded β-sheet corresponds to the thermolysin cleavage in the 121–128 loop that connects the second strand to the third. (C) Topology diagram of VSV M. β-strands are represented as arrows and α-helices as rectangles. Colour code is as in (A). This figure and Figure 3 were drawn with MOLSCRIPT (Kraulis, 1991) and Raster3D (Merritt and Bacon, 1997).
Results and discussion
Structure determination and description
M purified from virus grown on BHK cells was cleaved by thermolysin to obtain a resistant core Mth. This core, composed of two associated fragments [48–121 and 122 (or 123 or 124, depending on the thermolysin cleavage site) –229], was crystallized (Gaudier et al., 2001) and the structure was determined by the single isomorphous replacement with anomalous scattering method using a native crystal and a mercury derivative (data and phasing statistics are shown in Table I). The experimental maps calculated after SHARP and DM at 1.96 Å resolution enabled us to trace most of the polypeptide chain. This made it clear that the single mercury atom per asymmetric unit is not bound to the single M cysteine residue, but close to histidine 143 and threonine 80; this is due to an oxidation of the M single cysteine residue to cysteic acid, which makes this residue unavailable for complexation with methyl mercury, as shown by analysis of difference electron density maps following refinement. The refined model comprises M residues 58–121 and 128–227, and 115 water molecules; the crystallographic R factor is 20.6% (free R factor, 24%) at 1.96 Å resolution. Co ordinates have been deposited with the Protein Data Bank (PDB; accession code 1LG7).
Table I. Crystallographic statistics.
| Data collection | Native | MeHgCl | ||||
|---|---|---|---|---|---|---|
| Wavelength (Å) | 1.00 | 0.98 | ||||
| Resolution (Å) | 1.96 (2.01) | 2.6 (2.66) | ||||
| Reflections | 209 741 | 79 001 | ||||
| Unique reflections | 12 771 | 9425 | ||||
| Completeness (%) | 99.6 (95.3) | 91.8 (88.7) | ||||
| Rsym (%) | 6.9 (41) | 6.2 (18.3) | ||||
|
I/σ |
|
|
30 (3.5) |
|
11.6 (3.9) |
|
| Phasing |
|
|
|
|
|
|
| Riso (%) | 13 | |||||
| Phasing power | RCullis | FOM | ||||
| |
(ano) 1.27 |
(iso) 2.25 |
(ano) 0.88 |
(iso) 0.63 |
acentric 0.48 |
centric 0.40 |
| Refinement |
|
|
|
|
|
|
| No. of reflections in working set | 12 032 | |||||
| Rcryst (%) | 20.6 (23.9) | |||||
| Rfree (%) | 24.1 (32.7) | |||||
| Average B (Å2) | 29.7 | |||||
| Residues | 164 (out of 182) | |||||
| Bond length r.m.s.d. (Å) | 0.005 | |||||
| Bond angles r.m.s.d. (°) | 1.47 | |||||
| Water molecules | 115 | |||||
Mth is a single globular domain (Figure 1B and C) with dimensions 40 × 25 × 20 Å3. The N-terminal part is composed of a large five-stranded anti-parallel β-sheet (β1–β5) packed against two α-helices (α1 and α2); the C-terminal part comprises a small two-stranded anti-parallel β-sheet (β6 and β7) and an α-helix (α3). The N-terminal and C-terminal parts are connected by a 20 amino acid residue peptide without any defined secondary structure that links strand β5 to helix α3. We observed two kinds of interaction that give rise to a stable assembly of the N- and C-terminal parts of Mth: the three α-helices are packed together around a hydrophobic core, and strands β3 and β7 establish parallel β-sheet interactions. The cleavages by thermolysin between residues 121 and 124 take place in a surface loop delimited by prolines 120 and 129 that connects strands β2 and β3. This loop contains the hydrophobic peptide PAVLA that is involved in M self-assembly (Gaudier et al., 2001). Because of the multiple cleavage products of M due to the limited specificity of thermolysin, we could not determine whether residues 122–123 had been cleaved off the crystallized fragments or were present in the structure, but not sufficiently ordered to be visible in electron density maps.
