

Abstract
Correct cell cycle regulation and terminal mitosis are critical for nervous system development. The retinoblastoma (Rb) protein is a key regulator of these processes, as Rb–/– embryos die by E15.5, exhibiting gross hematopoietic and neurological defects. The extensive apoptosis in Rb–/– embryos has been attributed to aberrant S phase entry resulting in conflicting growth control signals in differentiating cells. To assess the role of Rb in cortical development in the absence of other embryonic defects, we examined mice with telencephalon-specific Rb deletions. Animals carrying a floxed Rb allele were interbred with mice in which cre was knocked into the Foxg1 locus. Unlike germline knockouts, mice specifically deleted for Rb in the developing telencephalon survived until birth. In these mutants, Rb–/– progenitor cells divided ectopically, but were able to survive and differentiate. Mutant brains exhibited enhanced cellularity due to increased proliferation of neuroblasts. These studies demonstrate that: (i) cell cycle deregulation during differentiation does not necessitate apoptosis; (ii) Rb-deficient mutants exhibit enhanced neuroblast proliferation; and (iii) terminal mitosis may not be required to initiate differentiation.
Keywords: apoptosis/cell cycle/central nervous system/differentiation/retinoblastoma
Introduction
Neurogenesis is a highly regulated developmental process in which cycling neural precursor cells withdraw from the cell cycle, differentiate into post-mitotic neurons and commence migration into cortical layers. Proper cell cycle regulation is critical during these initial phases of neuronal development. Precursor cell proliferation is restricted to the ventricular zone, and only following cell cycle withdrawal do these cells begin expressing early neuronal markers and commence migration into cortical layers (McConnell and Kaznowski, 1991; McConnell, 1995). The laminar structure of the cortex is formed by an inside-out pattern of migration, such that the earlier generated neurons form the deeper layers, and the later born neurons give rise to more superficial layers (Rakic, 1988; McConnell, 1995). It is believed that terminal mitosis initiates neuronal differentiation and migration of newly born neurons to their appropriate laminar fate (McConnell and Kaznowski, 1991; McConnell, 1995).
The tumor suppressor retinoblastoma (Rb) is a key regulator of the cell cycle and differentiation (Mulligan and Jacks, 1998; Lipinski and Jacks, 1999; Ferguson and Slack, 2001). While Rb interacts with many transcription factors (Morris and Dyson, 2001), the primary targets for Rb are members of the E2F family. When hypophosphorylated, Rb is active and able to bind to and repress E2F activity. Phosphorylation by cyclin-dependent kinases inactivates Rb, resulting in the release of E2Fs, which become able to bind DNA and transactivate genes related to DNA synthesis, differentiation and apoptosis (Muller et al., 2001; reviewed in Weinberg, 1995; Dyson, 1998; Nevins, 1998).
The importance of Rb as a regulator of the cell cycle and differentiation was shown in Rb knockouts, which die embryonically by E15, exhibiting widespread developmental defects (Clarke et al., 1992; Jacks et al., 1992; Lee et al., 1992, 1994). The severity of Rb deficiency varied by tissue, with the most prominent defects in the developing liver, lens and nervous system. Ectopic mitoses were observed in several brain regions, and massive cell death occurred throughout the central and peripheral nervous systems. Further examination revealed that the proliferating precursors in the ventricular zone appeared normal, but neuronal differentiation was impaired (Lee et al., 1994). Expression of a number of neuronal markers, including βII tubulin, and the neurotrophin receptors TrkA, TrkB and p75, was significantly decreased (Lee et al., 1994). Based on expression of a neuron-specific LacZ reporter gene, it was found that the requirement for Rb was immediately following commitment to a neuronal fate (Slack et al., 1998).
Although Rb null embryos exhibited dramatic neural defects, studies with chimeric animals revealed survival and differentiation of Rb-deficient cells in the central nervous system (CNS) (Maandag et al., 1994; Lipinski et al., 2001). Furthermore, mutant cells displayed comparable levels of ectopic S phase entry, in both chimeric and knockout embryos. Rb–/– cells from chimeric mice arrested in G2, prior to completion of the cell cycle, unlike cells from knockouts that exhibited ectopic mitoses. The absence of ectopic mitoses in chimeric mice appeared to account for the lack of apoptosis in these cells (Lipinski et al., 2001). In spite of their arrested state, the majority of Rb-deficient cells in chimeric mice seemed to differentiate normally (Lipinski et al., 2001). Based on these results, it was proposed that the neighboring wild-type cells could rescue the Rb–/– cells, possibly by providing survival factors (Lipinski et al., 2001). In contrast, Rb null cells from whole-embryo knockouts underwent complete ectopic division, followed by apoptosis (Lipinski et al., 2001).
Due to the widespread embryonic defects and early lethality of Rb–/– embryos prior to the completion of neurogenesis, we generated a tissue-specific knockout for Rb in the telencephalon. To produce conditional mutants in which Rb deletion is specific to the telencephalon, we have interbred mice carrying a floxed Rb allele (Marino et al., 2000) with mice in which cre recombinase (cre) was knocked into the Foxg1 locus (Hebert and McConnell, 2000). With these mice, we show the following. (i) Rb excision is virtually complete throughout the developing telencephalon. (ii) Rb-deficient brains exhibit ectopic cell divisions at a rate similar to whole-embryo knockouts. This is unlike Rb chimeric embryos in which cells become arrested in G2. (iii) Rb deficiency specific to the telencephalon is not associated with widespread apoptosis, despite ectopic cell division. Rb null neurons are able to survive and differentiate, unlike whole-embryo knockouts in which the majority of neurons are lost (Slack et al., 1998). (iv) Ectopically dividing cells express the early neuronal marker TuJ1, consistent with enhanced neuroblast proliferation in Rb-deficient brains. Taken together, these results demonstrate that Rb may be a key regulator of neural proliferation in the developing cortex, but may not be required for neuronal survival or the initiation of differentiation.
