

Abstract
Interactions with nucleoporins containing FxFG-repeat cores are crucial for the nuclear import of RanGDP mediated by nuclear transport factor 2 (NTF2). We describe here the 1.9 Å resolution crystal structure of yeast NTF2-N77Y bound to a FxFG-nucleoporin core, which provides a basis for understanding this interaction and its role in nuclear trafficking. The two identical FxFG binding sites on the dimeric molecule are formed by residues from each chain of NTF2. Engineered mutants at the interaction interface reduce the binding of NTF2 to nuclear pores and cause reduced growth rates and Ran mislocalization when substituted for the wild-type protein in yeast. Comparison with the crystal structure of FG-nucleoporin cores bound to importin-β and TAP/p15 identified a number of common features of their binding sites. The structure of the binding interfaces on these transport factors provides a rationale for the specificity of their interactions with nucleoporins that, combined with their weak binding constants, facilitates rapid translocation through NPCs during nuclear trafficking.
Keywords: FxFG repeats/NTF2/nuclear trafficking
Introduction
Transport of cargo macromolecules between the cell nucleus and cytoplasm occurs through nuclear pore complexes (NPCs), huge proteinaceous structures that span the nuclear envelope (reviewed by Bayliss et al., 2000a; Ryan and Wente, 2000; Kuersten et al., 2001; Stewart et al., 2001). The passage of cargo through NPCs is mediated by shuttling carrier molecules that bind their cargo in one compartment and release it in the other. For example, the nuclear import of proteins bearing a classical nuclear localization sequence (NLS) is mediated by importin-β, which binds its cargo in the cytoplasm through the importin-α adapter. The cargo–carrier complex is then translocated through the NPC. Once in the nucleus, RanGTP binds to importin-β and dissociates the cargo– carrier complex to release the cargo. Importin-β is recycled back to the cytoplasm in complex with RanGTP, where RanGAP promotes hydrolysis of the GTP bound to Ran, dissociating the importin–Ran complex and thus freeing importin-β for another round of nuclear protein import (reviewed by Görlich and Kutay, 1999; Bayliss et al., 2000a; Kuersten et al., 2001). Repeated cycles of this pathway will deplete RanGTP from the nucleus, and so cytoplasmic RanGDP has to be re-imported to be recharged with GTP by the nuclear Ran guanine nucleotide exchange factor (RanGEF), RCC1. The import of RanGDP is mediated by nuclear transport factor 2 (NTF2) (Ribbeck et al., 1998; Smith et al., 1998), which has served as a simple and specialized system in which to study the molecular principles underlying translocation (reviewed by Stewart, 2000; Ribbeck and Görlich, 2001).
NPCs are constructed from multiple copies of proteins termed collectively nucleoporins (Rout et al., 2000; Ryan and Wente, 2000). Yeast NPCs contain 30 different nucleoporins (Rout et al., 2000), whereas vertebrate NPCs, which are somewhat larger, are thought to contain on the order of 30–50 different nucleoporins (Vasu and Forbes, 2001). A striking feature of many nucleoporin sequences is the presence of large regions consisting of characteristic FG tandem sequence repeats based on hydrophobic Phe-rich cores separated by hydrophilic linkers (Rout and Wente, 1994; Ryan and Wente, 2000). The hydrophobic cores of FG repeats are highly conserved and contain one or two Phes. The most common repeat core sequence motifs are based on FG, GLFG or FxFG (where x is usually a small residue such as Ser, Gly or Ala). In contrast, the hydrophilic spacers that link successive cores are highly variable both in length and sequence, although they are generally rich in charged and polar residues and have few hydrophobic residues. Nucleoporins frequently have domains that contain many copies of these repeats. For example, there are 19 FxFG repeats in Nsp1p and 33 GLFG repeats in Nup116 (Rout and Wente, 1994).
Although the precise mechanism by which cargo– carrier complexes are translocated through NPCs remains controversial, there is an emerging consensus that interactions between carriers and FG nucleoporins are involved directly (Bayliss et al., 2000a; Ryan and Wente, 2000; Allen et al., 2001; Ribbeck and Görlich, 2001; Stewart et al., 2001). Most carrier molecules bind FG nucleoporins (Rexach and Blobel, 1995; Shah and Forbes, 1998; Ryan and Wente, 2000; Allen et al., 2001). Different carriers appear to bind specific nucleoporins and/or classes of nucleoporin preferentially (Shah and Forbes, 1998; Allen et al., 2001). For example, importin-β, its yeast homologue Kap95 and the mRNA export factor, TAP, bind both FxFG and GLFG nucleoporins (Rexach and Blobel, 1995; Bayliss et al., 2000b; Allen et al., 2001; Strawn et al., 2001), whereas NTF2 binds primarily FxFG nucleoporins (Paschal and Gerace, 1995; Clarkson et al., 1997). Mutants of NTF2 or importin-β in which the strength of FxFG repeat binding is reduced exhibit a correspondingly reduced rate of nuclear import (Bayliss et al., 1999, 2000b; Quimby et al., 2001; Ribbeck and Görlich, 2001).
NTF2 has been a powerful model system in which to study the molecular basis of translocation through NPCs on account of both its simplicity and its specialization (Stewart, 2000; Ribbeck and Görlich, 2001) as well as the availability of crystal structures of the molecule itself (Bullock et al., 1996) and its complex with RanGDP (Stewart et al., 1998). The interaction between NTF2 and FxFG nucleoporins, which is crucial in mediating the nuclear import of RanGDP (Bayliss et al., 1999), has been studied using both the vertebrate (rNTF2) and Saccharomyces cerevisiae (yNTF2) proteins. The yeast and rat proteins share 40% sequence identity and rNTF2 can functionally replace the essential yNTF2 protein (Corbett and Silver, 1996). Previous studies have identified a surface hydrophobic patch centred on Trp7 in rNTF2 (Phe5 in yNTF2) that interacts with FxFG repeats (Bayliss et al., 1999; Quimby et al., 2001). Site-directed mutagenesis in combination with protein–protein interaction studies and functional assays both in vivo and in vitro have established the importance of this residue in the interaction with FxFG nucleoporins (Bayliss et al., 1999; Ribbeck and Görlich, 2001; Quimby et al., 2001). However, although these studies have established a key residue in the NTF2 nucleoporin binding site, the precise structural basis of the interaction has remained obscure. Moreover, Ribbeck and Görlich (2001) found that wild-type rNTF2 passed through NPCs 125 times more quickly than green fluorescent protein (GFP), a molecule of similar size with no known role in nuclear trafficking, and suggested that the surface of rNTF2 has hydrophobic, translocation-promoting properties. Although they also showed that Trp7 contributed to translocation, it did not appear to be the sole determinant, because the W7R mutant of rNTF2 still passed through NPCs 30 times more quickly than GFP. Consistent with this observation, mutation of the corresponding residue, Phe5, in yNTF2 produces only a small effect on the in vivo function of yNTF2 (Quimby et al., 2001). Moreover, mutations that alter NTF2–nucleoporin interactions have been described that do not lie near or within the hydrophobic patch. These include the dominant-negative mutants, D23A (Lane et al., 2000) and N77Y (Quimby et al., 2001), both of which block nuclear transport when overexpressed. Thus it appears likely that, although Trp7 is a component of the rNTF2 FxFG binding site, the site itself is more extensive.
