

Abstract
Through the action of its membrane-bound type I receptors, transforming growth factor-β (TGF-β) elicits a wide range of cellular responses that regulate cell proliferation, differentiation and apoptosis. Many of the signaling responses induced by TGF-β are mediated by Smad proteins, but certain evidence has suggested that TGF-β can also signal independently of Smads. We found in mouse mammary epithelial (NMuMG) cells, which respond to TGF-β treatment in multiple ways, that TGF-β-induced activation of p38 MAP kinase is required for TGF-β-induced apoptosis, epithelial-to-mesenchymal transition (EMT), but not growth arrest. We further demonstrated that activation of p38 is independent of Smads using a mutant type I receptor, which is incapable of activating Smads but still retains the kinase activity. This mutant receptor is sufficient to activate p38 and cause NMuMG cells to undergo apoptosis. However, it is not sufficient to induce EMT. These results indicate that TGF-β receptor signals through multiple intracellular pathways and provide first-hand biochemical evidence for the existence of Smad-independent TGF-β receptor signaling.
Keywords: apoptosis/epithelial-to-mesenchymal transition/p38 MAP kinase/Smad/TGF-β receptor
Introduction
Transforming growth factor-β (TGF-β) and its related factors, including activins and bone morphological proteins (BMPs), exert diverse effects on a wide array of cellular processes ranging from proliferation and differentiation to apoptosis (reviewed in Massagué, 1998; Whitman, 1998). TGF-β itself is a well known potent growth inhibitor for cells of epithelial origin through its ability to downregulate the proto-oncogene c-myc and to activate transcription of cyclin-dependent kinase (CDK) inhibitors, such as p15ink4b and p21cip1 (Hannon and Beach, 1994; Datto et al., 1995; Reynisdottir et al., 1995). However, the molecular mechanism by which TGF-β exerts its apoptotic effect is still poorly understood. Overexpression of Bcl-2 blocks the TGF-β-induced apoptotic effect but does not suppress the antiproliferative activity of TGF-β, suggesting that these two cellular events are possibly mediated by divergent pathways (Selvakumaran et al., 1994). In addition, TGF-β has also been shown to promote tumorigenesis and increase metastasis (reviewed in Derynck et al., 2001; Wakefield and Roberts, 2002). Tumor cells often acquire resistance to the growth-inhibitory effect of TGF-β, but retain susceptibility to other TGF-β-elicited effects, such as induction of epithelial-to-mesenchymal transition (EMT) (Oft et al., 1996; Lehmann et al., 2000). EMT is a transient change in cell structure often associated with weaker cell–cell interactions, and acquisition of motile and invasive properties.
The signaling of the TGF-β family of growth factors is mediated by a heteromeric complex of two types of transmembrane serine/threonine kinase receptors. Binding of ligand to the receptor complex leads the type II receptor kinase to phosphorylate and thereby activate the type I receptor kinase. The activated type I receptor then phosphorylates receptor-activated Smads (R-Smads), e.g. Smad2 and Smad3 proteins in the TGF-β pathway, or Smad1, Smad5 or Smad8 in the BMP pathway. The specificity of a type I receptor kinase for an R-Smad is determined by the L45 loop between kinase subdomain IV and V (Feng and Derynck, 1997). Exchanging three diverged residues in the L45 loop between TGF-β and BMP type I receptors converts the TGF-β receptor to interact and activate the BMP pathway-specific R-Smads (Chen et al., 1998). After phosphorylation by the type I receptor kinase, the R-Smads bind to the related factor Smad4 and move into the nucleus. In the nucleus, this Smad complex associates with other transcription factors to activate transcription of target genes (reviewed in Zhang and Derynck, 1999; Massagué, 2000; ten Dijke et al., 2000; Wrana, 2000). Smad proteins have been shown to be directly involved in the transcriptional regulation of the promoters of p15ink4b, p21cip1 and c-myc (Feng et al., 2000; Pardali et al., 2000; Chen et al., 2001; Seoane et al., 2001; Yagi et al., 2002).
In addition to the Smad-mediated canonical TGF-β signaling pathway, evidence over the past few years suggests that TGF-β may signal through other pathways. For example, TGF-β can activate several mitogen-activated protein kinases (MAPKs), including extracellular signal-regulated kinases (Erks), c-Jun N-terminal kinases (JNKs) and p38 kinases (Hartsough and Mulder, 1995; Engel et al., 1999; Hanafusa et al., 1999; Hocevar et al., 1999; Sano et al., 1999; Bhowmick et al., 2001b). The effects of TGF-β on JNK, p38 and Erk MAPKs vary in kinetics and magnitude. In some cases, these kinases are activated by TGF-β with slow kinetics, indicating that these might be delayed, indirect effects, possibly resulting from Smad-dependent transcriptional response. In other cases, however, the activation is very rapid, suggesting that it might be through a direct post-translational modification. Despite all of these observations, the underlying mechanism and biological consequence of Erk, JNK or p38 activation are not clear.
Here we report that activation of p38 by TGF-β is required for the TGF-β-induced apoptosis and EMT in mouse mammary gland epithelial (NMuMG) cells. However, activation of p38 is not required for TGF-β-induced growth arrest. We have generated a mutant TGF-β type I receptor that is unable to interact with and phosphorylate Smads but still possesses the kinase activity. Using this mutant TGF-β type I receptor, we show that the TGF-β-mediated activation of p38 is independent of receptor-mediated Smad activation. Our results provide the first biochemical evidence for the existence of Smad-independent TGF-β receptor signaling. Furthermore, we found that although both TGF-β-mediated apoptosis and EMT require activation of p38, we are also able to dissociate these two responses. We found that Smad-independent TGF-β receptor signaling through p38 is sufficient to promote apoptosis, but not sufficient to cause an EMT response. Thus, three cellular events, growth arrest, apoptosis and EMT, induced by TGF-β, are mediated through distinct effector functions. These results have shed light on the ability of TGF-β to induce a plethora of diverse biological responses, and have also helped to explain why tumor cells, which often develop a resistance to TGF-β-induced growth inhibition, can retain an otherwise functional TGF-β signaling pathway.
Results
p38 is required for TGF-β-induced apoptosis and EMT but not for growth arrest
Cultured NMuMG cells undergo growth arrest and concomitant EMT in response to TGF-β treatment (Miettinen et al., 1994; Piek et al., 1999; Bhowmick et al., 2001a). However, overexpression of TGF-β1 in differentiating secretory mammary epithelium in transgenic mice causes premature programmed cell death (Jhappan et al., 1993). We found that NMuMG cells could also undergo apoptosis in response to TGF-β, but only do so in media with low serum and in the absence of insulin. The TGF-β-induced apoptosis was observed 9 h after stimulation and became most apparent around 24 h, and was marked by increased cell death (Figure 1A, left panel), release of histone-associated DNA fragments (Figure 1A, right panel) and activation of caspase-3 (Figure 1B). Pro longed serum deprivation also induced a late-onset apoptosis, which was accelerated by TGF-β treatment (Figure 1A). These observations suggest that NMuMG cells can be exploited as a useful model system to dissect the molecular pathways eliciting multiple TGF-β signaling responses.