Results
Generation of telencephalon-specific Rb knockouts
Whole-embryo Rb deletion results in embryonic lethality associated with neurological and hematological defects (Clarke et al., 1992; Jacks et al., 1992; Lee et al., 1992, 1994). To establish the role of Rb in cortical neurogenesis, it is imperative that the embryos survive during this developmental time period, and that the pleiotropic effects associated with Rb deficiency are eliminated. Conditional mutants were therefore generated in which Rb deletion is specific to the telencephalon. Animals in which cre was knocked into the Foxg1 locus (Hebert and McConnell, 2000) were interbred with mice carrying the floxed Rb allele (Marino et al., 2000). Foxg1 expression is detectable as early as E8–9, and shows peak levels at E17, restricted to the cerebral cortex, caudate putamen, hippocampus and dentate gyrus (Tao and Lai, 1992; Shimamura and Rubenstein, 1997). Thus cre-mediated excision of the floxed allele would occur in neural precursors at E9. This model system provides an excellent opportunity to examine the impact of Rb deficiency throughout the time course of telencephalon development, beginning at the time at which the first neurons are born.
Rb is completely deleted from the telencephalon of conditional mutants
To test the efficiency of cre-mediated recombination of a floxed allele, whole-mount LacZ analysis was performed on E13.5 embryos generated from Foxg1-cre mice crossed with Rosa26 reporter mice (Soriano, 1999). The Rosa 26R mice contain a transcriptional stop site flanked by loxP sites, located upstream of a β-galactosidase coding region. In the presence of cre, the stop site is excised, resulting in β-galactosidase transcription (Soriano, 1999). In contrast to control littermates, Foxg1-cre embryos displayed intense LacZ staining in the telencephalon, olfactory bulb, whisker barrels, dorsal midbrain and eye (Figure 1A). In the eye, it has been shown previously that cre expression is restricted to the lens and anterior (nasal) retina (Hebert and McConnell, 2000). Cross-sections revealed essentially ubiquitous LacZ expression in the telencephalon (Figure 1B), showing that cre-mediated recombination was virtually complete.

Fig. 1. Cre expression results in efficient recombination of floxed alleles. (A) Embryos from Foxg1-cre mice paired with Rosa26 reporter mice were taken at E13.5, whole mounted and stained with X-gal to visualize cre expression. (B) Robust LacZ staining was exhibited in the telencephalon of Foxg1-cre mice (coronal) (n = 4).
To assess deletion of the floxed Rb allele, PCR and western analyses were performed. First, PCR was used to detect the presence of Rb exon 19, using DNA isolated from the telencephalon of mutant and control littermates (Figure 2A). In the absence of cre (embryos 1 and 2), the floxed and wild-type alleles remain intact, as determined by 283 and 235 bp fragments, respectively. When cre-mediated recombination occurs (embryo 3), the floxed allele is deleted, resulting in a 260 bp band (Marino et al., 2000). The presence of only the middle, 260 bp band in the conditional mutant (embryo 3) suggested complete recombination (Figure 2A).

Fig. 2. Rb is completely deleted from the telencephalon of conditional mutants. (A) To detect the presence of Rb exon 19, DNA isolated from the telencephalon of E16.5 mutant and control embryos was subjected to PCR analysis. In the absence of cre, the floxed and wild-type alleles remain intact, as determined by 283 and 235 bp fragments, respectively. When cre-mediated recombination occurs, the floxed allele is deleted, resulting in a 260 bp band (Marino et al., 2000). The conditional mutant (embryo 3) exhibited only the middle 260 bp band, indicating that recombination had occurred, with no detection of the intact floxed allele (n = 3). (B) Rb and cre protein expression were assayed at E13.5 and E16.5 in tissue isolated from the telencephalon of mutant (homozygous Rb-F19 and heterozygous for Foxg1-cre) and control (double heterozygous at E13.5 and heterozygous for Rb-F19 at E16.5) embryos. As control, protein was also assayed from neural stem cells derived from wild-type and germline Rb–/– brains. Rb protein was undetectable in the mutant telencephalon (n = 4). (C) Protein was extracted from the telencephalon of E16.5 mutant and control littermates and subjected to EMSA. In contrast to the control (lane 1), the mutant extract exhibited increased levels of free E2F1/3 (lane 2). The uncomplexed E2F band was partially deleted upon the addition of an antibody to E2F1 (lane 4) or supershifted with antibodies specific for E2F3 (lane 6), and was completely deleted when antibodies to both E2F1 and E2F3 were added (lane 7). In the mutant extract, Rb–E2F complexes were undetectable (lane 2). The addition of the Rb antibody produced a supershift in littermate controls (arrowhead, lane 8) but not in extracts from the conditional Rb mutant (lane 9). Asterisks denote non-specific bands (n = 4).
Efficient Rb excision was confirmed further by western analysis. Tissue isolated from E13.5 and 16.5 mutant and control littermate telencephalon was assayed for Rb expression. Rb was undetectable in the mutant telencephalon (Figure 2B). Based on PCR and western analyses, our results show that cre-mediated excision of the floxed allele was virtually complete.
It was demonstrated previously that E2F activity is deregulated in whole-embryo Rb knockouts (Macleod et al., 1996; Callaghan et al., 1999). To determine whether E2F is deregulated in telencephalon-specific Rb knockouts, electrophoretic mobility shift assays (EMSAs) were performed. The telencephalon of E16.5 mutant and control littermates was examined for E2F DNA binding at E2F consensus sites (Figure 2C). Rb-deficient extracts exhibited increased levels of free E2Fs 1 and 3 (lane 2), which were completely supershifted when antibodies to both E2Fs 1 and 3 were added to the reaction (lane 7). While a supershift for Rb was readily detected in control tissue (lane 8 arrow), there was no Rb supershift in the tissue derived from the conditional knockout (lane 9). Thus, the conditional telencephalon-specific mutants reveal enhanced E2F DNA binding ability consistent with that previously shown in whole-embryo Rb deletions (Macleod et al., 1996; Callaghan et al., 1999).
