

Abstract
The interleukin-2 receptor α (IL-2Rα) chain is a component of high-affinity IL-2 receptors and thus is a key regulator of lymphocyte proliferation. Lineage-restricted and activation-dependent IL-2Rα transcription is controlled by four upstream positive regulatory regions (PRRs) and one downstream PRR. We now demonstrate that T-cell receptor (TCR) responsiveness requires both upstream sequences and an intronic region, PRRIV, previously identified as an IL-2 response element. Whereas IL-2 responsiveness requires Stat5 and HMG-I(Y) binding, TCR responsiveness of PRRIV requires two AP-1- and two NFAT-binding sites that bind Jun, Fos and NFAT family members in vitro and in vivo. Moreover, IL-2Rα induction is impaired in T lymphocytes from transgenic mice expressing a dominant-negative c-jun construct, or following treatment with cyclosporin A. Thus, our data indicate an important role for both AP-1 and NFAT proteins for TCR-induced IL-2Rα expression and establish that both upstream and intronic sequences mediate TCR responsiveness of the IL-2Rα gene. Moreover, our data reveal a previously unappreciated link between the TCR-mediated up-regulation of the IL-2 and IL-2Rα genes.
Keywords: AP-1/cyclosporin A/IL-2Rα/NFAT/TCR
Introduction
Interleukin-2 (IL-2) is a growth factor for antigen-activated lymphocytes (Lin and Leonard, 1997) and it also plays a critical role in antigen-induced cell death (Lenardo, 1991; Van Parijs et al., 1997, 1998; Refaeli et al., 1998). IL-2 acts on T cells via high-affinity IL-2 receptors which are composed of three transmembrane polypeptides, IL-2Rα, IL-2Rβ and the common cytokine receptor γ chain, γc (Lin and Leonard, 1997). Whereas dimerization of IL-2Rβ and γc is necessary and sufficient for IL-2 signaling (Nakamura et al., 1994; Nelson et al., 1994), IL-2Rα expression increases the affinity of IL-2 binding ∼100-fold, facilitating IL-2 responses at low physiological concentrations of this cytokine. Mutation of the IL-2Rα gene results in autoimmunity in patients (Sharfe et al., 1997) and mice (Willerford et al., 1995).
Expression of the IL-2Rα gene is tightly regulated at the transcriptional level. Previous studies have identified five positive regulatory regions (PRRs): PRRI (between nucleotides –276 and –244 relative to the major transcription initiation site of the human gene) (Cross et al., 1987, 1989; Bohnlein et al., 1988; Leung and Nabel, 1988; Ruben et al., 1988; Lin et al., 1990; Toledano et al., 1990); PRRII (–137 to –64 in human) (John et al., 1995); PRRIII (–3780 to –3703 in human and –1366 to –1319 in mouse) (Sperisen et al., 1995; John et al., 1996; Lecine et al., 1996); PRRIV/intronic IL-2 response element (herein designated as PRRIV) (+3389 to +3596 in human and +2539 to +2740 in mouse) (Kim et al., 2001); and PRRIV/CD28rE (herein designated as CD28rE) (–8689 to –8483 in human) (Yeh et al., 2001). PRRI, PRRII and CD28rE are required for mitogenic stimulation of the IL-2Rα gene, while both PRRIII and PRRIV are IL-2 response elements. Human PRRI binds to NF-κB1, c-Rel and serum response factor (SRF) (Ballard et al., 1988; Toledano et al., 1990), while PRRII binds the lymphoid/myeloid-specific Ets family protein, Elf-1, and the high mobility group protein, HMG-I(Y) (John et al., 1995). Intermolecular interactions between proteins that bind to PRRI and PRRII appear to result in a highly ordered stereospecific complex that regulates IL-2Rα promoter activity upon mitogenic stimulation (John et al., 1995). PRRIII can bind to Stat5 proteins, Elf-1, HMG-I(Y) and a GATA-1-like protein (Sperisen et al., 1995; John et al., 1996; Lecine et al., 1996) whereas PRRIV can bind Stat5 proteins and HMG-I(Y) (Kim et al., 2001). The CD28rE can bind to CREB and AP-1 proteins (Yeh et al., 2001). Two NFAT sites located at approximately –585 and –650 in the mouse gene were also reported to be important in the control of IL-2Rα promoter induction in T cells (Schuh et al., 1998).
Although PRRIV was identified previously as an IL-2 response element (Kim et al., 2001), we now show that this region also contains binding sites for AP-1 and NFAT transcription factors that mediate signaling after T-cell receptor (TCR)/CD3 stimulation. Thus, PRRIV appears to serve dual roles in mediating IL-2Rα regulation in response to TCR and IL-2 stimulation.
Results
AP-1 and NFAT bind to PRRIV in vitro
Although PRRIV was identified as an IL-2 response element (Kim et al., 2001), our analysis of the PRRIV sequence revealed potential consensus binding sites for AP-1 and NFAT. We therefore investigated factor binding to these sequences using electrophoretic mobility shift assays (EMSAs) and the potential role of TCR-mediated induction of the IL-2Rα gene. Nuclear extracts were prepared from EL4 cells that were unstimulated or stimulated with phorbol 12-myristate 13-acetate (PMA) plus ionomycin or from mouse splenocytes that were unstimulated or stimulated with anti-CD3 + anti-CD28 (αCD3/αCD28), and the extracts were assayed for factor binding using 32P-labeled wild-type or mutant probes spanning the AP-1 or NFAT sites (Figure 1A). When probes spanning the tetradecanoylphorbol acetate response elements, TRE1 or TRE2, were assayed, similar migrating complexes were generated with extracts from PMA + ionomycin (PI)-stimulated EL4 cells (Figure 1B, lanes 2 and 8) or αCD3/αCD28-stimulated mouse splenocytes (lanes 14 and 20) but not from unstimulated cells (lanes 1, 7, 13 and 19). Both the pan-Jun and pan-Fos AP-1 family antibodies blocked the formation of these complexes (lanes 3, 4, 9, 10, 15, 16, 21 and 22). Moreover, when the mtTRE1 or mtTRE2 probes, both of which have mutant AP-1-binding sites, were assayed, no complex was generated even with extracts from PI-stimulated EL4 cells (lanes 5, 6, 11 and 12) or αCD3/αCD28-stimulated mouse splenocytes (lanes 17, 18, 23 and 24).

