

Abstract
The Aspergillus nidulans protein kinase NIMA regulates mitotic commitment, while the human and Xenopus equivalents influence centrosome function. Two recessive, temperature-sensitive mutations in the Schizosaccharomyces pombe NIMA homologue, Fin1, blocked spindle formation at 37°C. One of the two spindle pole bodies (SPBs) failed to nucleate microtubules. This phenotype was reduced by accelerating mitotic commitment through genetic inhibition of Wee1 or activation of either Cdc25 or Cdc2. Polo kinase (Plo1) normally associates with the SPB of mitotic, but not interphase cells. cut12.s11 is a dominant mutation in an SPB component that both suppresses cdc25 mutants and promotes Plo1 association with the interphase SPB. Both cut12.s11 phenotypes were abolished by removing Fin1 function. Elevating Fin1 levels promoted Plo1 recruitment to the interphase SPB of wild-type cells and reduced the severity of the cdc25.22 phenotype. These data are consistent with Fin1 regulating Plo1 function during mitotic commitment. The fin1 mitotic commitment and spindle phenotypes resemble distinct nimA phenotypes in different systems and suggest that the function of this family of kinases may be conserved across species.
Keywords: cell cycle/mitosis/NIMA kinase/polo kinase/Schizosaccharomyces pombe
Introduction
It is well established that the kinase activity of the p34cdc2 cyclinB complex known as MPF regulates commitment to mitosis (Ohi and Gould, 1999). Once MPF has been activated, polo, aurora and NIMA-related kinases (Nrks) control most of the physical events of mitosis and cytokinesis (Nigg, 2001).
In interphase, Wee1 and related protein kinases phosphorylate and inhibit the p34cdc2 catalytic subunit of MPF. This inhibitory phosphate is removed by the phosphatase Cdc25. The balance between Wee1- and Cdc25-related activities controls mitotic commitment (Ohi and Gould, 1999). An initial trigger level of MPF promotes further activation of Cdc25 to drive commitment to mitosis (Hoffmann et al., 1993), so that the activity of the bulk of Cdc25 is actually dependent upon p34cdc2 activity (Kovelman and Russell, 1996). While direct phosphorylation of Cdc25 by MPF does contribute to positive feedback loop activation of Cdc25 (Izumi and Maller, 1993), the loop still exists in Xenopus extracts from which both cdc2 and cdk2 have been depleted (Izumi and Maller, 1995). Polo kinases are thought to act in the feedback loop, because Plk neutralizing antibodies block mitotic commitment in non-transformed human cells (Lane and Nigg, 1996) and the Xenopus Plk, Plx, was purified as a Cdc25 activating kinase (Kumagai and Dunphy, 1996). Plx influences the rate of commitment to mitosis rather than mitotic commitment per se (Abrieu et al., 1998). Although supporting evidence is currently scant, Polo may also downregulate Wee1-related kinases. Wee1 and the related kinase Myt1 are both hyperphosphorylated upon mitotic commitment, and the Saccharomyces cerevisiae Plk, Cdc5p, binds and inhibits the activity of the Wee1 kinase, Swe1p (Tang et al., 1993; Honda et al., 1995; Mueller et al., 1995; Bartholomew et al., 2001).
The behaviour of the Schizosaccharomyces pombe polo kinase, Plo1, in cut12.s11 mutants suggests that the association of polo kinase with the spindle pole may play a critical role in MPF activation. cut12/stf1+ encodes an essential component of the spindle pole body (SPB). In the recessive cut12.1 mutant only one of the two poles nucleates microtubules (Bridge et al., 1998). Dominant mutations in the same gene, called stf mutations (suppressor of twenty-five), enable cell division in the absence of Cdc25 (Hudson et al., 1990). Plo1 usually associates with mitotic, but not the interphase, SPBs (Bähler et al., 1998; Mulvihill et al., 1999). In stf1.1 mutants (referred to here as cut12.s11), Plo1 associates with SPBs throughout the cell cycle (Mulvihill et al., 1999). Thus, the cut12.s11 mutation confers mitotic behaviour to interphase Plo1.
In contrast to the conservation of function between species for polo- and aurora-related kinases, data do not currently suggest a similar conservation of function for the Nrks (reviewed in Nigg, 2001). nimA mutants were identified in a screen for conditional Aspergillus nidulans mutations that were unable to enter mitosis at the restrictive temperature (the never in mitosis phenotype; Morris, 1975). The nimA arrest point has all the features of interphase cells. The microtubules are cytoplasmic, spindles are absent and the chromosomes are not condensed (Oakley and Morris, 1983). Despite this apparent arrest before mitotic commitment, mitotic NIMA kinase activity is dependent upon prior activation of MPF (Ye et al., 1995).
The strength of the A.nidulans phenotype suggested that Nrks may play an important role in mitotic control in other systems. However, deletion of the S.cerevisiae and S.pombe Nrks suggested that this was not the case, as these cells survived (Jones and Rosamond, 1990; Krien et al., 1998). The functional data obtained to date for the human NIMA-related protein, Nek2, suggest that it regulates centrosome function, rather than mitotic commitment. Nek2 associates with the centrosome. Overproduction of Nek2 in interphase cells drives the premature separation of the two centrosomes (Fry et al., 1998). Similarly, Nek2B is required for the efficient centrosome maturation in Xenopus egg extracts (Fry et al., 2000; Uto and Sagata, 2000). Although overproduction of nimA also blocks spindle formation (Osmani et al., 1988a), the similarity of function between Nrks in different species remains in doubt (Nigg, 2001).
We have identified two temperature-sensitive, loss-of-function mutations in the S.pombe Nrk, fin1+, that could not form a mitotic spindle at 37°C. Genetic interactions suggested that Fin1 plays an important role in MPF activation during mitotic commitment. The recruitment of Plo1 to the interphase SPB of cut12.s11 mutants required Fin1 activity, while increasing fin1+ transcription mimicked the cut12.s11 phenotype. These data are discussed in terms of a role for Fin1 in the MPF amplification feedback loop.
Results
Isolation of sad44.1
We have screened for genes that are required for cor rect architecture of the mitotic spindle (A.Grallert, R.A.Craven, F.E.Stevens and I.M.Hagan, unpublished). SPBs were labelled with a Cut12/GFP fusion protein (Bridge et al., 1998) and conditional diploidizing mutants were isolated according to the methodology of Broek et al. (1991). Mutants that grew at 25°C but increased ploidy after one generation at 36°C were screened by DAPI and GFP fluorescence at 36°C to identify those in which spindle architecture was temperature sensitive. Immuno fluorescence microscopy then confirmed the sad– (spindle architecture defective) phenotype. Here we focus on sad44+. The severity of the sad44.1 spindle formation phenotype was reduced from 93 to 76% as the cut12.NEGFP allele was switched for a wild-type cut12+ gene during outcrossing. Re-introduction of cut12.NEGFP to an outcrossed sad44.1 mutation restored the spindle formation deficiency at 37°C to 93%.
sad44+ is allelic to fin1+
Screening a genomic library for sequences that could complement sad44.1 temperature sensitivity identified three different plasmids that contained the fin1+ gene. Crossing a sad44.1 strain to a fin1 deletion allele, fin1.Δ (Krien et al., 1998), showed no recombination between the ts– of sad44.1 and the ura4+ of fin1.Δ in 81 tetrads. There was a single amino acid change from glycine to glutamic acid at position 188 of the fin1+ open reading frame (ORF) in sad44.1. This residue lay within domain VIII (P + 1 loop) and is conserved in the majority of kinases. This established that sad44.1 is allelic to fin1 and so sad44.1 will be referred to as fin1.ts1 from here on.
Fin1 inactivation affects spindle formation and function
We were surprised to isolate the fin1.ts1 mutation in a screen for ts– mutations, because the fin1+ gene is not required for viability (Krien et al., 1998). We therefore compared the phenotypes of the fin1.ts1 and fin1.Δ strains after 3 h at 37°C. Both strains had mitotic defects (Figure 1A and B). In each case one spindle pole appeared to be nucleating a monopolar spindle (arrows in Figure 1A and B) whilst the other was either inactive or its nucleating capacity was greatly reduced. The defect was more severe in a fin1.ts1 (76% defective; Figure 1A and C) than fin1.Δ strain (26% defective; Figure 1B and C) at 37°C.

