

Abstract
Grim encodes a protein required for programmed cell death in Drosophila. The Grim N-terminus induces apoptosis by disrupting IAP blockage of caspases; however, N-terminally-deleted Grim retains pro apoptotic activity. We describe GH3, a 15 amino acid internal Grim domain absolutely required for its proapoptotic activity and sufficient to induce cell death when fused to heterologous carrier proteins. A GH3 homology region is present in the Drosophila proapoptotic proteins Reaper and Sickle. The GH3 domain and the homologous regions in Reaper and Sickle are predicted to be structured as amphipathic α-helixes. During apoptosis induction, Grim colocalizes with mitochondria and cytochrome c in a GH3-dependent but N-terminal- and caspase activity-independent manner. When Grim is overexpressed in vivo, both the N-terminal and the GH3 domains are equally necessary, and cooperate for apoptosis induction. The N-terminal and GH3 Grim domains thus activate independent apoptotic pathways that synergize to induce programmed cell death efficiently.
Keywords: apoptosis/BH3/cytochrome c/IAP/mitochondria
Introduction
Multicellular organisms eliminate unwanted or damaged cells by a regulated cell death process, whose alteration leads to either proliferative or degenerative disorders (Thompson, 1995). Cell death is particularly relevant in several critical processes of development, such as organ and tissue shaping during embryogenesis, tissue resorption during metamorphosis, and neuronal selection (Jacobson et al., 1997; Vaux and Korsmeyer, 1999). Cell death during development occurs in a temporally and spatially reproducible pattern that is genetically controlled, and is thus regarded as programmed cell death (PCD). Most PCD processes, independently of the origin of the death stimulus, are executed through a well-defined pattern of cellular and biochemical events known as apoptosis (Wyllie et al., 1980). Apoptosis involves the regulated function of a cellular machinery composed of inductor, inhibitor and executor molecules such as caspases and DNases that ultimately destroy cell contents (Green, 2000). Genetic analysis of PCD in the nematode Caenorhabditis elegans identified essential elements of the basic machinery for both the regulation and execution phases of apoptotic death, and showed for the first time their conservation throughout metazoan evolution (Metzstein et al., 1998). Endogenous and viral molecules that promote, inhibit or are required for apoptosis have been found to be conserved from nematodes to humans (Aravind et al., 1999).
At the cellular level, mitochondria are crucial for integrating death signals from very diverse sources, such as genotoxic damage, cytotoxic stress, growth or survival factor deprivation, glucocorticoids, heat shock and radiation. Following such stimuli, mitochondria trigger apoptosis by releasing to the cytosol cytochrome c and other proteins that activate the death program (Kroemer and Reed, 2000). A large number of molecules target the mitochondria to promote or inhibit induction of the apoptotic process. The most widespread and numerous family is the Bcl-2 homology group, which contains both pro- and anti-apoptotic molecules (Gross et al., 1999). Of the four homology blocks present among the Bcl-2 family members, the BH3 domain appears essential for proapoptotic member function; for some of them, BH3 is the only domain shared with the rest of the family (Kelekar and Thompson, 1998). BH3-containing proteins induce apoptosis either by altering the mitochondrial membrane directly or by counteracting the protective function of Bcl-2 family antiapoptotic members (Kelekar and Thompson, 1998; Gross et al., 1999; Zong et al., 2001).
Studies in the fly Drosophila melanogaster have uncovered four essential components of the genetic program controlling PCD, reaper, hid, sickle and grim, whose transcriptional activation precedes, induces and is necessary for PCD by apoptosis (White et al., 1994; Grether et al., 1995; Chen et al., 1996b; Christich et al., 2002; Srinivasula et al., 2002; Wing et al., 2002). The four genes map to a single genetic complex and function as death switches that are regulated mainly at the transcriptional level, although Hid is also regulated through phosphorylation by the MAPK pathway (Bergmann et al., 1998; Kurada and White, 1998). Ectopic activation of any of the four genes triggers apoptosis in otherwise viable cells (Grether et al., 1995; Chen et al., 1996b; Nordstrom et al., 1996; White et al., 1996; Christich et al., 2002; Srinivasula et al., 2002; Wing et al., 2002), and their inactivation prevents apoptosis of cells that would normally undergo PCD (White et al., 1994). Their activation not only mediates programmed apoptotic events during normal development, but reaper also mediates apoptosis induced in response to DNA damage (Nordstrom et al., 1996), acting as a direct transcriptional target of the p53 DNA damage response (Brodsky et al., 2000). This genetic complex, therefore, plays a pivotal role in integrating different death stimuli that lead to insect cell death by apoptosis (McCall and Steller, 1997; Abrams, 1999). In spite of their central role in Drosophila PCD, no homologue for any of these genes has yet been described in any other organism, although the four have been shown to activate death pathways in vertebrate cells (Evans et al., 1997; Clavería et al., 1998; McCarthy and Dixit, 1998; Haining et al., 1999; Srinivasula et al., 2002).
The products of these four genes show sequence similarity within the first 14 amino acids. The conserved N-terminus of Reaper, Hid, Grim and Sickle binds to members of the inhibitor of apoptosis protein (IAP) family, preventing their antiapoptotic activity (Vucic et al., 1997, 1998; McCarthy and Dixit, 1998; Wang et al., 1999; Lisi et al., 2000; Christich et al., 2002; Srinivasula et al., 2002). Flies deficient in DIAP1 suffer massive apoptosis early in development, suggesting that relief of the IAP protective effect could be sufficient for Reaper, Hid, Sickle and Grim to trigger cell death (Wang et al., 1999; Goyal et al., 2000). Whereas Hid requires the N-terminal domain to induce cell death efficiently in cultured cells (Vucic et al., 1998; Haining et al., 1999), Reaper and Grim can induce apoptosis in the absence of this domain in several experimental contexts (Chen et al., 1996a; Clavería et al., 1998; McCarthy and Dixit, 1998; Wing et al., 1998), suggesting that other regions of these proteins may have proapoptotic activity. The changes Grim and Reaper induce in cytochrome c display (Varkey et al., 1999) suggest that a mitochondrial death pathway might be relevant to the proapoptotic activity of these proteins. Here we report the identification of GH3, a novel domain required for Grim proapoptotic activity and sufficient to induce cell death. We show that the GH3 domain is required for Grim targeting to mitochondria and activates a proapoptotic pathway distinct from the IAP inhibition promoted by the N-terminal domain. Sequence homology between the GH3 domain and regions in Reaper (Wing et al., 2001) and Sickle (Christich et al., 2002; Srinivasula et al., 2002; Wing et al., 2002) suggests functional conservation of the domain between these three proapoptotic proteins. We propose that the N-terminal and GH3 Grim domains can induce apoptosis by triggering independent pathways that synergize to induce PCD, and may have variable relevance depending on cellular context.