Fig. 1. AP-1 and NFAT bind to PRRIV in vitro. (A) Sequences of the +2481/+2501 (TRE1), +2558/+2578 (TRE2), +2507/+2536 (NFAT1) and +2669/+2708 (NFAT2) oligonucleotides used in (B–D). For mutant (mt) probes, the mutations are shown in lower case; hyphens indicate wild-type sequences. (B) EMSAs using wild-type (TRE1, lanes 1–4 and 13–16; TRE2, lanes 7–10 and 19–22) or mutant probes (mtTRE1, lanes 5, 6, 17 and 18; mtTRE2, lanes 11, 12, 23 and 24) from PRRIV and nuclear extracts from untreated EL4 cells (lanes 1, 5, 7 and 11), from PI-treated EL4 cells (lanes 2–4, 6, 8–10 and 12), from untreated mouse splenocytes (lanes 13, 17, 19 and 23) or from αCD3/αCD28-treated mouse splenocytes (lanes 14–16, 18, 20–22 and 24). In lanes 3, 4, 9, 10, 15, 16, 21 and 22, EMSAs were performed in the presence of pan-Jun or pan-Fos antibodies, as indicated. (C) EMSAs using wild-type (NFAT1, lanes 1–6; NFAT2, lanes 9–14) or mutant probes (mtNFAT1, lanes 7 and 8; mtTRE2, lanes 15 and 16) from PRRIV and nuclear extracts from untreated EL4 cells (lanes 1–3, 7, lanes 9–11 and 15) or from PI-treated cells (lanes 4–6, 8, 12–14 and 16). In lanes 2, 3, 5, 6, 10, 11, 13,and 14, EMSAs were performed in the presence of antibodies to NFATp or NFATc, as indicated. (D) EMSAs using wild-type (NFAT1, lanes 1–8; NFAT2, lanes 11–18) or mutant probes (mtNFAT1, lanes 9 and 10; mtTRE2, lanes 19 and 20) from PRRIV and nuclear extracts from untreated mouse splenocytes (lanes 1, 9, 11 and 20) or from αCD3/αCD28-treated mouse splenocytes (lanes 2–8, 10, 12–18, and 20). In lanes 3–8 and 13–18, EMSAs were performed in the presence of antibodies to NFATp, NFATc, pan-Jun, pan-Fos, normal mouse IgG or normal rabbit IgG as indicated.
When the NFAT1 probe was assayed, two complexes were generated with extracts from unstimulated EL4 cells (Figure 1C, lane 1), while no complex was generated with the mtNFAT1 probe (lane 7). The formation of the two complexes was increased when the NFAT1 probe was assayed with extracts from PI-stimulated EL4 cells (lane 4), but the mtNFAT1 probe still exhibited no binding activity (lane 8). We confirmed the binding of NFATp and NFATc proteins to the NFAT1 probe by using anti-NFAT antibodies in EMSAs. An anti-NFATp antibody resulted in a supershift, producing a doublet of slower mobility (lanes 2 and 5), while the anti-NFATc antibody blocked the formation of both complexes (lanes 3 and 6). When the NFAT2 probe was assayed, two complexes were generated with extracts from unstimulated cells (lane 9), whereas no complex was generated with the mtNFAT2 probe (lanes 15 and 16). With extracts from PI-stimulated cells, the binding activity of the upper complex increased and two more slowly migrating complexes were generated (lane 12). We confirmed the binding of NFATp and NFATc proteins to the NFAT2 probe: the anti-NFATp antibody supershifted complexes to generate slower mobility complexes (lanes 10 and 13), whereas the anti-NFATc antibody blocked the formation of all complexes (lanes 11 and 14).
We also investigated factor binding to the NFAT1 and NFAT2 probes using nuclear extracts from normal murine splenocytes. When the NFAT probes were assayed, the formation of complexes was increased with extracts from αCD3/αCD28-stimulated mouse splenocytes (Figure 1D, lanes 2 and 12) as compared with the complexes formed with extracts from unstimulated cells (lanes 1 and 11), whereas no complex was detected with the mtNFAT probes even with extracts from αCD3/αCD28-stimulated mouse splenocytes (lanes 9, 10, 19 and 20). The complexes formed with the NFAT probes were evaluated for NFATp, NFATc or AP-1 using antibodies to each protein. We found that the anti-NFATc antibody almost completely blocked the formation of complexes (lanes 4 and 14), while the anti-NFATp antibody blocked the formation less efficiently (lanes 3 and 13). We also found that pan-Jun and pan-Fos family antibodies were capable of modifying the binding to two NFAT probes (lanes 5 and 6 versus lane 2; lanes 15 and 16 versus lane 12), with the anti-Fos antibody having a greater effect in each case. While the exact compositions of the NFAT-related complexes are not fully known, our data suggest that AP-1 components are involved in these complexes, particularly in the NFAT2 complex where the rabbit anti-Fos IgG markedly affected the complex (lane 16) but the control rabbit IgG did not (lane 18).
AP-1 and NFAT bind to PRRIV in vivo
To investigate further factor binding to PRRIV in vivo, we performed chromatin immunoprecipitation (ChIP) experiments. Chromatin was prepared from EL4 cells that were cross-linked with formaldehyde. DNA was sheared to an average size of 400 bp, and immunoprecipitations were performed with specific antisera. The complexes were harvested with magnetic beads, cross-linking was reversed and the DNA was then recovered and PCR amplified with primer pairs that separately amplified PRRIV and the IL-2Rα promoter region. Consistent with in vitro binding of AP-1 and NFAT proteins to PRRIV, treatment with PI induced an increase in occupancy of PRRIV by c-Jun, JunB, c-Fos, Fra-1, Fra-2, NFATp and NFATc proteins (Figure 2). The IL-2Rα promoter did not bind AP-1 family proteins but could bind NFATc and NFATp proteins. The lack of NFAT binding to the 3′-untranslated region (UTR) region indicated the specificity of its binding to PRRIV.

Fig. 2. AP-1 and NFAT bind to PRRIV in vivo. ChIP assays were performed using EL4 cells. DNA was purified and used as a template for PCRs with pairs of primers that amplify PRRIV, the IL-2Rα promoter or the 3′-UTR, as indicated.