The matrix proteins of several vesiculoviruses [VSV–Orsay and New Jersey strains, Chandipura virus, Piry virus and spring viremia carp virus (SVCV)] have been sequenced; an alignment of these sequences is presented in Figure 2. These proteins have a low identity with the VSV Orsay strain (∼60, 40, 40 and 20%, respectively) but a significant similarity (∼80, 50, 50 and 50%, respectively), as previously reported (Barge et al., 1996; Taylor et al., 1999). Twelve positions of conserved residues are on the surface of M and the corresponding total area is <5% of the total accessible surface area of the Mth fragment, demonstrating a very low degree of conservation of M surface among vesiculoviruses. By contrast, most of the conserved residues (22 out of 34) are buried in the structure of the protein and their conservation is due to folding or stability constraints. This strongly suggests that the VSV M fold is also conserved amongst all vesiculovirus matrix proteins.

Fig. 2. Sequence alignment of the vesiculovirus Ms from strains Orsay (DDJB/EMBL/GenBank accession No. J02428_3), New Jersey (accession No. M14553_1), Chandipura (accession No. AF128868_2), Piry (accession No. D26175_2) and spring viremia carp virus (accession No. U18101_3) was performed with Clustal_W (Thompson et al., 1994). Conserved and similar residues visible in the structure are dark and light coloured, respectively. Blue, buried residues (side chain or glycine Cα accessible surface <10 Å2); red, surface residues (side chain or glycine Cα accessible surface >10 Å2).
We have searched the PDB (Bernstein et al., 1977) for proteins that are structurally homologous to M, using the program DALI (Holm and Sander, 1993). No match of statistical significance was found, the best match being with a phospholipase a2 fragment (PDB accession code 1rlw), an Ig-like protein, and aligns only the five-stranded β-sheet of M. Therefore, the vesiculovirus matrix protein fold represents a new fold.
M temperature-sensitive mutants
VSV ts mutants have been extensively studied and classified in complementation groups. One of these groups is characterized by a default in budding: RNA and proteins are produced at normal levels, yet the production of viral particles is decreased ∼1000-fold at the non-permissive temperature (Flamand, 1970); these viruses are mutated in the M protein. Four of the mutations that have been characterized are located in the Mth core (Figure 3). One of these mutants, L111F, aggregates at the non-permissive temperature, as opposed to wild-type M or to a revertant (Lyles, 2000). This is accounted for by the structure; leucine 111 is surrounded by hydrophobic residues and there is no space to accommodate the aromatic ring of the mutant phenylalanine. Therefore, the structure of the ts mutant is expected to be less stable than that of wild-type M, as suggested by its biochemical behaviour. The structure suggests that two additional ts mutant M proteins have similar properties: mutation A133D substitutes a charged residue to an alanine located in a hydrophobic environment, and mutations F110S and K204T are substitutions that introduce hydrophilic groups in the hydrophobic environments of the phenylalanine side chain and the aliphatic part of the lysine side chain. Three of the four residues substituted in the three mutants listed so far are in the interior of the protein (their accessible surface is <10 Å2), so that a change at these positions is unlikely to affect directly the interaction of M with viral or cellular proteins; therefore, it is most likely that the ts phenotype of these three mutants is associated to a lower stability of M, as established in the case of the L111F substitution.

Fig. 3. VSV ts mutants in M that are deficient in budding. Each view is a close-up of the structure around a position where a ts mutation that confers budding deficiency to VSV has been identified; in each case the position of the mutation and the amino acid substitution are indicated, together with the most common designation of the mutant (in parentheses). Mutated residues (blue) and those that contact them (purple) are drawn as ball and stick models.
The substitution of the fourth ts mutant located in the Mth core involves residues on the surface of the protein. The mutant TsG31, with a K→E substitution at position 215, disrupts a hydrogen bond between the central β-sheet and the C-terminal small two-stranded β-sheet of the protein. Residue 215 is not conserved among vesiculoviruses (it is changed to a serine or a glutamine in other members of this family of viruses) so that it is unlikely to be involved in an interaction with a cellular partner that vesiculoviruses would have in common. Since the K215E mutation destabilizes the interaction of the two parts of the Mth core, it is likely that, in this case too, the ts phenotype is due to M destabilization.