Fig. 1. fin1 mutants have defects in spindle formation. The indicated strains were grown to early log phase in EMM2 at 25°C before the temperature of the culture was shifted to 37°C for 3 h. (A and B) Micrographs showing three images of the same field of cells shows from top to bottom microtubules, Sad1 and a combined DAPI DIC image. (A) fin1.ts1 cells at 37°C; (B) fin1.Δ cells at 37°C. Arrows indicate Sad1 staining of a functional SPB that is nucleating far more microtubules than the other. (C) The proportion of mitoses that exhibit spindle formation defects at the indicated temperatures for the haploid and diploid strains is indicated. (D) Mitotic profiles were scored in fin1.ts1 cells for 48 h after the temperature was changed from 25 to 37°C, and the proportion of those that had defective structure was calculated and plotted against time.
Because complete removal of the fin1+ gene from fin1.Δ cells did not affect viability, the severe phenotype of the fin1.ts1 suggested that fin1.ts1 was a dominant mutation. Unexpectedly however, the fin1.ts1/fin1+ heterozygous diploid had the same degree of spindle formation defect as a fin1.Δ/fin1+ diploid at 37°C (Figure 1C). Furthermore, integration of a wild-type copy of fin1+ under the control of its own promoter into the leu1 locus of a haploid fin1.ts1 strain effectively abolished the spindle formation defect conferred by the fin1.ts1 mutation (fin1.ts1 leu1::fin1+; Figure 1C). These data establish that, rather than being dominant, fin1.ts1 was a recessive, loss-of-function mutation and raised the question of why a recessive point mutation conferred a more severe defect than deletion of the same gene. Monitoring spindle formation in fin1.ts1 culture over 48 h showed that, although fin1.ts1 cells had a severe defect in the first division at 37°C, they adapted to the loss of Fin1 function. After 6 h at the restrictive temperature the severity of spindle defect in fin1.ts1 dropped towards the level seen in fin1.Δ (Figure 1D). We concluded that the severe phenotype of a fin1.ts1 mutant is a transitory response to loss of Fin1 function. This implied that fin1.Δ haploids adapted to loss of Fin1 after the first division of a germinating spore.
As both fin1.ts1 and fin1.Δ have constitutive spindle defects after adaptation, we asked whether the spindles in these surviving cells were still defective and whether the viability of these strains depended upon spindle checkpoint controls. The viability of both mutations at 25°C was greatly reduced when mad2+ was deleted (He et al., 1997) and neither mutant could grow at any temperature from 20 to 37°C if mph1+ or bub1+ was deleted (Bernard et al., 1998; He et al., 1998; Table I).
Table I. The mitotic checkpoint is required for viability of fin1 mutants.
| Strain | Viability at 25°C |
|---|---|
| mad2.Δ | ++++++ |
| mph1.Δ | ++++++ |
| Δbub1 | ++++++ |
| fin1.ts1 | ++++ |
| fin1.Δ | ++++++ |
| fin1.ts1 mad2.Δ | + |
| fin1.Δ mad2.Δ | ++ |
| fin1.ts1 mph1.Δ | Dead at all temperatures |
| fin1.Δ mph1.Δ | Dead at all temperatures |
| fin1.ts1 Δbub1 | Dead at all temperatures |
| fin1.Δ Δbub1 | Dead at all temperatures |
fin1.A5 delays mitotic commitment and blocks spindle formation
We were keen to establish whether the unusual transitory phenotype of fin1.ts1 was unique to this allele or whether it was a general feature of recessive conditional fin1 mutants. The A.nidulans nimA5 mutation changes Tyr91 to Asp (Pu et al., 1995). As this residue is conserved in all Nrks, we introduced the equivalent Y84N mutation in fin1 (fin1.A5) and expressed the gene from its native promoter at the leu1 locus in a fin1.Δ background (leu1::fin1.A5 fin1.Δ). leu1::fin1.A5 fin1.Δ cells entered mitosis, but arrested with the same spindle formation defect as fin1.ts1 (Figure 2B and C). At 37°C a diploid fin1.Δ leu1::fin1.A5/ fin1+ leu1+ strain had the same 26% deficiency in spindle structure as that of a fin1.ts1/fin1+ or a fin1.Δ/fin1+ diploid (Figures 1C and 2C). The spindle defect of a haploid leu1::fin1.A5 strain was suppressed by the inclusion of a wild-type gene at the fin1+ locus (Figure 2C). Thus, like fin1.ts1, fin1.A5 is a recessive, loss-of-function mutation. The reduction in the frequency of defective spindles upon prolonged incubation at 37°C of fin1.A5 also resembled that seen in fin1.ts1 (Figure 2D). There was a marked increase in cell length at commitment to mitosis during this extended incubation of fin1.A5 cells at 37°C (Figure 2A; Table II). This cdc phenotype was recessive as fin1.Δ leu1::fin1.A5/ fin1+ leu1+ heterozygous diploids divided at the same length as wild-type controls at 37°C (data not shown). While fin1.Δ cells also delay the timing of mitotic commitment (Table II; Krien et al., 1998), fin1.ts1 cells do not (Table II). We concluded that Fin1 was required for spindle formation and that the fin1.A5 phenotype may have indicated a function in cell cycle control.

Fig. 2. Temperature-sensitive defects in spindle formation and cell cycle progression in fin1.A5 cells. (A, B and D) fin1.A5 cells were grown to early log phase in EMM2 at 25°C before the temperature of the culture was shifted to 37°C for the times indicated. (A) DAPI/calcofluor staining of fin1.A5 at 37°C. Note the cell elongation 12 h after the temperature shift. (B) Micrographs showing three images of the same field of cells 3 h after the temperature shift. From left to right; microtubules, Sad1 staining and a combined DAPI DIC image. (C) The proportion of mitoses that exhibit spindle formation defects at the indicated temperatures for the haploid and diploid strains is indicated. (D) Mitotic profiles were scored in fin1.A5 cells at distinct time points for 24 h after the temperature shift, and the relative proportion of those that were defective was plotted against time.
Table II. Cell lengths upon commitment to mitosis.
| Length of dividing cell (µm), EMM2 | Length of dividing cell (µm), YES | |
|---|---|---|
| Wild type, 25°C | 11.5 ± 1.1 | 12.8 ± 0.78 |
| Wild type, 37°C, 3 h | 12.6 ± 0.82 | 13.8 ± 1.44 |
| fin1.Δ, 25°C | 12.6 ± 1.3 | 12.3 ± 1.1 |
| fin1.Δ, 37°C, 3 h | 14.6 ± 0.98 | 13.5 ± 1.1 |
| fin1.ts1, 25°C | 11.9 ± 0.94 | 13.3 ± 1.7 |
| fin1.ts1, 37°C, 3 h | 13.0 ± 1.63 | 13.8 ± 1.4 |
| leu1::fin1.A5 fin1.Δ, 25°C | 13.4 ± 1.3 | 13.1 ± 0.98 |
| leu1::fin1.A5 fin1.Δ, 37°C, 3 h | 14.7 ± 0.88 | 13.3 ± 0.68 |
| leu1::fin1.A5 fin1.Δ, 37°C, 6 h | 15.5 ± 1.1 | 14.9 ± 3.2 |
| leu1::fin1.A5 fin1.Δ, 37°C, 9 h | 16.1 ± 2.4 | 18.3 ± 3.6 |
| leu1::fin1.A5 fin1.Δ, 37°C, 12 h | 17.4 ± 3.3 | 19.2 ± 3.9 |
| wee1.6, 25°C | 8.0 ± 0.67 | 8.4 ± 0.74 |
| wee1.6, 37°C, 3 h | 7.9 ± 0.84 | 8.1 ± 0.97 |
| fin1.ts1 wee1.6, 25°C | 11.1 ± 1.4 | 10.3 ± 1.7 |
| fin1.ts1 wee1.6, 37°C, 3 h | 9.9 ± 1.6 | 10.7 ± 1.04 |
| pREP41fin1, no induction | 11.5 ± 1.1 | ND |
| pREP41fin1, 20 h induction | 12.7 ± 1.1 | ND |
| pREP1fin1, no induction | 12.1 ± 1.9 | ND |
| pREP1fin1, 20 h induction | 13.4 ± 2.1 | ND |
ND, not determined.