AP-1 and NFAT sites are important for PI-induced PRRIV activity
To clarify the functional significance and the relative contribution of each AP-1 and NFAT site in the PRRIV region, we performed site-directed mutagenesis and assayed the PI responsiveness of each mutant construct in EL4 cells (Figure 3). While the wild-type construct showed 4.7-fold PI inducibility in EL4 cells, selective mutation of the TRE1 (PRRIVm1), NFAT1 (PRRIVm4) or NFAT2 (PRRIVm5) sites diminished PI inducibility. Selective mutation of TRE2 (PRRIVm2), simultaneous mutation of TRE1 and TRE2 (PRRIVm3), of NFAT1 and NFAT2 (PRRIVm6) or of all the AP-1- and NFAT-binding sites (PRRIVm7) essentially abrogated PI inducibility. These results suggest that all the AP-1- and NFAT-binding sites are required for maximal PI inducibility. Interestingly, although two of the NFAT sites in PRRIV (NFAT1 and NFAT2) are vital, the third NFAT site (nucleotides 2776–2786; see Kim et al., 2001; Figure 2B) was not required (data not shown). We considered the possibility that the observed results were due at least in part to secondary effects of synthesized IL-2. However, the EL4 cells we used did not respond to IL-2 when we transfected PRRIVWT (Figure 3) or WT-PRRIVWT (Figure 4), excluding this possibility.

Fig. 3. Two AP-1 sites and two NFAT sites are required for the maximal TCR responsiveness of PRRIV. Mutations in the AP-1 sites and/or NFAT sites were made in the context of the +2350/+2820 murine IL-2Rα–luciferase reporter construct (PRRIVWT construct). Constructs were transfected into EL4 cells, followed by no stimulation or stimulation with PI.

Fig. 4. Multiple elements cooperate for maximal TCR responsiveness. Combinations of mutations in the upstream NFAT sites, the NF-κB site, AP-1 sites and the NFAT sites in PRRIV were introduced into the –2543 to +93/luciferase/+93 to +4533 murine IL-2Rα reporter construct (WT-PRRIVWT construct). Constructs were transfected into EL4 cells, and cells were then either not stimulated or stimulated with PI. Alternatively, constructs were transfected into normal mouse T cells followed by no stimulation or stimulation with anti-CD3 plus anti-CD28 antibodies. F.I., Fold induction; n.d., not done.
Both PRRI and PRRIV are required for maximal PI inducibility
The above results suggested that two NFAT sites and two AP-1 sites in PRRIV are PI response elements. Moreover, the NF-κB site and upstream NFAT sites have also been reported to be essential for maximal activity (Bohnlein et al., 1988; Leung and Nabel, 1988; Ruben et al., 1988; Cross et al., 1989; Lin et al., 1990; Toledano et al., 1990; Schuh et al., 1998). We therefore analyzed their relative contributions to PI-induced IL-2Rα promoter activity. The SphI–PstI murine IL-2Rα fragment (–2543 to +93) was inserted upstream of the luciferase reporter gene, and the adjacent PstI fragment (+93 to +4533) was subcloned downstream of the luciferase reporter gene (Figure 4, WT-PRRIVWT construct). Combinations of mutations were introduced into the previously identified upstream NFAT sites, the NF-κB site, the AP-1 sites and the downstream NFAT sites, and each mutant construct was assayed initially for PI responsiveness in EL4 cells (Figure 4). Note that the designation of mutations in PRRIV is identical to that used in Figure 3. The various intronic NFAT and AP-1 sites were mutated in the context of the wild-type upstream region (WT series), a mutant containing changes in the upstream NFAT sites (NFATm series), a mutant NF-κB site (NFκBm series) or an upstream region in which both the NFAT and NF-κB sites were mutated (NFAT/NFκBm series). While the wild-type construct (WT-PRRIVWT) showed ∼12.3-fold PI inducibility in EL4 cells, individual mutation of each site modestly decreased PI inducibility, and simultaneous mutation of all sites abrogated PI inducibility (NFAT/NFκBm-PRRIVm7, very bottom construct in Figure 4). The upstream NFAT sites appeared to play only a minor role in PI-induced IL-2Rα promoter activity, as mutation of these sites only slightly changed the inducibility when wild-type series constructs were compared with NFATm series constructs, or when the NFκBm series constructs were compared with NFAT/NFκBm series constructs. PRRIV appeared to elicit more potent TCR responsiveness than the upstream region containing NFAT and NF-κB sites, as the WT-PRRIVm7 construct in which two AP-1 sites and two intronic NFAT sites were mutated simultaneously showed only 2.6-fold PI inducibility, while the NFAT/NFκBm-PRRIVWT construct, in which simultaneous mutations were introduced into the two upstream NFAT sites and the NF-κB site, showed 4.1-fold induction. The NFAT/NFκBm series constructs, in which all upstream sites were mutated, showed inducibility similar to that of constructs shown in Figure 3.
Although it is well established that pharmacological treatment with both PMA and ionomycin mimics several TCR responses, we sought to confirm the responsiveness of PRRIV as well as the upstream region in a more physiological setting. We therefore transfected the –2543 to +93/luciferase/+93 to +4533 murine IL-2Rα reporter construct (WT-PRRIVWT in Figure 4) into normal mouse T cells and stimulated them with antibodies to CD3, CD28 or both CD3 and CD28 for 5 h. This relatively short treatment period was intended to minimize secondary effects resulting from IL-2 production. While stimulation with anti-CD3 showed 5.6-fold inducibility, stimulation with anti-CD28 alone did not confer any inducibility (data not shown) and combining anti-CD3 and anti-CD28 gave 5.9-fold inducibility (Figure 4, far right column, top construct), suggesting that the –2543 to +4533 region contains TCR response elements but not CD28 response elements. To assess the relative contribution of each responsive element, a series of mutant constructs were also evaluated (Figure 4). Simultaneous mutation of all sites (NFAT/NFκBm-PRRIVm7 construct) abrogated TCR inducibility (very bottom construct). As in EL4 cells, PRRIV appeared to mediate more potent TCR responsiveness than the upstream region that contains NFAT and NF-κB sites, as construct WT-PRRIVm7 (8th construct from the top of the figure) showed 1.8-fold TCR inducibility, while the NFAT/NFκBm-PRRIVWT construct (8th construct from the bottom of the figure) showed 2.7-fold induction. Taken together, the pattern of inducibility of each construct tested in normal mouse T cells stimulated with anti-CD3/anti-CD28 is similar to that seen in PI-stimulated EL4 cells. These data indicate that for maximal TCR-induced IL-2Rα promoter activity, in addition to the upstream region, the AP-1 and NFAT sites in PRRIV are also critical.