Membrane interaction
Membrane association of M has been demonstrated both in vitro (Zakowski et al., 1981) and in vivo (Chong and Rose, 1994). In order to define the regions of the structure that are involved in the interaction of M with phospholipids, we performed membrane floatation experiments. We used, on the one hand, negatively charged liposomes made with phosphatidylcholine and phosphatidylserine in identical quantities, meant to mimic the internal face of the plasma or VSV membranes (Zakowski et al., 1981), and, on the other hand, either M or the fragments of M that we have characterized, i.e. Mt and Mth cores (Figure 1A). Figure 4 summarizes our results. M binds strongly to liposomes (Figure 4A). This interaction is weakened in the presence of 700 mM NaCl (Figure 4B). This is consistent with previous data on M membrane association (Zakowski et al., 1981).
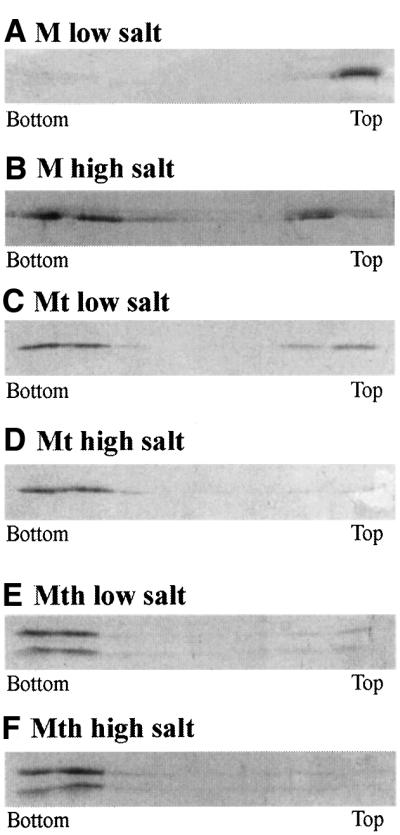
Fig. 4. Membrane floatation of M, Mt core and Mth core in the presence of PC:PS liposomes. Eight fractions were collected from each gradient and samples from each fraction were separated on SDS 14% PAGE and stained with Coomassie Blue.
The interaction of Mt with liposomes (Figure 4C) is weaker than that of M, which is consistent with the proposal that the lysine rich N-terminal part of the protein makes a major contribution to binding to negatively charged liposomes, and to interaction with the viral mem brane (Lenard and Vanderoef, 1990). As observed with M, the interaction of Mt with membranes is prevented in the presence of 700 mM NaCl, suggesting that this association also involves electrostatic interactions which, in M, come in addition to those of the N-terminal part.
Membrane association of Mth is hardly detectable (Figure 4E). The main difference between Mth and Mt is a cut in the hydrophobic pentapeptide Pro120–Ala124, which is embedded in a disordered loop on the surface of the protein. The membrane floatation results with Mth strongly suggest that this loop is involved in the asso ciation of Mt with membranes. The Pro120–Ala124 pentapeptide might contribute to Mt (or M) membrane association either by a direct interaction with the bilayer or by aggregating Mt (or M) proteins (Gaudier et al., 2001) that, individually, would bind more weakly to liposomes.
To summarize, three regions contribute to M binding to liposomes: its positively charged N-terminal part, which makes a major contribution, the 120–129 loop and, finally, a positively charged portion of Mth surface, which provides a smaller additional contribution. On the Mth surface, there are two main regions where the electrostatic potential is positive; the largest one is contributed by two arginine and three lysine residues, located at positions 76, 79, 117, 168 and 204, which surround the hydrophobic surface of residues Val84, Pro77, Ala118 and Tyr81 and are located close to the PAVLA hydrophobic peptide of the 120–129 loop (Figure 5). As discussed for a number of proteins (Bouma et al., 1999; Macedo-Ribeiro et al., 1999; Dessen et al., 2000), the combined presence of hydrophobic and basic residues suggests that this is a likely candidate for being the region responsible for Mth residual interaction with membranes. However, further studies are necessary to locate the exact membrane targeting sequence in Mth.