Crystal structures have been obtained of importin-β residues 1–442 complexed with a FxFG core (Bayliss et al., 2000b) and of a fragment of the TAP/p15 heterodimer, which has a similar structure to NTF2, complexed with a FG peptide (Fribourg et al., 2001). In both, the interaction interface involves primarily the Phe rings of the repeat core and hydrophobic residues on the surface of the transport factor. However, neither complex is directly analogous to that formed between NTF2 and FxFG nucleoporins. Although the TAP/p15 fragment is structurally homologous to NTF2, it is bound to a FG core, whereas NTF2 binds only FxFG cores. Conversely, the importin-β fragment is bound to a FxFG core, but is not structurally related to NTF2. Therefore, to address the precise nature of the NTF2–FxFG interaction, we have determined the crystal structure of yNTF2 in complex with a FxFG peptide. We describe here the crystal structures of both yNTF2-N77Y alone and also bound to a FxFG-repeat core. These structures have enabled us to evaluate the precise manner in which FxFG-nucleoporin cores interact with this nuclear transport factor at 1.9 Å resolution.
Results and discussion
Crystal structure of yNTF2-N77Y at 1.6 Å resolution
Although we have described previously trigonal crystals of wild-type yNTF2 complexed with a fragment of a FxFG repeat-containing nucleoporin (Bayliss et al., 2000c), merohedral twinning frustrated structure determination. However, we found that with ammonium sulfate, yNTF2-N77Y fortuitously crystallized in space group P21 and so was not subject to merohedral twinning. These crystals diffracted to 1.6 Å resolution and were solved by molecular replacement and refined to a final R-factor of 20.2% (Rfree = 22.3%) with good stereochemistry (Table I). The asymmetric unit contained four chains comprising two physiological NTF2 dimers and it was possible to model almost all of the yNTF2-N77Y sequence (Table I). As shown in Figure 1A, yNTF2-N77Y (red) closely resembled rNTF2 (blue). Large differences [>2 Å Cα root mean square deviation (r.m.s.d.)] were confined mostly to loop regions, which also differed most in sequence and often contained insertions (underlined in red, Figure 1B). The rNTF2 crystals were obtained using polyethylene glycol (PEG) and so, to check that these differences were not a result of the very different crystallization conditions used for rat and yeast NTF2, we also obtained crystals of rat NTF2 using ammonium sulfate (Table I) that diffracted to 1.6 Å resolution. The refined structure of rNTF2 in these crystals was not significantly different to that obtained in PEG (Bullock et al., 1996), confirming that the differences observed between the rat and yeast NTF2 structures were not due to different crystallization conditions.
Table I. Crystallographic data for NTF2 structures.
| yNTF2-N77Y native | yNTF2-N77Y + FxFG peptide | rNTF2 (NH4)2SO4 | rNTF2 D92N/D94N | ||
|---|---|---|---|---|---|
| Crystals |
|
|
|
|
|
| Space group | P21 | P21 | P212121 | P21 | |
| Lattice constants | a (Å) | 58.66 | 58.84 | 55.54 | 35.04 |
| b (Å) | 83.87 | 84.06 | 57.39 | 79.02 | |
| c (Å) | 61.50 | 61.79 | 86.89 | 42.14 | |
| |
β (°) |
115.86 |
116.00 |
90.00 |
104.36 |
| Data collection |
|
|
|
|
|
| Resolution (Å) | 46–1.6 | 53–1.9 | 36.3–1.6 | 18.1–2.3 | |
| Highest shell | 1.69–1.6 | 2.0–1.9 | 1.69–1.6 | 2.43–2.3 | |
| Observations | 353 243 | 266 201 | 234 160 | 18 416 | |
| Unique reflections | 66 296 | 39 224 | 36 958 | 7519 | |
| Completeness (%)a | 94 (69.5) | 92.4 (88.3) | 98.6 (93.5) | 96.6 (91.7) | |
| Multiplicitya | 3.4 (2.3) | 3.0 (3.4) | 3.9 (3.5) | 2.3 (2.2) | |
| Rmerge (%)a | 7.0 (32.1) | 7.0 (25.7) | 4.2 (19.0) | 4.5 (9.2) | |
|
I/σ(I)a |
|
5.2 (2.0) |
6.9 (2.7) |
24.6 (4.6) |
9.7 (7.4) |
| Refinement |
|
|
|
|
|
| Resolution range (Å) | 40–1.6 | 40–1.9 | 20–1.6 | 18–2.3 | |
| No. of residues/waters | 864/382 | 884/384 | 429/202 | 247/63 | |
| Chain A | 4–124 | 3–124 | 3–127 | 2–126 | |
| Chain B | 3–124 | 3–124 | 3–125 | 3–127 | |
| Chain C | 6–124 | 5–124 | n/a | n/a | |
| Chain D | 5–124 | 5–124 | n/a | n/a | |
| R-factor (Rfreeb) | 20.2 (22.3) | 19.7 (22.6) | 21.1 (23.8) | 22.2 (26.4) | |
| Bond length r.m.s. (Å) | 0.005 | 0.006 | 0.012 | 0.007 | |
| Bond angle r.m.s. (°) |
|
1.4 |
1.4 |
1.5 |
1.3 |
| Ramachandran plot |
|
|
|
|
|
| Most favoured (%) | 90.3 | 89.2 | 94.1 | 91.4 | |
| Allowed (%) | 7.6 | 9.0 | 4.5 | 7.3 | |
| Generously allowed (%) | 1.2 | 1.1 | 1.4 | 1.4 | |
| Forbidden (%) | 0.9c | 0.7c | 0.0 | 0.0 | |
| PDB accession code | 1gy7 | 1gyb | 1gy6 | 1gy5 |
aHighest resolution shell in parentheses.
bFree R-factor was computed using 5% of the data assigned randomly (Brünger, 1992).
cThe forbidden conformations adopted by Asp91 and Glu40 were supported by well-defined electron density. Asp91 located in a sharp turn and is equivalent to Asp92 in rNTF2, which also adopts a forbidden conformation (Bullock et al., 1996). Glu40 is part of a turn that, in yNTF2, is distorted from its rNTF2 conformation by the substitution of a conserved Gly by Thr41.

Fig. 1. Structural differences between yeast and rat NTF2. (A) Superposed Cα traces of rNTF2 (blue) and yNTF2-N77Y (red) showing the overall similarity between the two structures. Only the N-terminus (N), C-terminus (C) and the loop containing residues 91–96 differ between all four yNTF2-N77Y chains. (B) Structure-based sequence alignment of yNTF2 (Sc) and rNTF2 (rat). Conserved residues are shown with a black background and the variable regions are marked in red. (C) Fo – Fc omit electron density map (blue, contoured at 2σ), generated after simulated annealing of structure after subtraction of the FxFG peptide, showing a tube of density on the surface of yNTF2 (grey) around the FxFG peptide (yellow).
An aromatic residue near the N-terminus of the first α-helix of both vertebrate and yeast NTF2 has been implicated directly in binding to FxFG repeats (Bayliss et al., 1999; Quimby et al., 2001). Although this residue (Trp7) was well ordered in the rNTF2 structure (PDB 1OUN), the equivalent residue (Phe5) was less well ordered in yNTF2 and indeed in chain C the density was so poor that this residue could not be modelled reliably. The N-terminus of yNTF2-N77Y showed more variability, reflected in correspondingly higher B-factors, than the rest of the molecule. In addition, the conformation of a portion of the N-terminal helix (up to residue 11) was altered in chains C and D by crystal packing interactions. In summary, the yNTF2-N77Y structure was very similar to that of rNTF2, except at its N-terminus, which was less well ordered, and two loops where the sequence was poorly conserved.
