

Abstract
The cyclin-dependent kinase (CDK) inhibitors p21Cip1 and p27Kip1 are induced in response to anti-proliferative stimuli and block G1/S-phase progression through the inhibition of CDK2. Although the cyclin E–CDK2 pathway is often deregulated in tumors the relative contribution of p21Cip1 and p27Kip1 to tumorigenesis is still unclear. The MYC transcription factor is an important regulator of the G1/S transition and its expression is frequently altered in tumors. Previous reports suggested that p27Kip1 is a crucial G1 target of MYC. Our study shows that in mice, deficiency for p27Kip1 but not p21Cip1 results in decreased survival to retrovirally-induced lymphomagenesis. Importantly, in such p27Kip1 deficient lymphomas an increased frequency of Myc activation is observed. p27Kip1 deficiency was also shown to collaborate with MYC overexpression in transgenic lymphoma models. Thus, in vivo, the capacity of MYC to promote tumor growth is fully retained and even enhanced upon p27Kip1 loss. We show that in lymphocytes, MYC overexpression and p27Kip1 deficiency independently stimulate CDK2 activity and augment the fraction of cells in S phase, in support of their distinct roles in tumorigenesis.
Keywords: CDK2/lymphomagenesis/MYC/p21Cip1/p27Kip1
Introduction
Progression through the cell cycle is controlled by the sequential activation of a series of cyclin-dependent kinases (CDKs) through cyclin binding and defined phosphorylation (Morgan, 1995). CDK activity is impaired by a group of proteins termed CDK inhibitors (CKIs). CKI members have been assigned to two different families, which differ in structure and CDK specificity. The INK4 family members (p16INK4a, p15INK4b, p18INK4c and p19INK4d) specifically bind to the monomeric forms of CDK4 and CDK6 and inhibit cyclin D–CDK-associated kinase activity. A second group of proteins known as the Cip/Kip family is able to bind to both cyclin and CDK subunits and has its specificity extended to cyclin E–CDK2 and cyclin A–CDK2 complexes. The Cip/Kip family includes p21Cip1 (p21), p27Kip1 (p27) and p57Kip2 (Sherr and Roberts, 1999). Despite their analogous function, p21 and p27 are regulated differently and respond, at least in vitro, to very distinct stimuli such as p53-dependent DNA damage response or contact inhibition, respectively (Dulic et al., 1994; Polyak et al., 1994). It has been reported that p21 and p27 can act as positive regulators of cyclin D-dependent kinases by promoting the assembly of the cyclin/CDK complex (Cheng et al., 1998, 1999). This positive regulatory activity contrasts with what is thought to be the main function of p21 and p27: blocking S-phase entry by binding and hence inhibiting CDK2 active complexes (Dulic et al., 1994; Polyak et al., 1994; Pavletich, 1999).
Genetic alterations in regulators of G1-phase progression are frequently found in human cancers (reviewed in Sherr, 2000). Thus, the ability of p21 and p27 to induce G1 arrest suggests that they may also play a crucial role in transformation. In accordance with this notion low levels of p27 have been shown to correlate with poor prognosis in a range of human tumors, including lymphomas and leukemias (Chiarle et al., 2000; Yokozawa et al., 2000). Interestingly, no such correlation has been reported for p21. In mice, both p21 and p27 are ubiquitously expressed (Macleod et al., 1995; Nakayama et al., 1996). While p21–/– mice show no gross abnormalities (Deng et al., 1995), targeted disruption of p27 results in increased body size and enhanced proliferation (Fero et al., 1996; Kiyokawa et al., 1996; Nakayama et al., 1996). Yet, besides a high incidence of pituitary tumors after long latency periods, no predisposition to other tumors could be detected in p27–/– mice. More recently it was shown that deficiency for p27 increases susceptibility to tumorigenesis upon treatment with DNA-damaging agents (Fero et al., 1998) or when combined with the inactivation of a potent tumor suppressor, such as Pten (Di Cristofano et al., 2001). In addition, pronounced haplo-insufficiency of p27 for lung, pituitary and intestine tumor suppression has been reported (Fero et al., 1998). Surprisingly, and in contrast to the situation in humans, no indication for p27 tumor suppressor activity in murine lymphomagenesis has yet been reported, despite the disproportional enlargement of thymus and spleen in p27–/– mice.
The Myc proto-oncogene belongs to a family of transcription factors, which includes four other members (Nmyc1, Mycs, Bmyc and MYCL). The relevance of Myc is underscored by its involvement in key cellular processes such as proliferation, growth, differentiation and apoptosis. Expression of MYC is frequently deregulated in human tumors as a result of gene amplification, translocation, overexpression or mutation. Accordingly, transgenic studies in mice have demonstrated that MYC overexpression in skin or hematopoietic tissues predisposes to tumorigenesis (Dang, 1999; Eisenman, 2001; Nasi et al., 2001). The identification of MYC targets is a critical step for the understanding of its oncogenic properties and it has been the subject of intense study. Recently it was reported that p27 is an important target of MYC in the induction of proliferation. MYC is thought to overcome p27 inhibition by several mechanisms, such as direct induction of Cyclin D and, consequently, sequestration of the inhibitor (Bouchard et al., 1999; Perez-Roger et al., 1999), activation of the p27 degradation pathway (Muller et al., 1997; O’Hagan et al., 2000) and direct repression of the p27 promoter (Yang et al., 2001).
In the present study, we assessed the role of CKIs in lymphomagenesis and the relevance of p27 regulation by MYC in murine tumor development. Our analyses show that unlike p21, p27 is a strong tumor suppressor in retrovirally induced lymphomagenesis. Importantly, our data demonstrates that MYC overexpression and p27 deficiency strongly synergize in distinct lymphoma models. Thus, we provide convincing in vivo evidence for non-overlapping roles of MYC overexpression and loss of p27 in tumorigenesis. Furthermore, our data suggest that one of the components of the collaboration between the two is increased CDK2 activity with concomitant enhanced proliferation.
Results
Absence of p27 results in acceleration of retroviral-induced lymphomas
The putative tumor suppressor activity of p27 and p21 in lymphomagenesis was assessed through the analysis of tumor development in p21 and p27 knockout (KO) mice. Since both p21 and p27 are expressed in lymphoid tissues, although at different levels (data not shown), the relevance of these proteins for malignant transformation was estimated in single and double KO mice. Lymphomas were induced through infection of newborn mice with Moloney murine leukemia virus (MuLV), a slow transforming retrovirus, and the animals were sacrificed when moribund. Wild-type (WT) animals died from lymphomas from 60 days after infection onwards, and showed an average survival time of 122 days (Figure 1A). Whereas p21–/– and WT mice showed no significant difference in tumor survival upon MuLV infection, absence of p27 resulted in a clear acceleration in tumorigenesis (p < 0.00005). By 115 days after infection, all p27 deficient mice had died and their mean tumor survival was 74 days. Interestingly, compound p21–/–; p27–/– mutants behaved similarly to p27–/– mice, demonstrating that loss of p21 does not contribute to MuLV-induced tumor development either in p27–/– or WT backgrounds.

Fig. 1. Decreased survival of p27–/– mice with MuLV-induced lymphomagenesis. The percentage of surviving animals is plotted against the days after infection. (A) Incidence of MuLV-induced tumors in WT and CKI KO mice. (B) Tumor survival of p27+/– mice in the presence or absence of p21.