Fig. 5. Distribution of electrostatic potential on VSV M surface. Regions where the electrostatic potential is less than –10 kbT are red, while those where it is more than +10 kbT are blue (kb, Boltzmann constant; T, absolute temperature). A basic patch is clearly visible close to the PAVLA peptide and to a hydrophobic patch comprised of residues Val84, Pro77, Ala118 and Tyr81. This figure was drawn with GRASP (Nicholls et al., 1991).
The structures of negative-stranded virus matrix proteins
In addition to the structure of VSV M, crystal structures of domains of two negative single-stranded RNA virus matrix proteins have been determined: those of the influenza (Sha and Luo, 1997; Arzt et al., 2001) and Ebola viruses (Dessen et al., 2000). VSV M has been shown to play a pivotal role in viral assembly (Flamand, 1970; Lyles et al., 1996); the matrix proteins of the influenza or Ebola viruses are thought to do so, based, in particular, on the formation of particles that bud from cells transfected with the matrix protein alone (Gomez-Puertas et al., 2000; Timmins et al., 2001). As part of their function in virus assembly, these proteins bind to membranes or to negatively charged phospholipids (Zakowski et al., 1981; Zhang and Lamb, 1996; Ruigrok et al., 2000; Baudin et al., 2001). In addition to their involvement in budding, matrix proteins of negative single-stranded RNA viruses have functions that differ; for example, influenza M1 shuttles between the nucleus and the cytoplasm and is required for ribonucleoprotein transport from the nucleus (Bui et al., 2000), whereas VSV M inhibits the export of RNAs from the nucleus by binding to nuclear pores (Her et al., 1997; von Kobbe et al., 2000). Although their common biochemical properties and functions had led to the proposal that the negative-stranded RNA virus matrix proteins belong to the same family (Lenard, 1996), the sequences and folds of the domains of the three negative-stranded RNA virus matrix proteins whose structure has been determined differ. The difference of Ebola virus VP40 and VSV M folds is particularly intriguing because their genome organization suggests that rhabdoviruses and filoviruses share a common ancestor (Peters et al., 1996). Thus, the structural organization of negative-stranded RNA virus matrix proteins does not seem to be extensively constrained despite the functional similarities of these proteins.
In particular, two components of the budding processes of negative-stranded RNA viruses, membrane associa tion and the recruitment of cellular partners, share some features; as a consequence, the matrix proteins of negative-stranded RNA viruses have two characteristics in common. First, Ebola VP40 and VSV M share a PPXY motif that is known to be recognized by an E3 ubiquitin ligase (Harty et al., 2000, 2001); this motif has been shown to influence budding efficiency (Jayakar et al., 2000) through a mechanism that is shared by retroviruses (Craven et al., 1999) and still needs to be clarified. Secondly, all three matrix proteins self-associate and bind to membranes. VP40 forms defined oligomers (Scianimanico et al., 2000); VSV M and influenza M1 (Gomez-Puertas et al., 2000) form multimers or bind as multiple copies to the viral ribonucleoprotein complex. In all three cases, the sequences used for oligomerization or ribonucleoprotein binding are distinct from the major membrane binding domain (Kaptur et al., 1991; Scianimanico et al., 2000; Baudin et al., 2001). Furthermore, the structure of Ebola VP40 suggests that the link between its membrane binding and oligomerization domains is flexible (Scianimanico et al., 2000). Limited proteolysis results indicate that the link between the major membrane binding domain of VSV M (the N-terminal lysine-rich peptide) and its ribonucleoprotein binding domain (the Mt core) is also flexible (Kaptur et al., 1991); similar conclusions apply to influenza M1 (Baudin et al., 2001), and to the link between the capsid protein and the membrane targeting surface of MA in the Gag polyprotein of retroviruses (Matthews et al., 1996). This flexibility and the multiple copy number of these proteins allow, through an avidity effect, a strong membrane association of the virus core that will ultimately lead to particle budding.