Transactivation of PRRIV by AP-1 protein in vivo
To determine whether AP-1 proteins could transactivate PRRIV in vivo, we used a well-characterized dominant-negative transactivation mutant of c-jun, TAM-67. The TAM-67 mutant retains the leucine zipper domain necessary for dimerization with Jun and Fos family members and the DNA-binding domain, but the N-terminal region responsible for transactivation has been deleted (Brown et al., 1994). TAM-67 binds to consensus AP-1-binding sites as either a homodimer or a heterodimer with Jun and Fos family members, and specifically blocks AP-1-dependent transcription. Using EL4 cells stimulated with PI, we found that transfection of TAM-67 inhibited the activity of a murine IL-2Rα luciferase reporter construct (PRRIV-WT from Figure 3) in a dose-dependent manner (Figure 5A). At 30 µg of TAM-67, almost complete abrogation of PI inducibility was achieved (Figure 5A). When TAM-67 was co-transfected in increasing amounts together with the +2543/+93 and +93/+4533 murine IL-2Rα–luciferase reporter construct (WT-PRRIVWT in Figure 4) into EL4 cells and treated with PI, a dose-dependent inhibition of promoter activity was also observed, with maximum fold reduction at 10 or 30 µg of TAM-67 (Figure 5B). However, transfection of TAM-67 did not diminish activity of WT-m3 in which both AP-1 sites were mutated (Figure 5C, compare 5, 10 or 30 µg with 0 µg of TAM-67), confirming the specificity of the dominant-negative effect of TAM-67.
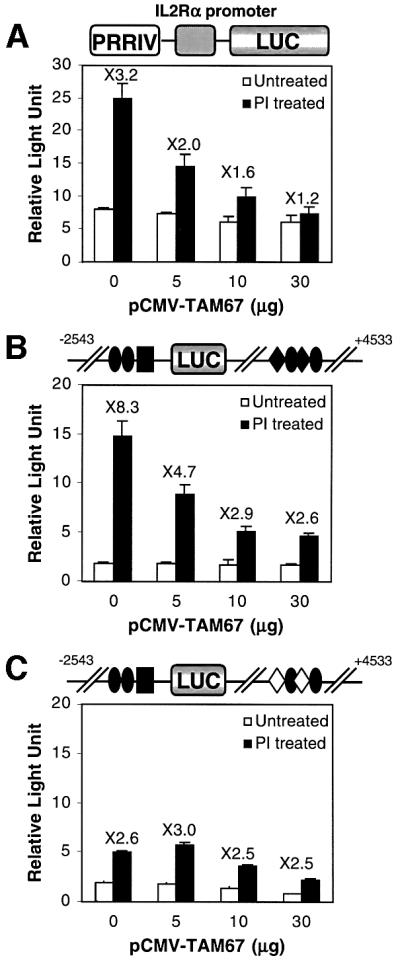
Fig. 5. The TAM-67 dominant-negative c-jun construct inhibits PRRIV activity. (A) EL4 cells were transfected with 10 µg of the PRRIVWT construct together with different amounts of pCMV-TAM67. (B) EL4 cells were transfected with 10 µg of WT-PRRIVWT construct together with different amounts of pCMV-TAM67. (C) EL4 cells were transfected with 10 µg of WT-PRRIVm3 construct, which has mutant AP-1 sites in PRRIV, together with different amounts of pCMV-TAM67. In (A–C), transfected cells were either not stimulated or stimulated with PI.
To assess further the degree to which TAM-67 affected anti-CD3-induced IL-2Rα expression, transgenic mice expressing TAM-67 were used (King et al., 1999). Because the transgenic mice express a much higher level of TAM-67 in the thymus than in the spleen, we used thymocytes for this study. Thymocytes from wild-type (WT) or TAM-67 transgenic mice were stimulated with anti-CD3 for 17 h and stained with Cy-chrome-labeled CD4, allophycocyanin (APC)-labeled CD8 and phycoerythrin (PE)-labeled CD25. As compared with wild- type, thymocytes from TAM-67 transgenic mice showed decreased expression of IL-2Rα in response to anti-CD3 in both CD4 SP (Figure 6A) and CD8 SP (Figure 6B) populations. Treatment with anti-CD3 did not significantly change the profile of IL-2Rα expression either in CD4–CD8– double-negative populations or in CD4+CD8+ double-positive populations (data not shown).

Fig. 6. IL-2Rα expression in TAM-67 transgenic mice. Thymocytes were stained with Cy-chrome-labeled CD4, APC-labeled CD8 and PE-labeled CD25, and analyzed by flow cytometry on a FACSort. Wild-type (WT) or TAM-67 mouse thymocytes were stimulated with anti-CD3 for 17 h and IL-2Rα expression was assayed in either CD4 SP (A) or CD8 SP (B) populations.
PI-inducible PRRIV activity is cyclosporin A sensitive
To establish further whether or not the PRRIV-NFAT sites bind an NFAT-related protein, we tested the ability of cyclosporin A (CsA) to inhibit PI-inducible PRRIV activity. EL4 cells were transiently transfected with the +2350/+2820 murine IL-2Rα–luciferase reporter construct (PRRIV-WT in Figure 3). While treatment of transfected cells with PI resulted in 4.1-fold induction, the addition of CsA almost abrogated this induction (Figure 7A), suggesting that the factor mediating the activity of PRRIV is calcineurin regulated. CsA sensitivity was also seen with the WT-PRRIVWT construct. Whereas treatment with PI showed 8.8-fold induction, CsA decreased this induciblity down to 2.4-fold (Figure 7B), a level similar to that seen in response to PI when EL4 cells were transfected with the NFATm-PRRIVm6 construct in which both upstream NFAT sites and intronic NFAT sites were mutated. Moreover, the NFATm-PRRIVm6 construct did not show any further decrease in fold induction when CsA was added (Figure 7C).
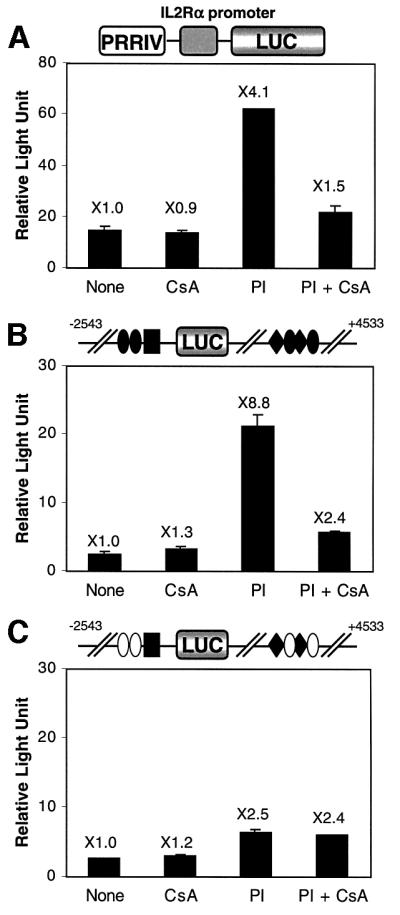
Fig. 7. Cyclosporin inhibits PRRIV activity. (A) EL4 cells were transfected with 10 µg of PRRIVWT construct. Transfected cells were incubated with medium, CsA, PI or PI + CsA. (B) EL4 cells were transfected with 10 µg of WT-PRRIVWT construct. Transfected cells were incubated with medium, CsA, PI or PI + CsA. (C) EL4 cells were transfected with 10 µg of NFATm-PRRIVm6 construct, which has mutant NFAT sites both upstream and in PRRIV. Transfected cells were incubated with medium, CsA, PI or PI + CsA.