Fig. 1. p38 is required for TGF-β-induced apoptotic but not antiproliferative responses. (A) Time course of TGF-β-induced apoptosis in NMuMG cells. The percentage of apoptotic cells was measured by Trypan Blue staining (left panel) and DNA fragmentation was quantified by the Cell Death Detection ELISA assay (right panel). (B) Inhibition of TGF-β-induced caspase-3 activation by the p38 kinase inhibitor SB203580. Cell lysates were harvested after incubation with or without TGF-β in the presence or absence of the indicated protein kinase inhibitor and blotted for full-length caspase-3 (inactive), cleaved caspase-3 (active), phospho-Akt and total Akt. (C) Inhibition of TGF-β-induced apoptosis by SB203580, a p38 kinase inhibitor. Cells were treated as in (B), then trypsinized and collected for apoptotic assay as in (A). (D) p38 kinase activity was not required for TGF-β-induced growth arrest.
To assess the involvement of different intracellular signaling pathways in the above TGF-β-induced responses, we made use of several small-molecule inhibitors to block endogenous protein kinases and monitored the resultant molecular changes. Among several inhibitors tested, we consistently observed that SB203580, a specific inhibitor of p38 kinase, completely blocked the TGF-β-induced caspase-3 activation (Figure 1B) and apoptosis (Figure 1C). In contrast, incubating NMuMG cells with the phosphatidylinositol 3-kinase (PI3K) inhibitor LY294002 augmented both serum deprivation and the TGF-β-induced apoptosis and caspase-3 activation (Figure 1B and C), concurring with the role of PI3K and its downstream target Akt as an anti-apoptotic signal (Brunet et al., 1999). A previous report showed that TGF-β could activate the PI3K/Akt pathway in NMuMG cells following 30 min of treatment, and the activation peaked at 2 h (Bakin et al., 2000). Consistent with this observation, we found that TGF-β modestly activated Akt during the first 3 h of treatment, but the activation was diminished thereafter (Figure 1B, see also Supplementary figure 1 available at The EMBO Journal Online). The low level of Akt activation may act to delay the TGF-β-induced apoptosis because blocking Akt activation by the PI3K inhibitor LY294002 accelerated the onset of apoptosis to 3 h following TGF-β treatment (Figure 1B and C).
In contrast to the apoptotic response, TGF-β-induced growth arrest was not affected by the p38 inhibitor SB203580. Treatment of NMuMG cells with increasing amounts of TGF-β gradually reduced the rate of DNA synthesis in these cells, as monitored by the [3H]thymi dine incorporation assay (Figure 1D). Addition of SB203580 did not alter the rate of thymidine incorporation, and thus the rate of DNA synthesis (Figure 1D).
The effect of the p38 kinase inhibitor on TGF-β-induced EMT was also examined. In the absence of TGF-β, NMuMG cells appeared as typical epithelial cells with actin cytoskeleton and E-cadherin arranged in a cortical pattern at cell–cell junctions (Figure 2A and B). Following 24–36 h of TGF-β treatment, this regular, cobblestone-like pattern gave way to a spindle-shaped fibroblast-like pattern with formation of actin stress fibers, downregulation and delocalization of E-cadherin from cell junctions (Figure 2C and D; Miettinen et al., 1994; Piek et al., 1999). Addition of the p38 kinase inhibitor SB203580 completely blocked EMT (Figure 2E–H), much as it did the TGF-β-induced apoptotic response. The TGF-β-induced EMT appeared to uniquely rely on the p38 pathway, since treating NMuMG cells with U0126, a MEK1/2 inhibitor, had no effect on EMT (Figure 2I–L).

Fig. 2. p38 is required for TGF-β-induced EMT. NMuMG cells were incubated with the p38 kinase inhibitor SB203580 (E–H) or MEK1/2 inhibitor U0126 (I–L) in the absence (A, B, E, F, I and J) or presence of TGF-β (C, D, G, H, K and L) for 36 h, followed by staining for the actin cytoskeleton and E-cadherin. Control cells in the absence of protein kinase inhibitor (A–D).
Taken together, these results indicate that p38 is required for the TGF-β-induced apoptosis and EMT in NMuMG cells, but not growth arrest.
TGF-β activates p38 through a type I receptor-mediated mechanism
Activation of p38 is mediated through phosphorylation at two residues, Thr180 and Tyr182, by upstream MAPK kinases (MKKs), such as MKK3 or MKK6 (reviewed in Ono and Han, 2000). The p38 pathway plays an essential role in regulating many cellular processes, such as inflammation, cell differentiation and death. In NMuMG cells, activation of p38 itself was sufficient to induce apoptosis (see Supplementary figure 2). To detect the activated p38, we used an anti-phospho-p38 antibody that only recognizes the dual phosphorylated p38. In NMuMG cells, activation of p38 was observed within 30 min of TGF-β stimulation and proceeded for at least 24 h (Figure 3A). Pre-incubation of NMuMG cells with the p38 inhibitor SB203580 completely blocked TGF-β-induced p38 activation. In contrast, SB203580 had no effect on the TGF-β-induced phosphorylation of Smad2 (Figure 3B). Interestingly, another member of the stress-activated protein kinases, JNK, was not activated following TGF-β treatment in NMuMG cells, although it was readily activated by anisomycin, a known activator for both JNK and p38 (Figure 3C). In addition to p38, two upstream MKKs, MKK3 and MKK6, were also activated by TGF-β, as evident in immunoblot analyses using phospho-specific antibodies against these two kinases (Figure 3D). In accordance with activation of p38 but not JNK, MKK4, the upstream kinase of JNK, was not activated by TGF-β (data not shown). These results indicate that TGF-β specifically activates the p38 branch of the MAPK pathway in NMuMG cells.

Fig. 3. TGF-β-induced p38 activation. (A) Time course of TGF-β-induced activation of p38 (left panel). SB203580 inhibited TGF-β-induced p38 activation (right panel). Cell lysates were harvested after incubation with or without TGF-β for the indicated times in the presence or absence of SB203580. (B) SB203580 did not inhibit TGF-β-induced Smad2 phosphorylation. Cell lysates were harvested after incubation with or without TGF-β for 1 h. (C) TGF-β did not activate JNK kinase, although JNK can be readily activated by anisomycin (Aniso) in NMuMG cells. Cell lysates were subjected to in vitro kinase assay for JNK activities using purified glutathione S-transferase–c-Jun as substrate following immunoprecipitation of JNK1. (D) TGF-β-induced activation of MKK3 and MKK6, the MKKs upstream of p38.