Materials and methods
Data collection and processing
Purification and crystallization of the Mth core of VSV Indiana (Orsay strain) have been reported elsewhere (Gaudier et al., 2001). Crystals are in space group P42(1)2 with unit cell dimensions a = b = 80.7 Å, c = 51.7 Å and one molecule per asymmetric unit. A single heavy atom derivative was obtained by soaking a native crystal in 1 mM MeHgCl, 30% PEG 2000 monomethylether, 200 mM AcNa and 20 mM phosphate pH 8.5 for 12 h at 18°C. Crystals were cryoprotected in harvesting buffer to which glycerol was added to a final 20% v:v ratio and kept frozen at 110 K for data collection. Data were collected at beamline BM30 of the European Synchrotron Radiation Facility (ESRF, Grenoble, France), using a MAR image plate detector. Native data were collected to 1.96 Å resolution at 1 Å. SIRAS data were collected up to 2.6 Å near the L III shell edge of mercury. Integration, scaling and merging of diffraction intensity measurements were performed with the programs Denzo and Scalepack (Otwinowsky and Minor, 1997).
Phase determination, model building, refinement and analysis
A single mercury site was identified with the program SOLVE (Terwilliger and Berendzen, 1999) and further refinement was performed with the program SHARP (de la Fortelle and Bricogne, 1997). Density modification was performed with SOLOMON (Abrahams and Leslie, 1996), as implemented in SHARP, considering a solvent content of 40%. Sequence assignment and model building were performed manually with the program TURBO-FRODO (Roussel and Cambillau, 1991) after extension of the calculated phases from 2.6 to 1.96 Å with DM (Cowtan and Zhang, 1999). Atomic coordinates refinement was performed by alternating use of CNS (Brünger et al., 1998) with rounds of manual rebuilding. The final model includes 164 residues (58–121 and 128–227) and 118 water molecules.
In vitro membrane association
A total of 1 mg of phosphatidylserine (PS) and 1 mg of phosphatidylcholine (PC) dissolved in organic solvents were dried in vacuo. The lipid film was suspended in 1 ml of 150 mM NaCl and 20 mM Tris–HCl pH 7.5, and the mixture was bath-sonicated for 20 min. One hundred micrograms of M, its Mt core or its Mth core were incubated with 200 µg of liposomes at room temperature for 30 min. The final volume was 500 µl. After addition of the protein, NaCl concentration was either 150 or 700 mM. The volume of the mix was adjusted to 1 ml, 80% sucrose in 20 mM Tris–HCl pH 7.5 and 150 or 700 mM NaCl, overlaid with 3 ml of 60% sucrose and 1 ml of 10% sucrose either in 150 or 700 mM NaCl, and 20 mM Tris–HCl pH 7.5. Gradients were spun overnight at 50 000 r.p.m. in a SW55 rotor. Six hundred microlitre fractions were collected, out of which 30 µl samples were analyzed by SDS–PAGE. Two types of experiment were performed: low salt, where proteins and liposomes were incubated at 150 mM NaCl and spun in 150 mM NaCl gradients, and high salt, where proteins and liposomes were incubated at 700 mM NaCl then spun in 700 mM NaCl gradients.
Acknowledgments
Acknowledgements
We thank Anne Flamand for support, Christine Maheu for the production of VSV and Alain Roussel for initial advice on model building. We gratefully acknowledge Richard Kahn’s help with the use of the BM30 beamline at ESRF. This work is supported by CNRS (UPR 9053 and UPR 9063) and by the ‘Programme de Recherche Fondamentale en Microbiologie, Maladies Infectieuses et Parasitaires’.