Telencephalon-specific Rb knockouts survive to birth
Whole-embryo Rb knockouts die by E15.5, associated with extensive developmental defects (Clarke et al., 1992; Jacks et al., 1992; Lee et al., 1992, 1994). To determine the survival rate of conditional mutants, mice homo zygous for the floxed allele [Rb-F19/F19] were interbred with [Foxg1-cre:Rb-F19+/–] double heterozygous mice. Embryonic survival was assessed in mice from FVB/N and 129SV genetic backgrounds at E13.5, 15.5, 16.5, 17.5, 18.5, 19.5 and P0. During embryogenesis, the frequency of telencephalon-specific mutants was consistent with the expected Mendelian frequency of 25%; however, conditional mutants died 10–20 min after birth, due to respiratory defects (data not shown). Thus, unlike animals carrying germline Rb mutations that die during mid-gestation, this conditional Rb knockout will provide the unique opportunity to study the role of Rb and cell cycle regulation throughout the embryonic time course of cortical development.
Telencephalon-specific Rb deficiency is compatible with neuronal survival
Widespread apoptosis throughout the CNS is a hallmark of the Rb-deficient embryo, such that, by E14.5, the majority of cells expressing neuronal markers are absent (Lee et al., 1994; Slack et al., 1998). This apoptotic phenotype was significantly diminished in Rb–/– chimeric mice and was attributed to rescue by surrounding wild-type cells (Maandag et al., 1994; Lipinski et al., 2001). To determine whether complete deletion of Rb in the telencephalon would affect neuronal survival, we examined the level of apoptosis in the conditional Rb knockouts. Telencephalon-specific mutants and control littermates, as well as germline Rb knockouts, were analyzed at E13.5 for TUNEL labeling (Figure 3). Unlike the whole-embryo knockout, massive apoptosis did not occur in the E13.5 conditional mutant telencephalon. There was, however, a slight 1.5-fold increase in TUNEL labeling in the mutant, in which 79.7 ± 14.0 cells/section were TUNEL positive relative to 51.0 ± 4.7 cells/section in control animals (Figure 3B). This phenotype is distinct from that observed in the whole-embryo knockout in which massive apoptosis is observed in the developing cortex (Figure 3Ac). Detection of neuronal degeneration by FluoroJade labeling (Schmued et al., 1997; Noraberg et al., 1999) yielded comparable results (data not shown). Cell death was examined further in later stage embryos (E15.5, 16.5, 17.5 and 19.5), with no detectable apoptosis over littermate controls (Figure 3B, and Ad and e). Consistent with the absence of apoptosis, neurons in the conditional Rb–/– telencephalon were able to differentiate and survive. Thus, in spite of deregulated E2F activity, telencephalon-specific Rb deficiency does not induce the massive neuronal cell death characteristic of germline Rb knockouts.

Fig. 3. Telencephalon-specific Rb deficiency does not produce widespread apoptosis. (A) To detect cell death, E13.5 conditional mutant and control littermates, as well as germline Rb knockouts, were sectioned (coronal) and assayed for TUNEL labeling. Unlike whole-embryo Rb knockouts (c), conditional Rb mutants (b) did not exhibit massive neuronal apoptosis. However, in the telencephalon, TUNEL-positive cells in the conditional mutant (b) were slightly higher than controls (a), with 79.7 ± 14.0 and 51.0 ± 4.7 cells/section, respectively (B). Cell death was examined further in later stage embryos (E15.5, 16.5, 17.5 and 19.5), with no detectable apoptosis over littermate controls (d and e). (n = 5). Error bars denote standard error. Bar = 100 µm.
Newborn Rb conditional mutant pups die perinatally due to respiratory difficulties. Since the pons and the medulla, which are known to function in respiration, revealed some cre-mediated recombination (Hebert and McConnell, 2000), we examined apoptosis in these regions. Embryos were sectioned at E13.5, 17.5 and 19.5, and assayed with FluoroJade and TUNEL to detect both necrotic and apoptotic forms of cell death. Although no differences were detected at E17.5 or 19.5, intense labeling was observed in the mutant lateral pons at E13.5 (data not shown), a region containing many critical nuclei for respiratory regulation (Dick et al., 1994; Jodkowski et al., 1994; Morrison et al., 1994). Cell death in this region was consistent with the perinatal lethality associated with respiratory difficulties in the conditional mutants.
Telencephalon-specific Rb-deficient progenitor cells undergo complete cell divisions
Since ectopic mitoses are characteristic of whole-embryo Rb knockouts, whereas chimeric Rb–/– mice exhibit growth arrest at G2, we determined whether telencephalon-specific Rb deficiency would lead to ectopic cell division. Females were injected with bromodeoxyuridine (BrdU; 100 µg/g body weight) at E15.5 of gestation. Embryos were removed 2 h later, and mutant and corresponding control littermates were assayed for BrdU incorporation by immunohistochemistry. The conditional mutant exhibited extensive BrdU incorporation, which extended beyond the ventricular zone (Figure 4Ab). The extent of BrdU labeling was similar to levels observed in whole-embryo Rb knockouts (data not shown). At later time points (E17.5, 19.5), however, ectopic proliferation in the conditional Rb mutants was reduced (data not shown). Because it was shown recently that cells in Rb chimeric embryos incorporated BrdU but arrested in G2 (Lipinski et al., 2001), we investigated whether the ectopically proliferating cells in the conditional knockouts were actually completing cell division. Sections were labeled with an antibody directed against the M phase marker phospho-histone H3 (PH3). In contrast to the control, the mutant exhibited abundant mitoses outside the ventricular zone (Figure 5). Thus, unlike the G2 arrest observed in Rb–/– chimeric mice, the telencephalon-specific knockout revealed progenitor cells undergoing complete cell divisions outside the ventricular zone.

Fig. 4. Telencephalon-specific Rb deficiency induces ectopic S phase entry. (A) Females at E15.5 were injected with BrdU and, 2 h later, mutant and control embryos were removed and assayed for BrdU incorporation immunohistochemically. The conditional mutants exhibited extensive BrdU incorporation outside the ventricular zone (horizontal). (B) Quantification revealed that BrdU-positive cells in the control and mutant did not differ within the ventricular zone (VZ), with 48.8 ± 1.6 and 48.8 ± 2.9 positive cells/section, respectively. In the intermediate zone (IZ) and cortical plate (CP), there were only 14.0 ± 1.2 BrdU-positive cells in the control, as compared with 72.1 ± 1.8 positive cells in the mutant. Quantification was performed by counting cells within a superimposed grid (n = 8). Error bars denote standard error. Bar = 50 µm.