Results
GH3, a novel Grim domain essential for Grim proapoptotic function in Drosophila SL2 cells
Secondary structure prediction of the Grim protein identified three regions with a very high probability of conforming to an α-helical structure. We termed these regions GH1, GH2 and GH3, for Grim Helix 1, 2 and 3 (Figure 1A). The GH3 domain showed similarity to a region in Reaper (Wing et al., 2001) and Sickle (Christich et al., 2002; Srinivasula et al., 2002; Wing et al., 2002) (Figure 1B), both also predicted to conform as an α-helix. The homology region spans the 15 amino acids predicted to conform as an α-helix in Grim, with two 5 amino acid regions of high similarity flanking a 5 amino acid central region with lesser homology (Figure 1B). Representation in a helical wheel projection of GH3 residues and of those in the Reaper and Sickle homology regions reveals the amphipathic nature of the predicted α-helices (Figure 1C).

Fig. 1. Sequence and structural similarity between Grim GH3 domain, Reaper and Sickle. (A) Grim protein sequence showing the three predicted α-helices GH1–GH3 (black boxes). (B) Sequence alignment of the Grim GH3 domain with Sickle and Reaper. Black boxes identify highly similar residues and grey boxes show those with lesser similarity. (C) Helical wheel projection diagram of the Grim GH3 domain (left) and corresponding Sickle and Reaper homology regions, in which hydrophobic residues are shown in black over grey and hydrophilic residues in white over black.
We tested whether GH3 was important for Grim proapoptotic function by assaying the cell killing ability of several Grim mutants altered in the GH3 domain in Drosophila SL2 cells (Figure 2A). As previously described (Chen et al., 1996b), wild-type (WT) Grim induced cell death when overexpressed in this assay (Figure 2B). In contrast, a Grim mutant form with a 13 amino acid deletion that removes the GH3 residues most reliably predicted to form an α-helix (Δ86–98, Figure 2A) only marginally induced apoptosis (Figure 2B). A 5′-shifted 11 amino acid deletion, such that the 3′ part of GH3 was respected (Δ83–93, Figure 2A), was less effective in eliminating the proapoptotic activity than the complete GH3 deletion (Figure 2B). An internal deletion removing four amino acids commonly deleted in the two larger deletions (Δ89–92, Figure 2A) also resulted in strong impairment of Grim killing ability, but to a lesser extent than the complete GH3 deletion (Figure 2B). We next tested the relevance of L89, a conserved residue within positions 89–92, whose hydrophobicity could be relevant for the amphipathic nature of the GH3 domain. A non-conservative replacement of L89 by glutamic acid (L89E, Figure 2A) impaired GH3 killing ability nearly to the same level as the Δ89–92 mutant (Figure 2B). Semi-conservative replacement of L89 by alanine (L89A, Figure 2A) had a mild effect on Grim proapoptotic function (Figure 2B). These results showed that the GH3 domain is required for Grim proapoptotic function and that L89 is a functionally relevant residue in the domain.
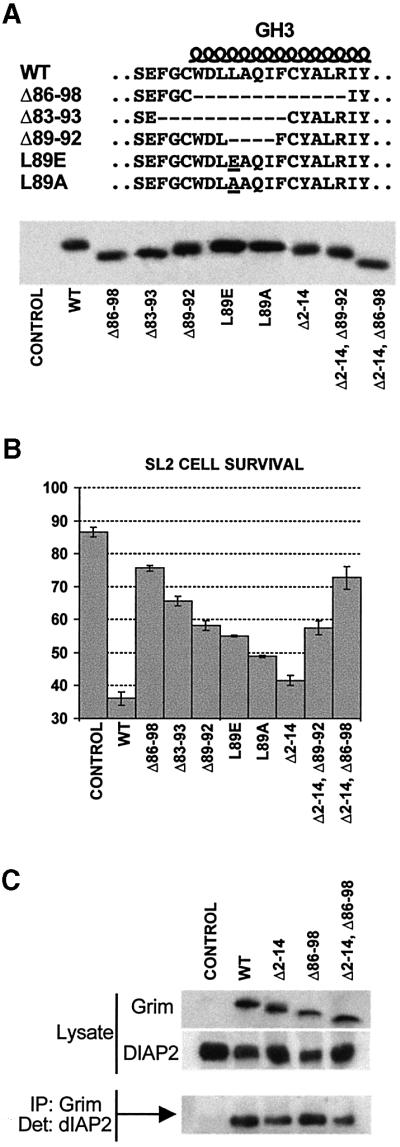
Fig. 2. Requirement of Grim N-terminal and GH3 domains for apoptosis induction and IAP binding. (A) Top, amino acid sequences of WT Grim GH3 and neighbouring residues, and of the mutant proteins synthesized. Bottom, western blot showing the stability of the various GH3 mutants and their combinations with the N-terminal deletion. (B) Bar diagram showing survival ratios of SL2 cells transfected with Grim and the various mutant forms. Survival ratios were calculated by comparing the ratio of LacZ-positive cells with and without copper induction; the result was expressed as a percentage. (C) Immuno precipitation showing the relevance of N-terminal and GH3 Grim domains for its association with DIAP2.
Since residual proapoptotic function was observed even in the most extreme case of Grim function impairment, we tested whether the N-terminal conserved domain was responsible for the residual killing activity. As previously described (Vucic et al., 1998), the N-terminal domain was unnecessary for Grim killing in SL2 cells (Δ2–14, Figure 2B). Combination of the N-terminal deletion with either the complete GH3 deletion (Δ2–14, Δ86–98), or the deletion of the four internal amino acids (Δ2–14, Δ89–92), did not augment Grim function impairment (Figure 2B). We therefore could not demonstrate the relevance of the N-terminal domain for Grim proapoptotic function in cultured SL2 cells.
Since N-terminally deleted Grim can still bind IAPs (Vucic et al., 1998), GH3 domain function could be related to Grim ability to bind IAPs and inhibit their protective function. We found that deletion of the Grim N-terminal domain leads to a slight reduction in its ability to bind DIAP2 (Figure 2C), however, deletion of the GH3 domain, either alone or in combination with the N-terminal deletion, does not impair Grim ability to bind DIAP2 (Figure 2C). We have obtained similar results with other viral and vertebrate IAPs (data not shown). These results suggest that GH3 activity is unrelated to the IAP inhibitory Grim activity.
The GH3 domain is sufficient to induce apoptosis in Drosophila SL2 cells
To determine whether the GH3 domain was just required to elicit Grim proapoptotic activity or could function as a proapoptotic motif itself, we fused a Grim fragment containing the GH3 domain to green fluorescent protein (GFP) as a carrier protein (GH3–GFP, Figure 3A) and determined the ability of this fusion protein to induce apoptosis in Drosophila SL2 cells. GH3–GFP induced cell death with the same efficiency as did the complete Grim–GFP (Grim–GFP, Figure 3A) fusion protein (Figure 3B). The cell death observed was specific to GH3 domain activity, since it was largely abolished by an L-to-E mutation in the residue equivalent to Grim L89 (GH3LE–GFP) (Figure 3B). A similar fusion using a Grim fragment containing the N-terminal domain (Nt-Grim–GFP, Figure 3A) led to low levels of cell death (Figure 3B). In all cases, cell death was rescued by coexpression with the baculoviral caspase inhibitor p35 (Figure 3B). The GH3 domain is therefore sufficient to trigger a specific proapoptotic route in SL2 cells.