The inhibition of the anti-CD3-inducible IL-2Rα expression by CsA was also evident in CD4 SP and CD8 SP thymocytes. While treatment of thymocytes with anti-CD3 resulted in increased expression of IL-2Rα, the addition of CsA almost completely abrogated IL-2Rα induction on both CD4 SP (Figure 8A) and CD8 SP (Figure 8B) populations. As expected, anti-CD3-mediated up-regulation of IL-2 was also inhibited by treatment with CsA (data not shown). Therefore, one might hypothesize that the decreased production of IL-2 might be the cause of the diminished IL-2Rα expression following treatment with anti-CD3 plus CsA. However, the effect of CsA on anti-CD3-inducible IL-2Rα expression was at least partially ‘direct’, as the addition of high dose (100 U/ml) IL-2 only partially reversed the inhibition by CsA (Figure 8).

Fig. 8. The effect of cyclosporin on IL-2Rα expression. Wild-type thymocytes were cultured in the presence of anti-CD3 and/or CsA, and/or IL-2 for 17 h, and then IL-2Rα expression was assayed in either CD4 SP (A) or CD8 SP (B) populations. MFI, mean fluorescence intensity.
Discussion
In this study, we have described a new TCR response element in the first intron of the IL-2Rα gene. This is within a response element, PRRIV, which was shown previously to be an IL-2 response element (Kim et al., 2001). We now have shown that PRRIV contains binding sites for AP-1 and NFAT and binds multiple Jun and Fos family proteins as well as NFATc and NFATp in vitro and in vivo. The ChIP assays only detect binding and cannot by themselves establish whether a given protein acts as a transcriptional activator or repressor when bound to a specific genomic region in vivo. However, several results support the hypothesis that AP-1 and NFAT family members are required for TCR-mediated transactivation of the IL-2Rα gene. First, site-specific mutageneses of the binding sites for AP-1 and/or NFAT significantly diminished IL-2Rα promoter activity. Our data suggest that TCR-mediated activation of the IL-2Rα promoter involves the cooperation between several transcription factors belonging to unrelated families, including NF-κB, AP-1 and NFAT. Secondly, the dominant-negative transactivation mutant of c-jun (TAM-67) repressesed TCR-mediated transactivation of PRRIV. Thirdly, in CD4 SP and CD8 SP thymocytes from transgenic mice expressing TAM-67, IL-2Rα expression in response to anti-CD3 was significantly decreased. Fourthly, CsA, an inhibitor of calcineurin that potently inhibits nuclear translocation of NFAT and NFAT-dependent transcription (Rao et al., 1997), markedly decreased TCR-mediated transactivation of PRRIV and IL-2Rα gene expression. Transient transfection experiments using luciferase reporter constructs showed that CsA could almost completely block PI-mediated IL-2Rα promoter activity, and this specific role of the NFAT-binding sites was confirmed using the NFATm-PRRIVm6 construct in which both upstream and intronic NFAT sites were mutated. It was reported previously that CsA prevented the concanavalin A-induced IL-2Rα expression in murine thymocytes (Gauchat et al., 1986). We observed that CsA diminished anti-CD3-inducible IL-2Rα expression in both CD4 SP and CD8 SP thymocytes, and this effect was at least partially direct as the addition of a high dose of IL-2 only partially reversed the inhibition by CsA. Consistent with our results, T lymphocytes from NFATp-deficient mice exhibit diminished IL-2Rα expression (Schuh et al., 1998). Schuh and co-workers previously described that binding sites for NFAT located at ∼585 and 650 upstream of the IL-2Rα promoter were important for NFATp-mediated transactivation of the IL-2Rα gene (Schuh et al., 1998). However, our results show that intronic NFAT-binding sites in PRRIV were also required, and that these sites appear to play an even more significant role than do the upstream NFAT-binding sites. Taken together, our results demonstrate that both theAP-1 and NFAT family members are important for TCR-mediated IL-2Rα expression. This suggests that the genes encoding IL-2Rα and IL-2 may be co-regulated in response to antigen-mediated T-cell activation. Such a coordinate regulation would help to ensure the availability of high affinity receptors for optimal responsiveness to IL-2.
It has been demonstrated that CD28 co-stimulation strongly amplifies IL-2Rα gene transcription. In a recent study, a CD28 response element was identified between –8689 and –8483 in the human IL-2Rα upstream region, and it was also shown that PRRI, PRRII and PRRIII do not respond to co-stimulation by CD28 (Yeh et al., 2001). To test if a CD28 response element is also located between –2543 and +4533 in the mouse gene, we transfected the WT-PRRIVWT construct into normal mouse T cells and analyzed the inducibility after stimulation with antibodies to CD3 and/or CD28. There was very little, if any, effect of anti-CD28 stimulation on the IL-2Rα promoter activity, indicating that neither PRRIV nor previously identified PRRs are CD28 response elements. When we compared the sequence of the human CD28 response element with the mouse IL-2Rα sequence up to –8047 bp, the furthest contiguous murine sequence data available in the Celera database, we did not find a homologous element, although such a sequence presumably might be located further upstream.
AP-1 proteins are also well known to play a central role in IL-2 regulation by binding to the functionally important AP-1 site in the IL-2 promoter (Jain et al., 1992b), as well as participating in the formation of transcriptionally active NFAT and NF-IL-2 (Ullman et al., 1991; Jain et al., 1992a). AP-1 has been suggested to play a role in preventing T-cell anergy, given that T-cell anergy occurs when the AP-1 level was reduced (Kang et al., 1992). We have now shown that AP-1 also plays a role in IL-2Rα transcription, in addition to its previously established role for transactivation of the IL-2 gene. Diminished AP-1 therefore might also decrease IL-2Rα expression, thus decreasing IL-2 responsiveness, and promoting anergy. Indeed, decreased IL-2Rα expression has been noted in anergic cells (Blackman et al., 1991; Grundstrom et al., 2000).