Fig. 5. Rb-deficient cells undergo complete ectopic mitoses. Sections were labeled with an antibody against the M phase marker phospho- histone H3 (PH3) at E16.5. In contrast to the control, the mutant exhibited widespread mitoses outside the ventricular zone (coronal). Boxed areas (A and B) are magnified (C and D) (n = 4). Bar = 25 µm.
Ectopically dividing Rb–/– cells express the early neuronal marker TuJ1
Although cortical gliogenesis normally begins at E18 (Cameron and Rakic, 1991; Levers et al., 2001), the cell type exhibiting ectopic proliferation was assessed. Tissue was, therefore, double-labeled for BrdU and cell-type specific markers, including nestin (progenitor cells), TuJ1 (early neurons) and glial fibrillary acidic protein (GFAP; astrocytes). Confocal microscopy of double-labeled cells revealed that cells ectopically incorporating BrdU did not express GFAP (data not shown). Several cells expressed nestin (data not shown) and the vast majority of ectopic BrdU-positive cells co-expressed TuJ1, an early pan-neuronal gene (Figure 6). Since TuJ1 labeling is cytoplasmic and BrdU is nuclear, serial confocal images are shown to demonstrate co-labeled cells. These results therefore indicate that ectopically dividing cells had committed to a neuronal fate and initiated differentiation.
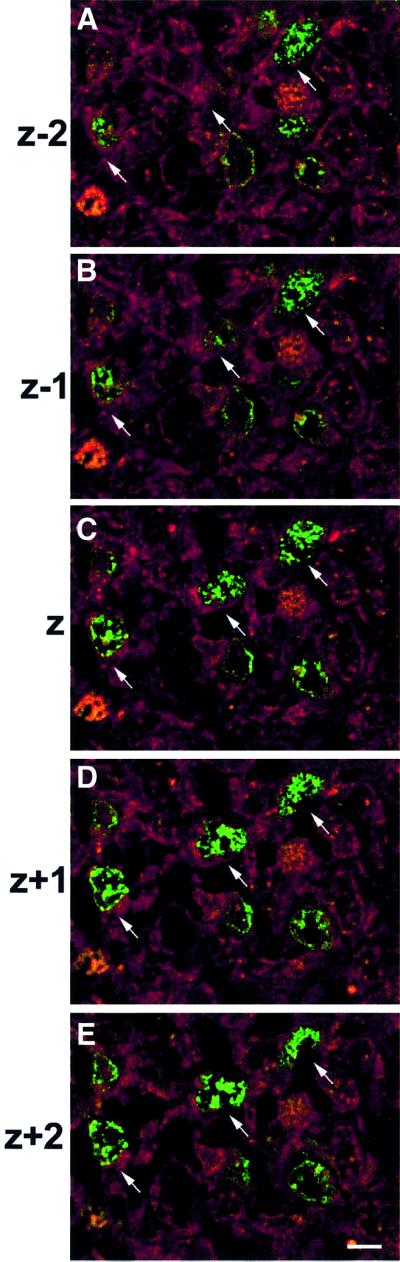
Fig. 6. Ectopically dividing Rb–/– cells expressed the early neuronal marker TuJ1. Females at E15.5 were injected with BrdU and, 2 h later, mutant and control embryos were removed, and double-labeled with cell type-specific markers (nestin, GFAP and TuJ1) and BrdU, then examined by confocal microscopy. Cells that had ectopically incorporated BrdU (green) were negative for GFAP (data not shown), but the vast majority of BrdU-positive cells co-expressed TuJ1, an early neuronal marker (red). Arrows point to representative cells. Serial confocal micrographs show sequential 0.7 µm levels through coronal sections, to demonstrate double labeling of cells (n = 4). Bar = 12.5 µm.
Telencephalon-specific mutants exhibit enhanced neurogenesis and aberrant cortical morphology
To determine whether the elevated proliferation in the conditional Rb mutant telencephalon would affect cortical development, we compared cortical morphology in mutants and littermate controls by cresyl violet staining (Figure 7). At E16.5, the average area of telencephalic sections was measured and compared between Rb mutant and littermate controls. Our results revealed that at E16.5, the conditional mutant telencephalic lobes were on average 30% larger than those of corresponding littermate controls (P < 0.01) (Figure 7B). A representative enlarged mutant telencephalon is shown (Figure 7Ab); however, it should be noted that some mutants did not differ from control littermates. Occasionally, Rb conditional mutants were observed with severely aberrant cortical development, for example large single-hemisphere telencephalic protrusions were observed at E16.5 (Figure 7Ad) and P0 (Figure 7Ae). These results demonstrate that Rb deficiency in the developing telencephalon may result in enhanced neurogenesis and, often, excessive cellularity.

Fig. 7. Telencephalon-specific mutants exhibit enhanced neurogenesis and aberrant cortical morphology. (A) At E16.5, many mutants exhibited enlarged telencephalic lobes compared with controls (coronal) (a and b), averaging an ∼30% increase over littermate controls (P < 0.01) (B). It should be noted that some Rb-deficient brains were found with no gross abnormalities, but occasionally mutants were observed with severely aberrant cortical development, for example large, single-hemisphere, telencephalic protrusions were observed at E16.5 (coronal) (Ad) and at P0 (sagittal, arrow points to protrusion) (Ae). E16.5 (n = 9), E17.5 (n = 7), E18.5 (n = 9), E19.5 (n = 7) and P0 (n = 11). Error bars denote standard error. Bar = 300 µm.
In summary, we have generated a conditional knockout, with complete Rb excision throughout the developing telencephalon, that survives until birth. Our results demonstrate that telencephalon-specific Rb mutants exhibit complete ectopic cell division of committed neuroblasts, based on the co-expression of BrdU and the early neuronal marker βIII tubulin. Thus, in Rb-deficient cortex, neurons are able to differentiate and survive, consistent with enhanced neurogenesis and increased cellularity. Taken together, these results demonstrate that Rb may be a key regulator of neuroblast proliferation; however, correct cell cycle regulation may not be required for neuronal differentiation and survival.