Crystal structure of yNTF2-N77Y complexed with a FxFG peptide at 1.9 Å resolution
P21 crystals of yNTF2-N77Y complexed to a FxFG peptide (DSGFSFGSK, modelled on Nsp1p) were obtained under similar conditions to those used for yNTF2-N77Y alone (albeit at higher ammonium sulfate concentration). These crystals diffracted to 1.9 Å resolution (Table I) and were solved by rigid body refinement of the native structure. Initial inspection of Fo – Fc difference electron density maps showed clear tubes of positive electron density with two protruding lobes that were located at the same position on the surface of all four chains. There were small changes to yNTF2 as a result of peptide binding and so, before building the FxFG peptide into the positive electron density, we first rebuilt and refined the NTF2 chains, after which the R-factor was reduced to 24.7% (Rfree = 27.6%). The only extended and significant positive difference density now remaining was located at the same position on the surface of all four chains, at the dimer interface close to Phe5, which was then modelled as FxFG peptide. In all four cases, the density of Phe side chains of the peptides was much clearer than their backbone density, indicating that the side chains in contact with NTF2 were probably more ordered than the backbone itself. Consistent with this interpretation, the location of the two Phe side chains was similar in all four chains, whereas the backbone conformation of the peptides was more variable. Moreover, the atoms with the lowest B-factors in the refined peptide were in the Phe aromatic rings. Addition of a FxFG to each NTF2 chain and 384 water molecules reduced the R-factor to 19.7% (Rfree = 22.6%), which was increased to 20.0% (Rfree = 23.1%) by removal of the FxFG peptide chains from the model. Furthermore, an omit electron density map showed a clear difference due to the peptide (Figure 1C). Only the five central residues (GFSFG) of the peptide, corresponding to the nucleoporin ‘core’, were visible. A similar pattern was seen with a FxFG-nucleoporin fragment bound to importin-β (Bayliss et al., 2000b) and was consistent with the residues in the peptide outside the FxFG core being unstructured and thus too mobile to produce clear electron density.
Interaction between NTF2 and FxFG-nucleoporin cores
As illustrated in Figure 2, the FxFG peptide (yellow) bound near the interface between the two chains of the yNTF2 dimer (light pink, light blue) at a stoichiometry of one FxFG core per symmetry-related site, equivalent to one FxFG core per yNTF2 chain. The location of the FxFG binding site on yNTF2 was different to the binding site for a FG peptide bound to the NTF2-like domain of the TAP/p15 heterodimer (Figure 2C) reported by Fribourg et al. (2001). Although there is a solvent-exposed hydrophobic patch on the surface of NTF2 at roughly the same location as the TAP FG binding site (Fribourg et al., 2001), this site was not occupied in the N77Y–FxFG structure. Instead, FxFG repeats bind to NTF2 across its homodimerization interface at a site that is well conserved among NTF2 homologues, but does not appear to be present in TAP or p15 homologues.

Fig. 2. FxFG-repeat cores bind to a hydrophobic depression at the dimer interface of yNTF2. (A) Stereo drawing showing how the FxFG peptide (chain E, yellow) binds to residues from both chains of the NTF2 dimer (chain A, red and chain B, blue). (B) The relative disposition of the two FxFG cores bound to the NTF2 dimer. The Tyr77 side chains are also shown in grey and three residues (Leu8, Phe12 and Tyr112) that may be involved in a tentative secondary site are in green. (C) The TAP/p15 dimer with bound FG peptide (Fribourg et al., 2001) in the same orientation, illustrating the different locations of the FG-nucleoporin binding sites on each molecule.
Figures 2A, 3A and 4A illustrate the contacts made between the FxFG peptide and yNTF2. The FxFG core (yellow) adopted a β conformation and was oriented to bury both its Phe side chains (labelled F1, F2) in a hydrophobic depression on the surface of the dimer, whereas the ‘x’ residue between the two Phes (a Ser in the structure solved) was oriented away from the NTF2 surface. This arrangement of the ‘x’ residue, so that its side chain does not make a contribution to the interaction, is consistent with its variability in different FxFG-nucleoporin sequences that are known to interact with NTF2 (examples include Gly, Ala, Ser, Lys and Val). The primary molecular contacts with the FxFG peptide involve side chains from both chains in the NTF2 dimer. From left to right in Figure 2A, in one chain (dark grey) the side chains of Pro76, Pro73 and Phe5 (red) contribute to the hydrophobic depression, whereas in the second chain (light grey) Glu34, Phe115, Met36, Gln45 and Gln43 (blue) contribute. Figure 3A shows a schematic representation of the interactions between NTF2 and the FxFG peptide. The aromatic rings of the FxFG repeat were primarily responsible for the intimate contact with yNTF2 (red flashes), although a putative H-bond (green) was also formed between Gln45 and the peptide backbone at residue ‘x’. The first Phe of the peptide, F1, interacted with Gln43(B), Gln45(B), Phe5(A) and Met36(B), whereas the second Phe, F2, interacted with Met36(B), Phe115(B), Pro73(A) and Glu34(B) (chain indicated in parentheses). The total surface area buried in the interaction was 611 Å2 for five core residues (GFSFG), which was comparable to 670 Å2 buried for the core in the FxFG–importin-β structure (Bayliss et al., 2000b). The only other substantial hydrophobic patch on the surface of NTF2 is associated with binding RanGDP (see Stewart et al., 1998) and so was unavailable for binding FG repeats during Ran import.
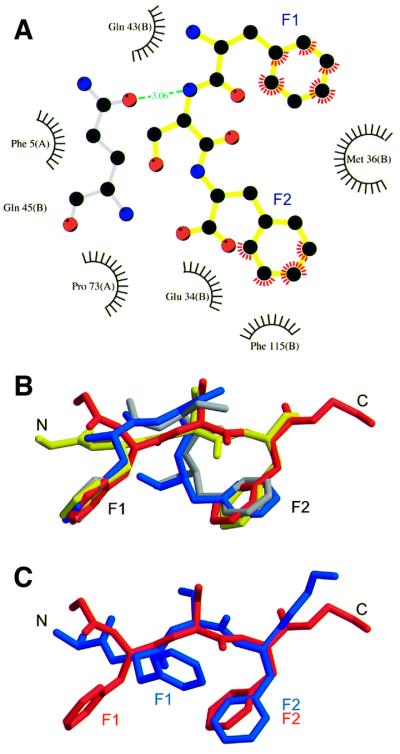
Fig. 3. FxFG–yNTF2 interface. (A) Schematic diagram of molecular contacts between FxFG-repeat core and yNTF2 dimer, adapted from the LIGPLOT output (Wallace et al., 1995). The FxFG core (yellow bonds between black C atoms, red O atoms and blue N atoms) contacts yNTF2 residues (black circles, flashes) primarily by its Phe-ring atoms (red flashes). A putative H-bond (green) is formed between Gln45 (grey bonds) and the FxFG main chain. (B) The backbone conformation of chains E (red) and G (yellow) are different from those seen with chains F (blue) and H (grey), but significantly the Phe rings of all four chains are in approximately the same position. (C) Chains E (red) and G most closely resemble the conformation of FxFG cores bound to importin-β (blue; Bayliss et al., 2000b).

Fig. 4. Surface representations comparing the binding of FG peptides to different transport factors. (A) yNTF2; (B) TAP/p15 (Fribourg et al., 2001); (C) importin-β (Bayliss et al., 2000b). In each case the Phe rings bind to hydrophobic depressions on the transport factor surface, but the overall structure of each site is different.