Defective spindle formation upon Fin1 overproduction
Krien et al. (1998) established that excessive fin1+ transcription from the strong nmt1+ promoter drove chromosome condensation in interphase cells, but their study did not address spindle architecture. We therefore monitored spindle architecture and chromosome morphology as the expression of multicopy plasmid borne fin1+ was induced from either the strong (nmt1 in pREP1), intermediate (nmt41 in pREP41) or weak (nmt81 in pREP81) versions of the repressible nmt1+ promoter (Maundrell, 1990; Basi et al., 1993). Spindle function was far more sensitive to increased expression of fin1+ than interphase chromosome condensation. Monopolar (Figure 3A) and metaphase (Figure 3B) spindles accumulated upon induction from all three promoters (Figure 3C), while condensed chromosomes in interphase cells were only seen after expression from the strongest nmt1 promoter (Figure 3C). Deletion of either mad2+ or bub1+ from the host cell abolished delay at the metaphase/anaphase transition arising from expression of pREP41fin1+ (Figure 4A) (He et al., 1997; Bernard et al., 1998). Many of the checkpoint-deficient anaphase cells were clearly elongating spindles despite considerable defects in chromatid separation (Figure 4B and C). This established that the accumulation of metaphase cells after induction of fin1+ was due to altered spindle function, rather than an inability to regulate this transition per se. We concluded that spindle formation was more sensitive to elevation or depletion of Fin1 levels than chromosome condensation.

Fig. 3. fin1+ overproduction induces defects in spindle function before it accumulates to a level that will induce chromosome condensation in interphase. Cells bearing pREP41fin1+ (fin1+ controlled by the nmt41 promoter) (A and C), pREP81fin1+ (fin1+ controlled by the nmt81 promoter) or pREP1fin1+ (fin1+ controlled by the nmt1 promoter) (C) were grown to early log phase in EMM2 + thiamine at 25°C before being washed and resuspended in thiamine-free EMM2 to induce gene expression. Cells were followed for 24 h. (A and B) Micrographs of pREP41fin1+ cells 16 h after induction. From left to right, microtubules, Sad1 and a combined DAPI DIC image. (C) The proportion of mitotic profiles that had either monopolar or metaphase spindles was plotted along with the proportion of total cells that had chromosome condensation in interphase.

Fig. 4. Metaphase arrest arising from fin1+ overproduction is due to an active spindle assembly checkpoint. Cultures of mad2.Δ or Δbub1 strains containing pREP41fin1+ were grown to early log phase in EMM2 + thiamine at 25°C before being washed and resuspended in thiamine-free EMM2 to induce gene expression. Cells were followed for 24 h. (A) The proportion of mitotic cells that had metaphase spindles was scored for each strain. (B and C) Micrographs of mad2.Δ pREP41fin1+ (B) or Δbub1 pREP41fin1+ (C) cells 20 h after induction. From left to right, microtubules, Sad1 and a combined DAPI DIC image.
Genetic interactions between fin1 mutants, and cut12/stf1 and mitotic regulators
Outcrossing the original fin1.ts1 cut12.NEGFP isolate indicated that the cut12.NEGFP allele enhanced the spindle formation deficiency conferred by fin1.ts1 (Figure 5A, B and D). As spindle formation is usually normal in cut12.NEGFP cells (Figure 5C), this interaction suggested a functional relationship between fin1 and cut12. To investigate this possibility further, we looked for genetic interactions between fin1 mutants and two different cut12 alleles. cut12.1 is a recessive temperature-sensitive mutation that confers a spindle formation defect at 36°C (Bridge et al., 1998). This defect is very similar to the fin1– spindle defect. cut12.1 reduces the restrictive temperature of cdc25.22 from 30 to 20°C. cut12.s11 is a dominant mutation that has normal spindle architecture but suppresses the mitotic commitment phenotypes of cdc25.22 and cdc25.Δ (Hudson et al., 1990; Bridge et al., 1998).
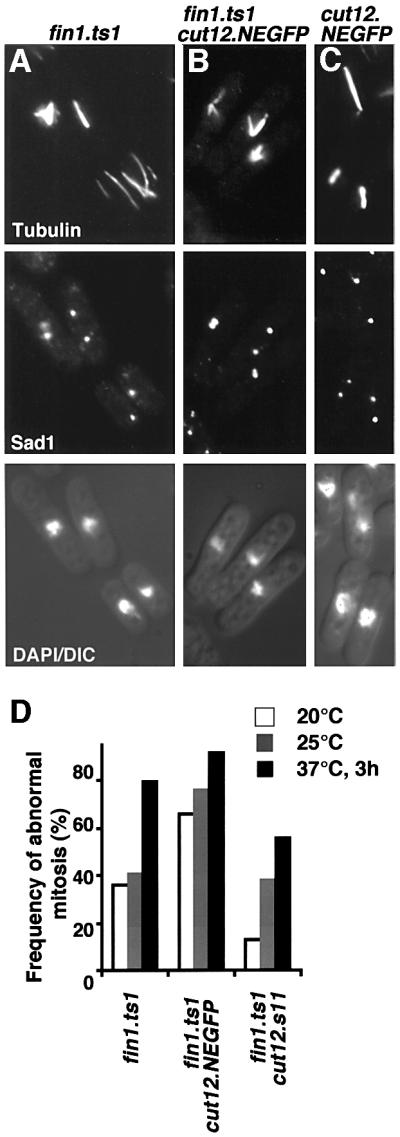
Fig. 5. The spindle formation defect of fin1.ts1 is exacerbated by cut12.NEGFP and suppressed by cut12.s11. (A–C) Cells were grown to mid-log phase in EMM2 at 25°C before processing for immunofluorescence microscopy. Micrographs showing three images of the same field of cells from top to bottom microtubules, Sad1 staining and a combined DAPI DIC image. (A) fin1.ts1; (B) fin1.ts1 cut12.NEGFP; (C) cut12.NEGFP. (D) The proportion of mitotic cells that had defects in spindle morphology at different temperatures for the indicated strains. Cells were grown overnight to mid-log phase at the indicated temperatures with the exception of 37°C cells, which were first grown at 25°C overnight to early log phase before the temperature was changed to 37°C for 3 h. For each strain >200 mitotic cells were counted.
Double mutants containing cut12.1 and either fin1.Δ or fin1.ts1 were dead at all temperatures (data not shown). In contrast, cut12.s11 reduced the severity of the fin1.ts1 spindle formation defect (Figure 5D, most apparent at 20°C). Thus, cut12 loss-of-function mutations exacerbated the fin1– phenotype, while gain-of-function mutations reduced the requirement for Fin1. Overexpression of fin1+ underscored the functional relationship between these two genes, as it both raised the restrictive temperature for cut12.1 from 30 to 34°C (Figure 6A) and mimicked the effect of the cut12.s11 in raising the restrictive temperature for cdc25.22 cells (Figure 6B).

Fig. 6. Increasing fin1+ gene dosage raises the restrictive temperature of cut12.1 and cdc25.22, while loss of fin1 function abolishes the ability of cut12.s11 to suppress cdc25.22. Five-fold dilution series of strains were spotted onto EMM2 and the plates were incubated at the temperatures indicated. (A) cut12.1 bearing pREP41fin1+, pREP81fin1+ or no plasmid; (B) cdc25.22 bearing empty vectors, pREP41fin1+ or pREP81fin1+. (C) The indicated strains without any plasmids.
The suppression of cdc25.22 following increased expression of fin1+ suggested that Fin1 may positively regulate commitment to mitosis. If this were the case, then independently promoting mitotic commitment should reduce the requirement for Fin1. Commitment to mitosis can be accelerated by abolishing the function of the mitotic inhibitor Wee1 or by mutations in cdc2 that make the protein somewhat independent of regulation by either Wee1 or Cdc25 (Thuriaux et al., 1978; Fantes, 1979; Russell and Nurse, 1987). When wee1.6, wee1.50, cdc2.1w or cdc2.3w were introduced into fin1.ts1 cells, the severity of the spindle formation defect was reduced (Figure 7A). Cdc25 is regulated, in part, by regulating the translation of cdc25+ message (Daga and Jimenez, 1999). Deletion of a control element in the 5′ untranslated region (UTR) of cdc25+ generates a hyperactive allele, cdc25.d1, that provides yet another way of accelerating commitment to mitosis (Daga and Jimenez, 1999). Expression of cdc25.d1 again reduced the extent of the spindle formation defect conferred by fin1.ts1 (Figure 7B).