Discussion
Rb deficiency in whole-embryo knockouts has been well characterized, with extensive apoptosis and differentiation defects in many tissues, including the nervous system (Clarke et al., 1992; Jacks et al., 1992; Lee et al., 1992, 1994). The Rb null apoptotic phenotype was shown to be rescued substantially in chimeric animals, which survive and show apparently normal cortical development (Maandag et al., 1994; Lipinski et al., 2001). Based on these studies, it was suggested that the Rb–/– cells in the chimeras may have been rescued by surrounding wild-type cells, possibly by a released survival factor (Maandag et al., 1994; Lipinski et al., 2001). The absence of complete cell division implied that survival of Rb-deficient cells in chimeras may result from cell cycle arrest at G2, prior to entering mitosis. In contrast, Rb null cells from whole-embryo knockouts undergo complete ectopic division, followed by apoptosis (Lipinski et al., 2001).
Using Rb conditional knockouts, we now show that in spite of a virtually full Rb deletion in the telencephalon, neurons were able to survive and initiate differentiation. Telencephalon-specific Rb mutants exhibited increased cell division leading to enhanced neurogenesis and, by E16.5, a significant increase in average telencephalon size was observed. While we have not yet examined regional development and lamination in the cortex, ectopic cell division and survival of neurons were characteristic features and were observed consistently in all conditional Rb knockouts. Gross cortical development exhibited some phenotypic variability, which could result from enhanced attrition, possibly due to limiting growth factors. This would be consistent with the slight increase in apoptosis observed at early time points (E13.5). In contrast to germline Rb knockouts, telencephalon-specific Rb deficiency did not induce massive neuronal fallout as previously reported in whole-embryo knockouts. Instead, brain tissue appeared morphologically normal. Neuronal loss observed in the CNS of germline knockouts may therefore result from an extrinsic defect, perhaps due to extensive liver degeneration and failed erythropoiesis. It is well established that hypoxia is a potent inducer of apoptosis, particularly in neurons (Gwag et al., 1995; Rosenblum, 1997; Banasiak and Haddad, 1998; Banasiak et al., 2000). Our results show that ectopic cell division in differentiating neuroblasts may not necessarily evoke a default apoptotic pathway due to conflicting signals, but that Rb-deficient neuroblasts can survive and initiate differentiation.
Consistent with our findings in conditional Rb mutants, primary neuronal cultures of Rb-deficient cells survive and differentiate, although terminal mitosis is delayed (Lee et al., 1994; Slack et al., 1998; Callaghan et al., 1999). Rb-deficient trigeminal, dorsal root ganglia (Lee et al., 1994) and cortical progenitor cells (Slack et al., 1998) were shown to be morphologically identical to wild-type cells, and were able to differentiate and express the neuronal marker MAP2 (Slack et al., 1998). Although p107 protein levels were up-regulated in Rb–/– cells in a presumably compensatory manner (Callaghan et al., 1999), Rb/p107 double null neural stem cells display neuronal differentiation and survival in vitro (J.L.Vanderluit and R.S.Slack, unpublished data).
Several lines of evidence have attributed the apoptotic phenotype in Rb-deficient mice to increased levels of free E2F and deregulated E2F activity. First, E2F protein levels, in both the free and complexed forms, were increased in the CNS of Rb-deficient embryos (Macleod et al., 1996; Callaghan et al., 1999). Secondly, E2F1 overexpression was found to induce apoptosis in several neuronal cell types (Azuma-Hara et al., 1999; Hou et al., 2000; O’Hare et al., 2000), and E2F1-deficient neurons were protected from certain apoptotic stimuli (Giovanni et al., 2000; Hou et al., 2000; O’Hare et al., 2000). Finally, the additional absence of E2F1 dramatically reduced p53-dependent apoptosis in Rb-deficient mice, associated with a down-regulation of the p53 pathway (Pan et al., 1998; Tsai et al., 1998). The role of Rb in apoptosis appears to be a protective function, while E2Fs, in particular E2F1, act as inducers of cell death.
Apoptosis, associated with Rb deficiency, is often explained by E2F’s ability to produce conflicting growth control signals. In this model, cell cycle gene transactivation, in the absence of mitogen or in a division-incompetent cell type, may instead trigger apoptosis. However, the dual abilities of E2F1 to induce proliferation and apoptosis have been shown to be separable (Phillips et al., 1997). Through the use of E2F1 deletion mutants, it was found that E2F1 requires transcriptional activity to promote proliferation, but it can initiate apoptosis in the absence of its transactivation domain and without inducing S phase entry (Phillips et al., 1997). In another study, mutation of one allele of E2F3 was able to suppress apoptosis but not ectopic proliferation in Rb knockouts (Ziebold et al., 2001). In addition, we have shown that levels of E2Fs 1 and 3 are elevated in the mutant telencephalon, similar to levels found in whole-embryo Rb knockouts. Together, these studies demonstrate that apoptosis is not necessarily induced as a default pathway of deregulated E2F activity.
In conclusion, we describe a conditional knockout with virtually complete Rb excision throughout the developing telencephalon. We demonstrate that telencephalon- specific Rb mutants survive throughout embryogenesis and exhibit neuronal survival, in spite of undergoing complete ectopic cell division. Our results indicate that the widespread cell death observed in the CNS of Rb–/– embryos is not likely to be due to deregulated cell division but probably occurs secondarily to other embryonic defects. Ectopically dividing cells expressed the early neuronal marker TuJ1, consistent with enhanced neurogenesis and increased cellularity in the developing Rb-deficient brain. Taken together, these results demonstrate that Rb is an important regulator of neuroblast proliferation, but may not be required for neuronal survival or differentiation.
Materials and methods
Mice
The floxed Rb-F19 mice were generously provided by Dr Anton Berns, of The Netherlands Cancer Institute. To generate the telencephalon-specific Rb conditional knockouts, floxed Rb-F19 nullizygous mice were bred with Foxg1-cre:Rb-F19 double heterozygous mice, to generate mutants (Foxg1-cre:Rb-F19–/–) at a frequency of 25%. Mice were maintained on CD1, FVB/N and 129SV uniform genetic backgrounds and individual colonies. Mice were genotyped by PCR, as described previously, using DNA extracted from tails or remaining embryonic tissue. For timed pregnancies, mice were bred, and the time of plug identification was counted as day 0.5.