References
- Abrahams J.P. and Leslie,A.G.W. (1996) Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr. D, 52, 30–42. [DOI] [PubMed] [Google Scholar]
- Arzt S., Baudin,F., Barge,A., Timmins,P., Burmeister,W.P. and Ruigrok,R.W. (2001) Combined results from solution studies on intact influenza virus M1 protein and from a new crystal form of its N-terminal domain show that M1 is an elongated monomer. Virology, 279, 439–446. [DOI] [PubMed] [Google Scholar]
- Barge A., Gagnon,J., Chaffotte,A., Timmins,P., Langowsky,J., Ruigrok,R. and Gaudin,Y. (1996) Rod-like shape of vesicular stomatitis virus matrix protein. Virology, 219, 465–470. [DOI] [PubMed] [Google Scholar]
- Baudin F., Petit,I., Weissenhorn,W. and Ruigrok,R.W. (2001) In vitro dissection of the membrane and RNP binding activities of influenza virus M1 protein. Virology, 281, 102–108. [DOI] [PubMed] [Google Scholar]
- Bergmann J. and Fusco,P. (1988) The M protein of vesicular stomatitis virus associates specifically with the basolateral membranes of polarized epithelial cells independently of the G protein. J. Cell Biol., 107, 1707–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein F.C., Koetzle,T.F., Williams,J.G.B., Meyer,E.F., Brice,M.D., Rodgers,J.R., Kennard,O., Shimanouchi,T. and Tasumi,M. (1977) The Protein Data Bank: a computer-based archival file for macromolecular structures. J. Mol. Biol., 112, 535–542. [DOI] [PubMed] [Google Scholar]
- Black B. and Lyles,D. (1992) Vesicular stomatitis virus matrix protein inhibits host cell-directed transcription of target genes in vivo. J. Virol., 66, 4058–4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black B., Brewer,G. and Lyles,D. (1994) Effect of vesicular stomatits virus matrix protein on host-directed translation in vivo. J. Virol., 68, 555–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondel D., Harmison,G. and Schubert,M. (1990) Role of matrix protein in cytopathogenesis of vesicular stomatitis virus. J. Virol., 64, 1716–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouma B., de Groot,P.G., van den Elsen,J.M., Ravelli,R.B., Schouten, A., Simmelink,M.J., Derksen,R.H., Kroon,J. and Gros,P. (1999) Adhesion mechanism of human β(2)-glycoprotein I to phospholipids based on its crystal structure. EMBO J., 18, 5166–5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Bui M., Wills,E.G., Helenius,A. and Whittaker,G.R. (2000) Role of the influenza virus M1 protein in nuclear export of viral ribonucleo proteins. J. Virol., 74, 1781–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong L. and Rose,J. (1993) Membrane association of functional vesicular stomatitis virus matrix protein in vivo. J. Virol., 67, 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong L. and Rose,J. (1994) Interaction of normal and mutant vesicular stomatitis virus matrix proteins with the plasma membrane and nucleocapsids. J. Virol., 68, 441–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinton G.M., Little,S.P., Hagen,F.S. and Huang,A.S. (1978) The matrix (M) protein of vesicular stomatitis virus regulates transcription. Cell, 15, 1455–1462. [DOI] [PubMed] [Google Scholar]
- Cowtan K.D. and Zhang,K.Y. (1999) Density modification for macromolecular phase improvement. Prog. Biophys. Mol. Biol., 72, 245–270. [DOI] [PubMed] [Google Scholar]
- Craven R.C., Harty,R.N., Paragas,J., Palese,P. and Wills,J.W. (1999) Late domain function identified in the vesicular stomatitis virus M protein by use of rhabdovirus-retrovirus chimeras. J. Virol., 73, 3359–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fortelle E. and Bricogne,G. (1997) Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol., 276, 472–494. [DOI] [PubMed] [Google Scholar]
- Dessen A., Volchkov,V., Dolnik,O., Klenk,H.D. and Weissenhorn,W. (2000) Crystal structure of the matrix protein VP40 from Ebola virus. EMBO J., 19, 4228–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamand A. (1970) Genetic study of vesicular stomatitis virus: classification of spontaneous thermosensitive mutants into comple mentation groups. J. Gen. Virol., 8, 187–195. [DOI] [PubMed] [Google Scholar]
- Flood E.A. and Lyles,D.S. (1999) Assembly of nucleocapsids with cytosolic and membrane-derived matrix proteins of vesicular stomatitis virus. Virology, 261, 295–308. [DOI] [PubMed] [Google Scholar]
- Gaudier M., Gaudin,Y. and Knossow,M. (2001) Cleavage of vesicular stomatitis virus matrix protein prevents self-association and leads to crystallization. Virology, 288, 308–314. [DOI] [PubMed] [Google Scholar]
- Gaudin Y., Barge,A., Ebel,C. and Ruigrok,R.W. (1995) Aggregation of VSV M protein is reversible and mediated by nucleation sites: implications for viral assembly. Virology, 206, 28–37. [DOI] [PubMed] [Google Scholar]