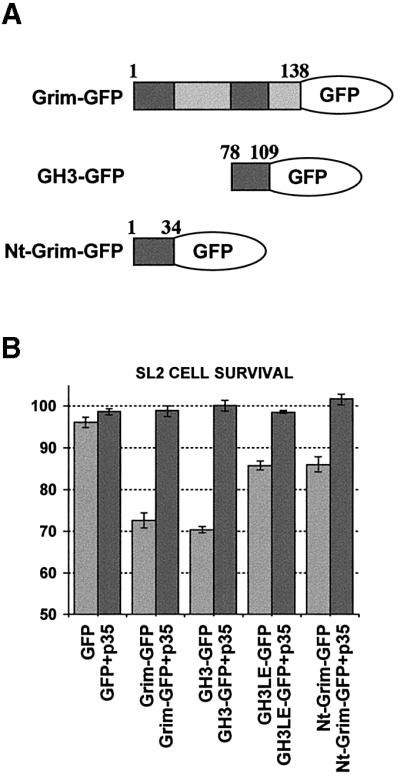
Fig. 3. The GH3 domain is sufficient to induce apoptosis in Drosophila SL2 cells. (A) Scheme shows the different GFP fusion proteins used, indicating the Grim amino acids included. (B) Bar diagram showing survival ratios of SL2 cells transfected with the different GFP fusion proteins with or without p35 coexpression. Survival ratios were calculated by comparing the ratio of LacZ-positive cells with and without copper induction; the result was expressed as a percentage.
Grim associates with mitochondria during apoptosis induction in Drosophila SL2 cells in a GH3-dependent and N-terminal-independent manner
We used immunocytochemistry to identify Grim subcellular localization in Drosophila SL2 cells. In phases previous to any obvious apoptotic phenotype, Grim generally showed a diffuse distribution in the cytoplasm, but also displayed rings of stronger Grim staining (Figure 4B and E). Grim rings are mitochondria-associated, as indicated by the presence of a mitochondrial matrix marker, mainly inside the rings, but also in colocalization with strong Grim staining (Figure 4A–F). Grim expression pattern was unchanged in cells exposed to zVAD.fmk or cotransfected with the caspase inhibitor p35 (data not shown). Grim colocalization with cytochrome c is similar to that observed with the mitochondrial matrix marker but, in addition, Grim and cytochrome c show extensive colocalization in larger dots (Figure 5A–F). These larger cytochrome c dots were not observed in untransfected cells, they increased in size and abundance as apoptosis progressed, and did not colocalize with a mitochondrial marker (data not shown). As previously described (Varkey et al., 1999), we thus found no substantial cytochrome c release to cytosol, instead, Grim promoted redistribution of cytochrome c signal in large dots in colocalization with Grim itself.

Fig. 4. Grim subcellular distribution associates with mitochondria in a GH3-dependent manner. Immunodetection of WT Grim or Δ86–98Grim (green), a mitochondrial marker (red), and their colocalization are shown as indicated. White arrowheads in (D–F) indicate regions of Grim–mitochondrial staining association.

Fig. 5. Grim subcellular distribution associates with cytochrome c in a GH3-dependent manner. Immunodetection of WT Grim or Δ86–98Grim (green), cytochrome c (red), and their colocalization are shown as indicated. White arrowheads in (D–F) indicate regions of Grim–cytochrome c staining association and/or colocalization; open arrowhead shows a region of strong Grim–cytochrome c staining and colocalization.
Grim subcellular localization was dependent on the presence of an intact GH3 domain. GH3-deficient Grim showed no obvious organization in rings associated with mitochondria and did not colocalize with either the mitochondrial marker or cytochrome c (Figures 4G–L and 5G–L). In contrast, deletion of Grim 2–14 amino acids did not alter subcellular distribution of the Grim protein, nor the changes induced in cytochrome c display (data not shown).
Subcellular localization of the different GFP fusion proteins correlated with the results obtained with the Grim deletion mutants. While GFP and Nt-Grim–GFP showed complete diffuse distribution throughout the entire cell (Figure 6D and E), GH3–GFP accumulated in dots or rings, some of which colocalized with mitochondria and cytochrome c (Figure 6A and B). Introduction of the L89E mutation in the GH3 domain (GH3LE–GFP) impaired localization of the fusion protein, which tended to distribute more diffusely than GH3–GFP (Figure 6F). Confirming that GH3–GFP reproduces part of the normal Grim expression pattern, GH3–GFP subcellular distribution partially overlapped that of WT Grim when both were cotransfected (Figure 6C). We conclude that the GH3 domain is sufficient to display at least part of Grim subcellular distribution.

Fig. 6. Subcellular distribution of GFP fused to different Grim domains. GFP alone (D) or fused to Grim regions containing GH3 (A–C), N-terminal (E) and GH3-L89E (F) domains are shown in green. Colocalization with mitochondria (red) is shown in (A), (D), (E) and (F), with cytochrome c (red) in (B), and with WT Grim (red) in (C).
Both GH3 and N-terminal domains are required and synergize in vivo for Grim proapoptotic activity
We analysed the relevance of the GH3 and N-terminal Grim domains by overexpressing the Grim mutants in transgenic flies using the Gal4-UAS system (Brand and Perrimon, 1993). To drive Grim expression, we used the GMR-Gal4 line, which targets expression to the eye disc, and the MS1096-Gal4 line, which directs expression to the wing imaginal disc. Overexpression of WT Grim with either driver caused total or partial lethality in all transgenic lines (Figure 7A and B). These results suggested that leaky expression from both promoters in vital tissues produced sufficient cell death to block development. Surviving adult flies overexpressing WT Grim displayed a considerable reduction in eye size with the GMR driver (Figure 8A and B), and wing agenesis plus notum reduction and elimination of macro- and microchaetae with the MS1096 driver (Figure 8I, J, Q and R).