There were 15 van der Waals contacts (Figure 3A) between the FxFG core and carbon or sulfur atoms of NTF2, seven of which involved Met36, which was buttressed against Phe115 from one chain and Pro73 of the other. The aliphatic side chain of Pro73 was also important in defining the hydrophobic binding patch and its rigidity probably helped define the precise shape of the site. Except for Phe5 and Gln45, there was little change in side chain conformation in the NTF2 residues involved in the interaction. Moreover, all the interacting side chains were well defined in the native structure, indicating that there was probably little loss of conformational entropy in NTF2 associated with binding. There may have been a loss of entropy associated with the FxFG core’s binding, but this cannot be assessed because there is no information about its conformation in solution. However, as FxFG cores bound to either NTF2 or importin-β have a β conformation, it is likely that they have this conformation in solution as well, and so any negative entropic effects would probably be restricted to the Phe side chains. The entropy associated with the release of ordered water from both the NTF2 binding site (where, for example, five strongly associated waters are lost on binding the FxFG peptide formed by A5, B43, B45) and the Phe rings of the FxFG core probably makes the dominant contribution to the binding energy. The way in which the Phe rings are encapsulated by aliphatic side chains at the interaction interface indicates that there may be π-electron interactions between the Phe rings and NTF2 (see Brandl et al., 2001). The involvement of these putative π interactions, together with Phe being the most hydrophobic amino acid and the geometry of the binding sites on transport factors, probably accounts for the invariable presence of Phe in the repeat cores. The residues involved in FxFG binding are generally conserved between species. Thus, Gln43 and Pro73 are absolutely conserved and residue 45 is almost always Gln. There are compensatory mutations at positions 5 and 36, so that residue 36 is Met when residue 5 is Phe, and Cys or Phe when it is Trp, thus retaining the general size of the hydrophobic patch.
The structure of the interface also accounts for NTF2’s preference for FxFG nucleoporins since the binding site contains separate pockets for each Phe ring. Repeat cores based on FG alone would therefore not be able to interact as efficiently. Although the GLFG core present in many nucleoporins (reviewed by Ryan and Wente, 2000) contains a second hydrophobic side chain, the separation between the two binding pockets on NTF2 together with the ridge between them formed by Met36 and Gln45, would impede the Leu and Phe side chains binding simultaneously. Moreover, the different conformation of the peptide backbone necessary to position both Phe and Leu side chains against NTF2 would prevent the formation of H-bonds by Gln45. Thus, the small hydrophilic ‘x’ residue in the repeat core serves both as a spacer and, by facing outwards (hence its hydrophilic character) helps to stabilize the β conformation of the FxFG core bound to NTF2. The location of the binding site also accounts for our failure to obtain a structure of rNTF2 bound to FxFG repeats because, in the rNTF2 crystals, the FxFG binding site was blocked by crystal-packing interactions.
The two FxFG cores bound to the NTF2 dimer (Figure 2A) were separated by 35 Å, a distance that could be spanned by the 15 residues found in Nsp1p linkers, for example. However, the two FxFG cores bound per dimer had the same orientation, so for consecutive FxFG cores to bind simultaneously, the linker could not lie simply along the direct path between the two sites. Thus, a single linker might not be sufficiently long to span between the two sites because the nucleoporin would have to deviate from its most direct path for cores to bind in the same orientation to both sites. However, since most FxFG nucleoporins contain a number of repeats, it would still be possible for NTF2 to bind two cores from the same nucleoporin simultaneously. The N77Y mutation introduces two hydrophobic Tyr side chains between the two FxFG sites and so probably functions to increase the hydrophobicity of the NTF2 surface and thus its affinity for the hydrophobic nucleoporin cores.
Structural changes in yNTF2 associated with peptide binding
Because the same crystal form of yNTF2-N77Y was used to obtain its structure in the absence and presence of peptide, differences between the yNTF2 chains in the two crystal structures were most probably associated with the binding of the peptide. There were two significant differences in backbone conformation between the native and peptide-bound structures. First, residues 121–124 of chain C moved dramatically (Cα r.m.s.ds of 4.1 Å for Tyr123 and 6.4 Å for Ser124). This movement appeared to be due to the FxFG peptide competing the C-terminus from its crystal contact binding site in the native structure and thus forcing it to adopt a different conformation, similar to that found with chain A. Although the crystal contacts involving the C-termini of chains A and C did not interfere directly with the binding of the FxFG core, they did appear to alter the conformation of the peptide backbone slightly by blocking the path of the peptide chain after the core sequence. Figure 3B illustrates the different FxFG conformations observed among the four chains bound to NTF2. The FxFG-repeat cores (chains F and H, blue and grey, respectively) bound to sites close to these crystal contacts adopted similar conformations, but differed somewhat from the conformation adopted by the cores at the other two sites (chains E and G, red and yellow, respectively) that were distant from crystal contacts and which adopted a backbone conformation closer to that observed in the importin-β complex (Figure 3C) (Bayliss et al., 2000b). In all instances the electron density for the peptide backbone was not as clear as for the two Phe side chains and the B-factors were higher, consistent with a degree of mobility in backbone conformation of FxFG repeat. However, although there were differences in backbone conformation between the four FxFG cores bound to NTF2, in each the location of the Phe rings was the same (Figure 3B). A second difference between native and peptide-bound NTF2 chains occurred at residues 107–112 of chain A. This difference was associated with two additional peaks of density close to a hydrophobic patch on the surface of this chain between the first helix of yNTF2 (residues 8–15) and the loop containing residues 107–112. Additionally, Leu8 adopted a different rotamer that brought its side chain in close contact with one of the peaks (green, Figure 2B). Although it was not possible to model a peptide chain into the difference peaks, they may correspond to the aromatic rings of a FxFG repeat. A secondary FxFG binding site at this location is consistent with the observation that the Y112A/F5A double mutant of yNTF2 exhibits a synergic reduction in binding to FxFG repeats compared with individual mutants (Quimby et al., 2001). However, this additional density was only observed on a single chain and so any identification with a secondary FxFG binding site must be considered tentative.
The presence of the peptide induced several minor changes at the primary FxFG binding site in all four NTF2 chains. The most obvious changes occurred at the N-terminus which, in yNTF2, was more disordered compared with the rest of the chain than in rNTF2 (see Bullock et al., 1996). Furthermore, in the absence of the FxFG peptide, the N-terminus of yNTF2-N77Y was different between the four chains in the asymmetric unit (although many of these differences appeared to derive from crystal contacts). In contrast, electron density maps for this region were clearer when the FxFG peptide was bound and residues could be modelled with much greater confidence. In the case of chain B, substantially lower B-factors were obtained, consistent with their becoming more ordered. In addition, the aromatic side chain of Phe5 moved closer to the ring of the peptide Phe F1. Gln45 adopted a different rotomer when the FxFG peptide was bound and this increased the intimacy of its contact with F1.