- Gomez-Puertas P., Albo,C., Perez-Pastrana,E., Vivo,A. and Portela,A. (2000) Influenza virus matrix protein is the major driving force in virus budding. J. Virol., 74, 11538–11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harty R.N., Brown,M.E., Wang,G., Huibregtse,J. and Hayes,F.P. (2000) A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: implications for filovirus budding. Proc. Natl Acad. Sci. USA, 97, 13871–13876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harty R.N., Brown,M.E., McGettigan,J.P., Wang,G., Jayakar,H.R., Huibregtse,J.M., Whitt,M.A. and Schnell,M.J. (2001) Rhabdoviruses and the cellular ubiquitin-proteasome system: a budding interaction. J. Virol., 75, 10623–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Her L., Lund,E. and Dahlberg,J. (1997) Inhibition of Ran guanosine triphosphate-dependant nuclear transport by the matrix protein of vesicular stomatitis virus. Science, 276, 1845–1848. [DOI] [PubMed] [Google Scholar]
- Holm L. and Sander,C. (1993) Protein structure comparison by alignment of distance matrices. J. Mol. Biol., 233, 123–138. [DOI] [PubMed] [Google Scholar]
- Jayakar H.R., Murti,K.G. and Whitt,M. (2000) Mutations in the PPPY motif of vesicular stomatitis virus matrix protein reduce virus budding by inhibiting a late step in virion release. J. Virol., 74, 9818–9827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaptur P.E., Rhodes,R.B. and Lyles,D.S. (1991) Sequences of the vesicular stomatitis virus matrix protein involved in binding to nucleocapsids. J. Virol., 65, 1057–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis P. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of proteins structures. J. Appl. Crystallogr., 24, 924–950. [Google Scholar]
- Lenard J. (1996) Negative-strand virus M and retrovirus MA proteins: all in a family? Virology, 216, 289–298. [DOI] [PubMed] [Google Scholar]
- Lenard J. and Vanderoef,R. (1990) Localization of the membrane-associated region of vesicular stomatitis virus M protein at the N terminus, using the hydrophobic, photoreactive probe 125I-TID. J. Virol., 64, 3486–3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyles D.S. (2000) Cytopathogenesis and inhibition of host gene expression by RNA viruses. Microbiol. Mol. Biol. Rev., 64, 709–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyles D.S., McKenzie,M., Kaptur,P., Grant,K. and Jerome,W. (1996) Complementation of M gene mutants of vesicular stomatitis virus by plasmid-derived M protein converts spherical extracellular particles into native bullet shapes. Virology, 217, 76–87. [DOI] [PubMed] [Google Scholar]
- Macedo-Ribeiro S. et al. (1999) Crystal structures of the membrane-binding C2 domain of human coagulation factor V. Nature, 402, 434–439. [DOI] [PubMed] [Google Scholar]
- Martinet C., Combard,A., Printz-Ane,C. and Printz,P. (1979) Envelope proteins and replication of vesicular stomatitis virus: in vivo effects of RNA+ temperature-sensitive mutations on viral RNA synthesis. J. Virol., 29, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews S., Mikhailov,M., Burny,A. and Roy,P. (1996) The solution structure of the bovine leukaemia virus matrix protein and similarity with lentiviral matrix proteins. EMBO J., 15, 3267–3274. [PMC free article] [PubMed] [Google Scholar]
- McCreedy J., McKinnon,K. and Lyles,D. (1990) Solubility of vesicular stomatitis virus M protein in the cytosol of infected cells or isolated from virions. J. Virol., 64, 902–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mebatsion T., Weiland,F. and Conzelmann,K.K. (1999) Matrix protein of rabies virus is responsible for the assembly and budding of bullet-shape particles and interacts with the transmembrane spike glycoprotein G. J. Virol., 73, 242–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melki R., Gaudin,Y. and Blondel,D. (1994) Interaction between tubulin and the viral matrix protein of vesicular stomatitis virus: possible implication in the viral cytopathic effect. Virology, 202, 339–347. [DOI] [PubMed] [Google Scholar]
- Merritt E.A. and Bacon,D.J. (1997) Raster3D: photorealistic molecular graphics. Methods Enzymol., 277, 505–524. [DOI] [PubMed] [Google Scholar]
- Newcomb W.W. and Brown,J.C. (1981) Role of the vesicular stomatitis virus matrix protein in maintaining the viral nucleocapsid in the condensed form found in native virions. J. Virol., 39, 295–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb W., Tobin,G., McGowan,J. and Brown,J. (1982) In vitro reassembly of vesicular stomatitis virus skeletons. J. Virol., 41, 1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls A., Sharp,K.A. and Honig,B. (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins, 11, 281–296. [DOI] [PubMed] [Google Scholar]
- Odenwald W.F., Arnheiter,H., Dubois-Dalcq,M. and Lazzarini,R.A. (1986) Stereo images of vesicular stomatitis virus assembly. J. Virol., 57, 922–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden J., Pal,R. and Wagner,R. (1986) Mapping regions of the matrix protein of vesicular stomatitis virus which bind to ribonucleocapsids, liposomes and monoclonal antibodies. J. Virol., 58, 860–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowsky Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–325. [DOI] [PubMed] [Google Scholar]
- Peters C.J., Sanchez,A., Rollin,P.E., Ksiazek,T.G. and Murphy,F.A. (1996) Filoviridæ: Marburg and Ebola Viruses. In Fields,B.N. (ed.), Virology. Raven, Philadelphia, PA, pp. 1161–1176.