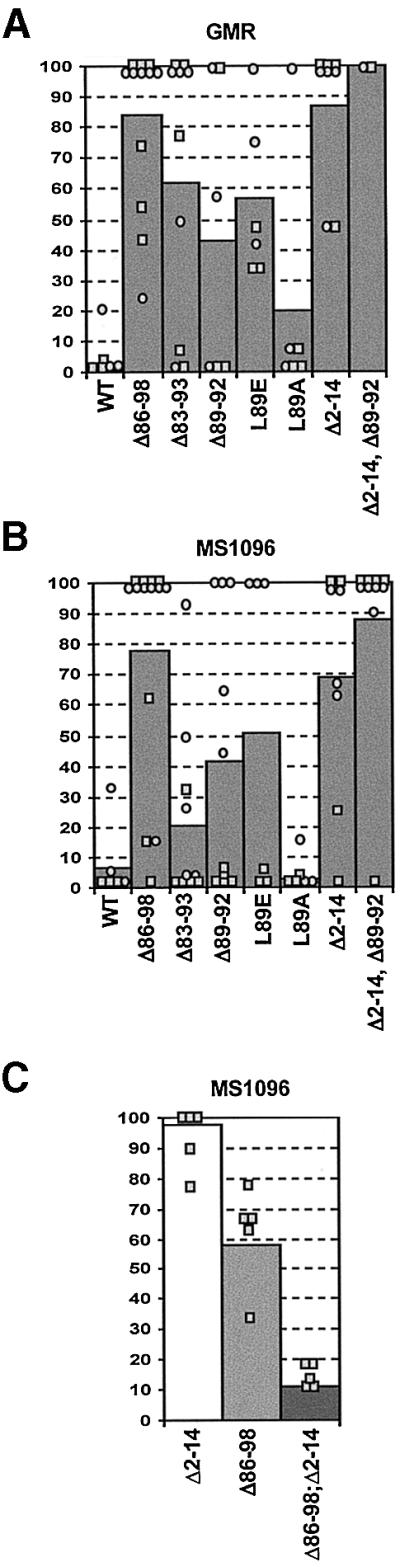
Fig. 7. Relevance of N-terminal and GH3 Grim domains during apoptosis induction in vivo. GMR-Gal4 (A) or MS1096-Gal4 (B) driver lines were crossed with several independent lines bearing UAS-Grim transgenes coding for the WT protein or the different mutant forms. Graphs show the viability of the independent transgenic lines analysed for each mutant. The GMR and MS1096 driver lines are located in the X chromosome. Due to hypertranscription of the male X chromosome, male viability was always lower than that of females; thus, male (squares) and female (circles) viability is shown independently. Bars represent the overall viability of each mutant, calculated by averaging results obtained with each independent line (males and females). Results were obtained from crosses at 18°C for GMR and 25°C for MS1096 lines. (C) Viability of transgenic male flies expressing Δ2–14 Grim or Δ86–98 Grim mutant proteins driven by MS1096-Gal4, alone or simultaneously, as indicated.

Fig. 8. Mutations in the GH3 and N-terminal domains rescue targeted tissue deletion by Grim in transgenic flies. Images show representative phenotypes induced in the eyes of GMR-Gal4;UAS-Grim (B–H), and the notum (J–P) and wings (R–U, W and X) of MS1096-Gal4;UAS-Grim female adult flies. Results correspond to overexpression of WT Grim and the distinct Grim mutants, as indicated. (V) Wing of an MS1096-Gal4;UAS-GrimΔ86–98;UAS-GrimΔ2–14 male adult fly. (A) GMR-Gal4 fly, (I and Q) WT notum and wing.
In contrast to these results, overexpression of a GH3-deleted Grim (Δ86–98) resulted in very low lethality (Figure 7A and B), as well as rescue of the eye, notum and wing phenotypes (Figure 8F, N and W). Elimination of amino acids 89–92 resulted in a lesser impairment of Grim killing ability, showing that the 3′ part of the GH3 domain is important for proapoptotic function (Figure 7A and B). In correlation with these results, the Δ89–92 mutant rescued the eye phenotype induced by WT Grim, but only partially rescued the more sensitive wing and notum phenotypes (Figure 8D, L and T). Elimination of amino acids 83–93, which extends the deletion N-terminal to the putative helical domain, did not increase the rescue observed with the 89–92 deletion, suggesting that residues 5′ of leucine 89 might be less important for proapoptotic function (Figure 7A and B; data not shown). The relevance of leucine 89 was again shown by the significant rescue of viability (Figure 7A and B) and of eye, notum and wing phenotypes (Figure 8E, M and U) in flies overexpressing the non-conservative L89E substitution. In contrast, the semi-conservative substitution L89A resulted in mild, but significant, impairment of grim death induction (Figure 7A and B) and targeted tissue deletion (Figure 8C, K and S).
In accordance with the results observed in cultured cells, mutations of the GH3 domain impaired Grim proapoptotic activity in transgenic flies. Deletion of the N-terminal domain, in contrast to the results observed in cultured cells, resulted in highly significant elimination of Grim proapoptotic function in vivo. Both viability (Figure 7A and B) and appearance of the tissues targeted by the GMR and MS1096 drivers (Figure 8G, O and X) were rescued by the 2–14 deletion to a level similar to that observed for the GH3 deletion. Simultaneous deletion of the N-terminus and amino acids 89–92 of the GH3 domain resulted in even lower lethality (Figure 7A and B) and fewer alterations in the targeted tissues than those induced by each deletion in isolation (Figure 8H and P).
To determine whether the N-terminal and GH3 Grim domains could function independently of each other, we tested whether the simultaneous expression of independent 2–14- and GH3-deleted Grim proteins could induce apoptosis. We generated flies carrying independent transgenes for 2–14 and 86–98 Grim deletion mutants driven by MS1096 expression. Whereas males carrying either protein alone showed little or no lethality (Figure 7C) and no alterations in wing development (data not shown), double transgenic males simultaneously expressing Δ2–14 and Δ86–98 Grim mutants displayed severe lethality and reduced wings (Figures 7C and 8V). Females did not display lethality in any situation, but frequently showed reduced wings in the double transgenics (data not shown), although not in single transgenics (Figure 8W and X). Functions of both the N-terminal and the GH3 domains are therefore essential for Grim activity in vivo and they independently activate specific death mechanisms that synergize to trigger apoptosis.
Discussion
We have defined a new domain essential for Grim proapoptotic function both in cultured cells and in vivo. Previous work showed that the 14 N-terminal amino acids conserved by Reaper, Hid, Sickle and Grim bind to and inhibit IAPs, an interaction that may lead to apoptosis induction (Vucic et al., 1997, 1998; McCarthy and Dixit, 1998; Wang et al., 1999; Goyal et al., 2000; Lisi et al., 2000; Christich et al., 2002; Srinivasula et al., 2002). Massive cell death in DIAP1-deficient flies suggested that IAP inhibition may suffice for Grim, Reaper, Sickle and Hid to induce apoptosis (Wang et al., 1999). Concurring with this, we observed that Grim cannot induce cell death efficiently in transgenic flies without the N-terminal domain. However, we also found that deletion of the internal GH3 domain in a Grim protein still retaining the N-terminal domain also eliminates most of the Grim proapoptotic activity, without affecting IAP binding ability. This suggests that complete elimination of IAPs by mutation is not equivalent to repression by Grim. Several situations may account for this possibility, for example, the cellular contents of IAPs, which may vary among different cell types or states, might exceed the levels Grim can counteract. Alternatively, IAPs may be confined in cellular compartments not accessible to Grim or, depending on the cellular context, they may be protected from Grim action by additional factors.
One possible explanation to our results would be that Grim requires the GH3 domain in order to function correctly in the IAP inhibitory pathway. Since the GH3 domain is not directly involved in binding IAPs, its hypothetical role in this pathway can only be postulated as an indirect role in allowing N-terminal domain action and IAP inhibition. In contradiction, however, deletion of the N-terminus does not eliminate Grim activity in cultured cells, but the GH3 domain is always required in this context (Clavería et al., 1998; McCarthy and Dixit, 1998; Vucic et al., 1998), suggesting a GH3 activity independent of the IAP inhibitory pathway. This conclusion is validated by the fact that apoptosis is induced by overexpression of the GH3 domain alone in SL2 cells, by the inability of IAP overexpression to block Grim activity in several in vitro and in vivo experimental systems (Clavería et al., 1998; Vucic et al., 1998; Wing et al., 1998), and by the observation that the GH3 domain is dispensable for Grim binding to IAPs. The transcomplementation observed between two independently-expressed Grim proteins, each deficient for one of the domains, is also best explained by an independent mode of action for each domain. Significant impairment of Grim function in vivo nonetheless requires elimination of both N-terminal and GH3 domains, suggesting that the two pathways must act in synergy to efficiently trigger apoptosis during endogenous PCD processes.