Whole-mount analysis
To determine the efficiency of cre-mediated recombination, whole-mount LacZ staining was performed by crossing Foxg1-cre mice with Rosa26 reporter mice. At E13.5, embryos were removed and fixed in 4% paraformaldehde (PFA) in 0.1 M NaH2PO4 pH 7.3 for 1 h at 4°C. The embryos were rinsed in wash solution (2 mM MgCl2, 0.01% sodium deoxycholate and 0.02% NP-40 in 0.1 M NaH2PO4 pH 7.3) for 3 × 30 min, then subjected to stain solution [1 mg/ml X-gal in dimethylsulfoxide (DMSO), 5 mM K3Fe (CN)6, 5 mM K4Fe (CN)6, 2 mM MgCl2, 0.01% sodium deoxycholate and 0.02% NP-40 in 0.1 M NaH2PO4 pH 7.3] for 2–24 h at 37°C. The embryos were subsequently washed three times for 5 min in 1× phosphate-buffered saline (PBS) and post-fixed for 24 h in 4% PFA in 0.1 M NaH2PO4 pH 7.3 at 4°C.
PCR analysis of recombination
To detect the presence of cre-mediated recombination, PCR analysis of Rb exon 19 was performed (Vooijs et al., 1998; Marino et al., 2000). DNA from the telencephalon of mutant and control embryos was isolated and extracted by phenol–chloroform. Primers for Rb212 (5′-GAAAG GAAAGTCAGGGACATTGGG-3′), Rb18 (5′-GGCGTGTGCATCA ATG-3′) and Rb19E (5′-CTCAAGAGCTCAGACTCATGG-3′) yielded 283 and 235 bp products for the unrecombined floxed and wild-type alleles, respectively, and a 260 bp band for the recombined floxed allele (Vooijs et al., 1998; Marino et al., 2000).
Western blot analysis
For western analysis, protein was harvested in lysis buffer A [50 mM HEPES pH 7.8, 250 mM KCl, 0.1 M EDTA, 0.1 M EGTA, 10% glycerol, 0.1% NP-40, 1 mM dithiothreitol (DTT), 0.5 mM phenylmethylsulfonyl fluoride (PMSF), 5 g/ml aprotinin, 2 g/ml leupeptin, 0.4 mM sodium vanadate] for 20 min on ice, followed by a 12 min centrifugation at 14 000 r.p.m. Protein was separated on a 10% polyacrylamide gel and transferred to a nitrocellulose membrane. After blocking overnight at 4°C in 5% skim milk, membranes were incubated in the primary antibody for 1–2 h at room temperature (mouse monoclonal anti-Rb, 1:250, PharMingen, 14001A or mouse monoclonal anti-cre, 1:500, Babco, MMS-106P). After three 5 min washes in TPBS (100 mM Na2HPO4, 100 mM NaH2PO4, 0.5 N NaCl, 0.1% Tween-20), membranes were incubated for 1 h at room temperature in the secondary horseradish peroxidase-conjugated antibody (anti-mouse HRP; Bio-Rad 170-6516). Blots were developed by chemiluminescence (ECL; Amersham Pharmacia Biotech) according to the manufacturer’s instructions.
EMSA (gel shift)
Total cell protein was extracted in lysis buffer (buffer A). A 10 µg aliquot of lysate was incubated with an excess of 32P-labeled double-stranded DNA probe (60 000 c.p.m./0.2 ng of DNA) containing a single E2F-binding site: 5′-GGATTTAAGTTTCGCGCCCTTTCTCAA-3′. The binding reaction (25 µl) was carried out at room temperature for 20 min in binding buffer [20 mM HEPES pH 7.6, 4% Ficoll, 2.5% MgCl2, 40 mM KCl, 0.1 mM EGTA, 0.5 mg/ml acetylated bovine serum albumin (BSA), 0.5 mM DTT and 100 ng of sonicated herring sperm DNA]. To control for binding specificity, a 100-fold excess of wild-type or mutant oligo (5′-GATTTAAGTTTCGATCCCTTTCTCAA-3′) was added to the binding reaction and incubated for 20 min prior to the addition of labeled probe. To identify the composition of the complexes, 1–2 µl of purified antibody were added to the reaction mixture. Complexes were resolved on a 5% gel run for 4 h, dried and visualized by autoradiography.
Tissue fixation and cryoprotection
Pregnant females were killed by a lethal injection of sodium pentobarbitol and embryos were removed and placed in 1× PBS. Embryos were fixed in 4% PFA/0.1 M phosphate buffer pH 7.4 for 1–2 days at 4°C. Tissue was rinsed three times in PBS, then subjected to sequential solutions of 12, 16 and 18% sucrose/0.1 M phosphate buffer for 1 day each at 4°C. Embryos were embedded in OCT (TissueTek 4583), frozen on liquid N2 and cut into 14 µm sections at –20°C on Superfrost slides (Fisher 12-550-15).
BrdU labeling and immunohistochemistry
Pregnant females were injected intraperitoneally with 100 µg/g body mass BrdU (Boehringer Mannheim 280879). Mice were killed and embryos harvested at different time points, and then fixed and cryoprotected as described above. For BrdU detection, sections were pre-treated in 1 M HCl for 10 min at 37°C, 0.2 M Na2B4O7 for 10 min at room temperature, then three 10 min rinses in PBS. Sections were incubated in mouse monoclonal anti-BrdU (1:50; Becton Dickinson) for 1–2 h at room temperature, rinsed three times in PBS for 10 min and then incubated in the appropriate secondary antibody (goat anti-mouse Alexa-488 or Alexa-594, 1:1000; Molecular Probes). Other antibodies used were TuJ1 (mouse monoclonal hybridoma supernatant, 1:50; Dr David Brown, University of Ottawa), GFAP (rabbit polyclonal, 1:400; Dako, AB986), nestin (mouse monoclonal, 1:200; RDI, 21714) and phospho-histone H3 (rabbit polyclonal, 1:200; Upstate Biotechnology, 06-570).