To determine which known component of the TGF-β pathway is responsible for the activation of p38, we examined the effect of dominant-negative mutants of TGF-β type I receptor, Smad3, and the inhibitory Smad, Smad7, in transfected NMuMG cells. Following transfection and immunoprecipitation of hemagglutinin (HA)-tagged p38, the activation of p38 was analyzed by phospho-p38 immunoblotting. Like its endogenous counterpart, the transfected p38 was also activated in response to TGF-β (Figure 4A). This activation could be blocked by TβRI(KR), a kinase-deficient mutant type I receptor, but not by Smad3ΔSSVS, the C-terminal truncated Smad3 that acts as a dominant-negative inhibitor in Smad-mediated TGF-β responses (Choy et al., 2000). The inhibitory Smad7, which suppresses TGF-β signaling by interfering with receptor-mediated activation of Smad2 and Smad3 (Hayashi et al., 1997; Nakao et al., 1997), also failed to block the p38 activation (Figure 4A). These results suggest that the TGF-β-mediated p38 activation relies on the kinase activity of TGF-β type I receptor but does not appear to involve a Smad-mediated mechanism.

Fig. 4. Dominant-negative TGF-β receptor but not dominant-negative Smad3 or inhibitory Smad, Smad7, blocks TGF-β-induced p38 activation and apoptosis. (A) TβRI(KR), a kinase-deficient mutant type I receptor, but not Smad3ΔSSVS, or inhibitory Smad, Smad7, inhibited TGF-β-mediated p38 activation. NMuMG cells, transfected with HA-p38 and the indicated expression plasmids, were incubated with or without TGF-β for 2 h, then TGF-β-mediated p38 activation was assayed by phospho-p38 blotting (top panel) following anti-HA immunoprecipitation of transfected p38. The levels of HA-p38 in the immunoprecipitates (middle panel) and FLAG-tagged Smads or receptor in total cell lysates (bottom panel) were shown as indicated. (B) TβRI(KR), Smad3ΔSSVS and Smad7 all inhibited the antiproliferative effect of TGF-β as measured by DNA synthesis. (C) TβRI(KR), but not Smad3ΔSSVS or Smad7, blocks TGF-β-induced apoptosis. Apoptosis was assayed as in Figure 1A. White bars, without TGF-β treatment; black bars, with TGF-β treatment. RI, TβRI.
We next examined the effect of TβRI(KR), Smad3ΔSSVS and Smad7 on TGF-β-induced apoptosis in NMuMG cells. Although expression of either Smad3ΔSSVS or Smad7 efficiently blocked TGF-β-mediated growth arrest (Figure 4B), the TGF-β-induced apoptotic response was not affected (Figure 4C). In contrast, TβRI(KR) blocked both TGF-β-induced apoptosis and growth arrest (Figure 4B and C). Interestingly, expression of Smad3ΔSSVS and Smad7, especially the latter, actually increased serum deprivation-induced apoptosis in the absence of TGF-β. This is consistent with previous reports that overexpression of Smad7 sensitizes various cells to cell death, suggesting that Smad7 may have other cellular functions in the absence of TGF-β signaling (Landstrom et al., 2000; Mazars et al., 2001).
Mutant TGF-β type I receptor retains kinase activity but is unable to activate the Smad pathway
To investigate the possibility that TGF-β activates the p38 pathway through a Smad-independent mechanism, we sought a mutant TGF-β type I receptor that is unable to activate Smads but retains kinase activity. Previous studies have indicated that the L45 loop between kinase subdomains IV and V of the type I receptor defines the signaling specificity (Feng and Derynck, 1997). Exchanging three diverged residues in the L45 loop between TGF-β and BMP type I receptors converts the TGF-β type I receptor to bind and activate BMP pathway-specific Smads (Chen et al., 1998). We speculate that replacing these three L45 loop residues with unrelated alanines would abolish the ability of the receptor to recognize any Smad as its substrate. Therefore, we introduced alanine to each of the N265, D267 and N268 positions in the L45 loop and analyzed the resultant TGF-β type I receptor, TβRImL45, for its ability to interact and phosphorylate Smads. Immediately following ligand binding, activated TGF-β receptors form a transient complex with Smad2 or Smad3, in which the C-terminal phosphorylation of Smad proteins occurs. This transient interaction can be detected using C-terminal-tagged Smads, as demonstrated previously (Zhang et al., 1996). To examine the ability of TβRImL45 to interact with Smad, we transfected receptor expression constructs along with C-terminally Myc-tagged Smad2 or Smad3 into COS-1 cells. After treating the transfected cells with [125I]TGF-β1 and chemical cross-linking cell surface [125I]TGF-β1 affinity-labeled receptors, Smad-bound recep tors were analyzed following immunoprecipitation of Smad using anti-Myc antibodies. Compared with the wild-type receptor, TβRImL45 showed complete loss of interaction with either Smad2 or Smad3 (Figure 5B, middle panel), despite the fact it was expressed at a level comparable to that of the wild-type receptor and was presented to the cell surface to bind [125I]TGF-β1 (Figure 5B, top panel).