- Petersen J.M., Her,L.S., Varvel,V., Lund,E. and Dahlberg,J.E. (2000) The matrix protein of vesicular stomatitis virus inhibits nucleocytoplasmic transport when it is in the nucleus and associated with nuclear pore complexes. Mol. Cell. Biol., 20, 8590–8601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel A. and Cambillau,C. (1991) Turbo Frodo. In Silicon Graphics Geometry. Silicon Graphics, Mountain View, CA.
- Ruigrok R.W., Barge,A., Durrer,P., Brunner,J., Ma,K. and Whittaker,G.R. (2000) Membrane interaction of influenza virus M1 protein. Virology, 267, 289–298. [DOI] [PubMed] [Google Scholar]
- Scianimanico S., Schoehn,G., Timmins,J., Ruigrok,R.H., Klenk,H.D. and Weissenhorn,W. (2000) Membrane association induces a conformational change in the Ebola virus matrix protein. EMBO J., 19, 6732–6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha B. and Luo,M. (1997) Structure of a bifunctional membrane–RNA binding protein, influenza virus matrix protein M1. Nature Struct. Biol., 4, 239–244. [DOI] [PubMed] [Google Scholar]
- Taylor A., Easton,A.J. and Marriott,A.C. (1999) Matrix protein of Chandipura virus inhibits transcription from an RNA polymerase II promoter. Virus Genes, 19, 223–228. [DOI] [PubMed] [Google Scholar]
- Terwilliger T.C. and Berendzen,J. (1999) Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr., 55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.D., Higgins,D.G. and Gibson,T.J. (1994) Clustal_W: improving the sensitivity of progressive multiple sequence align ment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res., 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmins J., Scianimanico,S., Schoehn,G. and Weissenhorn,W. (2001) Vesicular release of Ebola virus matrix protein VP40. Virology, 283, 1–6. [DOI] [PubMed] [Google Scholar]
- von Kobbe C., van Deursen,J.M., Rodrigues,J.P., Sitterlin,D., Bachi,A., Wu,X., Wilm,M., Carmo-Fonseca,M. and Izaurralde,E. (2000) Vesicular stomatitis virus matrix protein inhibits host cell gene expression by targeting the nucleoporin nup98. Mol. Cell, 6, 1243–1252. [DOI] [PubMed] [Google Scholar]
- Ye Z., Sun,W., Suryanarayana,K., Justice,P., Robinson,D. and Wagner,R. (1994) Membrane-binding domains and cytopathogenesis of the matrix protein of vesicular stomatitis virus. J. Virol., 68, 7386–7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakowski J.J., Petri,W.A.,Jr and Wagner,R.R. (1981) Role of matrix protein in assembling the membrane of vesicular stomatitis virus: reconstitution of matrix protein with negatively charged phospholipid vesicles. Biochemistry, 20, 3902–3907. [DOI] [PubMed] [Google Scholar]
- Zhang J. and Lamb,R.A. (1996) Characterization of the membrane association of the influenza virus matrix protein in living cells. Virology, 225, 255–266. [DOI] [PubMed] [Google Scholar]