Several lines of evidence point to the mitochondrial– cytochrome c pathway as the target of GH3 action. We have shown here that Grim associates with mitochondria in colocalization with cytochrome c and that this activity resides in the GH3 domain. In vertebrate cells, Grim targets the mitochondria (Clavería et al., 1998) and induces cytochrome c release in a GH3-dependent and N-terminal-, caspase- and IAP-independent manner (C.Clavería and M.Torres, in preparation), suggesting functional conservation of the pathway. Interestingly, during apoptosis induction by Grim and Reaper in flies, changes in cytochrome c display were observed, rather than its free release to the cytosol as in vertebrates (Varkey et al., 1999). We report here that Grim-expressing cells specifically show large cytoplasmic deposits of cytochrome c where Grim itself is present, but other mitochondrial markers are not. The changes observed in the distribution of cytochrome c may result from its relocation from mitochondria to hypothetical specialized cytoplasmic structures involved in apoptosis induction. Alternatively, the apoptosome might be formed in the vicinity of the mitochondria (Dorstyn et al., 2002), and cytochrome c deposits may constitute the remnants of damaged mitochondria, which have lost some of their constitutive components, but retain cytochrome c and Grim. The relevance of the cytochrome c proapoptotic pathway in Drosophila PCD is supported as well by the observation that elimination of Dark, a Drosophila homologue of Apaf-1 that mediates cytochrome c-primed apoptosis, impairs Reaper, Hid and Grim killing in flies (Rodriguez et al., 1999). Even though no cytochrome c free release appears to take place in Drosophila cells, it is possible that a mechanism homologous to that of vertebrate cells is activated, but from different subcellular compartments.
The involvement of the GH3 domain in a mitochondrial pathway and its predicted structure, an amphipathic α-helix, resemble the characteristics of the widespread proapoptotic BH3 domain (Muchmore et al., 1996; Chou et al., 1999; McDonnell et al., 1999). These similarities could be interpreted as functional homology between the two pathways; however, we have not been able to detect association between Grim and either mammalian (Bcl-2 and Bcl-xL) or insect (Debcl) Bcl-2-family members, as would be expected for a BH3-containing protein (C.Clavería and M.Torres, unpublished data). Rather than representing homologous proapoptotic pathways, BH3 and GH3 domains may have converged during evolution to a similar proapoptotic mechanism. Since BH3-containing proteins coexist in Drosophila (Brachmann et al., 2000; Colussi et al., 2000; Igaki et al., 2000; Zhang et al., 2000) with GH3-containing proteins, the two pathways may operate in alternative routes, or even cooperate in apoptosis induction, not only in Drosophila, but perhaps also in other species.
Two independent pathways may thus be triggered by Grim; an IAP inhibitory pathway activated by the N-terminal domain, and a mitochondrial–cytochrome c route activated by the GH3 domain. Either pathway could be alternatively or simultaneously promoted by Grim, and the relevance of each may depend on cellular context. The presence of a GH3 homology region in Reaper and Sickle suggests functional conservation of this domain in at least these other two Drosophila proapoptotic proteins (Wing et al., 2001, 2002; Christich et al., 2002; Srinivasula et al., 2002). In this context, it is important to consider that Reaper promotes cytochrome c release in a cell-free Xenopus egg extract and does not require the N-terminal domain for this function (Thress et al., 1999).
Although Reaper, Hid, Sickle and Grim induce specific apoptotic pathways in vertebrate cells, and in the fly participate in highly conserved routes, such as the p53 and Ras–MAPK pathways (Bergmann et al., 1998; Kurada and White, 1998; Brodsky et al., 2000), no homologue for these proteins has been yet identified in any other organism. The vertebrate Smac/Diablo protein may, however, represent a functional homologue of the IAP inhibitory pathway. Smac/Diablo can bind to and block the protective effect of IAPs (Du et al., 2000; Verhagen et al., 2000). However, it is unlikely that Smac/Diablo represent homologues of the mitochondrial–cytochrome c pathway. Database searches have failed to identify any protein with sequence similarity to the GH3 domain but, given the restricted sequence conservation among, for example, BH3 family members, this does not exclude conservation of this pathway. Whether vertebrate proapoptotic proteins exist that represent direct or functional homologues of the GH3 proapoptotic activity thus remains to be determined.
Materials and methods
Secondary structure prediction
The PHD programs (Rost, 1996) for secondary structure prediction were used to determine the regions with a high probability of having an α-helical conformation. The Predict Protein www server at Columbia University (http://cubic.bioc.columbia.edu/predictprotein/) was used to run the program. GH3 was composed of 15 amino acids, 11 of which showed the highest prediction reliability, two more exhibited a very high index and only the two C-terminal amino acids scored a moderate reliability index.
Expression vectors and site-directed mutagenesis
GrimΔ86–98, grimΔ89–92, grimL89A and grimL89E mutant forms were obtained with the QuickChange site-directed mutagenesis kit (Stratagene) using pcDNA3.1–grim as a template and amplifying it with the following primers: grimΔ86–98: 5′-CGATGACCATGTCGGAGTTTGGATGCATCTACAGCTACAGTTCGAGCC-3′ and 5′-GGCTCGAACTGTAG CTGTAGATGCATCCAAACTCCGACATGGTCATCG-3′; grimΔ89–92: 5′-GGATGCTGGGATCTTTTCTGCTACGCTC-3′ and 5′-GAGCGT AGCAGAAAAGATCCCAGCATCC-3′; grimL89A: 5′-GATGCTGG GATCTTGCGGCCCAGATCTTG-3′ and 5′-CAAGATCTGGGCCG CAAGATCCCAGCATC-3′; grimL89E: 5′-GATGCTGGGATCTTG AGGCCCAGATCTTG-3′ and 5′-CAAGATCTGGGCCTCAAGATC CCAGCATC-3′; the sequence was confirmed. GrimΔ2–14, Δ86–98 and grimΔ2–14, Δ89–92 compound mutants were obtained by replacement of the 228 N-terminal base pairs of grimΔ86–98 and grimΔ89–92 mutants with the corresponding sequence of grimΔ2–14 mutant (Clavería et al., 1998). WT grim, grimΔ2–14 and the different GH3 mutant form coding sequences were cloned into the pMT/V5-HisC Drosophila expression vector (Invitrogen), under the control of the inducible metallothionein (MT) promoter. WT grim and the mutant form coding sequences were also cloned in the pUAST vector (Brand and Perrimon, 1993) and in the pcDNA3.1 mammalian expression vector (Invitrogen).