In summary, we have now unraveled further some of the complexity of IL-2Rα gene regulation. Expression of this gene is controlled in a lineage-and activation-dependent fashion. Collectively, coupled with previous studies, it now appears that of the five positive regulatory elements that have been reported in the IL-2Rα gene, PRRI and PRRIV are important for TCR-mediated regulation whereas PRRIII and PRRIV are important for IL-2-mediated regulation of the gene (Figure 9). The intronic PRRIV therefore contributes to both TCR-mediated and IL-2-mediated induction of this gene. Moreover, our current data suggest a greater degree of shared elements for TCR-mediated induction of the IL-2 and IL-2Rα genes than was appreciated previously.

Fig. 9. Scheme showing how PRRI, PRRII and PRRIV are regulated for optimal TCR activation whereas PRRIII and PRRIV are regulated for IL-2 inducibility. The CD28-responsive enhancer (CD28rE) is also shown.
Materials and methods
Cell culture
The cell line EL4 was maintained at 37°C in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 100 U/ml penicillin G and 100 µg/ml streptomycin.
Antibodies
Antibodies specific for c-Jun, JunB, JunD, c-Fos, FosB, Fra-1 and Fra-2 as well as pan-Jun and pan-Fos family antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Antibodies to NFATc and NFATp were from Affinity Bioreagents, Inc. (Golden, CO).
Plasmid constructs
To generate the murine PRRIV luciferase reporter construct (PRRIV-WT), we subcloned the murine –135 to +93 IL-2Rα promoter fragment 5′ to the luciferase gene between the XhoI and HindIII sites in the polylinker of the pGL3-Basic luciferase reporter vector (Promega, Madison, WI). The PCR fragment containing murine PRRIV (+2350 to +2820) was then subcloned between the KpnI and SacI sites in the polylinker upstream of the promoter fragment. Site-directed mutagenesis of this PRRIV-WT plasmid was performed using the QuikChange kit (Stratagene, La Jolla, CA). Two mutagenic primers, mTRE1 (+2473 to +2508; 5′-GAT TCAATTTCTAGCCAATTCATAAGGCAGATTTTC-3′) and mTRE2 (+2554 to +2589; 5′-CAAGGGATGTATTAGTTGACCCGGGTGTTT TTAGTC-3′) were used, respectively, to introduce the indicated (underlined) 2 bp changes into the TRE1 and TRE2 sites of murine PRRIV. Analogously, primers for mutating NFAT, mNFAT1 (+2498 to +2545; 5′-GGCAGATTTTCATTTTGAAAATTTAAGAAGCCACC ATTTAAAATTGTG-3′) and mNFAT2 (+2668 to +2703; 5′-CCA CCTTTCTTAGAACTGCTTAAACTCTGAGCTGTC-3′), were used, respectively, to introduce the indicated (underlined) 3 bp changes into the NFAT1 and NFAT2 sites of murine PRRIV. Mutant primers mNFAT3 (–667 to –632; 5′-GCTAGACTTAAAATCTATCATTGC AGCTGTAAACAC-3′) and mNFAT4 (-596 to –562; 5′-CCCACA CCCATGATACTATGAATCGTGCATCAGAG-3′) were used to introduce the indicated 2 bp changes into the upstream NFAT sites of the murine IL-2Rα gene. Primer mNF-κB (–279 to –245; 5′-CCCTCCTGC CGCGGCACTCAATCCCCCTTTCCTTG-3′) was used to introduce the indicated 3 bp changes into the NF-κB site of murine PRRI.
The pCMV-TAM67 plasmid encoding a dominant-negative mutant variant of c-jun (Brown et al., 1994) was a gift of M.J.Birrer.
Transient transfections and luciferase assays
Transient transfections of EL4 cells were performed by the DEAE–dextran technique (Sompayrac and Danna, 1981). In each case, 5 × 106 cells in logarithmic growth phase were transfected with 10 µg of supercoiled test plasmid and 40 ng of pRL-SV40 as a transfection efficiency control; cells were then allowed to recover for 24 h at 37°C. Transfected cells were stimulated with either medium alone or 10 ng/ml of PMA plus 1 µg/ml of ionomycin for 18 h, and the cells were harvested and analyzed for luciferase activity using an Analytical Luminescence Laboratory luminometer, Monolight® 2010, and the Dual luciferase assay system kit (Promega, Madison, WI).
Transient transfections of normal murine T cells were performed by electroporation (Eder et al., 1998). C57BL/6 mice were sacrificed at 4–6 weeks of age, and single-cell suspensions of spleens were prepared, followed by anti-mouse IgG antibody panning to remove B cells. The isolated splenocytes were cultured in 2 ng/ml of PMA and 1 µg/ml of ionomycin. After 2 days, the cells were washed, resuspended in fresh medium containing 10 U/ml of IL-2 and cultured for one additional day. Prior to transfection, the T-cell blasts were washed in medium and resuspended at 60 × 106 cells/ml in RPMI medium containing 20 mM HEPES and 4 mM glutamine. A 0.2 ml aliquot of the cell suspension was mixed with 10 µg of plasmid DNA in an electroporation cuvette (0.4 cm electrode gap; Bio-Rad). Electroporation was performed at 960 µF and 250 V using a Bio-Rad Gene Pulser. The cells were then transferred into complete medium, cultured at 37°C for 3 h and aliquoted into 24-well plates, half of which had been coated with anti-CD3 antibody (2 µg/ml) and supplemented with 2 µg/ml of anti-CD28 antibody. After 5 h, the cells were harvested and analyzed for luciferase activity.
Electrophoretic mobility shift assays
Nuclear extracts were prepared as described (Schreiber et al., 1989) from untreated EL4 cells or cells that had been treated with 10 ng/ml of PMA plus 1 µg/ml of ionomycin for 4 h at 37°C. EMSAs were performed as described previously (John et al., 1996) by using glycerol-containing 5% polyacrylamide gels (29:1) containing 0.5× Tris-borate-EDTA buffer. For supershifting assays, nuclear extracts were pre-incubated for 10 min with pan-Jun or pan-Fos rabbit polyclonal IgG (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), or with NFATc or NFATp mouse monoclonal antibodies (Affinity Bioreagents, Inc., Golden, CO). Normal mouse IgG and normal rabbit IgG (Upstate, Lake Placid, NY) were used as negative controls in supershift assays. Oligonucleotide sequences from PRRIV used as probes are shown in Figure 1.