Haplo-insufficiency for p27 in tumor suppression has been previously described in ENU- and γ-radiation-induced tumor models (Fero et al., 1998). Thus, tumor survival of p27 heterozygous mice, in the different p21 genetic contexts, was also examined (Figure 1B; data not shown). As shown in Figure 1B, MuLV-induced tumors developed slightly faster in p27 heterozygous mice compared with WT, but this acceleration only became significant when p27 heterozygosity was combined with p21 deficiency (p = 0.0055). p21–/–; p27+/– mice were significantly less predisposed than p21–/–; p27–/– animals (p < 0.00005) to MuLV-induced lymphomagenesis and therefore, showed intermediate survival compared with WT (or p21–/–) and p27–/– animals. No loss of the p27 wild-type allele could be detected in p21–/–; p27+/– tumors (data not shown). In addition, all the p21–/–; p27+/– lymphomas analyzed (n = 20) expressed p27 protein, albeit at varying levels (data not shown). Thus, p27 but not p21 shows tumor suppressor activity in MuLV-induced lymphomas. However, when p27 levels are reduced, p21 loss accelerates tumorigenesis, indicating that in lymphoid cells p27 haplo-insufficiency for tumor suppression is conditioned by p21 status.
Increased frequency of Myc activation in p27–/– MuLV-induced lymphomas
Slow transforming retroviruses are capable of activating cellular proto-oncogenes by insertional mutagenesis, and the analysis of proviral integration sites has enabled the identification of a range of genes contributing to lymphoma development, such as Myc, Pim1 and Gfi1 (Jonkers and Berns, 1996). The acceleration in MuLV-induced lymphomagenesis observed in p27–/– mice suggests that regulation of p27 may be a key factor in tumor onset/development. To determine whether the known MuLV-induced tumor pathways were activated in the absence of p27, Southern blotting analysis of tumor DNA was performed using specific probes for Myc (Myc and NMyc1), Pim1, Gfi1 and Cyclin D2. Table I summarizes the frequency of activation of these oncogenes for each genotype. In the absence of p27 the frequency of activation of the Myc genes was increased at least 2-fold compared with WT. In contrast, proviral insertions affecting Pim1, a strong collaborator of MYC in MuLV-induced lymphomagenesis (van Lohuizen et al., 1989b, 1991; Verbeek et al., 1991), were less frequent in p27–/– than in WT tumors. Northern blotting analysis of tumor RNA showed low levels of Pim1 expression in p27–/– lymphomas (data not shown), arguing for a diminished requirement for the activation of the kinase in this background. GFI1 is a frequent collaborator of MYC and PIM in lymphomagenesis (Scheijen et al., 1997) and its activation was found at similar rates in all the genetic backgrounds analyzed. Cyclin D2 activation (Tremblay et al., 1992; Hanna et al., 1993) was observed, although at low frequency, in both WT and p21–/– tumors but was absent in p27–/– lymphomas. Deficiency for p21 did not affect the frequency of activation of the commonly tagged oncogenes in either the p27–/– or WT background. Interestingly, the frequency of Myc activation in p21–/–; p27+/– lymphomas was intermediate to those in the WT and p27 KO groups in accordance with the observed tumor survival. This indicates that absence of p27 collaborates with Myc overexpression in MuLV-induced lymphomagenesis.
Table I. Frequency of proviral integrations near common activated genes in MuLV-induced lymphomas.
| Genotype | Oncogene activation (%) |
|||
|---|---|---|---|---|
| Myc | Pim1 | Gfi1 | Cyclin D2 | |
| WT (n = 38) | 32 | 21 | 26 | 3 |
| p21–/– (n = 41) | 32 | 29 | 26 | 5 |
| p21–/–; p27+/– (n = 58) | 53 | 17 | 29 | 4 |
| p27–/– (n = 37) | 74 | 11 | 29 | 0 |
| p21–/–; p27–/– (n = 40) | 69 | 10.5 | 32 | 0 |
Flow cytometric analysis (FACS) of B- (B220, IgM, IgD) and T-cell (TCRαβ, CD4, CD8) specific markers (Table II) demonstrated that the majority of MuLV-induced tumors were of T-cell origin. However, WT and CKI KO lymphomas differed somewhat with respect to their primary location and cellular markers. While 48% of the WT tumors analyzed were CD4+/CD8+ thymic lymphomas, <10% of the p27–/– tumors belonged to that group. Conversely, the majority of p27–/– lymphomas grew in peripheral lymphoid tissues and were TCRαβ+/CD4+, a group virtually absent in WT tumors. p21–/– lymphomas were approximately in between WT and p27–/– with respect to their location and marker profile.
Table II. Tumor profiles of MuLV lymphomas from CKI KO mice (%).
| Genotype | T cell (TCRαβ) |
B cell |
Other |
||||
|---|---|---|---|---|---|---|---|
| CD4–CD8– | CD4+CD8+ | CD4+CD8– | CD4–CD8+ | Oligoclonal | B220+ | TCRαβ–B220– | |
| WT (n = 23) | 22 | 48 | 0 | 17 | 4 | 4 | 4 |
| p21–/– (n = 30) | 17 | 33 | 30 | 7 | 13 | 0 | 0 |
| p27–/– (n = 25) | 0 | 8 | 80 | 4 | 0 | 0 | 8 |
| p21–/–; p27–/– (n = 29) | 7 | 7 | 79 | 3 | 3 | 0 | 0 |
Collaboration between Myc overexpression and p27 loss in spontaneous T-cell lymphomas
We (Jonkers et al., 1997) and others (Morello et al., 1989) have generated transgenic mice in which the Myc oncogene is driven from the Class I H2K promoter (H2K-Myc mice). In the H2K-Myc transgenic line employed here, the Myc transgene is primarily expressed in lymphoid organs, in a mosaic fashion and at rather low levels (data not shown). Nonetheless, all transgenic animals develop lymphomas after an average latency period of ∼180 days. Tumors localize mainly to thymus and spleen. The relevance of p27 for the induction/growth of such spontaneous Myc-overexpressing tumors was determined by monitoring tumor survival of mice carrying the H2K-Myc transgene on the p27–/– background. We could not detect any effects of p21 deficiency in tumor survival of H2K-Myc mice, even in the p27+/– background. However, the number of mice generated for each genotype was not sufficiently high to draw conclusions for each of the independent genotypes (data not shown). Therefore, regardless of their p21 status, survival was compared between two groups of transgenic mice with or without p27 (Figure 2A). The H2K-Myc; p27–/– group is composed of mice lacking both copies of p27, while the H2K-Myc control group consists of mice that have at least one WT allele of p27. No significant difference in survival could be detected within each group (p > 0.25). p21–/–; p27–/– mice were also monitored and only one out of 21 animals succumbed from a lymphoma during the observation period. Importantly, when p27 deficiency was combined with the H2K-Myc transgene, a significant acceleration in tumor development was observed (p < 0.00005) resulting in a decrease of ∼70 days in mean survival.

Fig. 2. p27 deficiency accelerates spontaneous lymphomagenesis in Myc-transgenic mice. The percentage of surviving animals is plotted against their age. (A) Survival to spontaneous lymphomas in H2K-Myc mice in the presence or absence of p27. Lymphoma incidence in p21–/–; p27–/– animals is also depicted. (B) Tumor latency in Eµ-Myc mice in the presence or absence of p27.