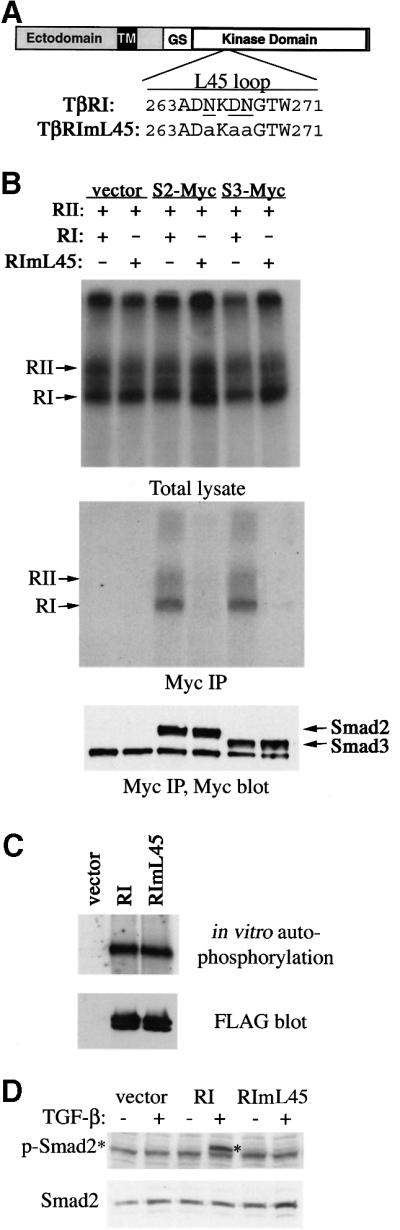
Fig. 5. Generation of a mutant TGF-β type I receptor that no longer activates Smad. (A) Schematic representation of TβRI and amino acid sequence alignment of L45 loops of the wild-type and mutated type I receptor. The three residues that differ between the TGF-β and BMP type I receptors are underlined. Mutations in TβRImL45 are shown in lower case. (B) L45 loop-mutated type I receptors can no longer associate with Smad2 or Smad3. COS-1 cells were transfected with expression plasmids for FLAG-tagged RII and RI, and for C-terminal Myc-tagged Smad2 or Smad3. [125I]TGF-β binding and cross-linking allowed the detection of receptor complex at the cell surface (top panel). Immunoprecipitation using Myc antibody showed co-precipitation of Smad2 or Smad3 with ligand-bound RII and wild-type RI, but not with mutated RI (middle panel). Smad2 and Smad3 levels were assessed by anti-Myc blotting (lower panel). (C) In vitro kinase activity of type I receptors. Transfected type I receptors in COS-1 cells were immunoprecipitated using anti-FLAG antibodies and subjected to in vitro kinase assay using [γ-33P]ATP and SDS–PAGE. (D) RIBL17 cells expressing the TβRI or TβRImL45 were stimulated with TGF-β for 1 h and C-terminal serine phosphorylation of Smad2 was examined by blotting of cell lysates using the anti-phospho-Smad2 antibody (upper panel). The level of Smad2 protein was analyzed by anti-Smad2 blotting (lower panel). RI, TβRI; RII, TβRII.
To determine the ability of TβRImL45 to phosphorylate Smads, we first tested whether this mutant receptor still possesses kinase activity. Using an in vitro kinase assay analyzing the ability of receptors to undergo autophosphorylation, we found that TβRImL45 displayed strong kinase activity at levels similar to that of the wild-type receptor (Figure 5C). To determine the ability of TβRImL45 to phosphorylate Smad2, we expressed this mutant receptor in RIBL17 cells, a type I receptor-deficient cell line (Cárcamo et al., 1994). These cells were normally incapable of phosphorylating Smad2 in response to TGF-β (Figure 5D). Introducing the wild-type but not the mutant TβRImL45 type I receptor restored the TGF-β-induced phosphorylation of endogenous Smad2 in RIBL17 cells (Figure 5D). Thus, despite retaining an active kinase domain, TβRImL45 is unable to phosphorylate Smad2.
In agreement with its inability to interact with and activate Smads, TβRImL45 is also incapable of eliciting Smad-dependent downstream TGF-β-induced transcriptional responses. We tested the ability of TβRImL45 to activate several reporter assays commonly used to measure the Smad-dependent transcriptional responses. In contrast to the wild-type receptor, TβRImL45 was unable to reconstitute the TGF-β-induced transcriptional responses from a Smad- and AP-1-dependent reporter, 3TP-lux (Wrana et al., 1992) (Figure 6A); a Smad3- and Smad4-dependent reporter, 4×SBE-luc (Zawel et al., 1998) (Figure 6B); or a FAST-1 binding site-containing Smad2-dependent reporter, 3×ARE-luc (Chen et al., 1996; Hayashi et al., 1997) (Figure 6C). In addition, TβRImL45 was also unable to activate reporters derived from promoters of the CDK inhibitor, p15ink4 and p21wwp, -113p15-luc (Figure 6D) and p21p-luc (Figure 6E), respectively. Activation of these two reporters has been shown to correlate with cell cycle arrest (Datto et al., 1995; Li et al., 1995). To rule out the possibility that the three alanine substitutions in the L45 loop rendered this receptor able to mediate a BMP-like response, we tested the ability of TβRImL45 to activate a BMP-specific 15×GCCG-luc reporter (Kusanagi et al., 2000). In this case, neither the wild-type TβRI nor mutant TβRImL45 receptor was able to activate the BMP-responsive promoter in response to TGF-β (Figure 6F), although expression of a constitutively active BMP type I receptor, Alk6(QD), markedly increased the luciferase activity. These results indicated that TβRImL45 is not only incapable of interacting with and phosphorylating Smads, but is also incapable of eliciting the Smad-mediated downstream TGF-β signaling responses. Therefore, this mutant receptor, TβRImL45, provides a very useful tool to address Smad-independent TGF-β receptor signaling.

Fig. 6. TβRImL45 is inactive in TGF-β- and Smad-mediated transcription response. Luciferase reporter assays of wild-type or mutant TβRImL45 in RIBL17 cells. White bars, without TGF-β treatment; black bars, with TGF-β treatment. Values are relative to control vector-transfected cells in the absence of TGF-β. RI, TβRI.
TβRImL45 is sufficient to activate p38 and promote apoptosis independently of receptor-mediated Smad activation
To investigate whether TGF-β could activate p38 independently of Smad activation, we first examined the ability of TβRImL45 to activate p38 in type I receptor-deficient RIBL17 cells. The activities of the co-transfected HA-tagged p38 were determined by in vitro kinase assay using purified ATF-2 as substrate. In this experiment, both wild-type TβRI and TβRImL45 were able to restore the TGF-β-induced activation of p38 (Figure 7A). We then tested the activation of p38 by TβRImL45 in NMuMG cells. To avoid activating the endogenous TGF-β receptors of NMuMG cells through ligand stimulation, we introduced a second activating mutation in the TβRImL45. This mutation was first reported in the constitutively active receptor TβRI(TD) (Wieser et al., 1995). Thus, the combined mutant type I receptor, TβRImL45(TD), should have a constitutively active kinase domain but be deficient in Smad binding and activation. Indeed, TβRImL45(TD) was unable to activate Smad-mediated transcriptional responses, as measured using both 3TP-Lux and 4×SBE-Luc reporters (Figure 7B), but it was able to activate p38 as potently as the original TβRI(TD) (Figure 7C). As shown in Figure 7D, TβRImL45(TD) activated p38 in a dose-dependent manner. In contrast, a kinase-deficient version of this mutant receptor TβRImL45(KR) did not activate p38 (Figure 7C). These results indicate that TGF-β-induced activation of p38 is independent of receptor-mediated Smad activation and occurs through a Smad-independent, alternate TGF-β receptor signaling pathway.