Engineered mutations that reduce the binding of NTF2 to NPCs
The functional importance of the FxFG binding site identified in the crystal structure of the yNTF2–FxFG complex was explored by mutating Glns 43 and 45 to Asp (Q43/45D mutant). In pull-down experiments (Clarkson et al., 1996) this mutant retained wild-type affinity for RanGDP and was a dimer by gel filtration. However, when the Q43/45D mutant was expressed in yeast as the only functional copy of the essential NTF2 protein, these cells grew more slowly than wild-type cells (Figure 5A). Western blots confirmed that, in these assays, mutant and wild-type proteins were expressed to equivalent levels. To examine the impact of the Q43/45D mutation on targeting of NTF2 to the nuclear pore in vivo, we examined the localization of wild-type NTF2–GFP and Q43/45D NTF2–GFP. As reported previously (Quimby et al., 2001), wild-type NTF2–GFP was concentrated at the nuclear rim. In contrast, Q43/45D NTF2–GFP was localized diffusely throughout the cell (Figure 5B), in the same way as seen with other NTF2 mutants that show decreased interactions with nucleoporins (Quimby et al., 2001). Finally, we tested whether the Q43/45D mutation affected nuclear Ran import, the physiological function of NTF2 (Ribbeck et al., 1998; Smith et al., 1998). Q43/45D NTF2 mutant cells showed an increased cytoplasmic concentration of Ran–GFP (Figure 5C) compared with wild-type NTF2, consistent with Ran nuclear import being impeded by the reduced binding affinity of the mutant NTF2 for nucleoporins. Taken together, these results underline the physiological importance of the NTF2/nucleoporin interface identified in the crystal structure and also demonstrate that Gln43 and Gln45 are important in binding nucleoporins to wild-type NTF2, thus confirming that the binding site identified in the crystals was not an artefact associated with the N77Y mutation.
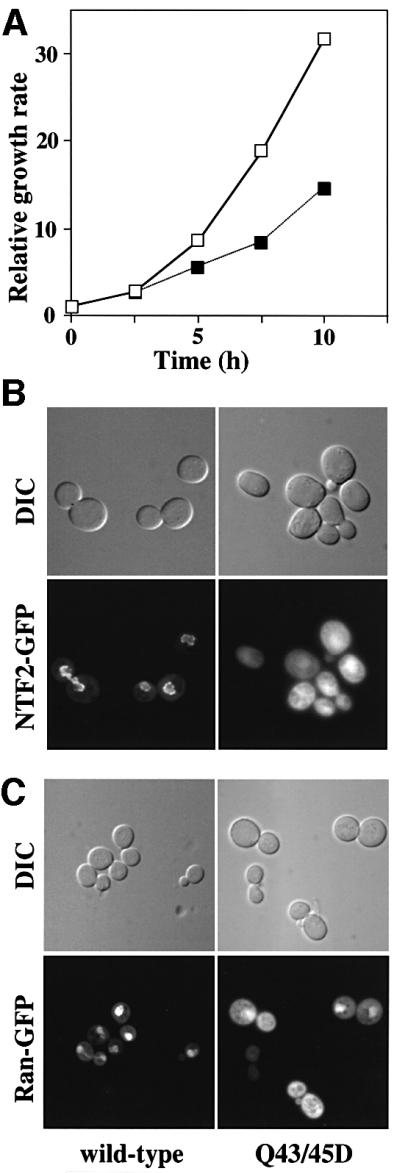
Fig. 5. Q43/45D yNTF2 shows reduced cell growth rate and mislocalization of NTF2 and Ran. (A) When the Q43/45D mutant was expressed in yeast as the only functional copy of NTF2 (filled squares), these cells grew more slowly than wild-type cells (open squares). (B) Localization of GFP fusions to NTF2. Wild-type NTF2 showed primarily the typical punctate nuclear envelope staining characteristic of its binding to NPCs (see Quimby et al., 2001), whereas the Q43/45D mutant showed diffuse staining throughout the cells. (C) With wild-type NTF2, Ran–GFP was localized to the nucleus, whereas with Q43/45D-NTF2 a considerable proportion of the Ran–GFP was mislocalized to the cytoplasm.
Putative FxFG binding site on rat NTF2
Although it has not yet been possible to obtain a crystal structure of an rNTF2–FxFG complex, it is likely that the interaction is very similar to the yNTF2–FxFG interaction. This idea was supported by the crystal contact interactions in two rNTF2 crystal structures where a Phe126 side chain packed into a pocket composed of Trp7, Gln45, Gln47 and Cys38, analogous to the yNTF2 pocket that bound F1 of the FxFG peptide. One example was in the crystal structure of D92N/D94N rNTF2, which shows reduced binding to RanGDP compared with wild-type rNTF2 (Clarkson et al., 1997), and crystallizes in a crystal form different from that of wild-type rNTF2. We solved the structure of P21 D92N/D94N rNTF2 crystals (which have very different lattice parameters from the yNTF2-N77Y P21 crystals, see Table I) to 2.3 Å resolution by molecular replacement. As shown in Figure 6A, in these crystals the side chain of Phe126 (red) is sandwiched between Gln45 and Gln47, causing both residues to adopt different rotamers (yellow) from those seen in the wild-type structure (blue), as well as producing a slight movement of the Trp7 side chain towards the inserted Phe126 aromatic ring. An analogous crystal-packing interaction was also observed in the rNTF2-W7A structure (Bayliss et al., 1999). The rotamers adopted by Gln45 and Gln47 in both structures were a close match to those seen in the yNTF2-N77Y structure. As shown in Figure 6B, the position and conformation of the FxFG core bound to yNTF2 is compatible with binding to the analogous site on rNTF2. Consistent with the essential features of the FxFG binding sites of rNTF2 and yNTF2 being retained, previous studies have shown that the W7A mutation reduced the strength of the rNTF2–FxFG interaction and that rNTF2-W7A mediated nuclear import of RanGDP less efficiently than wild type (Bayliss et al., 1999). However, neither this mutation nor the more aggressive W7R mutation fully eliminated the shuttling of rNTF2 (Ribbeck and Görlich, 2001).
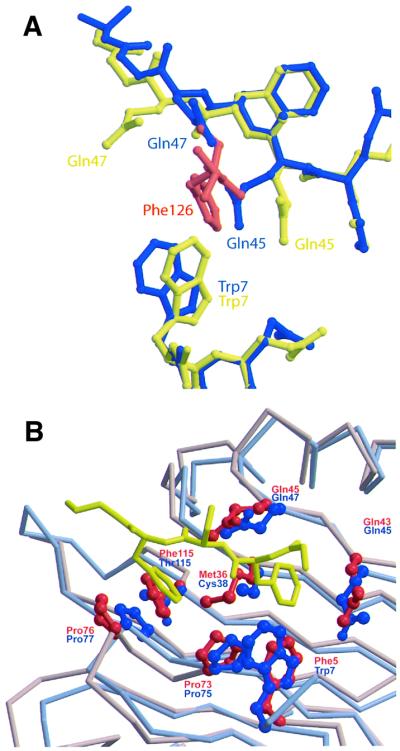
Fig. 6. Putative FxFG binding site on rNTF2. (A) Comparison of FxFG binding pocket in wild-type (blue) and D92N/D94N crystal forms (yellow). Crystal lattice interactions in the structure of the D92N/D94N rNTF2 mutant (yellow) include the insertion of a Phe side chain (red) between Gln45 and Gln47. Rotamers adopted by Gln45 and Gln47 in the D92N/D94N structure (yellow) are similar to those found in the yNTF2-N77Y structure, but different from wild-type rNTF2 (blue). In addition, Trp7 moves closer to the inserted Phe aromatic ring. (B) FxFG core (yellow) fits on to the surface of rNTF2 structure (blue), which superposes closely with yNTF2 (red). Rotamers of rNTF2 Gln45 and Gln47 vary between rNTF2 structures. Those shown in the figure are from the structure of rNTF2 bound to RanGDP, which closely match those observed in crystals of D92N/D94N rNTF2, rNTF2-W7A or yNTF2-N77Y.