Unlike MuLV-treated mice, H2K-Myc transgenics develop lymphoma/leukemia from different cell lineages. The majority of the tumors carried cell surface markers corresponding to different stages of T-cell development, but tumors of B-cell and myeloid origin were also found. No major differences could be detected between H2K-Myc control and H2K-Myc; p27–/– mice with respect to tumor location (data not shown) and marker profile (Table III). Therefore, this genetic model shows that overexpression of Myc synergizes with p27 loss in tumorigenesis, regardless of the differentiation stage of the target cell population.
Table III. Tumor profiles of H2K-Myc mice in the presence or absence of p27 (%).
| Genotype | Myeloid T-/B-cell | Immature T-cell | Mature T-cell | Immature B-cell | Mature B-cell |
|---|---|---|---|---|---|
| H2K-Myc control (n = 21) | 14 | 36 | 23 | 23 | 4 |
| H2K-Myc; p27–/– (n = 16) | 16 | 42 | 26 | 16 | 0 |
Synergy between Myc overexpression and p27 deficiency in B-cell lymphomagenesis
In order to assess whether the collaboration between MYC overexpression and p27 deficiency can be extended to B cells, p27–/– (and p21–/–; p27–/–) mice were crossed to Eµ-Myc-transgenic animals (Verbeek et al., 1991). The Eµ-Myc construct targets overexpression of Myc to the B-cell lineage by the presence of the Eµ immunoglobulin heavy chain enhancer at its promoter (Adams et al., 1985; Langdon et al., 1986). These transgenic mice developed primarily clonal B-cell lineage tumors similar to the ones previously reported (Adams et al., 1985; Verbeek et al., 1991). As described for the H2K-Myc mice, Eµ-Myc animals were assembled in two groups, either p27 proficient (p27+/+ and p27+/–) or lacking p27, regardless of their p21 status. Within the groups, no significant variation in tumor survival was observed when p21 status was taken into consideration (p > 0.2) however, the number of animals used from each subgroup was relatively small (data not shown). The two groups showed a significant difference (p < 0.00005) in tumor survival. The combination of p27 deficiency and Myc overexpression led to an acceleration in tumor development as evidenced by the decrease of 40 days in mean survival (Figure 2B). In both transgenic groups lymphomas developed mainly in the lymph nodes and spleen. Yet in Eµ-Myc; p27–/– mice, the simultaneous involvement of both tissues was seen more frequently (46% versus 30%), probably as a result of more aggressive tumor growth. Tumors from Eµ-Myc transgenic animals were analyzed by FACS and classified according to Iritani and Eisenman (1999) (Table IV). p27 deficiency did not alter the tumor spectrum seen in Eµ-Myc mice and only a minor variation in the frequency of each differentiation stage was observed. Hence, p27 deficiency collaborates with Myc overexpression in both T- and B-cell lymphomagenesis, independently of their differentiation stage.
Table IV. Tumor profiles of Eµ-Myc mice in the presence or absence of p27 (%).
| Genotype | Large Pre B-cell | Small Pre B-cell | Immature B-cell | Mature B-cell | Mature T-cell |
|---|---|---|---|---|---|
| Eµ-Myc control (n = 19) | 21 | 10.5 | 37 | 21 | 10.5 |
| Eµ-Myc; p27–/– (n = 14) | 22 | 7 | 64 | 7 | 0 |
Increased proliferation in pre-neoplastic Eµ-Myc; p27–/– spleens
To elucidate the nature of the collaboration observed in lymphomagenesis a comparative analysis of pre-neoplastic features of Eµ-Myc and Eµ-Myc; p27–/– mice was carried out. For this analysis the Eµ-Myc mice were favored over the H2K-Myc since their phenotypic characteristics have been well defined and were shown to be highly reproducible (Langdon et al., 1986; Iritani and Eisenman, 1999). In these studies the spleens of 2-week-old mice were analyzed, as frequently older Eµ-Myc; p27–/– mice already showed onset of lymphomagenesis. At this stage the body weight of p27–/– mice was only slightly increased, while hyperplasia of thymus and spleen was already evident. Eµ-Myc animals also showed splenic hyperplasia, which became more profound in compound Eµ-Myc; p27–/– mice (data not shown). Although some variation was observed, overall the genotypes analyzed showed clear differences in morphology, proliferation and FACS profiles. Representative data from independent experiments are shown in Figure 3. Haematoxylin–eosin staining of sections (Figure 3A) from p27–/– spleens showed expansion of the white pulp when compared with WT, in line with observations previously reported for older p27–/– mice (Fero et al., 1996). Eµ-Myc spleens consisted largely of an expanded homogeneous blast population and less differentiated follicular structures. The Eµ-Myc phenotype was accentuated by p27–/– deficiency, suggesting an additional boost of proliferation of blast cells in this genotype. Propidium iodide (PI) staining of cell suspensions confirmed the enhanced proliferation of Eµ-Myc; p27–/– splenocytes, showing a substantial increase in the cycling cell population (S+ G2/M) (Figure 3B). Interestingly, the increase in proliferation seen in Eµ-Myc; p27–/– splenocytes roughly equalled the combined individual effects of Eµ-Myc and p27–/– each as compared with WT. No significant difference in apoptosis could be detected between the two transgenic groups by PI staining. However, since the values observed for Eµ-Myc samples were rather low it is possible that significant differences in apoptosis will only become evident later in development.

Fig. 3. Pre-neoplastic phenotype of Eµ-Myc p27–/– spleens. Representative illustration of data collected from multiple groups of 2-week-old mice from all the genotypes indicated at the top. (A) Haematoxylin–eosin-stained sections from spleens. The scale specified is maintained in all panels. (B) Flow cytometric analysis of the DNA content of freshly isolated splenocytes stained with PI. PI uptake and relative cell number are shown by the horizontal and vertical axes, respectively. For each panel, the percentage of cells in the distinct phases of the cell cycle is shown in the upper right corner. The horizontal bar represents the percentage of proliferating cells (S+ G2/M) for each genotype. (C and D) Flow cytometric analysis of splenocytes after staining for B220/TCRαβ (C) or IgM/B220 (D). The percentage of cells in each quadrant is indicated above the panels. In (D), the horizontal axis mean value for the B220+/IgM+ population (top right) is 360, 300, 470 and 900, from left to right.
The surface marker profile in the spleen of the transgenic mice was also determined (Figure 3C and D). p27–/– mice differ from WT mainly by an increased B220–/TCRαβ– population, possibly the result of an increase in the number of hematopoietic progenitor cells. As expected, Eµ-Myc splenocytes showed a decrease in T-cells (TCRαβ+), accompanied by a notable enlargement of the B-cell lineage (B220+, Figure 3C). In these transgenic animals, B220+ cells were mainly at the pre-B cell stage (IgM–) and only a small portion had differentiated into mature B cells (B220+high/IgM+low) (Figure 3D). Interestingly, p27 deficiency slightly aggravated the Eµ-Myc phenotype. This is suggested by the increased representation of a B220–/TCRαβ– population at the expense of mature lymphocytes. Furthermore, despite the high abundance of B220+ (mainly pre-B) cells in spleens of Eµ-Myc; p27–/– mice, the mature B-cell population was replaced by less mature B cells (B220+low/IgM+high). No major difference could be detected between the transgenic genotypes in the distribution of myeloid cell markers (data not shown). Therefore, these data indicate that in Eµ-Myc; p27–/– transgenic mice, the increased susceptibility to lymphomagenesis results from increased proliferation in early stages of the B-cell lineage.