Fig. 7. TGF-β type I receptor activates p38 independently of Smad activation. (A) TβRImL45 activated p38 in response to TGF-β. RIBL17 cells expressing HA-p38 and the wild-type or mutant receptor were stimulated with TGF-β for 30 min, and activities of p38 were examined by in vitro kinase assay using purified ATF-2 as substrate. (B) Transcriptional assays of constitutively active TβRI(TD) or TβRImL45(TD) in NMuMG cells using 3TP-Lux (open bar) or 4×SBE-Luc (striped bar) reporters. (C) Constitutively active TβRImL45(TD), but not kinase-deficient TβRImL45(KR), activated p38. NMuMG cells were transfected with HA-p38 and the indicated expression plasmids. Activities of transfected p38 were measured by phospho-p38 or phospho-ATF-2 blotting after an in vitro kinase reaction using ATF-2 as substrate following immunoprecipitation of transfected HA-p38. (D) TβRImL45(TD) activated p38 in a dose-dependent manner. Experiments were performed as in (C). The levels of HA-p38 in the immunoprecipitates and FLAG-tagged receptors in total cell lysates were shown as indicated. RI, TβRI.
To examine the ability of TβRImL45(TD) to induce apoptosis and EMT, we generated pools of NMuMG cells expressing transfected TβRI(TD) or TβRImL45(TD). A plasmid encoding yellow fluorescent protein (YFP) was included in the transfection for selecting positively transfected cells by fluorescence-activated cell sorting (FACS). Expressing TβRI(TD), but not TβRImL45(TD), in these cells caused Smad2 phosphorylation (Figure 8A, lower panel). However, both TβRI(TD) and TβRImL45 (TD) induced apoptosis comparing to the control vector (Figure 8B). Interestingly, only constitutively active TβRI(TD) resulted in EMT, whereas none of the pools for TβRImL45(TD) showed complete EMT even when the expression level of TβRImL45(TD) was much higher than TβRI(TD) (Figure 8C). This result suggests that while the p38 pathway activated by TβRImL45 is sufficient in inducing apoptosis, a fully fledged EMT response requires additional input from the TGF-β pathway and possibly requires a Smad-mediated response. This is consistent with the observation that Smads in the TGF-β pathway synergize with TGF-β receptor to regulate EMT (Piek et al., 1999).

Fig. 8. TβRImL45(TD) is sufficient to promote apoptosis but not sufficient for EMT. (A) Generation of pools of NMuMG cells that express constitutively active wild-type TβRI(TD) or mutant TβRImL45(TD). Upper panel: expression level of FLAG-tagged TβRI(TD) and TβRImL45(TD); lower panel: TβRI(TD), but not TβRImL45(TD), induced phosphorylation of Smad2. Control: cell lysates of COS-1 cells transfected with FLAG-tagged TβRI. (B) Both constitutively active wild-type receptor and constitutively active mutant type I receptor-promoted apoptosis. Apoptosis was assayed as in Figure 1A. (C) TβRI(TD), but not TβRImL45(TD), promoted EMT. One microgram of each plasmid DNA was used in (A) and (B). For (C), 1 µg of TβRI(TD) or 4 µg of TβRImL45(TD) were used.
Discussion
Recent insight into cellular signal transduction mechanisms from a variety of studies has given rise to the appreciation that cells respond to extracellular environmental cues through a web of intertwined molecular actions, rather than a linear pathway to elicit a diverse array of responses (Hunter, 2000; Jordan et al., 2000). Using a mutant TGF-β type I receptor that lacks the ability to activate Smads, but retains kinase activity, we have demonstrated the existence of a Smad-independent TGF-β receptor signaling pathway. In this non-canonical, Smad-independent pathway, activation of p38 is required for the TGF-β-induced apoptosis and EMT, but not for the TGF-β-mediated growth inhibition. This mutant receptor is sufficient to activate p38 and cause NMuMG cells to undergo apoptosis. However, it is not sufficient to induce EMT, suggesting that signaling input from the Smad-mediated pathway is also required to generate a fully fledged EMT response. Thus, three distinct responses of TGF-β, growth arrest, apoptosis and EMT, use different downstream signaling pathways, employing either a Smad-dependent or a Smad-independent mechanism, or both. These results provide an explanation for the ability of TGF-β to induce diverse biological responses.
The generation of the Smad-independent TGF-β type I receptor provides a useful tool to dissect the molecular mechanisms underlying different functions of TGF-β receptor signaling. Previous studies have indicated that the TGF-β receptor-activated Smad protein complex is directly involved in the antiproliferative effect of TGF-β by binding to promoters of CDK inhibitors, p15ink4b, p21cip1 and the proto-oncogene c-myc, in cooperation with other transcription factors (Feng et al., 2000; Pardali et al., 2000; Chen et al., 2001; Seoane et al., 2001; Yagi et al., 2002). Consistent with previous observations, the mutant TGF-β receptor we generated, acting in a Smad-independent manner, was unable to activate the p15ink4b and p21cip1 promoters.
In serum-free medium, TGF-β treatment induces apoptosis in NMuMG cells. Prolonged serum deprivation eventually also causes cell death, but the apoptosis specifically triggered by TGF-β is clearly seen within 24 h. Nevertheless, the TGF-β-induced apoptosis in NMuMG cells has a delayed onset compared with the apoptotic response induced by other death-promoting stimuli. These slow kinetics can be attributed to the activation of the PI3K/Akt cell survival pathway during the early stage of TGF-β treatment. We have observed a modest Akt activation in NMuMG cells, as have other research groups (Bakin et al., 2000; Shin et al., 2001), but the activated Akt diminished after 4 h (see Supplementary figure 1). Apparently, this low level of transient activation of the Akt pathway is not sufficient to trigger the reversal of a late-stage robust apoptotic program. Indeed, the TGF-β-induced apoptosis occurred at a much earlier time after pretreatment of NMuMG cells with LY294002, an inhibitor of PI3K that also inhibits Akt activation (Figure 1C). In contrast to the TGF-β-induced antiproliferative effect, activation of the mutant receptor that acts independently of Smads is sufficient to cause NMuMG cells to undergo apoptosis.
Recently, DAXX, a protein previously found to associate with Fas receptor, has been reported to directly associate with TGF-β type II receptor, and is implicated in the TGF-β-mediated cell death through activation of JNK in B-cell lymphomas and mouse hepatocytes (Perlman et al., 2001). However, DAXX is unlikely to account for TGF-β-induced cell death in all types of cells. In NMuMG cells, no TGF-β-induced JNK activation was detected, and DAXX is mainly localized in the nucleus (data not shown). Nevertheless, since both JNK and p38 belong to the same family of stress-activated MAPKs, and both play important roles in apoptotic responses (Davis, 2000). We do not preclude JNK from playing a role in TGF-β-induced apoptosis in other cell types. It is possible that by activating either JNK or p38, TGF-β promotes its apoptotic response. Our studies also do not exclude the possibility that TGF-β-activated JNK or p38 converges on Smads, in parallel to the direct effect of JNK and p38, to reach the maximal magnitude of TGF-β-induced apoptosis.