The grim stop codon was replaced by an XhoI site with the QuickChange site-directed mutagenesis kit using pcDNA3.1-grim as a template and amplifying it with the primers: 5′-CCTCCAA GGAGAACCTCGAGTCCGAGCTCGGTACC-3′ and 5′-GGTACCG AGCTCGGACTCGAGGTTCTCCTTGGAGG-3′. This mutant form was fused to the N-terminus of EGFP (Grim–GFP), a variant of wild-type GFP, by cloning it in the pEGFP-N1 vector (Clontech). Fragments encoding grim amino acids 78–109, containing the grim GH3 or grim GH3L89E domains, flanked by EcoRI and BamHI sites, were obtained by PCR using pcDNA3.1–grim or pcDNA3.1–grimL89E as templates and amplifying them with the primers: 5′-GTGAATTCGCCACCA TGACCATGTCGGAGTTTGGATG-3′ and 5′-CTGGATCCGGTTG ACGCTGGCTCGAACT-3′. These fragments were fused to the N-terminus of EGFP (GH3–GFP and GH3LE–GFP). A fragment encoding the first 34 grim amino acids was fused to the N-terminus of EGFP (Nt-Grim–GFP). GFP and the sequence-confirmed GFP fusion proteins were subcloned into the pMT/V5-HisC Drosophila expression vector.
The baculovirus p35-coding sequence was cloned in the pMT/V5-HisC Drosophila expression vector. The Drosophila DIAP2-coding sequence was excised from pHSP70PLVI+Epi-D-iap2 (Vucic et al., 1997) and N-terminally fused to a Flag tag by cloning it in the pCMV–Tag2A mammalian expression vector (Stratagene).
Cell culture and death assays
Drosophila Schneider L2 (SL2) cells were maintained in Drosophila expression system (DES) expression medium (Invitrogen) supplemented with 10% fetal bovine serum and transfected with Cellfectin reagent (Gibco-BRL), as recommended by the manufacturer. For cell death assays, cells were cotransfected with pMT/V5-HisC empty vector (control) or pMT/V5-HisC–grim (WT or mutants) inducible expression vector together with pAc5.1/V5-His/lacZ (Invitrogen) constitutive expression vector at a 4:1 molar ratio. After 24 h, cells were divided into two pools, one of which was treated with 0.3 mM CuSO4 for 30 h. Cells were then fixed in 0.2% glutaraldehyde, washed in phosphate-buffered saline (PBS) and X-Gal-stained following standard protocols.
For GFP fusion protein cell death assays, Drosophila SL2 cells were cotransfected with pMT/V5-HisC–GFP (GFP or GFP fusion proteins), with pMT/V5-HisC empty vector or pMT/V5-HisC–p35, and with pAc5.1/V5-His/lacZ at 3:3:1 molar ratio. Cell death assays were performed as above.
NIH 3T3 fibroblasts were cultured in Dulbecco’s modified Eagle’s medium (Bio-Whittaker) supplemented with 10% newborn calf serum and transfected using Lipofectamine plus reagent (Gibco-BRL), as recommended by the manufacturer.
Western blotting and immunoprecipitation
To test the stability of the Grim protein and its mutant forms, NIH 3T3 fibroblasts were transfected with pcDNA3.1 empty vector or pcDNA3.1–grim (WT or mutants) expression vector. Cells were treated with 50 µM zVAD.fmk (Bachem) and, after 20 h, they were lysed in 0.2% NP-40 isotonic lysis buffer with freshly added protease inhibitors, then proteins were eluted and analysed by SDS–PAGE. Western blotting analyses were performed using rabbit anti-grim IgG (Clavería et al., 1998) and HPR-conjugated goat anti-rabbit IgG (Dako) and developed by enhanced chemiluminescence (ECL; Amersham Pharmacia Biotech).
For immunoprecipitations, NIH 3T3 fibroblasts were transfected with pcDNA3.1 empty vector or pcDNA3.1–grim (WT or mutants) expression vector together with pCMV–Tag2A–DIAP2 expression vector at a 1:1 molar ratio. Cells were treated with 50 µM zVAD.fmk (Bachem) and, after 20 h, they were lysed in 0.2% NP-40 isotonic lysis buffer with freshly added protease inhibitors. Equal quantities of cell lysates were incubated with an affinity-purified fraction of rabbit anti-Grim IgG (Clavería et al., 1998) (overnight, 4°C), mixed with 25 µl of a 1:1 slurry of gammabind G–Sepharose (Amersham Pharmacia Biotech) and incubated (1 h, 4°C). The Sepharose beads were washed twice in 0.2% NP-40 lysis buffer and twice in 50 mM Tris–HCl pH 7.5 washing buffer before proteins were eluted and analysed in SDS–PAGE. Western blotting analyses were performed using anti-Flag M2 antibody (Stratagene) and horseradish peroxidase (HPR)-conjugated goat anti-mouse IgG (Dako) or rabbit anti-grim IgG (Clavería et al., 1998) and HPR-conjugated goat anti-rabbit IgG (Dako) and developed by ECL (Amersham Pharmacia Biotech).
Immunofluorescence
Drosophila SL2 cells were transfected with pMT/V5-HisC–grim (WT or mutants) inducible expression vectors. After 24 h, cells were plated on poly-dl-lysine (Sigma)-coated coverslips and treated with 0.3 mM CuSO4 for 4 h. Cells were then fixed in 4% paraformaldehyde/PBS (15 min, room temperature) and washed twice in PBS. Cells were permeabilized in 0.2% Triton X-100/PBS for 15 min, washed in PBS, preincubated in 10% goat serum/PBS for 1 h and successively incubated with the primary and secondary antibodies for 1 h each, with three 5 min washes in 0.1% Tween-20/PBS between incubations.
Drosophila SL2 cells were transfected with pMT/V5-HisC–GFP (GFP or GFP fusion proteins) or cotransfected with pMT/V5-HisC–grim- and pMT/V5-HisC–GH3–GFP-inducible expression vectors. After 24 h, cells were plated on poly-dl-lysine-coated coverslips and treated with 0.3 mM CuSO4 for 7 h. Immunofluorescence was performed as above. Optical sections were obtained using an Ar-Kr laser and TCS-NT Leica confocal imaging systems.
Grim was detected with rabbit anti-grim IgG (Clavería et al., 1998), mitochondria with human anti-mitochondrial serum (Clavería et al., 1998), and cytochrome c with mouse anti-cytochrome c monoclonal antibody (4 µg/ml, clone 6H2.B4; Pharmingen). Secondary antibodies used were goat anti-rabbit IgG-Alexa 488, goat anti-rabbit IgG-Cy3, biotinylated goat anti-mouse IgG, streptavidin-Cy3 and goat anti-human IgG-Cy3 (all from Jackson ImmunoResearch).
Drosophila stocks
The strains used in this study, w1118, MS1096-Gal4, GMR-Gal4, w, If/CyO; TM2 Ubx/MKRS, y; SM6aTM6b/+ and w; TM6b/MKRS are described in FlyBase (http://fly.ebi.ac.uk:7081/).