Increased CDK2 activity in Eµ-Myc; p27–/– pre-neoplastic splenocytes
In an attempt to elucidate the molecular mechanisms underlying the phenotype reported here, pre-neoplastic Eµ-Myc; p27–/– splenocytes were analyzed for expression of cell cycle proteins known to interact with p27. Immunoblots of protein lysates from splenocytes of WT, p27–/–, Eµ-Myc and Eµ-Myc; p27–/– 2-week-old mice were stained with antibodies against cyclin E, cyclin A, CDK2 and p27. As shown in Figure 4A, no major difference in the levels of expression of either cyclin could be detected between p27+/+ and p27–/– transgenic mice in a population of cells composed of at least 60% B220+ cells (see Figure 3C). In contrast, the absence of p27 in Eµ-Myc transgenic mice resulted in a significant increase in the abundance of the faster-migrating CDK2 phosphorylated active form (CDK2-P) (Gu et al., 1992) (Figure 4A). The increased kinase activity of CDK2 in Eµ-Myc; p27–/– samples was further confirmed by its enhanced capacity to phosphorylate histone H1 (HH1) following immunoprecipitation (IP) (Figure 4B). Cyclin E and cyclin A IP complexes from Eµ-Myc; p27–/– lysates showed similar levels of kinase activity (data not shown).
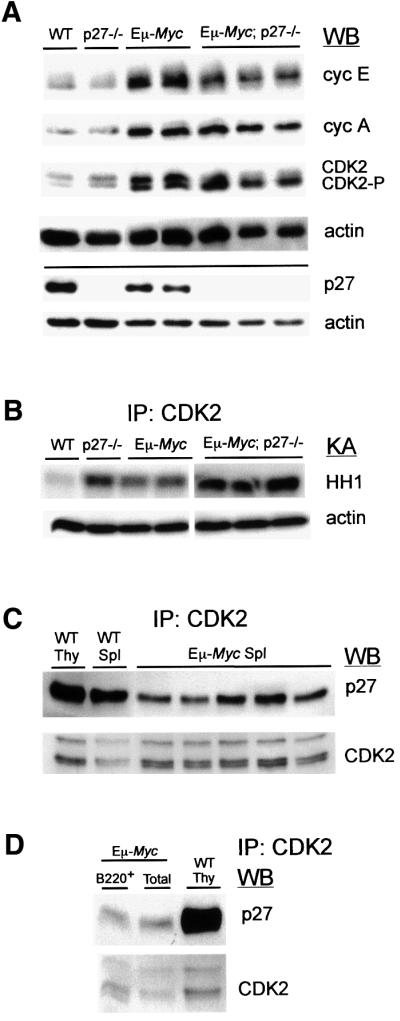
Fig. 4. p27 deficiency enhances CDK2 activity in Eµ-Myc pre-neoplastic splenocytes. Representative data from multiple groups of 2-week-old mice is depicted. (A) Splenocytes from mice of the indicated genotypes were analyzed by western blotting (WB) using antibodies against cyclin E (cycE), cyclin A (cycA), CDK2, p27 and actin (loading control). (B) Upper panel: CDK2 immunoprecipitation (IP) of protein lysates from splenocytes was followed by kinase assay (KA) for histone H1 (HH1)-associated kinase activity. The genotypes analyzed are depicted at the top. Lower panel: expression of actin in the same extracts. (C) CoIP analysis of CDK2-bound p27 from Eµ-Myc and WT splenocytes (Spl) or WT thymocytes (Thy). The amount of p27 and CDK2 present in the same blot is shown. (D) Protein lysates from the three left Eµ-Myc samples depicted in (C) were pooled and CDK2–p27 CoIP analysis was carried out either on total (Total) or positively selected B220+ splenocytes (B220+). WT thymic lysates were used as control.
Importantly, p27 was expressed at substantial levels in Eµ-Myc splenocytes (Figure 4A), suggesting that at least a fraction of p27 retains its inhibitory role in the Eµ-Myc background. To substantiate this, we determined whether p27 could be found in complex with CDK2 in Eµ-Myc splenocytes. As reported previously (Russo et al., 1996; Pavletich, 1999), when bound to CDK2, p27 causes conformational changes in the catalytic cleft of the kinase, preventing ATP binding and thus, activity. Protein lysates from WT thymocytes, WT splenocytes and Eµ-Myc pre-malignant splenocytes were incubated with CDK2 antibody, immunoprecipitated and the levels of p27 bound to CDK2 were determined by western blotting analysis. As shown in Figure 4C, a significant amount of p27 was found to bind and therefore inhibit CDK2 in Eµ-Myc pre-neoplastic samples. Since we have used lysates from total splenocytes in these co-immunoprecipitation studies (CoIP), we then asked whether active p27 could also be found in B220+ Eµ-Myc extracts. For that purpose, B220+ cells from Eµ-Myc splenocytes were isolated and the purity of the selected population (>99%) was confirmed by FACS analysis (data not shown). Isolation of B220+ cells was relatively inefficient and for CoIP analysis the use of pooled extracts was required. In Figure 4D the levels of p27 recovered following CDK2-IP are compared for pools of total and B220+ Eµ-Myc splenocytes. No significant difference could be observed between these cell populations. Thus, our data indicate that in Eµ-Myc pre-malignant splenocytes p27 is still an inhibitor of CDK2, and consequently the disruption of p27 in the Eµ-Myc background leads to increased CDK2 activity.
p27 expression and activity are differentially regulated in Myc-overexpressing tumors
In order to determine whether changes in p27 levels were selected for during lymphomagenesis we compared Eµ-Myc pre-malignant splenocytes and tumors for the expression of this inhibitor (Figure 5A). The Eµ-Myc samples shown here are the same depicted in Figure 4C. Although a substantial variation between tumors was observed, p27 protein was detected in all samples analyzed. However, in general tumors showed slightly reduced levels of p27 in comparison to the levels seen in pre-neoplastic tissues. When we assayed for CDK2-bound p27 levels in those tumors, a significant variation in binding was seen. While in some tumors p27 activity was impaired, in others the functional inhibitor was retained (Figure 5B). Pre-malignant and tumor tissues of Eµ- Myc mice were also compared for CDK2 expression (Figure 5A). Although no major differences could be observed, pre-neoplastic splenocytes showed slightly higher levels of CDK2-P. The same type of analysis was carried out in Eµ-Myc; p27–/– tissues where CDK2-P was significantly more abundant in pre-malignant samples (data not shown).

Fig. 5. Differential expression and activity of p27 in Eµ-Myc and WT MuLV-induced tumors. (A) Western blot (WB) analysis of the expression of p27, CDK2 and actin (loading control) in Eµ-Myc pre-neoplastic splenocytes (Spl) and tumors. WT splenocytes and thymocytes (Thy) were also analyzed. (B) CDK2–p27 co-immunoprecipitation (IP) analysis of the first six Eµ-Myc tumor samples shown in (A). The amount of p27 and CDK2 present in the same blot is shown. (C) CD4+/CD8+ thymic lymphomas from WT MuLV-infected mice were analyzed for p27 expression by WB. Protein lysates from WT and p27–/– (KO) thymus were used as control. Lymphomas with MuLV proviral integrations in the Myc loci are indicated by asterisks.