Fig. 7. Acceleration of mitotic commitment reduces the spindle defect of fin1 mutants. (A) Cells were grown overnight to mid-log phase at 25°C (no shading) or were at 25°C overnight to early log phase before the temperature was changed to 37°C for 3 h (black). For each strain in (A) and (B), >200 mitotic cells were counted. (B) fin1.ts1 cells containing pREP3xcdc25.d1 were grown to mid-log phase in EMM2 containing thiamine, washed in thiamine-free medium, resuspended at a lower density and 13 h later the culture was split in two and the temperature of one half was shifted to 37°C (37°C) for 3 h whilst the other was maintained at 25°C (25°C). The bar chart shows the proportion of mitotic cells that had defects in spindle morphology. In all strains the expression of the cdc25.d1 allele reduced the severity of the spindle defect and the strength of suppression correlated with the strength of expression.
Fin1 function is required for the premature recruitment of Plo1 to the SPB by cut12.s11
The genetic interactions in the previous section suggested that the functions of Fin1 and Cut12 may be related: both molecules are required for spindle formation; accentuated function in either gene reduces the defects arising from deficiencies in the other; simultaneously reducing the function of both kills the cell; and manipulation of either can suppress the cdc25.22 mutation. We therefore asked whether the suppression of cdc25.22 by cut12.s11 was dependent upon Fin1 function, and found that it was. Introduction of either fin1.ts1 or fin1.Δ to a cut12.s11 cdc25.22 mutant background abolished the growth of cdc25.22 above 30°C that had been conferred by the cut12.s11 mutation (Figure 6C).
Polo kinase is proposed to act during the MPF-amplifying feedback loop that activates Cdc25 and downregulates Wee1. cut12.s11 may bypass cdc25.22 by influencing polo kinase function in this feedback loop. There is no discrete localization of the S.pombe polo kinase, Plo1, during interphase; upon mitotic commitment it is recruited to the SPB (Bähler et al., 1998; Mulvihill et al., 1999). Strong Plo1 staining is seen until anaphase. In cut12.s11 mutant cells, strong Plo1 SPB staining is seen in interphase as well as mitotic cells (Mulvihill et al., 1999). This premature recruitment of Plo1 to the SPB is apparent when scoring the number of single versus double dots of Plo1 staining. Single dots represent interphase or early mitotic SPBs, while two dots represent the SPBs at a range of stages of mitosis. Strong association of Plo1 with the SPB is the earliest mitotic event recorded to date (Mulvihill et al., 1999). This strong association persists through to the start of anaphase when the staining intensity reduces. Plo1 associates with a single SPB structure for the same amount of time as it takes to proceed from this stage to anaphase. This means that the ratio of cells with a single spot of Plo1 to those with two spots is around 1 in a wild-type strain (Figure 8). In a cut12.s11 mutant this value increases to 2.8 because the increase in the association of Plo1 with the interphase SPB increases the number of single spots.

Fig. 8. Premature recruitment of Plo1 to the SPB of cut12.s11 cells depends upon Fin1 function. Cells were grown to mid-log phase in EMM2 at 25°C and either immediately processed for anti-Plo1 immunofluorescence, or processed 3 h after the temperature of the culture had been changed to 37°C. The ratio of cells with one spot of Plo1 SPB staining versus the number of cells with two spots of Plo1 SPB staining was scored for the strains indicated on the graph (see text for details). The graph is the result of four independent experiments, and 200 cells were counted for each strain in each experiment.
We were able to use the one to two spot assay to ask whether Fin1 was also required for the premature recruitment of Plo1 to the interphase SPB in cut12.s11. The ratio of one versus two spots of Plo1 staining was reduced from the cut12.s11 level of 2.8 to the wild-type level of 0.92 at both 25°C and 37°C when fin1+ was deleted (Figure 8). More significantly, the temperature sensitivity in Fin1 function conferred by the fin1.ts1 mutation was mirrored by temperature-dependent association of Plo1 with the interphase SPB in cut12.s11 fin1.ts1 cells (Figure 8). It associated with the SPB at 25°C when Fin1 was working, but did not associate at 37°C when Fin1 was defective.
As Fin1 was required for spindle formation (Figures 1 and 2), it was possible that compromising Fin1 function in the previous cycle led to an inherited change in SPB structure that affected Plo1 recruitment in the subsequent cell cycle. In this case the altered behaviour of the cut12.s11 SPB in a fin1– background was a secondary consequence of the role of Fin1 in mitosis rather than a reflection of a genuine function for Fin1 during interphase. We therefore assessed the effect of Fin1 function upon the association of Plo1 with the SPB of mutant cells that had been returned to the cell cycle following G1 arrest at the permissive temperature. In order to avoid the confusion in the analysis arising from the normal association of Plo1 with the SPB that accompanies mitotic commitment, cells were stopped from progressing through the cell cycle beyond S phase by inclusion of the drug hydroxyurea (HU). This assesment therefore addresses Plo1 recruitment to the SPB independently of mitotic defects in the previous, or current, cell cycles.
cut12.s11 and cut12.s11 fin1.ts1 cells that had been arrested in G1 phase at 25°C by removal of a nitrogen source for 12 h cells were resuspended in nitrogen-rich medium containing HU and incubated at the 25°C for 3 h. The temperature of the culture was shifted to 37°C to inactivate Fin1 at this 3-h time point and the culture was monitored for a further 2 h. The frequency of cells with a single spot of Plo1 on the nucleus for cut12.s11 cells is represented by light blue diamonds in all experiments shown in Figure 9, while cut12.s11 fin1.ts1 cells with a single spot of Plo1 on the nucleus are represented by red triangles. The grey shading represents a period at 25°C, and the white background a period at 37°C. At 25°C, in either strain, ∼80% of the cells had Plo1 on the SPB (Figure 9A). The number of cells with Plo1 on the interphase SPB dramatically dropped in the fin1.ts1 cut12.s11 double mutant (red line) after the temperature of the culture was shifted to 37°C, but remained largely unaffected in the single cut12.s11 mutant (light blue line). For further controls see Supplementary data available at The EMBO Journal Online.

Fig. 9. Plo1 association with the SPB in a cut12.s11 mutant is dependent upon Fin1 activity. (A) Cells were grown to mid-log phase in EMM2 at 25°C before being washed three times and resuspended in nitrogen-free EMM2 at 25°C. Twelve hours later cells were pelleted once more and resuspended in EMM2 + HU. The nitrogen-free medium induced G1 phase arrest, whereas the nitrogen-rich medium induced cells to return to the cell cycle, where the HU then induced cell cycle arrest in S phase. The areas of the graphs with a white background indicate the periods at 37°C, whilst the grey background indicates periods at 25°C. The point of temperature shifts are indicated by arrows below the x-axes (blue for a shift down to 25°C, red for a shift up to 37°C). The number of spots of Plo1 on the nucleus was scored for each strain as indicated in the key. cut12.s11 mutants show a normal checkpoint arrest in HU (data not shown). Note that premature recruitment of Plo1 in stf1.1 does not result in septation. G1 cells were resuspended in nitrogen-rich EMM2 medium containing HU at 25°C for 3 h and then the temperature was shifted to 37°C. (B and C) G1 cells were resuspended nitrogen-rich EMM2 medium containing HU at 37°C and incubated for a further 3 h at 37°C. The culture was then split in two. One half was maintained at 37°C (B), while the temperature of the other was dropped to 25°C (C). Each half was incubated at its respective temperature for a further 2 h.
The experiment was repeated with both mutants but this time the temperature was increased at the start of the experiment (t = 0) rather than at the 3 h time point (Figure 9B and C). Each culture was then split into two after 3 h; one half was maintained at 37°C while the other half was returned to 25°C. The temperature shift to 37°C at time 0 caused Plo1 to leave the interphase SPBs in the double mutant cut12.s11 fin1.ts1 (Figure 9B, red), but not single cut12.s11 (Figure 9B, light blue) mutant. Plo1 re-associated with the SPBs, from which it had been excluded by the temperature shift at time 0, when Fin1 function was restored in the double mutant culture by returning the temperature to 25°C (Figure 9C, red).
We concluded that Fin1 was required for both cut12.s11 suppression of cdc25.22 and the premature association of Plo1 with the SPB of interphase cut12.s11 cells. Moreover, the data show that the association of Plo1 with the SPB was dynamic and indicated that Fin1 could influence interphase events (i.e. it could function in interphase as well as during mitosis).