Common features of the interaction between nuclear transport factors and FxFG-nucleoporin cores
We investigated the structural basis of FG-nucleoporin recognition by comparing three different transport factors (NTF2, TAP/p15 and importin-β) encompassing two different folds (Figure 2B and C and Figure 4). In all three structures, the interactions appear to involve primarily the FG cores, with little contribution from the linkers. When bound to transport factors, the FxFG-repeat cores adopt an essentially β conformation (see Figure 3B) in which the aromatic Phe side chains are oriented to point inwards and so become buried in the hydrophobic groove on the transport factor, whereas the generally small and hydrophilic ‘x’ residue between the two Phes points outwards towards the solvent. The Gly residue in the FxFG-repeat core accommodates a tight turn at the end of the β-segment of the core that, in the case of importin-β, is crucial in avoiding contact with other regions of the transport factor (Bayliss et al., 2000b). However, although this form of interaction appears to be fundamental to the interaction between transport factors and FxFG nucleoporins, in some instances, it might be strengthened by additional interactions with specific linker sequences. Such an explanation would be consistent, for example, with the stronger binding observed between importin-β and Nup153 compared with other nucleoporins (Shah and Forbes, 1998; Ben-Efraim and Gerace, 2001). Each transport factor presents a site for FG binding located between two different structural elements (the two chains of the dimer for NTF2; the β-sheet and first α-helix of TAP, and the A-helices of HEAT repeats 5 and 6 for importin-β) that form a primarily hydrophobic surface depression (Figure 4). Although it can accommodate the aromatic rings of the two Phes in the repeat core, the depression is not sufficiently deep or wide to also accommodate the peptide backbone, which instead rests on the top of it. The depression is lined with a combination of large hydrophobic side chains (Phe, Trp, Leu) and the aliphatic portions of longer polar or even charged side chains (such as Gln and Glu), leaving cavities that can accommodate Phe side chains. Furthermore, the aliphatic portions of the longer polar and/or charged residues form hydrophobic interactions with the Phe rings, while their hydrophilic ends can form H-bonds with the peptide backbone of the repeat core. In addition to the common features involved in FG-core recognition identified by comparing binding to different nuclear transport factors, it is also instructive to compare bona fide FG binding sites with other hydrophobic patches on the molecules that do not seem to bind. Thus, the three NTF2-like domains of different transport factors (NTF2, TAP and p15) each have two surface hydrophobic patches that could form putative FG binding sites. However, these sites differ fundamentally in their recognition of FG repeats. Neither of NTF2’s FxFG binding sites (which are identical because NTF2 is a homodimer) are found in the TAP/p15 heterodimer. One equivalent location on TAP/p15 is blocked by a difference in secondary structure (the extended first α-helix of p15 that is two turns longer than the equivalent helix in NTF2 or TAP), whereas the other contains none of the residues conserved between NTF2 homologues (e.g. the two Gln residues that are not found in p15 and the two Pro residues that are not found in TAP). Likewise, the FG binding site of TAP is not conserved in NTF2, where the equivalent location is sterically blocked by the side chain of Tyr112 and the backbone of residues 78 and 79. Overall, although the general features of the FG binding site of transport factors may be similar, there are significant differences in how they recognize FG-repeat cores.
The structures of transport factors bound to FG repeats provide clues about the specificity of these functionally important interactions. Different FG cores are unlikely to adopt exactly the same shape, and so the fit of a core to its binding site on a transport factor is a key determinant of affinity. For example, although the NTF2 FG binding site fits FxFG cores well (Figure 4A), this site is unable to accommodate the cores from GLFG repeat nucleoporins well because, whereas the two hydrophobic pockets are well placed for both Phe side chains of an FxFG repeat to fit simultaneously, they are too far apart to accommodate both the Leu and Phe of a GLFG-repeat core simultaneously. This is consistent with the observation that although NTF2 has a micromolar affinity for FxFG repeats, it does not bind GLFG repeats (Clarkson et al., 1997). In contrast, the FG binding site in TAP (Figure 4B) only appears to have a single Phe-shaped hole and appears to bind only a single Phe side chain (Fribourg et al., 2001). This is consistent with TAP’s observed wide specificity for nucleoporin repeats (see, for example, Strawn et al., 2001).
In summary, although the structure of the FG-nucleoporin binding sites are different between NTF2, TAP/p15 and importin-β, in each transport factor the molecular recognition fundamental to this interaction is based primarily on one or two Phe side chains of the FG-repeat core, with only minor contributions from its peptide backbone.
FxFG nucleoporins in nuclear trafficking
The decreased growth rate and Ran mislocalization seen in yeast in which the Q43/45D mutant replaced wild-type NTF2 (Figure 5) confirmed the physiological importance of the NTF2–nucleoporin interaction. However, the interaction interface between the FxFG core and both importin-β and NTF2 is comparatively small. Indeed, the interactions seem to serve primarily to shield the exposed hydrophobic surfaces of carrier and FG repeats from bulk solvent. However, to enable nuclear trafficking to proceed sufficiently rapidly, it is crucial that the interaction between nucleoporins and transport factors be weak (Bayliss et al., 1999, 2000a; Chaillan-Huntington et al., 2000; Quimby et al., 2001; Ribbeck and Görlich, 2001; Stewart et al., 2001). The small size of these interfaces raises the question of how transport factors specifically recognize nucleoporin FG repeats rather than general hydrophobic surfaces. First, the FG binding sites have evolved to recognize a Phe side chain in either the context of a tight turn where the Gly adopts a conformation forbidden to all other amino acids (in the case of importin-β and TAP–NTF2), or in the context of the sequence FxF in the case of yNTF2. A further enhancement to specificity arises from the multiple independent FG binding sites on each transport factor, which complement the many FG repeats in each nucleoporin domain. Multiple weak sites may contribute to more efficient trafficking because, to pass from one nucleoporin to the next, a transport factor must initially release an FG repeat from only one of its sites, which is freed to bind an adjacent nucleoporin, whilst still maintaining an interaction with FG repeats at its other site(s). Thus the activation energy of moving from one nucleoporin to the next is minimized, a crucial prerequisite for trafficking to occur at almost diffusion-limited rates (Ribbeck and Görlich, 2001).
In summary, we have determined the crystal structure of yNTF2-N77Y bound to a FxFG peptide and provided a structural basis for understanding this interaction, which is fundamental for the function of NTF2 in the nuclear import of RanGDP. The FxFG binding site on NTF2 contains residues from both chains of the dimeric molecule and some side chains change their conformation to encapsulate the Phe rings of the repeat core. Thus, although Phe5 (Trp7 in vertebrate NTF2) is a central component of the binding site, there are also extensive contributions made by Glu34, Met36, Gln43, Gln45, Pro73 and Phe115. Moreover, comparison with the crystal structure of FxFG-nucleoporin-repeat cores bound to importin-β indicates that both types of nuclear transport factor bind to FxFG cores in similar ways that involve a hydrophobic surface depression in which the two Phe rings of the core become buried, with generally only minor contributions from the peptide backbone of the core, which is located on the top of the groove. However, the binding site on importin-β is somewhat deeper, consistent with its binding FxFG nucleoporins more strongly. Overall, the structure of the binding interfaces provides a rationale for the specificity of their interaction with nucleoporins, and also provides a basis for the rapid translocation required to move transport factors and cargoes through the NPC in the course of nuclear transport.