Finally, we analyzed WT MuLV-induced CD4+/CD8+ lymphomas to assess the effect of proviral integrations in the Myc loci on p27 expression (Figure 5C). As seen for Eµ-Myc tumors, a strong variation in p27 expression was observed in WT MuLV-induced lymphomas with a significant part of the tumors showing markedly reduced levels of p27. Down-regulation of p27 was observed both in the presence and absence of proviral integrations in the Myc loci; however, at the terminal stage Myc overexpression is also frequently seen in the latter group (data not shown). When we compared WT and p27–/– (Eµ-Myc and MuLV-induced) tumors for CDK2 expression no consistent differences could be found between the two groups (data not shown). Thus, our data suggest that in the early stages of tumorigenesis, next to a strong selective pressure for Myc overexpression there is an additional selective pressure for loss of CDK2 inhibition by p27.
Discussion
The relevance of p27 for cell proliferation is most notably exemplified by the increased body size of p27-deficient mice (Fero et al., 1996; Kiyokawa et al., 1996; Nakayama et al., 1996). This characteristic is most apparent in lymphoid tissues where disproportional hyperplasia is observed. However, in contrast to other tissues, no tumor suppressor activity of p27 was noted in lymphoid cells (Fero et al., 1998). Interestingly, p21-deficient mice show no aberrant size or spontaneous tumor predisposition (Deng et al., 1995) in spite of the fact that p21 and p27 exhibit similar biochemical activity. Therefore, we have studied the effect of both deficiencies in a well-defined lymphoma model in which subtle predisposition for tumorigenesis can be scored effectively. We show here that loss of p27 but not p21 accelerates lymphomagenesis in mice upon infection with MuLV. Moreover, we demonstrate that in vivo functional overlap between p21 and p27 is very limited as p27–/– and p21–/–; p27–/– mice showed a comparable tumor predisposition. Only on a p27 hemizygous background, p21 levels became limiting, indicating that haplo-insufficiency for p27 is partly compensated by p21. In this respect it would be of interest to monitor p21 levels in tissues for which haplo-insufficiency of p27 in tumor suppression has been described.
Recent reports have suggested that the capacity of MYC to override the inhibitory function of p27 is an important element of its oncogenic potential (Vlach et al., 1996; Bouchard et al., 1999; Perez-Roger et al., 1999; O’Hagan et al., 2000; Yang et al., 2001). Our analyses show that loss of p27 does not reduce the dependence on MYC overexpression for lymphoid transformation. Moreover and contrary to expectation, Myc activation was at least 2-fold more frequent on a p27–/– background than in WT mice. In a similar study, increased frequency of Myc activation in p27–/– mice was also observed (Hwang,H.C., Martins,C.P., Bronkhorst,Y., Randel,E., Berns,A., Fero,M. and Clurman,B.E., submitted).
The strong collaboration between p27 loss and MYC overexpression was further supported by the pattern of oncogene activation detected in the tumors. For instance, all genotypes showed a comparable frequency of Gfi1 activations, arguing against an increase in the number of target cells in p27–/– mice. In addition, the decreased requirement for Pim1 activation in p27–/– lymphomas is likely to reflect a partial functional overlap between PIM1 overexpression and p27 inactivation, both in transformation and collaboration with MYC. In agreement with this notion, a role for PIM1 in the G1/S transition was reported (Mochizuki et al., 1999) by the identification of CDC25A, a positive regulator of CDK2 (Blomberg and Hoffmann, 1999), as one of the few PIM1 targets known to date. Although at low frequency, Cyclin D2 insertions were detected in WT tumors always in combination with Myc activation but could not be found in the p27-deficient background. This indicates that in WT mice selective pressure for overexpression of both Cyclin D2 and MYC can occur within the same cell. The absence of Cyclin D2 mutations in p27–/– tumors (even in the presence of p21) argues that Cyclin D2 overexpression and p27 loss confer a similar selective advantage. This indicates that in lymphomagenesis Cyclin D2 overexpression may be required for its involvement in the sequestration of p27 (Cheng et al., 1999).
The cooperation between MYC and p27 loss in lymphoma development was independently confirmed in distinct lymphoid compartments by the analysis of compound Myc transgenic/CKI KO mice. Tumor survival was clearly decreased in H2K-Myc; p27–/– and Eµ-Myc; p27–/– mice, with p27 loss having little effect on the tumor spectrum. Thus, our data show that in lymphoid cells the synergism between Myc overexpression and p27 loss is lineage and developmental stage independent.
The incidence of CD4+ MuLV-induced lymphomas was greatly increased in p27–/– mice. The cause for this variation remains unclear but a similar tendency is observed in p21–/– mice, although to a lesser extent. Since p21–/– lymphomas showed otherwise a WT phenotype, these data indicate that tumor pathways are conserved in distinct lymphoid cells, as is also evident from our transgenic studies. Unlike p21, p27 has been shown to play a key role in the proliferation of WT lymphocytes (Nourse et al., 1994; Mohapatra et al., 2001). In addition, expansion of activated/memory CD4+ splenic T cells (Zhang et al., 2000) and enhanced T-cell activation in response to mitogenic stimuli (Fero et al., 1996; Zhang et al., 2000; Mohapatra et al., 2001) is seen in p27–/– mice. Thus, p27 deficiency seems to favor the expansion of the lymphoid compartment and in this respect resembles the effects of MYC overexpression, well documented in transgenic mouse studies (Adams et al., 1985; Langdon et al., 1986; Morello et al., 1989).
Interestingly, our studies of pre-neoplastic Eµ-Myc; p27–/– mice provided a strong indication for a relevant role of p27 in the proliferation of the B-cell lineage. More surprisingly, our analysis of Eµ-Myc; p27–/– splenocytes shows that p27 loss exacerbates the MYC overexpression phenotype. This was evidenced by the decrease in B-cell differentiation and increased proliferation seen in Eµ-Myc; p27–/– splenocytes. The early onset of lymphomagenesis as well as the lower incidence of fully differentiated tumors in these compound mice further supported this notion. The absence of detectable variation in apoptosis between cells from the different genotypes argues for enhanced proliferation as the cause for the synergism observed.
CDK2 activity promotes proliferation, and regulation of this kinase is the main known function of p27 (Pavletich, 1999; Sherr and Roberts, 1999). Importantly, our data show that in pre-leukemic Eµ-Myc splenocytes CDK2 activity is increased compared with WT despite the presence of p27, but further enhanced upon loss of this inhibitor. Moreover, we show that Eµ-Myc splenocytes express active p27 (CDK2 bound), thus arguing that absence of the inhibitor is the direct cause for the enhanced CDK2 activity seen in Eµ-Myc; p27–/– samples. Hence, our data strongly suggest that enhanced CDK2 activity is an important component of the synergism reported here.
Besides showing collaboration between Myc overexpression and p27 deficiency in lymphomagenesis, our study also demonstrates that in Eµ-Myc tumors there is selective pressure to surpass inhibition by p27. Accord ingly, part of these tumors expressed low levels of the inhibitor or evidence for p27 degradation. Interestingly, some Eµ-Myc tumors showed substantial levels of CDK2-bound p27 indicating that these tumors harbor mutations that promote proliferation despite the high levels of active p27 present. How growth arrest can be prevented in these cells in the presence of CDK2/p27 complexes is not known but a similar effect was observed in fibroblasts upon overexpression of E1A (Alevizopoulos et al., 1998). Our data also suggest that Eµ-Myc (and Eµ-Myc; p27–/–) tumor samples show a somewhat diminished requirement for high levels of CDK2-P compared with pre-malignant tissues, possibly reflecting a lower proliferation index.