Determination of cell death
TUNEL staining. Sections were incubated for 1 h at 37°C with 75 µl of mixture (Roche Diagnostics, Mississauga, Ontario) consisting of 0.5 µl of terminal deoxynucleotide transferase (TdT), 0.95 µl of biotin-16-dUTP, 6.0 µl of CoCl2, 15.0 µl of 5× TdT buffer, and 52.55 µl of distilled water. After three washes in 4× SSC buffer, sections were incubated with Alexa 488–streptavidin (1:1000; Molecular Probes) for 1 h at room temperature.
FluoroJade staining. To evaluate neuronal degeneration including both apoptotic and necrotic modes of cell death, FluoroJade staining was used (Schmued et al., 1997; Noraberg et al., 1999). Sections were dried on a slide warmer, then dipped in alcoholic formalin pH 10 for 15 s. Following a 1 min rinse in dH2O, sections were treated in 0.06% KMnO4 for 15 min. After a brief rinse in dH2O, sections were incubated in 0.001% FluoroJade solution in 0.1% acetic acid for 30 min at 4°C. Following three washes in dH2O, sections were air dried and mounted with Permount.
Microscopy
Sections were examined by a Zeiss Axioskop 2 fluorescence microscope, and visualized by a Sony Power HAD 3CCD color video camera with Northern Eclipse software. Confocal images were generated with a Bio-Rad 1024 confocal microscope using a standard two-channel configuration and a ×60 1.4 NA oil immersion objective.
Acknowledgments
Acknowledgements
We are indebted to Dr Anton Berns for providing us with the floxed Rb mice prior to publication. We thank Katherine Bisby for excellent technical assistance, Andrew Ridsdale for assistance with confocal microscopy, Dr Sean Cregan for critical review of this manuscript, and Dr Donald Wang for anatomical examinations. This work was funded by a CIHR grant to R.S.S. K.L.F. is a recipient of a CIHR studentship; J.L.V. a CSN fellowship; and D.S.P., V.A.W. and R.S.S. are CIHR scholars. D.S.P. is a Glaxo Wellcome professor.
References
- Azuma-Hara M., Taniura,H., Uetsuki,T., Niinobe,M. and Yoshikawa,K. (1999) Regulation and deregulation of E2F1 in postmitotic neurons differentiated from embryonal carcinoma P19 cells. Exp. Cell Res., 251, 442–451. [DOI] [PubMed] [Google Scholar]
- Banasiak K.J., Xia,Y. and Haddad,G.G. (2000) Mechanisms underlying hypoxia-induced neuronal apoptosis. Prog. Neurobiol., 62, 215–249. [DOI] [PubMed] [Google Scholar]
- Banasiak K.L. and Haddad,G.G. (1998) Hypoxia-induced apoptosis: effect of hypoxic severity and role of p53 in neuronal cell death. Brain Res., 29, 295–305. [DOI] [PubMed] [Google Scholar]
- Callaghan D.A., Dong,L., Callaghan,S.M., Hou,Y.X., Dagnino,L. and Slack,R.S. (1999) Neural precursor cells differentiating in the absence of Rb exhibit delayed terminal mitosis and deregulated E2F1 and 3 activity. Dev. Biol., 207, 257–270. [DOI] [PubMed] [Google Scholar]
- Cameron R.S. and Rakic,P. (1991) Glial cell lineage in the cerebral cortex: a review and synthesis. Glia, 4, 124–137. [DOI] [PubMed] [Google Scholar]
- Clarke A.R., Maandag,E.R., van Roon,M., van der Lugt,N.M.T., van der Valk,M., Hooper,M.I., Berns,A. and te Reile,H. (1992) Requirement for a functional Rb-1 gene in murine development. Nature, 359, 328–330. [DOI] [PubMed] [Google Scholar]
- Dick T.E., Bellingham,M.C. and Richter,D.W. (1994) Pontine respiratory neurons in anesthetized cats. Brain Res., 636, 259–269. [DOI] [PubMed] [Google Scholar]
- Dyson N. (1998) The regulation of E2F by pRb-family proteins. Genes Dev., 12, 2245–2262. [DOI] [PubMed] [Google Scholar]
- Ferguson K.L. and Slack,R.S. (2001) The Rb pathway in neurogenesis. Neuroreport, 12, 55–62. [DOI] [PubMed] [Google Scholar]
- Giovanni A., Keramaris,E., Morris,E.J., Hou,S.T., O’Hare,M., Dyson,N., Robertson,G.S., Slack,R.S. and Park,D.S. (2000) E2F1 mediates death of B-amyloid-treated cortical neurons in a manner. J. Biol. Chem., 275, 11553–11560. [DOI] [PubMed] [Google Scholar]
- Gwag B.J., Lobner,D., Koh,J.Y., Wie,M.B. and Choi,D.W. (1995) Blockade of glutamate receptors unmasks neuronal apoptosis after oxygen–glucose deprivation in vitro. Neuroscience, 68, 615–619. [DOI] [PubMed] [Google Scholar]
- Hebert J. and McConnell,S.K. (2000) Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev. Biol., 222, 296–306. [DOI] [PubMed] [Google Scholar]
- Hou S.T. et al. (2000) The transcription factor E2F1 modulates apoptosis of neurons. J. Neurochem., 75, 91–100. [DOI] [PubMed] [Google Scholar]
- Jacks T., Fazeli,A., Schmitt,E.M., Bronson,R.T., Goodell,M.A. and Weinberg,R.A. (1992) Effects of an Rb mutation in the mouse. Nature, 359, 295–300. [DOI] [PubMed] [Google Scholar]
- Jodkowski J.S., Coles,S.K. and Dick,T.E. (1994) A ‘pneumatic centre’ in rats. Neurosci. Lett., 172, 67–72. [DOI] [PubMed] [Google Scholar]
- Lee E.H.P., Chang,C.Y., Hu,N., Wang,Y.C.J., Lai,C.C., Herrup,K., Lee,W.H. and Bradley,A. (1992) Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature, 359, 288–294. [DOI] [PubMed] [Google Scholar]
- Lee E., Hu,N., Yuan,S.S.F., Cox,L.A., Bradley,A., Lee,W. and Herrup,K. (1994) Dual roles of the retinoblastoma protein in cell cycle regulation and neuron differentiation. Genes Dev., 8, 2008–2021. [DOI] [PubMed] [Google Scholar]