Overproduction of fin1+ promoted the association of Plo1 with the interphase SPB of wild-type cells
We next asked whether the effect of Fin1 upon the recruitment of Plo1 to the SPB was a specific feature of the cut12.s11 mutant background, or whether Fin1 could influence the SPB association of Plo1 in wild-type cells. We reasoned that increasing Fin1 levels might recruit Plo1 to the interphase SPB. fin1+ expression was induced from the nmt81 and nmt41 promoters of pREP81 and pREP41 (Basi et al., 1993) and we monitored Plo1 association with the SPB. Again, to rule out any confusion arising from the association of Plo1 that accompanies normal commitment to mitosis, HU was added to a portion of each culture 5 h before the time point indicated as ‘HU arrest’ on the graphs in Figure 10A. The very low level of basal transcription from the nmt81 promoter (Forsburg, 1993) forced Plo1 to associate with interphase SPBs before expression was induced. Induction of transcription enhanced this association and the basal transcription from the nmt41 promoter of pREP41 was sufficient to drive maximal association of Plo1 with the SPB (Figure 10A and B) of cells that had the interphase microtubule configuration of an S-phase-arrested culture (Figure 10C).

Fig. 10. Modest increases in fin1+ transcription recruits Plo1 to the interphase SPB of wild-type cells. (A) Cells bearing pREP81fin1+ or pREP41fin1+ were grown to early log phase in EMM2 + thiamine at 25°C before being washed and resuspended in EMM2 to induce gene expression. HU (10 mM) was added to a portion of the culture 5 h before each time point indicated on the graph, and this portion was then processed for immunofluorescence microscopy to detect Plo1 5 h later at the time shown on the graph. Thus, where indicated, the values indicated by a black diamond or a grey square on each graph are for an S phase arrested population. The percentage of total cells with any Plo1 staining is shown on the graph. Wild-type cells arrest in S phase without any Plo1 on the SPB (grey squares), while increasing expression of fin1+ promotes association of Plo1 with the SPB. (B) Micrographs from the uninduced (t = 0) pREP41fin1+ strain showing a Plo1 localization (top) and a DAPI DIC image (bottom). (C) Micrographs from the pREP41fin1+ culture in (B) showing, from left to right, microtubules, Sad1 and a combined DAPI DIC image. No mitotic cells were seen in 200 cells counted.
Discussion
We have described two mutations in the non-essential fin1+ gene that conferred a temperature-sensitive inability to form a mitotic spindle. One of these alleles resembled the null allele in displaying the additional phenotype of cell cycle delay. These two recessive, conditional mutations conferred a much more severe spindle formation defect than that arising from complete loss of the gene. The strong phenotype in the conditional mutants is probably an initial response to the loss of Fin1 function, after which the cells adapt, because the severity of the defect in both alleles declined towards fin1.Δ levels with prolonged incubation at 37°C. The mechanism by which cells adapt to the loss of function is unclear at present; however, it is unlikely to arise from the accumulation of suppressor mutations in the strain as the same suppressor would have to arise spontaneously in at least 40% of the population within one generation. It is most likely, therefore, that the organization of control networks enables cells to adapt to losing Fin1 function. Future genetic screens will identify how cells compensate for the loss of Fin1.
Despite the recessive nature of the fin1.Δ, fin1.ts1 and fin1.A5 mutations, heterozygous diploids which combine any of these alleles with a wild-type allele exhibited a 26% deficiency in spindle formation at 37°C. The greater sensitivity of diploids to a reduction in fin1+ gene dosage may reflect the greater challenges faced by the diploid spindle. Differences in the demands of chromosome segregation between haploid and diploid spindles are highlighted by the fact that haploid cells lacking the spindle checkpoint protein Bub1 grow without any apparent difficulty, whereas diploid Δbub1 cells rely heavily upon this gene for survival (Bernard et al., 1998). For fin1+, these challenges would be greater at 37°C than 25°C as fin1.Δ fin1+ heterozygotes have essentially normal spindle function at the lower temperature. Sensitivity to gene dosage may be a common feature for Nrks in different systems, as the number of wild-type nimA nuclei in nimAΔ/nimA heterokaryons is disproportionately high (Ye et al., 1998).
Fin1 and spindle formation
The nucleation capacity of one of the two SPBs was compromised in fin1.ts1 mutants at the restrictive temperature. A similar phenotype has been reported in A.nidulans when the nimA.5 mutation was combined with a mutation in the APC component BIME (Osmani et al., 1988b, 1991; Peters et al., 1996). The bimE7 mutation increased the level of NIMA protein (Ye et al., 1998) and so, presumably, increased NIMA activity at a critical location, or above a critical threshold, to enable mitotic commitment but not spindle formation. Spindle formation defects are also seen upon overproduction of nimA (Osmani et al., 1988a). The shared spindle phenotype raises the possibility that NIMA and Fin1 perform similar functions in each system. If this is the case then the complete cell cycle arrest of nimA5 mutants suggests that A.nidulans has a greater dependency upon Nrk function than S.pombe.
Related function for Fin1 and Cut12?
Genetic manipulations established a strong link between Fin1 and the SPB component Cut12. The recessive cut12.1 mutation blocks spindle formation in a manner that is indistinguishable from the fin1– spindle formation defect, while dominant mutations in the same gene, such as cut12.s11, suppress cdc25.22 (Hudson et al., 1990; Bridge et al., 1998). Combining cut12.1 with fin1.ts1 or fin1.Δ was lethal, while cut12.s11 reduced the severity of the fin1– spindle formation. Conversely, increasing fin1+ expression reduced the severity of the cut12.1 defect (although it could not suppress cut12Δ; data not shown). Furthermore, increasing fin1+ expression mimicked the phenotype of cut12.s11 in both raising the restrictive temperature of cdc25.22 and in promoting the premature recruitment of Plo1 to the interphase SPB.
How could cut12.s11 or overproduction of fin1+ suppress Cdc25?
The only requirement for Cdc25 is to counteract Wee1 as inhibition of Wee1 suppresses cdc25.22 and cdc25.Δ mutants (Fantes et al., 1979; Russell and Nurse, 1986). Wee1 inhibition during mitotic commitment correlates with increased phosphorylation of the protein (Tang et al., 1993; Honda et al., 1995; Mueller et al., 1995). This phosphorylation is probably achieved by a feedback loop kinase(s) which is triggered by the initial activation of MPF (Hoffmann et al., 1993). Considerable data suggest that polo kinases act as a Cdc25 feedback loop kinase (Nigg, 2001). While the analysis of Wee1 feedback loop kinase is less extensive, the budding yeast polo kinase, Cdc5p, does bind and inhibit the Wee1 homologue Swe1p, suggesting that polo kinases could both inhibit Wee1 and activate Cdc25 during mitotic commitment (Bartholomew et al., 2001). If this were the case, manipulations that activate feedback loop kinases such as polo would suppress cdc25 mutations, because they would inhibit Wee1 without the need for the priming activation of MPF, which is dependent upon Cdc25.
Both the cut12.s11 mutation and fin1+ overexpression suppress cdc25.22 and promote the premature recruitment of Plo1 to the interphase SPB. As Plo1 is usually only found on mitotic SPBs (Bähler et al., 1998; Mulvihill et al., 1999), these two manipulations result in mitotic behaviour of Plo1 in interphase cells. As mitotic behaviour of Plo1 could be indicative of an activate feedback loop. These two manipulations may therefore suppress cdc25.22 by inappropriately activating the feedback loop. Fin1 and Cut12 are likely to act in a common pathway, because Fin1 is required for both cut12.s11 phenotypes. Inactivating Fin1 blocks the suppression of cdc25.22 by cut12.s11, and stops Plo1 from associating with the interphase SPB in cut12.s11 mutants. Given that Fin1 associates with the SPB from late G2 through to the end of mitosis (Krien et al., 2002) it is possible that these two proteins physically interact. However, we have been unable to detect an interaction in a yeast two-hybrid assay (data not shown).
A number of the phenotypes are consistent with a function for Fin1 in feedback loop control of mitotic commitment: fin1+ is not an essential gene. In the absence of fin1+, cells rely upon the spindle assembly checkpoint to survive. The checkpoint may halt mitosis long enough for MPF activity to accumulate to permit full mitotic commitment; fin1.Δ cells transit the G2/M boundary more slowly than wild-type cells (Krien et al., 1998). This is reminiscent of the consequences of depleting Plx from Xenopus extracts—the rate of mitotic commitment is slow, but cells eventually enter mitosis (Abrieu et al., 1998); wee1, cdc25 or cdc2 mutants, which accelerate commitment to mitosis by reducing the reliance upon correct activation of the mitotic switch, also reduce the spindle formation defect of fin1 mutants.