Unlike for pre-neoplastic tissues, no significant difference for CDK2-P levels could be detected between WT and p27–/– Eµ-Myc tumors. Similar data was obtained when MuLV-induced tumors from WT and p27–/– mice were compared for CDK2 expression (data not shown), despite the different levels of p27 seen in WT tumors. Hence, overall our data indicate that at the neoplastic stage cells have already compensated for the differences imposed by the presence of p27. Accordingly, the analysis of proviral insertion sites from WT MuLV-induced tumors might lead to the identification of new mechanisms that help evading inhibition by p27. It follows that upon combined Myc overexpression and p27 deficiency, cells are able to bypass this additional selective stage in tumor development, in agreement with the accelerated tumorigenesis seen in mice harboring both mutations.
Our data suggest that a similar collaboration might be observed in other tissues that rely mostly on p27 for keeping CDK2 in check. Conversely, the synergy might be lost in cells where other CDK2 inhibitors are present or activated upon p27 loss, as previously reported for mouse embryonic fibroblasts (MEFs) (Coats et al., 1999). In agreement with this, we have shown that in MEFs the capacity of MYC to induce proliferation is not affected by loss of p27 (Berns et al., 2000). Given the fact that p21 is expressed in lymphocytes this also implies that in vivo p21 does not act as a strong CDK2 inhibitor in these cells under the conditions described here.
Our data provide the first in vivo evidence for enhanced oncogenic activity of MYC in the absence of p27 and underscores the non-redundant roles of MYC overexpression and p27 loss in lymphomagenesis. Since Myc overexpression is associated with the development of malignancies both in mice and humans, it will be interesting to determine whether the synergy described here can also be observed in man. Furthermore, it will be interesting to monitor other tumors for the same synergistic interaction.
Materials and methods
Mice, tumor induction and tissue isolation
Animals were kept under conventional barrier maintenance, and food pellets (Hope Farms) and water (pH 2.7–2.8) were administrated ad libitum. Genetic background effects were minimized by the generation of all genotypes (including WT) from crosses between p27–/– (Fero et al., 1996) and p21–/– (Deng et al., 1995) (for MuLV infection) or p21–/–; p27–/– and Eµ-Myc (founder line 186) (Verbeek et al., 1991) /H2K-Myc (Jonkers et al., 1997) transgenic mice (analysis of Myc transgenics). MuLV-induced tumors were generated by intra-peritoneal injection of newborn mice (2–3 days old) with 50 µl of 104–105 infectious units of MuLV clone 1A (Jaenisch et al., 1975). MuLV-infected animals and transgenic animals were killed when terminally ill and only animals with lymphoma/leukemia or older than 8 months were considered for survival curves. Statistical analysis for tumor survival was performed with the Kaplan–Meier log-rank test and p-values below 0.05 were considered significant. For genotyping, proviral tagging and biochemistry, tissues were collected and immediately frozen (–80°C). Tail and tumor DNA was isolated (van der Putten et al., 1979; Laird et al., 1991) and used for genotyping. Mice were genotyped as described previously excluding the following modifications (primers: 5′–3′): p27-KO band: K3, N1 (Fero et al., 1996); WT band: WTF (GCCTGGCTCTGCTCCATTTGAC), WTR (CTCTCCACCTCCTGCCATTC) (400 bp). p21-E2F (GACAAGAGGCCCAGTACTTCCTC); E3R (CAATCTGCGCTTGGAGTGATAG) PGKF (GCAGCCTCTGTTCCACATACAC). WT band: E2F/E3R (700 bp), KO band PGKF/E3R (250 bp). For pre-malignancy studies, four distinct groups of 14- to 17-day-old animals were analyzed, each containing at least two mice per genotype. Spleens were isolated and fixed (histology) or prepared as cell suspensions for immediate staining (FACS) and freezing (protein analysis).
Proviral tagging
For Southern analysis, 10 µg of genomic DNA were digested with restriction enzymes as recommended by the supplier, separated in a 0.6% agarose gel, transferred to a nylon membrane and hybridized to 32P-labeled probes. For northern analysis, 10 µg of total RNA were isolated (TRIzol Reagent; Gibco-BRL), separated on 1% agarose–formaldehyde gel and transferred to nitrocellulose. The following probes were used: MuLV U3 LTR (Cuypers et al., 1984), MycN (3 kb PstI–PstI fragment) (van Lohuizen et al., 1989a), Myc (2.6 kb XbaI–HindIII fragment) (Selten et al., 1984), Gfi1 (1.4 kb BglII–EcoRV fragment) (Scheijen et al., 1997), Cyclin D2 (480 bp PCR fragment. Primers: CD2F, CCGAGCAGACACCTAGGGCG; CD2R, CGTGTGTGCTCTCTACCTTCG) and Pim1 (0.9 kb BamHI–BamHI fragment) (Cuypers et al., 1984).
Flow cytometry and PI staining
For FACS analysis of tumor and normal tissues, cell suspensions were obtained by pressing the tissue between nylon sheets and resuspending in PBS enriched with 2% serum (2% S-PBS). Erythrocytes were depleted by blood lysis buffer treatment (pre-malignant spleens; Sigma) and lympho cytes were isolated by Ficoll gradient (splenic tumors; Amersham) For antibody staining, 1 × 106 cells were incubated for 40 min at 4°C with saturating amounts of monoclonal antibodies in 2% S-PBS. Cells were then washed twice in 2% S-PBS and, when required, incubated with streptavidin-APC (SA-APC). The following antibodies were used: CD3ε (CT-CD3), CD4 (YTS 191.1), Mac-1 (M1/70.15) (Caltag), CD8 (53–6.7), TCRαβ (H57-597), IgM (G53-238), IgD (11-26c.2a), CD45R/B220 (RA3-6B2), Gr-1 (RB6-8C5) and SA-APC (PharMingen). For cell cycle analysis, 1 × 106 splenocytes were stained with PI as described previously (Berns et al., 2000) immediately after isolation. FACS analysis was performed on a Becton-Dickinson FACSCalibur flow cytometer. Data were analyzed with CELLQuest (Becton Dickinson).
Histological analysis
Tissues were isolated and fixed in formalin for at least 24 h. Fixed samples were dehydrated, embedded in Histowax and cut into 4 µm sections. Sections were stained with hematoxylin and eosin, and images were captured with a Color CCD-camera (Axiocam; Zeiss).