Using the L45 loop mutant type I receptor TβRImL45, we found that both Smad-dependent and -independent mechanisms were required for the TGF-β-induced EMT. Bhowmick et al. (2001a) reported that TGF-β could activate RhoA in NMuMG cells, and overexpression of a mutant RhoA or mutant Rho-associated kinase ROCK inhibited EMT in a dominant-negative manner. These observations have implicated the RhoA-dependent signaling pathway in TGF-β-mediated EMT. We also observed the activation of RhoA by TGF-β in NMuMG cells (data not shown), but it is not clear whether RhoA functions in parallel or in sync with p38 in inducing EMT. Neither is it clear whether RhoA is activated by a Smad-dependent or -independent mechanism. However, without Smad activation, TGF-β receptor activation could not induce the EMT. These results indicate that both Smad-dependent and -independent signaling from the TGF-β receptor are required for the TGF-β-induced EMT. This conclusion is consistent with the report from another group that Smad proteins synergize with the activated TGF-β receptor to promote EMT (Piek et al., 1999).
At present, it is not clear how the type I TGF-β receptor activates the p38 pathway. The ability of the TGF-β type I receptor to activate the p38 most likely requires the kinase activity of the receptor. The kinase-deficient TGF-β type I receptor could no longer activate the p38 and acted as a dominant-negative inhibitor of the TGF-β-mediated p38 activation. Several lines of evidence indicate that the Rho family of small GTPases can mediate JNK and possibly p38 activation by direct interaction with several members of the MEKK group and the members of the mixed-lineage protein kinase group of MAPKKK (Fanger et al., 1997). Further studies regarding a potential involvement of MAPKKK and the Rho family of small GTPases in TGF-β-mediated activation of p38 may provide additional insight into the diversity of TGF-β signaling pathways.
The current findings that both TGF-β-mediated EMT and apoptosis, but not growth arrest, require a Smad-independent signaling pathway are consistent with the tumor-promoting role of TGF-β. Tumor cells often develop a resistance to TGF-β-induced growth inhibition, but retain an otherwise functional TGF-β signaling pathway (reviewed in Derynck et al., 2001; Wakefield and Roberts, 2002). TGF-β exerts its positive influence on tumor development through its effects on tumor cell invasion and changes in the tumor microenvironment. The EMT of tumor cells has an important role in tumor metastasis and invasiveness (Thiery and Chopin, 1999). In addition, TGF-β can also promote tumor progression through inducing apoptosis in nearby lymphocytes, thereby enabling the tumor cells to evade and destroy the immune system.
Finally, the existence of different signaling pathways other than Smad, downstream of the TGF-β receptor, resembles the Wingless/Wnt signaling pathway, which, like the TGF-β signaling pathway, also play a role in both development and tumorigenesis. Recent studies in both invertebrates and vertebrates indicate that known com ponents of the canonical Wingless/Wnt signaling pathway can function independently of Armadillo/β-catenin to establish planar cell polarity through a JNK or RhoA signaling cascade (McEwen and Peifer, 2000). Our study indicates that TGF-β signaling pathways can also use multiple intracellular signaling cascades. Further dissection of downstream targets of the TGF-β receptor by genetic and biochemical means will deepen our understanding of mechanisms that regulate TGF-β signaling.
Materials and methods
Expression plasmids
Expression plasmids for C-terminal FLAG-tagged wild-type TβRI, kinase-deficient TβRI(KR) (Feng et al., 1995) and constitutively active TβRI(TD) (Choy and Derynck, 1998) were used as parental plasmids to create L45 loop-mutated type I receptors using PCR-based approaches. Expression plasmids encoding C-terminal FLAG-tagged type II receptor TβRII, C-terminal Myc-tagged Smad2 and Smad3, N-terminal FLAG-tagged Smad7 and dominant-negative Smad3ΔSSVS have been described previously (Zhang et al., 1996; Choy et al., 2000). Plasmids for HA-tagged p38 were kindly provided by Dr Z.-G.Liu (NCI/NIH).
Cell culture
The mutant mink lung epithelial cells (RIBL17) provided by Dr J.Massagué, COS-1 cells (CRL1650, ATCC) and mouse mammary gland epithelial NMuMG cells (CRL1636; ATCC) were cultured as described previously (Cárcamo et al., 1994; Piek et al., 1999). Unless stated otherwise, cells were transfected using LipofectAmine plus (Invitrogen).
Reagents
The PI-3 kinase inhibitor LY294002 was from Cell Signaling Technology and used at 50 µM; the p38 kinase inhibitor SB203580 and MEK1/2 inhibitor U0126 were from Calbiochem and used at 10 µM. Antibodies against phospho-p38, p38, phospho-c-Jun, JNK1, phospho-MKK3/6, MKK3, phospho-Akt, Akt, cleaved caspase-3 (17 kDa) and full-length caspase-3 were purchased from Cell Signaling Technology. Anti-MKK6 antibody (sc1991) was from Santa Cruz Biotech. Rabbit anti- phospho-Smad2 antibody (Faure et al., 2000) was kindly provided by Drs P.ten Dijke and C.-H.Heldin (Ludwig Institute for Cancer Research, Sweden). Anti-Smad2 antibody was from Transduction Laboratory.
Immunoprecipitation, in vitro kinase reaction and [125I]TGF-β cross-linking of receptor proteins
Immunoprecipitations and in vitro kinase assays were performed as described previously (Feng and Derynck, 1997) using anti-FLAG antibody-conjugated beads (Sigma), or anti-HA (Covance; HA11), anti-Myc (Santa Cruz; 9E10) or anti-JNK1 monoclonal antibodies absorbed in protein G–Sepharose (Pharmacia). For receptor autophosphorylation, 5 µCi of [γ-33P]ATP (5000 µCi/mmol; NEN) were included in the reaction. For JNK or p38 kinase activities, 2 µg of purified c-Jun or ATF-2 (New England Biolab) were used as substrates, respectively, and 1 mM ATP was included in the reaction. After 30 min incubation, the reaction was stopped by adding an equal volume of 2× SDS sample buffer and subjected to autoradiography or phospho-c-Jun or phospho-ATF-2 blotting after electrophoresis. Cross-linking of cell surface TGF-β receptors with [125I]TGF-β1 (NEN) was carried out as described previously (Zhang et al., 1996).
Thymidine incorporation assay
For DNA synthesis inhibition assays, cells (2–4 × 104) plated in 24-well plates were incubated with or without different concentrations of TGF-β for 24 h; then 5 µCi/ml [3H]thymidine (NEN) was added during the last 3 h of incubation. The incorporation of acid-insoluble [3H]thymidine was measured as described previously (Zhang et al., 1996).