The bypass of cdc25.22 when Plo1 is prematurely recruited to the SPB suggests that association of Plo1 with the SPB may be a critical step in regulating mitotic commitment. As Fin1 can influence this step, it may regulate the affinity of Plo1 for the SPB (Figure 11A). Alternatively Fin1 may modify the SPB to promote Plo1 recruitment (Figure 11B), perhaps by modifying Cut12 function. Either way Plo1 is brought into a context in which it promotes MPF activation (Figure 11C). Because polo kinase regulates many events that are likely to be associated with the SPB (such as APC activation and cytokinesis; Nigg, 2001), it may bind to different SPB components to regulate different mitotic events. The step that we postulate here could be one of several means by which Plo1 associates with the mitotic SPB. The nature of the interaction between Fin1 and Plo1 remains to be established, but it is worth noting that overproduction of fin1+ in HU arrested cells does not affect bulk Plo1 activity, or alleviate the spindle formation defects of plo1.ts mutants (data not shown). Furthermore, Plo1 associates with the SPB in fin1.ts monopolar spindles (data not shown).

Fig. 11. Fin1, Plo1 and mitotic commitment. Fin1 either modifies the Plo1/regulatory subunit (A) or it modifies the SPB (B) to promote Plo1 association with the SPB (C). (D) SPB associated Plo1 promotes entry into mitosis by activating Cdc25 and inhibiting Wee1. P represents phosphate on Y15 and T14 of Cdc2.
NIMA and commitment to mitosis
The relationship between NIMA and MPF is consistent with a function in the feedback loop. Mitotic activation of NIMA is dependent upon prior activation of MPF (Ye et al., 1995). MPF directly phosphorylates NIMA in vitro in a manner that mimics the modification of NIMA during mitotic commitment in vivo (Ye et al., 1995). NIMA kinase activity peaks before MPF activity peaks at metaphase (Ye et al., 1995). Finally, although NIMA acts after MPF, the cells remain in interphase when NimA function is compromised (Oakley and Morris, 1983). This last fact could argue against a feedback loop function. If NIMA acts in the feedback loop it might be expected that, like Plx (Abrieu et al., 1998), it would alter the rate rather than the extent of mitotic commitment. However, the need to divide multiple nuclei in a single cell compartment could be a driving force to make a control that is peripheral to mitotic commitment in many systems, and central to mitotic commitment in a syncitial filamentous fungus. If, as Cut12 data suggest is true in S.pombe, events at the SPB play an important part in mitotic commitment in A.nidulans, it would be critical to co-ordinate the waves of MPF amplifying loop emerging from each SPB of the multi-nucleated cell compartments. The activity of the amplification loop that would propagate the trigger signals is a logical point at which to interpose such co-ordination. Thus, NIMA A.nidulans may have been co-opted to become a rate limiting step in mitotic commitment while Fin1 remains a fine tuning control in S.pombe.
Common functions for all Nrks?
The fin1– phenotypes reported here identify some common themes between Nrks in different organisms. Mutation of both nimA and fin1 affects the timing of mitotic commitment. The spindle formation phenotypes seen with fin1 or overexpression of nimA are reminiscent of the slow centrosome maturation seen upon depletion of Nek2B from Xenopus extracts and the centrosome splitting induced by injection of Nek2 into human cells (Osmani et al., 1988a; Fry et al., 1998, 2000; Uto and Sagata, 2000). Furthermore, if the prime function of the S.cerevisiae Nrk Kin3p is also to regulate Wee1 and Cdc25 equivalents, it would explain why KIN3+ is not an essential gene (Jones and Rosamond, 1990). Phosphorylation of Tyr19 of Cdc28p is only essential in S.cerevisiae when cytokinesis or morphogenesis are defective (Lew, 2000). Thus, Nrk function in diverse systems may have more in common than has been appreciated previously.
Materials and methods
Cell culture and strains
The strains used in this study are listed in Table III. Cells were cultured in yeast extract media (YES), or appropriately supplemented EMM2 with or without nitrogen minimal media as described in Moreno et al. (1991). Thiamine (4 µM) was added to minimal EMM2 media to repress nmt promoters (Maundrell, 1990; Basi et al., 1993). HU was added from an aqueous 1 M stock solution to a final concentration of 10 mM.
Table III. Strains used in this study.
| Strain | Source/reference | |
|---|---|---|
| IH365 | leu1.32 ura4.D18 h– | Laboratory stock |
| IH571 | cut12.NEGFP leu1.32 ura4.D18 h– | Bridge et al. (1998) |
| IH646 | cdc2.1w h– | Thuriaux et al. (1976) |
| IH666 | cut12.s11 leu1.32 h+ | Hudson et al. (1990) |
| IH740 | cdc25.22 cut12.s11 leu1.32 ura4.D18 h+ | Laboratory stock |
| IH754 | cut12.1 ade6.704 ura4.D18 h– | Bridge et al. (1998) |
| IH776 | wee1.50 ura4.D18 h– | Thuriaux et al. (1976) |
| IH1307 | mad2::ura4+ leu1.32 ura4.D18 h– | He et al. (1997) |
| IH1308 | ura4.D18 h– | Laboratory stock |
| IH1361 | cut12.NEGFP mam2 leu1.32 ura4.D18 his1.102 h90 | This study |
| IH1363 | mph1::ura4+ leu1.32 ura4.D18 h– | He et al. (1998) |
| IH1545 | cdc2.3w leu1.32 ura4.D18 h– | Thuriaux et al. (1976) |
| IH1688 | fin1.ts1 ura4.D18 leu1.32 h– | This study |
| IH1705 | fin1.ts1 ura4.D18 leu1.32 his2 h+ | This study |
| IH1754 | wee1.6 h+ | Thuriaux et al. (1976) |
| IH1824 | fin1.Δ ura4.D18 leu1.32 h+ | Krien et al. (1998) |
| IH1865 | fin1.ts1 cdc25.22 leu1.32 ura4.D18 h– | This study |
| IH1911 | fin1.Δ cdc25.22 leu1.32 ura4.D18 h– | This study |
| IH1912 | fin1.ts1 cut12.s11 leu1.32 ura4.D18 h– | This study |
| IH1915 | fin1.ts1 cut12.NEGFP leu1.32 ura4.D18 h– | This study |
| IH1917 | fin1.ts1 wee1.50 leu1.32 h– | This study |
| IH1951 | fin1.ts1 dma1::LEU2 leu1.32 ura4.D18 h– | This study |
| IH1953 | fin1.Δ mad2::ura4+ leu1.32 ura4.D18 | This study |
| IH1960 | cdc25.22 ade6.M216 leu1.32 ura4.D18 h– | Fantes (1981) |
| IH1964 | Δbub1::LEU2 leu1.32 ura4.D18 ade6.M216 h+ | Bernard et al. (1998) |
| IH1978 | fin1.Δ ura4.D18 leu1.32 h– | This study |
| IH1980 | fin1.Δ cut12.NEGFP leu1.32 ura4.D18 | This study |
| IH1981 | fin1.ts1 cdc2.1w ura4.D18 h– | This study |
| IH1982 | fin1.ts1 cdc2.3w leu1.32 ura4.D18 h– | This study |
| IH1987 | fin1.ts1 mad2::ura4+ leu1.32 ura4.D18 h– | This study |
| IH2031 | fin1.Δ cut12.s11 leu1.32 ura4.D18 h– | This study |
| IH2049 | fin1.ts1 wee1.6 leu1.32 | This study |
| IH2051 | fin1.Δ dma1::LEU2 leu1.32 ura4.D18 | This study |
| IH2061 | fin1.ts1 cdc25.22 cut12.s11 leu1.32 ura4.D18 h– | This study |
| IH2062 | fin1.Δ cdc25.22 cut12.s11 leu1.32 ura4.D18 h– | This study |
| IH2249 | fin1.Δ ura4.D18 h+ | This study |
| IH2273 | leu1::fin1.A5:ura4+ ura4.d18 h– | This study |
| IH2274 | leu1::fin1.A5:ura4+ fin1.Δ ura4.d18 h– | This study |
| IH2285 | leu1.32 ura4.D18 h– + pREP1fin1+ | This study |
| IH2286 | leu1.32 ura4.D18 h– + pREP41fin1+ | This study |
| IH2287 | fin1.ts1 ura4.D18 leu1.32 his2 h+ pREP3Xcdc25.d1 | This study |
| IH2288 | fin1.Δ ura4.D18 leu1.32 h– pREP3Xcdc25.d1 | This study |
| IH2330 | leu1.32 ura4.D18 h– pREP81fin1+ | This study |
| IH2331 | cut12.1 leu1.32 ura4.D18 his2 h+ pREP81fin1+ | This study |
| IH2332 | cut12.1 ade6.704 ura4.D18 h– pREP41fin1+ | This study |