CoIP and kinase assays
Splenocytes were frozen at –80°C as a cell pellet, subsequently lysed in buffer (50 mM Tris, 150 mM NaCl, 20 mM EDTA, 0.5% NP-40) supplemented with protease and phosphatase inhibitors, kept on ice for 45 min and centrifuged at 14 000 r.p.m. for 15 min at 4°C. Protein concentration was determined with the Bio-Rad protein assay and for western blotting 50 µg of lysate were separated by SDS–PAGE and blotted into nitrocellulose, which was then blocked in 20 mM Tris, 140 mM NaCl, 0.1% Tween-20 (TBST pH 7.6), 5% dried milk for at least 2 h at 4°C. Membranes were incubated in TBST, 1% dried milk with the antibody at the appropriate dilution for 2–3 h at room temperature and after three washes the antibody was detected using horseradish peroxidase-linked secondary antibodies and enhanced chemiluminescence (ECL; Amersham). Kinase assays were carried out as described previously (Gil-Gomez et al., 1998) following incubation of 50 µg of protein lysate with CDK2, cyclin A or cyclin E antibodies and using histone H1 as substrate. For CoIP studies, 200 µg of cell lysate were incubated with CDK2 antibody and IP was followed by WB with p27 and CDK2 antibodies, as described above. For the analysis of CDK2–p27 immunocomplexes from pooled extracts (Figure 4D), the amounts of protein lysate used were 100 µg (pooled samples) and 200 µg (WT thymus). Antibodies used were: cyclin E (M-20), cyclin A (C-19), CDK2 (M2) (Santa Cruz), p27 (K25020) (Transduction Laboratories) and actin (MAB1501) (Chemicon).
Acknowledgments
Acknowledgements
We would like to thank P.Leder and J.Roberts for generously supplying the p21–/– and p27–/– mice, respectively. We also thank N.Bosnie and F.van der Ahé for help with the retrovirus injections. We thank R.Beijersbergen, J.Jonkers, K.Quon and H.Hwang for critical reading of the manuscript. This work was funded by a Portuguese grant from the Fundação Gulbenkian (PGDBM) and PRAXIS XXI.
References
- Adams J.M., Harris,A.W., Pinkert,C.A., Corcoran,L.M., Alexander,W.S., Cory,S., Palmiter,R.D. and Brinster,R.L. (1985) The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature, 318, 533–538. [DOI] [PubMed] [Google Scholar]
- Alevizopoulos K., Catarin,B., Vlach,J. and Amati,B. (1998) A novel function of adenovirus E1A is required to overcome growth arrest by the CDK2 inhibitor p27Kip1. EMBO J., 17, 5987–5997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berns K., Martins,C., Dannenberg,J.H., Berns,A., te Riele,H. and Bernards,R. (2000) p27kip1-independent cell cycle regulation by MYC. Oncogene, 19, 4822–4827. [DOI] [PubMed] [Google Scholar]
- Blomberg I. and Hoffmann,I. (1999) Ectopic expression of Cdc25A accelerates the G1/S transition and leads to premature activation of cyclin E- and cyclin A-dependent kinases. Mol. Cell. Biol., 19, 6183–6194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard C. et al. (1999) Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J., 18, 5321–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M., Sexl,V., Sherr,C.J. and Roussel,M.F. (1998) Assembly of cyclin D-dependent kinase and titration of p27Kip1 regulated by mitogen-activated protein kinase kinase (MEK1). Proc. Natl Acad. Sci. USA, 95, 1091–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M., Olivier,P., Diehl,J.A., Fero,M., Roussel,M.F., Roberts,J.M. and Sherr,C.J. (1999) The p21Cip1 and p27Kip1 CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J., 18, 1571–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarle R. et al. (2000) Increased proteasome degradation of cyclin-dependent kinase inhibitor p27 is associated with a decreased overall survival in mantle cell lymphoma. Blood, 95, 619–626. [PubMed] [Google Scholar]
- Coats S., Whyte,P., Fero,M.L., Lacy,S., Chung,G., Randel,E., Firpo,E. and Roberts,J.M. (1999) A new pathway for mitogen-dependent cdk2 regulation uncovered in p27Kip1-deficient cells. Curr. Biol., 9, 163–173. [DOI] [PubMed] [Google Scholar]
- Cuypers H.T., Selten,G., Quint,W., Zijlstra,M., Maandag,E.R., Boelens,W., van Wezenbeek,P., Melief,C. and Berns,A. (1984) Murine leukemia virus-induced T-cell lymphomagenesis: integration of proviruses in a distinct chromosomal region. Cell, 37, 141–150. [DOI] [PubMed] [Google Scholar]
- Dang C.V. (1999) c-Myc target genes involved in cell growth, apoptosis and metabolism. Mol. Cell. Biol., 19, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C., Zhang,P., Harper,J.W., Elledge,S.J. and Leder,P. (1995) Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell, 82, 675–684. [DOI] [PubMed] [Google Scholar]
- Di Cristofano A., De Acetis,M., Koff,A., Cordon-Cardo,C. and Pandolfi,P.P. (2001) Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat. Genet., 27, 222–224. [DOI] [PubMed] [Google Scholar]
- Dulic V., Kaufmann,W.K., Wilson,S.J., Tlsty,T.D., Lees,E., Harper,J.W., Elledge,S.J. and Reed,S.I. (1994) p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell, 76, 1013–1023. [DOI] [PubMed] [Google Scholar]
- Eisenman R.N. (2001) Deconstructing myc. Genes Dev., 15, 2023–2030. [DOI] [PubMed] [Google Scholar]
- Fero M.L. et al. (1996) A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis and female sterility in p27Kip1-deficient mice. Cell, 85, 733–744. [DOI] [PubMed] [Google Scholar]
- Fero M.L., Randel,E., Gurley,K.E., Roberts,J.M. and Kemp,C.J. (1998) The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature, 396, 177–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Gomez G., Berns,A. and Brady,H.J. (1998) A link between cell cycle and cell death: Bax and Bcl-2 modulate Cdk2 activation during thymocyte apoptosis. EMBO J., 17, 7209–7218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y., Rosenblatt,J. and Morgan,D.O. (1992) Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 and Tyr15. EMBO J., 11, 3995–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna Z., Jankowski,M., Tremblay,P., Jiang,X., Milatovich,A., Francke,U. and Jolicoeur,P. (1993) The Vin-1 gene, identified by provirus insertional mutagenesis, is the cyclin D2. Oncogene, 8, 1661–1666. [PubMed] [Google Scholar]
- Iritani B.M. and Eisenman,R.N. (1999) c-Myc enhances protein synthesis and cell size during B lymphocyte development. Proc. Natl Acad. Sci. USA, 96, 13180–13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch R., Fan,H. and Croker,B. (1975) Infection of preimplantation mouse embryos and of newborn mice with leukemia virus: tissue distribution of viral DNA and RNA and leukemogenesis in the adult animal. Proc. Natl Acad. Sci. USA, 72, 4008–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers J. and Berns,A. (1996) Retroviral insertional mutagenesis as a strategy to identify cancer genes. Biochim. Biophys. Acta, 1287, 29–57. [DOI] [PubMed] [Google Scholar]