Materials and methods
Protein expression and purification
Expression plasmids encoding wild-type rNTF2, D92N/D94N rNTF2 and yNTF2-N77Y have been described previously (Kent et al., 1996; Clarkson et al., 1997; Quimby et al., 2001). All NTF2 proteins were purified (essentially as in Kent et al., 1996) by ion-exchange chromatography and gel filtration. Protein used for the growing of crystals was over 99% pure by SDS–PAGE. FF1 peptide (sequence DSGFSFGSK) at >95% purity was produced by solid-phase synthesis.
Protein crystallization
All NTF2 crystals were grown at 18°C by vapour diffusion. Native yNTF2-N77Y crystals were grown in 8 µl hanging drops composed of 4 µl of drop buffer (100 mM ammonium acetate pH 6.5, 1.6 M ammonium sulfate) and 4 µl of yNTF2-N77Y protein at 14 mg/ml suspended over reservoir buffer containing 100 mM ammonium acetate pH 6.5 and 1.12 M ammonium sulfate. FxFG-bound yNTF2-N77Y crystals were grown in 8 µl hanging drops composed of 4 µl of drop buffer (100 mM ammonium acetate pH 6.5, 1.6 M ammonium sulfate), 4 µl of protein mix (yNTF2-N77Y protein at 7 mg/ml and 12.5 mM FF1 peptide) suspended over reservoir buffer comprising 100 mM ammonium acetate pH 6.5, 1.52 M ammonium sulfate. Diffraction-quality crystals of yNTF2-N77Y in both native and FxFG-bound forms were obtained by microseeding with yNTF2-N77Y crystals immediately before sealing the wells. Wild-type rNTF2 crystals were grown in 6 µl hanging drops composed of 3 µl of buffer (100 mM ammonium acetate pH 5.5, 1.2 M ammonium sulfate) and 3 µl of rNTF2 (at 10 mg/ml). D92N/D94N rNTF2 crystals were grown using a reservoir solution containing 8% PEG 8000 (Hampton), 100 mM pH 4.5 Na acetate buffer, 50 mM MgCl2 using 8 µl hanging drops containing 2.8 mg/ml protein and half-strength reservoir buffer. yNTF2-N77Y crystals (± FF1) were soaked briefly in well buffer plus 25% glycerol before flash-freezing in liquid nitrogen. Native 1.6 Å and FxFG-bound 1.9 Å yNTF2-N77Y data were collected at beamline ID14 EH2 (ESRF, Grenoble) using 0.933 Å wavelength radiation and a MAR CCD detector. D92N/D94N-rNTF2 crystals were first soaked in reservoir buffer plus 25% glycerol for several minutes before freezing at 100K in a cryostream. A 2.3 Å resolution native dataset was collected from an in-house rotating anode source using CuKα radiation (wavelength 1.542 Å) and a MAR 300 detector. Wild-type rNTF2 crystals were soaked briefly in well buffer plus 25% glycerol before freezing at 100 K in a cryostream. A 1.6 Å dataset was collected at beamline ID14 EH1 (ESRF, Grenoble) using 0.934 Å wavelength radiation and a MAR CCD detector.
Structure determination and refinement
Data reduction and scaling for all crystals were performed using MOSFLM and the CCP4 suite of programs (CCP4, 1994). CNS (Brünger et al., 1998) was used to obtain molecular replacement solutions for yNTF2-N77Y and wild-type rNTF2 and for their refinement. The D92N/D94N-rNTF2 structure was solved by molecular replacement using AMORE (CCP4, 1994) and refined using REFMAC (CCP4). The 2.3 Å structure of NTF2 (Bullock et al., 1996; PDB 1OUN) was used as an initial model in all cases, although its sequence was adjusted for the yNTF2-N77Y native structure. yNTF2-N77Y complexed with FxFG peptide was solved by rigid-body refinement of the native yNTF2-N77Y structure, which gave an initial R-factor of 31.5%. Model rebuilding was carried out using O (Jones et al., 1991). Final refinement statistics are shown in Table I. Structure illustrations were produced using LIGPLOT (Wallace et al., 1995), MOLSCRIPT (Kraulis, 1991), BOBSCRIPT (Esnouf, 1997), RASTER3D (Merritt and Bacon, 1997) and PYMOL (Warren L.DeLano, PyMOL Molecular Graphics System on World Wide Web URL http://www.pymol.org). Structure alignments were produced using LSQKAB (CCP4, 1994). Protein co-ordinates have been deposited with the Protein Database (PDB) (Table I).
Yeast manipulation
All DNA manipulations were performed according to standard methods (Sambrook et al., 1989) and all media were prepared by standard procedures (Rose et al., 1990). The wild-type (PSY580) and NTF2 deletion strains (ACY114) used in this study have been described previously (Corbett and Silver, 1996). Site-directed mutagenesis was performed on pBS-NTF2 (pAC240) using the QuickChange PCR-based mutagenesis kit from Stratagene (La Jolla, CA). To determine growth rates, cells were grown in SD-Leu overnight at 30°C and diluted 1:1000 into SD-Leu medium. Cells were then counted every 2.5 h. Ran and NTF2 were localized in vivo as described previously (Quimby et al., 2001). To localize NTF2, wild-type and mutant NTF2–GFP fusion proteins were transformed into the NTF2 deletion strain, ACY114, maintained by a wild-type copy of GSP1 (pAC78). To localize Ran, scRan–GFP was transformed into ACY114 expressing each of the mutant alleles of NTF2 as the only copy of NTF2. The GFP fusion proteins were localized by viewing the GFP signal directly in living cells through a GFP optimized filter (Chromo Technology) using an Olympus BX60 epifluorescence microscope equipped with a Photometrics Quantix digital camera.
Acknowledgments
Acknowledgements
We are most grateful to our many colleagues in Cambridge and Atlanta, and especially to Helen Kent, Trevor Littlewood, Phil Evans and Andrew Leslie for their many helpful suggestions, comments and criticisms. We also thank Stephanie Monaco for assistance at ESRF, David Owen for peptide synthesis and Gabriel Wong for protein preparation. R.B. is a Research Fellow of Trinity College, Cambridge. This work was supported in part by grants from the Human Frontiers Science Program (RGP 0386/2001-MR) to M.S. and A.H.C., by a grant from the NIH to A.H.C. and by an NRSA to B.B.Q.