Chromatin immunoprecipitation
ChIP assays were performed essentially as described (Kim et al., 2001). EL4 cells were either left unstimulated or stimulated with 10 ng/ml of PMA plus 1 µg/ml of ionomycin for 4 h at 37°C, followed by cross-linking with formaldehyde. Formaldehyde-treated nuclear lysates were subjected to immunoprecipitation with various antibodies. DNA fragments present in the immunoprecipitates were amplified with primers which specifically recognize PRRIV, the IL-2Rα promoter or the 3′-UTR of the murine IL-2Rα gene. The primers used in this study were as follows: MCH1_1 (designated ‘MCH’ for mouse ChIP oligos), 5′-GACAGACTGTCAACAGATTC-3′ (+2458 to +2477) and MCH1_2, 5′-GGATTTGATTACAGAATTCC-3′ (+2744 to +2725) were used to amplify PRRIV. MCH2_1, 5′-GGCAGGGAATCCCCCTTTCC-3′ (–267 to –248) and MCH2_2, 5′-CAGTATATTGGGTCAACCCC-3′ (+95 to +76) were used to amplify the IL-2Rα promoter. MCH3_1, 5′-AGA GCAGAAGAACCATCTAG-3′ and MCH3_2, 5′-AGTGCTGAGTTT TACTTGGG-3′ were used to amplify the 3′-UTR.
Analysis of transgenic mice expressing TAM-67
TAM-67 transgenic mice were obtained from Dr Jonathan Ashwell and evaluated at 6–8 weeks of age. All experiments were performed under protocols approved by the National Institutes of Health Animal Use and Care Committee and followed the National Institutes of Health guidelines ‘Using Animals in Intramural Research’. Single-cell suspensions from thymus were prepared and thymocytes (1 × 106/ml) were stimulated in plates coated with 2 µg/ml of 2C11 anti-CD3ε monoclonal (PharMingen, San Diego, CA) on RPMI 1640 containing 10% FBS, 2 mM glutamine and antibiotics for 18 h. Thymocytes were stained with Cy-chrome-labeled CD4, APC-labeled CD8 or PE-labeled CD25 (all from PharMingen) and analyzed using a FACSort® with CELLQuest software (Becton Dickinson, San Jose, CA).
Acknowledgments
Acknowledgements
We thank Jonathan Ashwell for providing TAM67 transgenic mice, Michael J.Birrer for providing the TAM67 construct, Rosanne Spolski, Jian-Xin Lin, Katsutoshi Ozaki, Susan John, Hai-Hui Xue, Julie Bollenbacher, John Kelly and other members of our laboratory for helpful advice and discussion, and Jian-Xin Lin, Keji Zao, Ronald H.Schwartz and Ronald N.Germain for critical comments.
References
- Ballard D.W., Bohnlein,E., Lowenthal,J.W., Wano,Y., Franza,B.R. and Greene,W.C. (1988) HTLV-I tax induces cellular proteins that activate the κB element in the IL-2 receptor α gene. Science, 241, 1652–1655. [DOI] [PubMed] [Google Scholar]
- Blackman M.A., Finkel,T.H., Kappler,J., Cambier,J. and Marrack,P. (1991) Altered antigen receptor signaling in anergic T cells from self-tolerant T-cell receptor β-chain transgenic mice. Proc. Natl Acad. Sci. USA, 88, 6682–6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnlein E., Lowenthal,J.W., Siekevitz,M., Ballard,D.W., Franza,B.R. and Greene,W.C. (1988) The same inducible nuclear proteins regulates mitogen activation of both the interleukin-2 receptor-α gene and type 1 HIV. Cell, 53, 827–836. [DOI] [PubMed] [Google Scholar]
- Brown P.H., Chen,T.K. and Birrer,M.J. (1994) Mechanism of action of a dominant-negative mutant of c-Jun. Oncogene, 9, 791–799. [PubMed] [Google Scholar]
- Cross S.L., Feinberg,M.B., Wolf,J.B., Holbrook,N.J., Wong-Staal,F. and Leonard,W.J. (1987) Regulation of the human interleukin-2 receptor α chain promoter: activation of a nonfunctional promoter by the transactivator gene of HTLV-I. Cell, 49, 47–56. [DOI] [PubMed] [Google Scholar]
- Cross S.L., Halden,N.F., Lenardo,M.J. and Leonard,W.J. (1989) Functionally distinct NF-κB binding sites in the immunoglobulin κ and IL-2 receptor α chain genes. Science, 244, 466–469. [DOI] [PubMed] [Google Scholar]
- Eder A.M., Dominguez,L., Franke,T.F. and Ashwell,J.D. (1998) Phosphoinositide 3-kinase regulation of T cell receptor-mediated interleukin-2 gene expression in normal T cells. J. Biol. Chem., 273, 28025–28031. [DOI] [PubMed] [Google Scholar]
- Gauchat J.F., Khandjian,E.W. and Weil,R. (1986) Cyclosporin A prevents induction of the interleukin 2 receptor gene in cultured murine thymocytes. Proc. Natl Acad. Sci. USA, 83, 6430–6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundstrom S., Dohlsten,M. and Sundstedt,A. (2000) IL-2 unresponsiveness in anergic CD4+ T cells is due to defective signaling through the common γ-chain of the IL-2 receptor. J. Immunol., 164, 1175–1184. [DOI] [PubMed] [Google Scholar]
- Jain J., McCaffrey,P.G., Valge-Archer,V.E. and Rao,A. (1992a) Nuclear factor of activated T cells contains Fos and Jun. Nature, 356, 801–804. [DOI] [PubMed] [Google Scholar]
- Jain J., Valge-Archer,V.E. and Rao,A. (1992b) Analysis of the AP-1 sites in the IL-2 promoter. J. Immunol., 148, 1240–1250. [PubMed] [Google Scholar]
- John S., Reeves,R.B., Lin,J.X., Child,R., Leiden,J.M., Thompson,C.B. and Leonard,W.J. (1995) Regulation of cell-type-specific interleukin-2 receptor α-chain gene expression: potential role of physical interactions between Elf-1, HMG-I(Y) and NF-κB family proteins. Mol. Cell. Biol., 15, 1786–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John S., Robbins,C.M. and Leonard,W.J. (1996) An IL-2 response element in the human IL-2 receptor α chain promoter is a composite element that binds Stat5, Elf-1, HMG-I(Y) and a GATA family protein. EMBO J., 15, 5627–5635. [PMC free article] [PubMed] [Google Scholar]
- Kang S.M., Beverly,B., Tran,A.C., Brorson,K., Schwartz,R.H. and Lenardo,M.J. (1992) Transactivation by AP-1 is a molecular target of T cell clonal anergy. Science, 257, 1134–1138. [DOI] [PubMed] [Google Scholar]