| IH2333 | cdc25.22 ade6.M216 leu1.32 ura4.D18 h– pREP81fin1+ | This study |
| IH2334 | cdc25.22 ade6.M216 leu1.32 ura4.D18 h– pREP41fin1+ | This study |
| IH2335 | mad2Δ::ura4+ leu1.32 ura4.D18 ade6 h– pREP41fin1+ | This study |
| IH2336 | Δbub1:: ura4+ leu1.32 ura4-DS/E his1.102 ade6.M216 pREP41fin1+ | This study |
| IH2337 | leu1::fin1+:ura4+ fin1.ts1 ura4.d18 h– | This study |
| IH2338 | fin1.ts1/fin1+ leu1.32/leu1+ ura1.d18/ura4+ ade6.M210/ade6/M216 h–/h– | This study |
| IH2339 | fin1.Δ/fin1+ leu1.32/leu1+ ura4.d18/ura4+ ade6.M210/ade6/M216 h–/h– | This study |
| IH2340 | leu1::fin1.A5:ura4+/leu1+ fin1.Δ/fin1+ ura4.d18/ura4+ ade6.M216/ade6.M210 h–/h– | This study |
| IH2350 | leu1.32/leu1+ ura4.d18/ura4+ ade6.M216/ade6.M210 h–/h– | This study |
Molecular genetics
fin1.ts1-suppressing plasmids were cloned from a genomic library (Barbet and Carr, 1992) using standard procedures (Moreno et al., 1991). The QuickChange Mutagenesis (Stratagene) approach along with appropriate primers was used for all site directed mutagenesis. The use of the SMART RACE kit (Clontech) confirmed that the ATG codon that was assigned to fin1 in Krien et al. (1998) corresponded to the first ATG of the fin1+ message. This ATG was mutated to an NdeI site and plasmid pfin1.1, which extended from the Pst1 site 185 bp 5′ to this ATG to the XbaI site that lies 627 bp 3′ to the stop codon. This mutagenesis generated pfin1.ATG*. The 3108 bp NdeI–XbaI fragment from pfin1.ATG* was subcloned into pREP81 pREP41 and pREP1 (Basi et al., 1993) to generate pREP81fin1+, pREP41fin1+ and pREP1fin1+, respectively. Note that pREP1fin1 differed from nmt::fin1 used by Krien et al. (1998) by not having the 83-bp UTR between the nmt1+ promoter and the initiator ATG that is present in nmt::fin1. Five independent PCRs amplified the fin1 gene from fin1.ts1. These reactions were pooled and sequenced. Repeating this procedure identified the same single mutation. Nucleotide 564 of the fin1+ ORF in pfin1.1 was changed from T to A to generate fin1.A5. The PstI–SacI fragment from the resultant plasmid pfin1.A5 or pfin1.1 was inserted between the PstI and SacI sites of pINTA (Petersen et al., 2001) to generate pfin1.A5int or pfin1+int, respectively. The NotI fragment from each plasmid was independently transformed into IH1308, and ura+ leu– colonies were isolated.
Indirect immunofluorescence microscopy
Standard anti-α-tubulin immunofluorescence procedures were used to stain spindles (Hagan and Yanagida, 1995). For detection of Plo1, glutaraldehyde was omitted from the fixation step and fixation was with 1.4% formaldehyde for 5 min. This fixation differs from that in Mulvihill et al. (1999). With the fixation used here, Plo1 is found on all cut12.s11 interphase SPBs, but not on wild-type interphase SPBs. Anti-Sad1 AP9.2, TAT1 (anti-α–tubulin) and anti-Plo1 antibodies were used as described previously (Woods et al., 1989; Hagan and Yanagida, 1995; Mulvihill et al., 1999). DAPI (4,6-diamidine-2-phenyl-indole) and calcofluor staining were according to Moreno et al. (1991). Images and cell length measurements were obtained using a Quantix camera (Photometrics) with Metamorph software (Universal Imaging). More than 200 dividing cells were measured for each strain for the cell length measurements in Table II.
Supplementary data
Supplementary data for this paper are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank Tony Carr, Keith Gull, Hiro Ohkura and Juan Jimenez for reagents, Matthew O’Connell and Michael Krien for reagents and communicating results prior to publication, and Fiona MacIver for technical assistance. This work was supported by Cancer Research UK.
References
- Abrieu A., Brassac,T., Galas,S., Fisher,D., Labbe,J.C. and Doree,M. (1998) The Polo-like kinase Plx1 is a component of the MPF amplification loop at the G2/M-phase transition of the cell cycle in Xenopus eggs. J. Cell Sci., 111, 1751–1757. [DOI] [PubMed] [Google Scholar]
- Bähler J., Steever,A.B., Wheatley,S., Wang,Y.L., Pringle,J.R., Gould,K.L. and McCollum,D. (1998) Role of polo kinase and Mid1p in determining the site of cell division in fission yeast. J. Cell Biol., 143, 1603–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbet N., Muriel,W.J. and Carr,A.M. (1992) Versatile shuttle vectors and genomic libraries for use with Schizosaccharomyces pombe. Gene, 114, 59–66. [DOI] [PubMed] [Google Scholar]
- Bartholomew C.R., Woo,S.H., Chung,Y.S., Jones,C. and Hardy,C.F. (2001) Cdc5 interacts with the Wee1 kinase in budding yeast. Mol. Cell. Biol., 21, 4949–4959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basi G., Schmid,E. and Maundrell,K. (1993) TATA box mutations in the Schizosaccharomyces pombe nmt1 promoter affect transcription efficiency but not the transcription start point or thiamine repressibility. Gene, 123, 131–136. [DOI] [PubMed] [Google Scholar]
- Bernard P., Hardwick,K. and Javerzat,J.-P. (1998) Fission yeast Bub1 is a mitotic centromere protein essential for the spindle checkpoint and the preservation of correct ploidy through mitosis. J. Cell Biol., 143, 1775–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridge A.J., Morphew,M., Bartlett,R. and Hagan,I.M. (1998) The fission yeast SPB component Cut12 links bipolar spindle formation to mitotic control. Genes Dev., 12, 927–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broek D., Bartlett,R., Crawford,K. and Nurse,P. (1991) Involvement of p34cdc2 in establishing the dependency of S phase on mitosis. Nature, 349, 388–393. [DOI] [PubMed] [Google Scholar]
- Daga R.R. and Jimenez,J. (1999) Translational control of the cdc25 cell cycle phosphatase: a molecular mechanism coupling mitosis to cell growth. J. Cell Sci., 112, 3137–3146. [DOI] [PubMed] [Google Scholar]
- Fantes P.A. (1979) Epistatic gene interactions in the control of cell division in fission yeast. Nature, 279, 428–430. [DOI] [PubMed] [Google Scholar]
- Forsburg S.L. (1993) Comparison of Schizosaccharomyces pombe expression systems. Nucleic Acids Res., 21, 2955–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry A.M., Meraldi,P. and Nigg,E.A. (1998) A centrosomal function for the human Nek2 protein kinase, a member of the NIMA family of cell cycle regulators. EMBO J., 17, 470–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry A.M., Descombes,P., Twomey,C., Bacchieri,R. and Nigg,E.A. (2000) The NIMA-related kinase X-Nek2B is required for efficient assembly of the zygotic centrosome in Xenopus laevis. J. Cell Sci., 113, 1973–1984. [DOI] [PubMed] [Google Scholar]
- Hagan I. and Yanagida,M. (1995) The product of the spindle formation gene sad1+ associates with the fission yeast spindle pole body and is essential for viability. J. Cell Biol., 129, 1033–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X., Patterson,T.E. and Sazer,S. (1997) The Schizosaccharomyces pombe spindle checkpoint protein Mad2p blocks anaphase and genetically interacts with the anaphase-promoting complex. Proc. Natl Acad. Sci. USA, 94, 7965–7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X., Jones,M.H., Winey,M. and Sazer,S. (1998) Mph1, a member of the Mps1-like family of dual specificity protein kinases, is required for the spindle checkpoint in S. pombe. J. Cell Sci., 111, 1635–1647. [DOI] [PubMed] [Google Scholar]