- Levers T.E., Edgar,J.M. and Price,D.J. (2001) The fates of cells generated at the end of neurogenesis in developing mouse cortex. J. Neurobiol., 48, 265–277. [DOI] [PubMed] [Google Scholar]
- Lipinski M.M. and Jacks,T. (1999) The retinoblastoma gene family in differentiation and development. Oncogene, 18, 7873–7882. [DOI] [PubMed] [Google Scholar]
- Lipinski M.M., Macleod,K.F., Williams,B.O., Mullaney,T.L., Crowley,D. and Jacks,T. (2001) Cell-autonomous and non-cell-autonomous functions of Rb tumor suppressor in developing central nervous system. EMBO J., 20, 3402–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maandag E.R., van der Valk,M., Vlaar,M., Feltkamp,C., O’Brien,J., van Roon,M., van der Lugt,N., Berns,A. and te Riele,H.T. (1994) Developmental rescue of an embryonic-lethal mutation in the retinoblastoma gene in chimeric mice. EMBO J., 13, 4260–4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod K.F., Hu,Y. and Jacks,T. (1996) Loss of Rb activates both p53-dependent and independent cell death pathways in the developing mouse nervous system. EMBO J., 15, 6178–6188. [PMC free article] [PubMed] [Google Scholar]
- Marino S., Vooijs,M., van der Gulden,H., Jonkers,J. and Berns,A. (2000) Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev., 14, 994–1004. [PMC free article] [PubMed] [Google Scholar]
- McConnell S.K. (1995) Constructing the cerebral cortex: neurogenesis and fate determination. Neuron, 15, 761–768. [DOI] [PubMed] [Google Scholar]
- McConnell S.K. and Kaznowski,C.E. (1991) Cell cycle dependence of laminar determination in developing neocortex. Science, 254, 282–285. [DOI] [PubMed] [Google Scholar]
- Morris E.J. and Dyson,N.J. (2001) Retinoblastoma protein partners. Adv. Cancer Res., 82, 1–54. [DOI] [PubMed] [Google Scholar]
- Morrison S.F., Cravo,S.L. and Wilfehrt,H.M. (1994) Pontine lesions produce apneusis in the rat. Brain Res., 652, 83–86. [DOI] [PubMed] [Google Scholar]
- Muller H. et al. (2001) E2Fs regulate the expression of genes involved in differentiation, development, proliferation and apoptosis. Genes Dev., 15, 267–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan G. and Jacks,T. (1998) The retinoblastoma gene family: cousins with overlapping interests. Trends Genet., 14, 223–229. [DOI] [PubMed] [Google Scholar]
- Nevins J.R. (1998) Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ., 9, 585–593. [PubMed] [Google Scholar]
- Noraberg J., Kristensen,B.W. and Zimmer,J. (1999) Markers for neuronal degeneration in organotypic slice cultures. Brain Res. Brain Res. Protoc., 3, 278–290. [DOI] [PubMed] [Google Scholar]
- O’Hare M.J., Hou,S.T., Morris,E.J., Cregan,S.P., Xu,Q., Slack,R.S. and Park,D.S. (2000) Induction and modulation of cerebellar granule neuron death by E2F-1. J. Biol. Chem., 18, 25358–25364. [DOI] [PubMed] [Google Scholar]
- Pan H., Yin,C., Dyson,N.J., Harlow,E., Yamasaki,L. and van Dyke,T. (1998) Key roles for E2F1 in signaling p53-dependent apoptosis and in cell division within developing tumors. Mol. Cell, 2, 283–292. [DOI] [PubMed] [Google Scholar]
- Phillips A.C., Bates,S., Ryan,K.M., Helin,K. and Vousden,K.H. (1997) Induction of DNA synthesis and apoptosis are separable functions of E2F-1. Genes Dev., 11, 1853–1863. [DOI] [PubMed] [Google Scholar]
- Rakic P. (1988) Specification of cerebral cortical areas. Science, 241, 170–176. [DOI] [PubMed] [Google Scholar]
- Rosenblum W.I. (1997) Histopathologic clues to the pathways of neuronal death following ischemia/hypoxia. J. Neurotrauma, 14, 313–326. [DOI] [PubMed] [Google Scholar]
- Schmued L.C., Albertson,C. and Slikker,W.J. (1997) Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res., 751, 37–46. [DOI] [PubMed] [Google Scholar]
- Shimamura K. and Rubenstein,J.L.R. (1997) Inductive interactions direct early regionalization of the mouse forebrain. Development, 124, 2709–2718. [DOI] [PubMed] [Google Scholar]
- Slack R.S., El-Bizri,H., Wong,J., Belliveau,D.J. and Miller,F.D. (1998) A critical temporal requirement for the pRb family during neuronal determination. J. Cell Biol., 140, 1497–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet., 21, 70–71. [DOI] [PubMed] [Google Scholar]
- Tao W. and Lai,E. (1992) Telencephalon-restricted expression of BF-1, a new member of the HNF-3/fork head gene family, in the developing rat brain. Neuron, 8, 957–966. [DOI] [PubMed] [Google Scholar]
- Tsai K.Y., Hu,Y., Macleod,K.F., Crowley,D., Yamasaki,L. and Jacks,T. (1998) Mutation of E2F-1 suppresses apoptosis and inappropriate S phase entry and extends survival of Rb-deficient mouse embryos. Mol. Cell, 2, 293–304. [DOI] [PubMed] [Google Scholar]
- Vooijs M., van der Valk,M., te Riele,H. and Berns,A. (1998) Flp-mediated tissue-specific inactivation of the retinoblastoma tumor suppressor gene in the mouse. Oncogene, 17, 1–12. [DOI] [PubMed] [Google Scholar]
- Weinberg R.A. (1995) The retinoblastoma protein and cell cycle control. Cell, 81, 323–330. [DOI] [PubMed] [Google Scholar]
- Ziebold U., Reza,T., Caron,A. and Lees,J.A. (2001) E2F3 contributes both to the inappropriate proliferation and to the apoptosis arising in Rb mutant embryos. Genes Dev., 15, 386–391. [DOI] [PMC free article] [PubMed] [Google Scholar]