- Kim H.P., Kelly,J. and Leonard,W.J. (2001) The basis for IL-2-induced IL-2 receptor α chain gene regulation: importance of two widely separated IL-2 response elements. Immunity, 15, 159–172. [DOI] [PubMed] [Google Scholar]
- King L.B., Tolosa,E., Lenczowski,J.M., Lu,F., Lind,E.F., Hunziker,R., Petrie,H.T. and Ashwell,J.D. (1999) A dominant-negative mutant of c-Jun inhibits cell cycle progression during the transition of CD4(–)CD8(–) to CD4(+)CD8(+) thymocytes. Int. Immunol., 11, 1203–1216. [DOI] [PubMed] [Google Scholar]
- Lecine P., Algarte,M., Rameil,P., Beadling,C., Bucher,P., Nabholz,M. and Imbert,J. (1996) Elf-1 and Stat5 bind to a critical element in a new enhancer of the human interleukin-2 receptor α gene [published erratum appears in Mol. Cell. Biol. 1997 Apr; 17(4): 2351]. Mol. Cell. Biol., 16, 6829–6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenardo M.J. (1991) Interleukin-2 programs mouse αβ T lymphocytes for apoptosis. Nature, 353, 858–861. [DOI] [PubMed] [Google Scholar]
- Leung K. and Nabel,G.J. (1988) HTLV-1 transactivator induces interleukin-2 receptor expression through an NF-κB-like factor. Nature, 333, 776–778. [DOI] [PubMed] [Google Scholar]
- Lin B.B., Cross,S.L., Halden,N.F., Roman,D.G., Toledano,M.B. and Leonard,W.J. (1990) Delineation of an enhancerlike positive regulatory element in the interleukin-2 receptor α-chain gene. Mol. Cell. Biol., 10, 850–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.X. and Leonard,W.J. (1997) Signaling from the IL-2 receptor to the nucleus. Cytokine Growth Factor Rev., 8, 313–332. [DOI] [PubMed] [Google Scholar]
- Nakamura Y., Russell,S.M., Mess,S.A., Friedmann,M., Erdos,M., Francois,C., Jacques,Y., Adelstein,S. and Leonard,W.J. (1994) Heterodimerization of the IL-2 receptor β- and γ-chain cytoplasmic domains is required for signalling. Nature, 369, 330–333. [DOI] [PubMed] [Google Scholar]
- Nelson B.H., Lord,J.D. and Greenberg,P.D. (1994) Cytoplasmic domains of the interleukin-2 receptor β and γ chains mediate the signal for T-cell proliferation. Nature, 369, 333–336. [DOI] [PubMed] [Google Scholar]
- Rao A., Luo,C. and Hogan,P.G. (1997) Transcription factors of the NFAT family: regulation and function. Annu. Rev. Immunol., 15, 707–747. [DOI] [PubMed] [Google Scholar]
- Refaeli Y., Van Parijs,L., London,C.A., Tschopp,J. and Abbas,A.K. (1998) Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity, 8, 615–623. [DOI] [PubMed] [Google Scholar]
- Ruben S., Poteat,H., Tan,T.H., Kawakami,K., Roeder,R., Haseltine,W. and Rosen,C.A. (1988) Cellular transcription factors and regulation of IL-2 receptor gene expression by HTLV-I tax gene product. Science, 241, 89–92. [DOI] [PubMed] [Google Scholar]
- Schreiber E., Matthias,P., Muller,M.M. and Schaffner,W. (1989) Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res., 17, 6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuh K., Twardzik,T., Kneitz,B., Heyer,J., Schimpl,A. and Serfling,E. (1998) The interleukin 2 receptor α chain/CD25 promoter is a target for nuclear factor of activated T cells. J. Exp. Med., 188, 1369–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharfe N., Dadi,H.K., Shahar,M. and Roifman,C.M. (1997) Human immune disorder arising from mutation of the α chain of the interleukin-2 receptor. Proc. Natl Acad. Sci. USA, 94, 3168–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sompayrac L.M. and Danna,K.J. (1981) Efficient infection of monkey cells with DNA of simian virus 40. Proc. Natl Acad. Sci. USA, 78, 7575–7578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperisen P., Wang,S.M., Soldaini,E., Pla,M., Rusterholz,C., Bucher,P., Corthesy,P., Reichenbach,P. and Nabholz,M. (1995) Mouse interleukin-2 receptor α gene expression. Interleukin-1 and interleukin-2 control transcription via distinct cis-acting elements. J. Biol. Chem., 270, 10743–10753. [DOI] [PubMed] [Google Scholar]
- Toledano M.B., Roman,D.G., Halden,N.F., Lin,B.B. and Leonard,W.J. (1990) The same target sequences are differentially important for activation of the interleukin 2 receptor α-chain gene in two distinct T-cell lines. Proc. Natl Acad. Sci. USA, 87, 1830–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullman K.S., Flanagan,W.M., Edwards,C.A. and Crabtree,G.R. (1991) Activation of early gene expression in T lymphocytes by Oct-1 and an inducible protein, OAP40. Science, 254, 558–562. [DOI] [PubMed] [Google Scholar]
- Van Parijs L., Biuckians,A., Ibragimov,A., Alt,F.W., Willerford,D.M. and Abbas,A.K. (1997) Functional responses and apoptosis of CD25 (IL-2Rα)-deficient T cells expressing a transgenic antigen receptor. J. Immunol., 158, 3738–3745. [PubMed] [Google Scholar]
- Van Parijs L., Refaeli,Y., Lord,J.D., Nelson,B.H., Abbas,A.K. and Baltimore,D. (1999) Uncoupling IL-2 signals that regulate T cell proliferation, survival and Fas-mediated activation-induced cell death. Immunity, 11, 281–288. [DOI] [PubMed] [Google Scholar]
- Willerford D.M., Chen,J., Ferry,J.A., Davidson,L., Ma,A. and Alt,F.W. (1995) Interleukin-2 receptor α chain regulates the size and content of the peripheral lymphoid compartment. Immunity, 3, 521–530. [DOI] [PubMed] [Google Scholar]
- Yeh J.H., Lecine,P., Nunes,J.A., Spicuglia,S., Ferrier,P., Olive,D. and Imbert,J. (2001) Novel CD28-responsive enhancer activated by CREB/ATF and AP-1 families in the human interleukin-2 receptor α-chain locus. Mol. Cell. Biol., 21, 4515–4527. [DOI] [PMC free article] [PubMed] [Google Scholar]