In vivo overexpression experiments
Transgenic UAS lines of flies were generated using pUAST plasmids containing grim or its mutant forms to transform w1118 embryos (Ashburner, 1989). For phenotypic analysis associated to the overexpression of WT or mutated Grim forms, males of each UAS line (denoted hereafter as UAS-G*) were crossed individually with females of the balancer stock w, If/CyO; TM2 Ubx/MKRS (cross 1), which carries dominant mutations allowing unambiguous identification of chromosomes 2 and 3. For analysis of UAS-G* lines inserted at chromosome 2, progeny males of the genotype w, UAS-G*/Cyo were crossed with GMR-Gal4 females, whereas w, UAS-G*/If males were crossed with MS1096-Gal4 females. Larvae were raised at 18 or 25°C. Adult phenotypes were scored in experimental males (GMR-Gal4; UAS-G*/+ and MS1096-Gal4; UAS-G*/+) or females (GMR-Gal4/+; UAS-G*/+ and MS1096-Gal4/+; UAS-G*/+). Control flies are distinguished as they carry the CyO or If markers. For UAS-G* lines inserted at chromosome 3, progeny males from cross 1 of the w, UAS-G*/MKRS genotype were crossed with GMR:Gal4 females, and w, UAS-G*/TM2 Ubx males with MS1096–Gal4 females. In this case, experimental females are of the GMR-Gal4/+; UAS-G*/+ and MS1096-Gal4/+; UAS-G*/+ genotype; experimental males are GMR-Gal4; UAS-G*/+ and MS1096-Gal4; UAS-G*/+, and control flies carried the MKRS or Ubx markers. Viability of experimental females or males is expressed as a percentage relative to the number of sibling control flies.
Acknowledgments
Acknowledgements
We thank Juan Modolell and Cathy Mark for helpful comments on the manuscript, and Giovanna Giovinazzo and Marisol Obrero for their help during this work. We thank the L.Miller laboratory for the DIAP2 plasmid. M.T. and C.C. are supported by grant CICYT-SAF2000-0160 from the Spanish Ministry of Science and Technology and grant CAM-08.6/0032.1/2000 from the Comunidad Autónoma de Madrid (CAM). C.C. is supported by a predoctoral fellowship from the Fundación Ramón Areces. A fellowship to E.C. from the CAM is acknowledged. E.C. and S.C. are supported by grants from the CAM (08.5/0030/2000) and Dirección General de Investigación Científica y Técnica (PB98-0682) to J.Modolell and an institutional grant from Fundación Ramón Areces to the Centro de Biología Molecular Severo Ochoa. The Department of Immunology and Oncology was founded and is supported by the Spanish Council for Scientific Research and by the Pharmacia Corporation.
References
- Abrams J.M. (1999) An emerging blueprint for apoptosis in Drosophila. Trends Cell Biol., 9, 435–440. [DOI] [PubMed] [Google Scholar]
- Aravind L., Dixit,V.M. and Koonin,E.V. (1999) The domains of death: evolution of the apoptosis machinery. Trends Biochem. Sci., 24, 47–53. [DOI] [PubMed] [Google Scholar]
- Ashburner M. (1989) Drosophila: A Laboratory Handbook. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 1017–1063.
- Bergmann A., Agapite,J., McCall,K. and Steller,H. (1998) The Drosophila gene hid is a direct molecular target of Ras-dependent survival signaling. Cell, 95, 331–341. [DOI] [PubMed] [Google Scholar]
- Brachmann C.B., Jassim,O.W., Wachsmuth,B.D. and Cagan,R.L. (2000) The Drosophila bcl-2 family member dBorg-1 functions in the apoptotic response to UV-irradiation. Curr. Biol., 10, 547–550. [DOI] [PubMed] [Google Scholar]
- Brand A.H. and Perrimon,N. (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development, 118, 401–415. [DOI] [PubMed] [Google Scholar]
- Brodsky M.H., Nordstrom,W., Tsang,G., Kwan,E., Rubin,G.M. and Abrams,J.M. (2000) Drosophila p53 binds a damage response element at the reaper locus. Cell, 101, 103–113. [DOI] [PubMed] [Google Scholar]
- Chen P., Lee,P., Otto,L. and Abrams,J. (1996a) Apoptotic activity of REAPER is distinct from signaling by the tumor necrosis factor receptor 1 death domain. J. Biol. Chem., 271, 25735–25737. [DOI] [PubMed] [Google Scholar]
- Chen P., Nordstrom,W., Gish,B. and Abrams,J.M. (1996b) grim, a novel cell death gene in Drosophila. Genes Dev., 10, 1773–1782. [DOI] [PubMed] [Google Scholar]
- Chou J.J., Li,H., Salvesen,G.S., Yuan,J. and Wagner,G. (1999) Solution structure of BID, an intracellular amplifier of apoptotic signaling. Cell, 96, 615–624. [DOI] [PubMed] [Google Scholar]
- Christich A., Kauppila,S., Chen,P., Sogame,N., Ho,S.I. and Abrams,J.M. (2002) The damage-responsive Drosophila gene sickle encodes a novel IAP binding protein similar to but distinct from reaper, grim and hid. Curr. Biol., 12, 137–140. [DOI] [PubMed] [Google Scholar]
- Clavería C., Albar,J.P., Serrano,A., Buesa,J.M., Barbero,J.L., Martínez-A, C. and Torres,M. (1998) Drosophila grim induces apoptosis in mammalian cells. EMBO J., 17, 7199–7208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colussi P.A., Quinn,L.M., Huang,D.C., Coombe,M., Read,S.H., Richardson,H. and Kumar,S. (2000) Debcl, a proapoptotic Bcl-2 homologue, is a component of the Drosophila melanogaster cell death machinery. J. Cell Biol., 148, 703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorstyn L., Read,S., Cakouros,D., Huh,J.R., Hay,B.A. and Kumar,S. (2002) The role of cytochrome c in caspase activation in Drosophila melanogaster cells. J. Cell Biol., 156, 1089–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C., Fang,M., Li,Y., Li,L. and Wang,X. (2000) Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell, 102, 33–42. [DOI] [PubMed] [Google Scholar]
- Evans E.K., Kuwana,T., Strum,S.L., Smith,J.J., Newmeyer,D.D. and Kornbluth,S. (1997) Reaper-induced apoptosis in a vertebrate system. EMBO J., 16, 7372–7381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal L., McCall,K., Agapite,J., Hartwieg,E. and Steller,H. (2000) Induction of apoptosis by Drosophila reaper, hid and grim through inhibition of IAP function. EMBO J., 19, 589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green D.R. (2000) Apoptotic pathways: paper wraps stone blunts scissors. Cell, 102, 1–4. [DOI] [PubMed] [Google Scholar]
- Grether M.E., Abrams,J.M., Agapite,J., White,K. and Steller,H. (1995) The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev., 9, 1694–1708. [DOI] [PubMed] [Google Scholar]
- Gross A., McDonnell,J.M. and Korsmeyer,S.J. (1999) BCL-2 family members and the mitochondria in apoptosis. Genes Dev., 13, 1899–1911. [DOI] [PubMed] [Google Scholar]