- Jonkers J., Korswagen,H.C., Acton,D., Breuer,M. and Berns,A. (1997) Activation of a novel proto-oncogene, Frat1, contributes to progression of mouse T-cell lymphomas. EMBO J., 16, 441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyokawa H. et al. (1996) Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27Kip1. Cell, 85, 721–732. [DOI] [PubMed] [Google Scholar]
- Laird P.W., Zijderveld,A., Linders,K., Rudnicki,M.A., Jaenisch,R. and Berns,A. (1991) Simplified mammalian DNA isolation procedure. Nucleic Acids Res., 19, 4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langdon W.Y., Harris,A.W., Cory,S. and Adams,J.M. (1986) The c-myc oncogene perturbs B lymphocyte development in Eµ-myc transgenic mice. Cell, 47, 11–18. [DOI] [PubMed] [Google Scholar]
- Macleod K.F., Sherry,N., Hannon,G., Beach,D., Tokino,T., Kinzler,K., Vogelstein,B. and Jacks,T. (1995) p53-dependent and independent expression of p21 during cell growth, differentiation and DNA damage. Genes Dev., 9, 935–944. [DOI] [PubMed] [Google Scholar]
- Mochizuki T., Kitanaka,C., Noguchi,K., Muramatsu,T., Asai,A. and Kuchino,Y. (1999) Physical and functional interactions between Pim-1 kinase and Cdc25A phosphatase. Implications for the Pim-1-mediated activation of the c-Myc signaling pathway. J. Biol. Chem., 274, 18659–18666. [DOI] [PubMed] [Google Scholar]
- Mohapatra S., Agrawal,D. and Pledger,W.J. (2001) p27Kip1 regulates T cell proliferation. J. Biol. Chem., 276, 21976–21983. [DOI] [PubMed] [Google Scholar]
- Morello D., Lavenu,A., Bandeira,A., Portnoi,D., Gaillard,J. and Babinet,C. (1989) Lymphoproliferative syndrome associated with c-myc expression driven by a class I gene promoter in transgenic mice. Oncogene Res., 4, 111–125. [PubMed] [Google Scholar]
- Morgan D.O. (1995) Principles of CDK regulation. Nature, 374, 131–134. [DOI] [PubMed] [Google Scholar]
- Muller D., Bouchard,C., Rudolph,B., Steiner,P., Stuckmann,I., Saffrich,R., Ansorge,W., Huttner,W. and Eilers,M. (1997) Cdk2-dependent phosphorylation of p27 facilitates its Myc-induced release from cyclin E/cdk2 complexes. Oncogene, 15, 2561–2576. [DOI] [PubMed] [Google Scholar]
- Nakayama K., Ishida,N., Shirane,M., Inomata,A., Inoue,T., Shishido,N., Horii,I. and Loh,D.Y. (1996) Mice lacking p27Kip1 display increased body size, multiple organ hyperplasia, retinal dysplasia and pituitary tumors. Cell, 85, 707–720. [DOI] [PubMed] [Google Scholar]
- Nasi S., Ciarapica,R., Jucker,R., Rosati,J. and Soucek,L. (2001) Making decisions through Myc. FEBS Lett., 490, 153–162. [DOI] [PubMed] [Google Scholar]
- Nourse J., Firpo,E., Flanagan,W.M., Coats,S., Polyak,K., Lee,M.H., Massagué,J., Crabtree,G.R. and Roberts,J.M. (1994) Interleukin-2-mediated elimination of the p27Kip1 cyclin-dependent kinase inhibitor prevented by rapamycin. Nature, 372, 570–573. [DOI] [PubMed] [Google Scholar]
- O’Hagan R.C., Ohh,M., David,G., de Alboran,I.M., Alt,F.W., Kaelin,W.G.,Jr and DePinho,R.A. (2000) Myc-enhanced expression of Cul1 promotes ubiquitin-dependent proteolysis and cell cycle progression. Genes Dev., 14, 2185–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavletich N.P. (1999) Mechanisms of cyclin-dependent kinase regulation: structures of Cdks, their cyclin activators and Cip and INK4 inhibitors. J. Mol. Biol., 287, 821–828. [DOI] [PubMed] [Google Scholar]
- Perez-Roger I., Kim,S.H., Griffiths,B., Sewing,A. and Land,H. (1999) Cyclins D1 and D2 mediate myc-induced proliferation via sequestration of p27Kip1 and p21Cip1. EMBO J., 18, 5310–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K., Kato,J.Y., Solomon,M.J., Sherr,C.J., Massagué,J., Roberts,J.M. and Koff,A. (1994) p27Kip1, a cyclin–Cdk inhibitor, links transforming growth factor-β and contact inhibition to cell cycle arrest. Genes Dev., 8, 9–22. [DOI] [PubMed] [Google Scholar]
- Russo A.A., Jeffrey,P.D., Patten,A.K., Massagué,J. and Pavletich,N.P. (1996) Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A–Cdk2 complex. Nature, 382, 325–331. [DOI] [PubMed] [Google Scholar]
- Scheijen B., Jonkers,J., Acton,D. and Berns,A. (1997) Characterization of pal-1, a common proviral insertion site in murine leukemia virus-induced lymphomas of c-myc and Pim-1 transgenic mice. J. Virol., 71, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selten G., Cuypers,H.T., Zijlstra,M., Melief,C. and Berns,A. (1984) Involvement of c-myc in MuLV-induced T cell lymphomas in mice: frequency and mechanisms of activation. EMBO J., 3, 3215–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr C.J. (2000) The Pezcoller lecture: cancer cell cycles revisited. Cancer Res., 60, 3689–3695. [PubMed] [Google Scholar]
- Sherr C.J. and Roberts,J.M. (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev., 13, 1501–1512. [DOI] [PubMed] [Google Scholar]
- Tremblay P.J., Kozak,C.A. and Jolicoeur,P. (1992) Identification of a novel gene, Vin-1, in murine leukemia virus-induced T-cell leukemias by provirus insertional mutagenesis. J. Virol., 66, 1344–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Putten H., Terwindt,E., Berns,A. and Jaenisch,R. (1979) The integration sites of endogenous and exogenous Moloney murine leukemia virus. Cell, 18, 109–116. [DOI] [PubMed] [Google Scholar]
- van Lohuizen M., Breuer,M. and Berns,A. (1989a) N-myc is frequently activated by proviral insertion in MuLV-induced T cell lymphomas. EMBO J., 8, 133–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Lohuizen M., Verbeek,S., Krimpenfort,P., Domen,J., Saris,C., Radaszkiewicz,T. and Berns,A. (1989b) Predisposition to lymphomagenesis in pim-1 transgenic mice: cooperation with c-myc and N-myc in murine leukemia virus-induced tumors. Cell, 56, 673–682. [DOI] [PubMed] [Google Scholar]
- van Lohuizen M., Verbeek,S., Scheijen,B., Wientjens,E., van der Gulden,H. and Berns,A. (1991) Identification of cooperating oncogenes in Eµ-myc transgenic mice by provirus tagging. Cell, 65, 737–752. [DOI] [PubMed] [Google Scholar]
- Verbeek S., van Lohuizen,M., van der Valk,M., Domen,J., Kraal,G. and Berns,A. (1991) Mice bearing the Eµ-myc and Eµ-pim-1 transgenes develop pre-B-cell leukemia prenatally. Mol. Cell. Biol., 11, 1176–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlach J., Hennecke,S., Alevizopoulos,K., Conti,D. and Amati,B. (1996) Growth arrest by the cyclin-dependent kinase inhibitor p27Kip1 is abrogated by c-Myc. EMBO J., 15, 6595–6604. [PMC free article] [PubMed] [Google Scholar]
- Yang W. et al. (2001) Repression of transcription of the p27Kip1 cyclin-dependent kinase inhibitor gene by c-Myc. Oncogene, 20, 1688–1702. [DOI] [PubMed] [Google Scholar]
- Yokozawa T. et al. (2000) Prognostic significance of the cell cycle inhibitor p27Kip1 in acute myeloid leukemia. Leukemia, 14, 28–33. [DOI] [PubMed] [Google Scholar]
- Zhang S., Lawless,V.A. and Kaplan,M.H. (2000) Cytokine-stimulated T lymphocyte proliferation is regulated by p27Kip1. J. Immunol., 165, 6270–6277. [DOI] [PubMed] [Google Scholar]