Transcriptional assay
RIBL17 or NMuMG cells were transfected using Fugene 6 (Roche Molecular Biochemicals) in 6-well plates. Plasmid pTK-RL (Promega), which expresses Renilla luciferase under the control of the TK promoter, was included in all transfections to normalize transfection efficiency. For each transfection, 0.5 µg of each expression plasmid, 0.5 µg of luciferase reporter plasmid and 10 ng of pRL-TK plasmid were used. The total plasmid concentration was kept constant and, when needed, vector DNA was added. Twelve hours after transfection, cells were incubated with or without 5 ng/ml TGF-β for 18–20 h before harvest. Luciferase activities were measured in a MicroLumatPlus luminometer (Perkin-Elmer) using the dual luciferase reporter assay system (Promega).
Apoptosis assay
NMuMG cells plated in 6-well plates were incubated with or without 5 ng/ml TGF-β in 0.2% FBS medium without insulin for 24 h or for the time period as indicated in Figure 1. When needed, various protein kinase inhibitors were added 30 min before TGF-β stimulation. The cells were then trypsinized and combined with their medium to include the detached dead cells. Each sample was either stained with Trypan Blue (Bio-Whittaker) and counted with a hemacytometer for dead cells (blue) or subjected to Cell Death Detection ELISA assay (Roche Molecular Biochemicals) for the quantitative determination of DNA fragmentation using both anti-histone and peroxidase-conjugated anti-DNA antibodies. For each treatment, duplicate experiments were repeated at least three times.
Transdifferentiation and fluorescence staining
NMuMG cells seeded on 2-well chamber slides were stimulated with 5 ng/ml TGF-β in the presence of 10% FBS. The high serum concentration in the medium protects cells from apoptosis. After 36 h of stimulation, cells were fixed with 4.5% paraformaldehyde and permeabilized in 0.2% Triton X-100. The actin cytoskeleton was stained using fluorescent or Texas Red-conjugated phalloidin (Molecular Probe). E-cadherin or transfected FLAG-tagged TβRI was stained first with anti-E-cadherin (Transduction Lab) or anti-FLAG antibodies, then incubated with fluorescent or Texas Red-conjugated secondary antibodies and analyzed by fluorescence microscopy.
Cell sorting
In the experiments requiring cell sorting, a YFP plasmid, pEYFP-N1 (Clontech), was added to other plasmids during transfection at a ratio of 1:20. Forty-eight hours later, cells were sorted according to their YFP expression levels by a FACS Vantage cell sorter (Becton and Dickinson). The sorted (fluorescent) cells were plated into 24-well plates at 20 000 cells per well or into chamber slides and allowed to recover from the procedure overnight before further analysis.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We are grateful to Dr Z.-G.Liu for providing us with many reagents and helpful suggestions for our study. We also would like to thank Drs J.Massagué, K.Miyazono, P.ten Dijke, C.-H.Heldin, J.Han, M.Greenberg, X.F.Wang and B.Vogelstein for kindly providing various reagents, Drs L.Samelson and S.Y.Cheng for critically reading the manuscript, and Dr R.Derynck for suggestions. M.C.H. was a recipient of an NIH Summer Cancer Research Training Award from Carnegie Melon University.
References
- Bakin A.V., Tomlinson,A.K., Bhowmick,N.A., Moses,H.L. and Arteaga,C.L. (2000) Phosphatidylinositol 3-kinase function is required for transforming growth factor β-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem., 275, 36803–36810. [DOI] [PubMed] [Google Scholar]
- Bhowmick N.A., Ghiassi,M., Bakin,A., Aakre,M., Lundquist,C.A., Engel,M.E., Arteaga,C.L. and Moses,H.L. (2001a) Transforming growth factor-β1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell, 12, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick N.A., Zent,R., Ghiassi,M., McDonnell,M. and Moses,H.L. (2001b) Integrin β1 signaling is necessary for transforming growth factor-β activation of p38 MAPK and epithelial plasticity. J. Biol. Chem., 276, 46707–46713. [DOI] [PubMed] [Google Scholar]
- Brunet A. et al. (1999) Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell, 96, 857–868. [DOI] [PubMed] [Google Scholar]
- Cárcamo J., Weis,F.M.B., Ventura,F., Wieser,R., Wrana,J.L., Attisano,L. and Massagué,J. (1994) Type I receptors specify growth-inhibitory and transcriptional responses to transforming growth factor β and activin. Mol. Cell. Biol., 14, 3810–3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.R., Kang,Y. and Massagué,J. (2001) Defective repression of c-myc in breast cancer cells: a loss at the core of the transforming growth factor β growth arrest program. Proc. Natl Acad. Sci. USA, 98, 992–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Rubock,M.J. and Whitman,M. (1996) A transcriptional partner for MAD proteins in TGF-β signaling. Nature, 383, 691–696. [DOI] [PubMed] [Google Scholar]
- Chen Y.G., Hata,A., Lo,R.S., Wotton,D., Shi,Y., Pavletich,N. and Massagué,J. (1998) Determinants of specificity in TGF-β signal transduction. Genes Dev., 12, 2144–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy L. and Derynck,R. (1998) The type II transforming growth factor (TGF)-β receptor-interacting protein TRIP-1 acts as a modulator of the TGF-β response. J. Biol. Chem., 273, 31455–31462. [DOI] [PubMed] [Google Scholar]
- Choy L., Skillington,J. and Derynck,R. (2000) Roles of autocrine TGF-β receptor and Smad signaling in adipocyte differentiation. J. Cell Biol., 149, 667–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datto M.B., Yu,Y. and Wang,X.F. (1995) Functional analysis of the transforming growth factor β responsive elements in the WAF1/Cip1/p21 promoter. J. Biol. Chem., 270, 28623–28628. [DOI] [PubMed] [Google Scholar]
- Davis R.J. (2000) Signal transduction by the JNK group of MAP kinases. Cell, 103, 239–252. [DOI] [PubMed] [Google Scholar]
- Derynck R., Akhurst,R.J. and Balmain,A. (2001) TGF-β signaling in tumor suppression and cancer progression. Nat. Genet., 29, 117–129. [DOI] [PubMed] [Google Scholar]
- Engel M.E., McDonnell,M.A., Law,B.K. and Moses,H.L. (1999) Interdependent SMAD and JNK signaling in transforming growth factor-β-mediated transcription. J. Biol. Chem., 274, 37413–37420. [DOI] [PubMed] [Google Scholar]