- Hoffmann I., Clarke,P.R., Marcote,M.J., Karsenti,E. and Draetta,G. (1993) Phosphorylation and activation of human Cdc25-C by Cdc2 cyclin-B and its involvement in the self-amplification of MPF at mitosis. EMBO J., 12, 53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda R., Tanaka,H., Ohba,Y. and Yasuda,H. (1995) Mouse p87wee1 kinase is regulated by M-phase specific phosphorylation. Chromosome Res., 3, 300–308. [DOI] [PubMed] [Google Scholar]
- Hudson J.D., Feilotter,H. and Young,P.G. (1990) stf1: non-wee mutations epistatic to cdc25 in the fission yeast Schizo saccharomyces pombe. Genetics, 126, 309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi T. and Maller,J.L. (1993) Elimination of cdc2 phosphorylation sites in the cdc25 phosphatase blocks initiation of M-phase. Mol. Biol. Cell, 4, 1337–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi T. and Maller,J.L. (1995) Phosphorylation and activation of the Xenopus Cdc25 phosphatase in the absence of Cdc2 and Cdk2 kinase activity. Mol. Biol. Cell, 6, 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones R.G.L. and Rosamond,J. (1990) Isolation of a novel protein kinase encoding gene from yeast by oligonucleotide probing. Gene, 90, 87–92. [DOI] [PubMed] [Google Scholar]
- Kovelman R. and Russell,P. (1996) Stockpiling of Cdc25 during a DNA replication checkpoint arrest in Schizosaccharomyces pombe. Mol. Cell. Biol., 16, 86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krien M.J., Bugg,S.J., Palatsides,M., Asouline,G., Morimyo,M. and O’Connell,M.J. (1998) A NIMA homologue promotes chromatin condensation in fission yeast. J. Cell Sci., 111, 967–976. [DOI] [PubMed] [Google Scholar]
- Krien M.J.E., West,R.R., John,U.P., Koniaras,K., McIntosh,J.R. and O’Connell,M.J. (2002) The fission yeast NIMA kinase Fin1p is required for spindle function and nuclear envelope integrity. EMBO J., 21, 1713–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A. and Dunphy,W.G. (1996) Purification and molecular cloning of Plx1, a Cdc25-regulatory kinase from Xenopus egg extracts. Science, 273, 1377–1380. [DOI] [PubMed] [Google Scholar]
- Lane H.A. and Nigg,E.A. (1996) Antibody microinjection reveals an essential role for human polo-like kinase 1 (Plk1) in the functional maturation of mitotic centrosomes. J. Cell Biol., 135, 1701–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew D.J. (2000) Cell-cycle checkpoints that ensure coordination between nuclear and cytoplasmic events in Saccharomyces cerevisiae. Curr. Opin. Genet. Dev., 10, 47–53. [DOI] [PubMed] [Google Scholar]
- Maundrell K. (1990) nmt1 of fission yeast: a highly transcribed gene completely repressed by thiamine. J. Biol. Chem., 265, 10857–10864. [PubMed] [Google Scholar]
- Moreno S., Klar,A. and Nurse,P. (1991) Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol., 194, 795–823. [DOI] [PubMed] [Google Scholar]
- Morris N.R. (1975) Mitotic mutants of Aspergillus nidulans. Genet. Res., 26, 237–254. [DOI] [PubMed] [Google Scholar]
- Mueller P.R., Coleman,T.R. and Dunphy,W.G. (1995) Cell cycle regulation of a Xenopus Wee1-like kinase. Mol. Biol. Cell, 6, 119–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvihill D.P., Petersen,J., Ohkura,H., Glover,D.M. and Hagan,I.M. (1999) Plo1 kinase recruitment to the spindle pole body and its role in cell division in Schizosaccharomyces pombe. Mol. Biol. Cell, 10, 2771–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg E.A. (2001) Mitotic kinases as regulators of cell division and its checkpoints. Nature Rev. Mol. Cell. Biol., 2, 21–32. [DOI] [PubMed] [Google Scholar]
- Nurse P., Thuriaux,P. and Nasmyth,K. (1976) Genetic control of the cell division cycle in the fission yeast Schizosaccharomyces pombe. Mol. Gen. Genet., 146, 167–178. [DOI] [PubMed] [Google Scholar]
- Oakley B.R. and Morris,N.R. (1983) A mutation in Aspergillus nidulans that blocks the transition from interphase into prophase. J. Cell Biol., 96, 1155–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohi R. and Gould,K.L. (1999) Regulating the onset of mitosis. Curr. Opin. Cell Biol., 11, 267–273. [DOI] [PubMed] [Google Scholar]
- Osmani A.H., O’Donnell,K. Pu,R.T. and Osmani,S.A. (1991) Activation of the NIMA protein kinase plays a unique role during mitosis that cannot be bypassed by absence of the BIME checkpoint. EMBO J., 10, 2669–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmani S.A., Pu,R.T. and Morris,N.R. (1988a) Mitotic induction and maintenance by overexpression of a G2-specific gene that encodes a potential protein kinase. Cell, 53, 237–244. [DOI] [PubMed] [Google Scholar]
- Osmani S.A., Engle,D.B., Doonan,J.H. and Morris,N.R. (1988b) Spindle formation and chromatin condensation in cells blocked at interphase by mutation of a negative cell cycle control gene. Cell, 52, 241–251. [DOI] [PubMed] [Google Scholar]
- Peters J.M., King,R.W., Hoog,C and Kirschner,M.W. (1996) Identification of BIME as a subunit of the anaphase-promoting complex. Science, 274, 1199–1201. [DOI] [PubMed] [Google Scholar]
- Petersen J., Paris,J., Willer,M., Philippe,M. and Hagan,I.M. (2001) The S. pombe Aurora related kinase Ark1 associates with mitotic structures in stage dependent manner and is required for chromosome segregation. J. Cell Sci., 114, 4371–4384. [DOI] [PubMed] [Google Scholar]
- Pu R.T., Xu,G., Wu,L., Vierula,J., O’Donnell,K., Ye,S.X. and Osmani,S.A. (1995) Isolation of a functional homolog of the cell cycle specific NIMA protein kinase of Aspergillus nidulans and functional analysis of conserved residues. J. Biol. Chem., 270, 18110–18116. [DOI] [PubMed] [Google Scholar]
- Russell P. and Nurse,P. (1986) cdc25+ functions as an inducer in the mitotic control of fission yeast. Cell, 45, 145–153. [DOI] [PubMed] [Google Scholar]
- Russell P. and Nurse,P. (1987) Negative regulation of mitosis by wee1+, a gene encoding a protein kinase homolog. Cell, 49, 559–567. [DOI] [PubMed] [Google Scholar]
- Tang Z., Coleman,T.R. and Dunphy,W.G. (1993) Two distinct mechanisms for negative regulation of the Wee1 protein kinase. EMBO J., 12, 3427–3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuriaux P., Nurse,P. and Carter,B. (1978) Mutants altered in the control co-ordinating cell division with cell growth in the fission yeast Schizosaccharomyces pombe. Mol. Gen. Genet., 161, 215–220. [DOI] [PubMed] [Google Scholar]
- Uto K. and Sagata,N. (2000) Nek2B, a novel maternal form of Nek2 kinase, is essential for the assembly or maintenance of centrosomes in early Xenopus embryos. EMBO J., 19, 1816–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A., Sherwin,T., Sasse,R., Macrae,T.H., Baines,A.J. and Gull,K. (1989) Definition of individual components within the cytoskeleton of Trypanosoma brucei by a library of monoclonal-antibodies. J. Cell Sci., 93, 491–500. [DOI] [PubMed] [Google Scholar]
- Ye S.X., Xu,G., Pu,R.T., Fincher,R.R., McGuire,S.L., Osmani,A.H. and Osmani,S.A. (1995) The NIMA protein kinase is hyperphosphorylated and activated downstream of p34cdc2/cyclinB: coordination of two mitosis promoting kinases. EMBO J., 14, 986–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye S.X., Fincher,R.R., Tang,A., Osmani,A.H. and Osmani,S.A. (1998) Regulation of the anaphase-promoting complex/cyclosome by BIMAAPC3 and proteolysis of NIMA. Mol. Biol. Cell, 9, 3019–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]