References
- Allen N.P., Huang,L., Burlingame,A. and Rexach,M. (2001) Proteomic analysis of nucleoporin interacting proteins. J. Biol. Chem., 276, 29268–29274. [DOI] [PubMed] [Google Scholar]
- Bayliss R., Ribbeck,K., Akin,D., Kent,H.M., Feldherr,C.M., Görlich,D. and Stewart,M. (1999). Interaction between NTF2 and xFxFG-containing nucleoporins is required to mediate nuclear import of RanGDP. J. Mol. Biol., 293, 579–593. [DOI] [PubMed] [Google Scholar]
- Bayliss R., Corbett,A.H. and Stewart,M. (2000a) The molecular mechanism of transport of macromolecules through nuclear pore complexes. Traffic, 1, 448–456. [DOI] [PubMed] [Google Scholar]
- Bayliss R., Littlewood,T.D. and Stewart,M. (2000b) Structural basis for the interaction between FxFG nucleoporin repeats and importin-β in nuclear trafficking. Cell, 102, 99–108. [DOI] [PubMed] [Google Scholar]
- Bayliss R., Kent,H.M., Corbett,A.H. and Stewart,M. (2000c) Crystallization and initial X-ray diffraction characterization of complexes of FxFG nucleoporin repeats with nuclear transport factors. J. Struct. Biol., 131, 240–247. [DOI] [PubMed] [Google Scholar]
- Ben-Efraim I. and Gerace,L. (2001) Gradient of increasing affinity of importin-β for nucleoporins along the pathway of nuclear import. J. Cell Biol., 152, 411–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandl M., Weiss,M.S., Jabs,A., Suhnel,J. and Hilgenfeld,R. (2001) C-H…π interactions in proteins. J. Mol. Biol., 307, 357–377. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. (1992) Free R value: a novel statistical quality for assessing the accuracy of crystal structures. Nature, 355, 472–474. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 50, 905–921. [DOI] [PubMed] [Google Scholar]
- Bullock T.L., Clarkson,W.D., Kent,H.M. and Stewart,M. (1996) The 1.6 Å resolution crystal structure of nuclear transport factor 2 (NTF2). J. Mol. Biol., 260, 422–431. [DOI] [PubMed] [Google Scholar]
- Chaillan-Huntington C., Villa-Braslavsky,C., Kuhlman,J. and Stewart,M. (2000) Dissecting the interactions between NTF2, RanGDP and the nucleoporin xFxFG repeats. J. Biol. Chem., 275, 5874–5879. [DOI] [PubMed] [Google Scholar]
- Clarkson W.D., Kent,H.M. and Stewart, M. (1996) Separate binding sites on nuclear transport factor 2 (NTF2) for GDP-Ran and the phenylalanine-rich repeat regions of nucleoporins p62 and Nsp1p. J. Mol. Biol., 263, 517–524. [DOI] [PubMed] [Google Scholar]
- Clarkson W.D., Corbett,A.H., Paschal,B.M., Kent,H.M., McCoy,A.J., Gerace,L., Silver,P.A. and Stewart,M. (1997) Nuclear protein import is decreased by engineered mutants of nuclear transport factor 2 (NTF2) that do not bind GDP Ran. J. Mol. Biol., 272, 716–730. [DOI] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Corbett A.H. and Silver,P.A. (1996) The NTF2 gene encodes an essential, highly conserved protein that functions in nuclear transport in vivo. J. Biol. Chem., 271, 18477–18484. [DOI] [PubMed] [Google Scholar]
- Esnouf R.M. (1997) An extensively modified version of MolScript that includes greatly enhanced colouring capabilities. J. Mol. Graph. Model., 15, 132–136. [DOI] [PubMed] [Google Scholar]
- Fribourg S., Braun,I.C., Izaurralde,E. and Conti,E. (2001) Structural basis for the recognition of a nucleoporin FG repeat by the NTF2-like domain of the TAP/p15 mRNA nuclear export factor. Mol. Cell, 8, 645–656. [DOI] [PubMed] [Google Scholar]
- Görlich D. and Kutay,U. (1999) Transport between the cell nucleus and the cytoplasm. Annu. Rev. Cell Dev. Biol., 15, 607–660. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.-Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron density maps and for locating errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kent H.M., Clarkson,W.D., Bullock,T.L. and Stewart,M. (1996) Crystallization and preliminary X-ray diffraction analysis of nuclear transport factor 2. J. Struct. Biol., 116, 326–329. [DOI] [PubMed] [Google Scholar]
- Kraulis P.J. (1991) MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Kuersten S., Ohno,M. and Mattaj,I.W. (2001) Nucleocytoplasmic transport: ran, beta and beyond. Trends Cell Biol., 11, 497–503. [DOI] [PubMed] [Google Scholar]
- Lane C.M., Cushman,I. and Moore,M.S. (2000) Selective disruption of nuclear import by a functional mutant nuclear carrier protein. J. Cell Biol., 151, 321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt E.A. and Bacon,D.J. (1997) Raster3D: photorealistic molecular graphics. Methods Enzymol., 277, 505–524. [DOI] [PubMed] [Google Scholar]
- Paschal B.M. and Gerace,L. (1995) Identification of NTF2, a cytosolic factor for nuclear import that interacts with nuclear pore protein p62. J. Cell Biol., 129, 925–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quimby B.B., Leung,S.W., Bayliss,R., Harreman,M.T., Thirumala,G., Stewart,M. and Corbett,A.H. (2001) Functional analysis of the hydrophobic patch on nuclear transport factor 2 involved in interactions with the nuclear pore in vivo. J. Biol. Chem., 276, 38820–38829. [DOI] [PubMed] [Google Scholar]
- Rexach M. and Blobel,G. (1995) Protein import into nuclei: association and dissociation reactions involving transport substrate, transport factors, and nucleoporins. Cell, 83, 683–692. [DOI] [PubMed] [Google Scholar]
- Ribbeck K. and Görlich,D. (2001) Kinetic analysis of translocation through nuclear pore complexes. EMBO J., 20, 1320–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribbeck K., Lipowsky,G., Kent,H.M., Stewart,M. and Görlich,D. (1998) NTF2 mediates nuclear import of Ran. EMBO J., 17, 6587–6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose M.D., Winston,F. and Hieter,P. (1990) Methods in Yeast Genetics: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Rout M.P. and Wente,S.R. (1994) Pores for thought: nuclear pore complex proteins. Trends Cell Biol., 4, 357–365. [DOI] [PubMed] [Google Scholar]
- Rout M.P., Aitchison,J.D., Suprapto,A., Hjertaas,K., Zhao,Y. and Chait,B.T. (2000) The yeast nuclear pore complex: composition, architecture, and transport mechanism. J. Cell Biol., 148, 635–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan K.J. and Wente,S. (2000) The nuclear pore complex: a protein machine bridging the nucleus and cytoplasm. Curr. Opin. Cell Biol., 12, 361–371. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Shah S. and Forbes,D. (1998) Separate nuclear import pathways converge on the nucleoporin Nup153 and can be dissected with dominant-negative inhibitors. Curr. Biol., 8, 1376–1386. [DOI] [PubMed] [Google Scholar]
- Smith A., Brownawell,A. and Macara,I.G. (1998) Nuclear import of Ran is mediated by the transport factor NTF2. Curr. Biol., 8, 1403–1406. [DOI] [PubMed] [Google Scholar]
- Stewart M. (2000) Insights into the molecular mechanism of nuclear trafficking using nuclear transport factor 2 (NTF2). Cell Struct. Funct., 25, 217–225. [DOI] [PubMed] [Google Scholar]
- Stewart M., Kent,H.M. and McCoy,A.J. (1998) Structural basis for molecular recognition between nuclear transport factor 2 (NTF2) and the GDP-bound form of the Ras-family GTPase Ran. J. Mol. Biol., 277, 635–646. [DOI] [PubMed] [Google Scholar]
- Stewart M., Baker,R.P., Bayliss,R., Clayton,L., Grant,R.P., Littlewood,T. and Matsuura,Y. (2001) Molecular mechanism of translocation through nuclear pore complexes during nuclear protein import. FEBS Lett., 498, 145–149. [DOI] [PubMed] [Google Scholar]
- Strawn L.A., Shen,T. and Wente,S.R. (2001) The GLFG regions of Nup116p and Nup110p serve as binding sites for both Kap95p and Mex67p at the nuclear pore complex. J. Biol. Chem., 276, 6445–6452. [DOI] [PubMed] [Google Scholar]
- Vasu S.K. and Forbes,D.J. (2001) Nuclear pores and nuclear assembly. Curr. Opin. Cell Biol., 13, 363–375. [DOI] [PubMed] [Google Scholar]
- Wallace A.C., Laskowski,R.A. and Thornton,J.M. (1995) LIGPLOT: A program to generate schematic diagrams of protein–ligand interactions. Protein Eng., 8, 127–134. [DOI] [PubMed] [Google Scholar]