- Haining W.N., Carboy-Newcomb,C., Wei,C.L. and Steller,H. (1999) The proapoptotic function of Drosophila Hid is conserved in mammalian cells. Proc. Natl Acad. Sci. USA, 96, 4936–4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaki T., Kanuka,H., Inohara,N., Sawamoto,K., Nunez,G., Okano,H. and Miura,M. (2000) Drob-1, a Drosophila member of the Bcl-2/CED-9 family that promotes cell death. Proc. Natl Acad. Sci. USA, 97, 662–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson M.D., Weil,M. and Raff,M.C. (1997) Programmed cell death in animal development. Cell, 88, 347–354. [DOI] [PubMed] [Google Scholar]
- Kelekar A. and Thompson,C.B. (1998) Bcl-2-family proteins: the role of the BH3 domain in apoptosis. Trends Cell Biol., 8, 324–330. [DOI] [PubMed] [Google Scholar]
- Kroemer G. and Reed,J.C. (2000) Mitochondrial control of cell death. Nat. Med., 6, 513–519. [DOI] [PubMed] [Google Scholar]
- Kurada P. and White,K. (1998) Ras promotes cell survival in Drosophila by downregulating hid expression. Cell, 95, 319–329. [DOI] [PubMed] [Google Scholar]
- Lisi S., Mazzon,I. and White,K. (2000) Diverse domains of THREAD/DIAP1 are required to inhibit apoptosis induced by REAPER and HID in Drosophila. Genetics, 154, 669–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall K. and Steller,H. (1997) Facing death in the fly: genetic analysis of apoptosis in Drosophila. Trends Genet., 13, 222–226. [DOI] [PubMed] [Google Scholar]
- McCarthy J.V. and Dixit,V.M. (1998) Apoptosis induced by Drosophila reaper and grim in a human system. Attenuation by inhibitor of apoptosis proteins (cIAPs). J. Biol. Chem., 273, 24009–24015. [DOI] [PubMed] [Google Scholar]
- McDonnell J.M., Fushman,D., Milliman,C.L., Korsmeyer,S.J. and Cowburn,D. (1999) Solution structure of the proapoptotic molecule BID: a structural basis for apoptotic agonists and antagonists. Cell, 96, 625–634. [DOI] [PubMed] [Google Scholar]
- Metzstein M.M., Stanfield,G.M. and Horvitz,H.R. (1998) Genetics of programmed cell death in C. elegans: past, present and future. Trends Genet., 14, 410–416. [DOI] [PubMed] [Google Scholar]
- Muchmore S.W. et al. (1996) X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature, 381, 335–341. [DOI] [PubMed] [Google Scholar]
- Nordstrom W., Chen,P., Steller,H. and Abrams,J.M. (1996) Activation of the reaper gene during ectopic cell killing in Drosophila. Dev. Biol., 180, 213–226. [DOI] [PubMed] [Google Scholar]
- Rodriguez A., Oliver,H., Zou,H., Chen,P., Wang,X. and Abrams,J.M. (1999) Dark is a Drosophila homologue of Apaf-1/CED-4 and functions in an evolutionarily conserved death pathway. Nat. Cell Biol., 1, 272–279. [DOI] [PubMed] [Google Scholar]
- Rost B. (1996) PHD: predicting one-dimensional protein structure by profile based neural networks. Methods Enzymol., 266, 525–539. [DOI] [PubMed] [Google Scholar]
- Srinivasula S.M. et al. (2002) sickle, a novel Drosophila death gene in the reaper/hid/grim region, encodes an IAP-inhibitory protein. Curr. Biol., 12, 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson C.B. (1995) Apoptosis in the pathogenesis and treatment of disease. Science, 267, 1456–1462. [DOI] [PubMed] [Google Scholar]
- Thress K., Evans,E.K. and Kornbluth,S. (1999) Reaper-induced dissociation of a Scythe-sequestered cytochrome c-releasing activity. EMBO J., 18, 5486–5493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varkey J., Chen,P., Jemmerson,R. and Abrams,J.M. (1999) Altered cytochrome c display precedes apoptotic cell death in Drosophila. J. Cell Biol., 144, 701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaux D.L. and Korsmeyer,S.J. (1999) Cell death in development. Cell, 96, 245–254. [DOI] [PubMed] [Google Scholar]
- Verhagen A.M., Ekert,P.G., Pakusch,M., Silke,J., Connolly,L.M., Reid,G.E., Moritz,R.L., Simpson,R.J. and Vaux,D.L. (2000) Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell, 102, 43–53. [DOI] [PubMed] [Google Scholar]
- Vucic D., Kaiser,W.J., Harvey,A.J. and Miller,L.K. (1997) Inhibition of reaper-induced apoptosis by interaction with inhibitor of apoptosis proteins (IAPs). Proc. Natl Acad. Sci. USA, 94, 10183–10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucic D., Kaiser,W.J. and Miller,L.K. (1998) Inhibitor of apoptosis proteins physically interact with and block apoptosis induced by Drosophila proteins HID and GRIM. Mol. Cell. Biol., 18, 3300–3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.L., Hawkins,C.J., Yoo,S.J., Muller,H.A. and Hay,B.A. (1999) The Drosophila caspase inhibitor DIAP1 is essential for cell survival and is negatively regulated by HID. Cell, 98, 453–463. [DOI] [PubMed] [Google Scholar]
- White K., Grether,M.E., Abrams,J.M., Young,L., Farrell,K. and Steller,H. (1994) Genetic control of programmed cell death in Drosophila. Science, 264, 677–683. [DOI] [PubMed] [Google Scholar]
- White K., Tahaoglu,E. and Steller,H. (1996) Cell killing by the Drosophila gene reaper. Science, 271, 805–807. [DOI] [PubMed] [Google Scholar]
- Wing J.P., Zhou,L., Schwartz,L.M. and Nambu,J.R. (1998) Distinct cell killing properties of the Drosophila reaper, head involution defective and grim genes. Cell Death Differ., 5, 930–939. [DOI] [PubMed] [Google Scholar]
- Wing J.P., Schwartz,L.M. and Nambu,J.R. (2001) The RHG motifs of Drosophila Reaper and Grim are important for their distinct cell death-inducing abilities. Mech. Dev., 102, 193–203. [DOI] [PubMed] [Google Scholar]
- Wing J.P., Karres,J.S., Ogdahl,J.L., Zhou,L., Schwartz,L.M. and Nambu,J.R. (2002) Drosophila sickle is a novel grim-reaper cell death activator. Curr. Biol., 12, 131–135. [DOI] [PubMed] [Google Scholar]
- Wyllie A.H., Kerr,J.F. and Currie,A.R. (1980) Cell death: the significance of apoptosis. Int. Rev. Cytol., 68, 251–306. [DOI] [PubMed] [Google Scholar]
- Zhang H., Huang,Q., Ke,N., Matsuyama,S., Hammock,B., Godzik,A. and Reed,J.C. (2000) Drosophila pro-apoptotic Bcl-2/Bax homologue reveals evolutionary conservation of cell death mechanisms. J. Biol. Chem., 275, 27303–27306. [DOI] [PubMed] [Google Scholar]
- Zong W.X., Lindsten,T., Ross,A.J., MacGregor,G.R. and Thompson,C.B. (2001) BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev., 15, 1481–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]