- Fanger G.R., Johnson,N.L. and Johnson,G.L. (1997) MEK kinases are regulated by EGF and selectively interact with Rac/Cdc42. EMBO J., 16, 4961–4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure S., Lee,M.A., Keller,T., ten Dijke,P. and Whitman,M. (2000) Endogenous patterns of TGFβ superfamily signaling during early Xenopus development. Development, 127, 2917–2931. [DOI] [PubMed] [Google Scholar]
- Feng X.H. and Derynck,R. (1997) A kinase subdomain of transforming growth factor-β (TGF-β) type I receptor determines the TGF-β intracellular signaling specificity. EMBO J., 16, 3912–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X.H., Filvaroff,E.H. and Derynck,R. (1995) Transforming growth factor-β (TGF-β)-induced down-regulation of cyclin A expression requires a functional TGF-β receptor complex. Characterization of chimeric and truncated type I and type II receptors. J. Biol. Chem., 270, 24237–24245. [DOI] [PubMed] [Google Scholar]
- Feng X.H., Lin,X. and Derynck,R. (2000) Smad2, Smad3 and Smad4 cooperate with Sp1 to induce p15Ink4B transcription in response to TGF-β. EMBO J., 19, 5178–5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanafusa H., Ninomiya-Tsuji,J., Masuyama,N., Nishita,M., Fujisawa,J., Shibuya,H., Matsumoto,K. and Nishida,E. (1999) Involvement of the p38 mitogen-activated protein kinase pathway in transforming growth factor-β-induced gene expression. J. Biol. Chem., 274, 27161–27167. [DOI] [PubMed] [Google Scholar]
- Hannon G.J. and Beach,D. (1994) p15INK4B is a potential effector of TGF-β-induced cell cycle arrest. Nature, 371, 257–261. [DOI] [PubMed] [Google Scholar]
- Hartsough M.T. and Mulder,K.M. (1995) Transforming growth factor β activation of p44MAPK in proliferating cultures of epithelial cells. J. Biol. Chem., 270, 7117–7124. [DOI] [PubMed] [Google Scholar]
- Hayashi H. et al. (1997) The MAD-related protein Smad7 associates with the TGFβ receptor and functions as an antagonist of TGFβ signaling. Cell, 89, 1165–1173. [DOI] [PubMed] [Google Scholar]
- Hocevar B.A., Brown,T.L. and Howe,P.H. (1999) TGF-β induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J., 18, 1345–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T. (2000) Signaling—2000 and beyond. Cell, 100, 113–127. [DOI] [PubMed] [Google Scholar]
- Jhappan C., Geiser,A.G., Kordon,E.C., Bagheri,D., Hennighausen,L., Roberts,A.B., Smith,G.H. and Merlino,G. (1993) Targeting expression of a transforming growth factor β1 transgene to the pregnant mammary gland inhibits alveolar development and lactation. EMBO J., 12, 1835–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan J.D., Landau,E.M. and Lyengar,R. (2000) Signaling networks: the origins of cellular multitasking. Cell, 103, 193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusanagi K., Inoue,H., Ishidou,Y., Mishima,H.K., Kawabata,M. and Miyazono,K. (2000) Characterization of a bone morphogenetic protein-responsive Smad-binding element. Mol. Biol. Cell, 11, 555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landstrom M., Heldin,N.E., Bu,S., Hermansson,A., Itoh,S., ten Dijke,P. and Heldin,C.H. (2000) Smad7 mediates apoptosis induced by transforming growth factor β in prostatic carcinoma cells. Curr. Biol., 10, 535–538. [DOI] [PubMed] [Google Scholar]
- Lehmann K., Janda,E., Pierreux,C.E., Rytomaa,M., Schulze,A., McMahon,M., Hill,C.S., Beug,H. and Downward,J. (2000) Raf induces TGFβ production while blocking its apoptotic but not invasive responses: a mechanism leading to increased malignancy in epithelial cells. Genes Dev., 14, 2610–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.M., Nichols,M.A., Chandrasekharan,S., Xiong,Y. and Wang,X.F. (1995) Transforming growth factor β activates the promoter of cyclin-dependent kinase inhibitor p15INK4B through an Sp1 consensus site. J. Biol. Chem., 270, 26750–26753. [DOI] [PubMed] [Google Scholar]
- Massagué J. (1998) TGF-β signal transduction. Annu. Rev. Biochem., 67, 753–791. [DOI] [PubMed] [Google Scholar]
- Massagué J. (2000) How cells read TGF-β signals. Nat. Rev. Mol. Cell Biol., 1, 169–178. [DOI] [PubMed] [Google Scholar]
- Mazars A., Lallemand,F., Prunier,C., Marais,J., Ferrand,N., Pessah,M., Cherqui,G. and Atfi,A. (2001) Evidence for a role of the JNK cascade in Smad7-mediated apoptosis. J. Biol. Chem., 276, 36797–36803. [DOI] [PubMed] [Google Scholar]
- McEwen D.G. and Peifer,M. (2000) Wnt signaling: moving in a new direction. Curr. Biol., 10, R562–R564. [DOI] [PubMed] [Google Scholar]
- Miettinen P.J., Ebner,R., Lopez,A.R. and Derynck,R. (1994) TGF-β induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J. Cell Biol., 127, 2021–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao A. et al. (1997) Identification of Smad7, a TGFβ-inducible antagonist of TGF-β signalling. Nature, 389, 631–635. [DOI] [PubMed] [Google Scholar]
- Oft M., Peli,J., Rudaz,C., Schwarz,H., Beug,H. and Reichmann,E. (1996) TGF-β1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev., 10, 2462–2477. [DOI] [PubMed] [Google Scholar]
- Ono K. and Han,J. (2000) P38 signal transduction pathway: activation and function. Cell Signal., 12, 1–13. [DOI] [PubMed] [Google Scholar]
- Pardali K., Kurisaki,A., Moren,A., ten Dijke P., Kardassis,D. and Moustakas,A. (2000) Role of Smad proteins and transcription factor Sp1 in p21Waf1/Cip1 regulation by transforming growth factor-β. J. Biol. Chem., 275, 29244–29256. [DOI] [PubMed] [Google Scholar]
- Perlman R., Schiemann,W.P., Brooks,M.W., Lodish,H.F. and Weinberg,R.A. (2001) TGF-β-induced apoptosis is mediated by the adapter protein Daxx that facilitates JNK activation. Nat. Cell Biol., 3, 708–714. [DOI] [PubMed] [Google Scholar]
- Piek E., Moustakas,A., Kurisaki,A., Heldin,C.H. and ten Dijke,P. (1999) TGF-β type I receptor/ALK-5 and Smad proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J. Cell Sci., 112, 4557–4568. [DOI] [PubMed] [Google Scholar]
- Reynisdottir I., Polyak,K., Iavarone,A. and Massagué,J. (1995) Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-β. Genes Dev., 9, 1831–1845. [DOI] [PubMed] [Google Scholar]
- Sano Y., Harada,J., Tashiro,S., Gotoh-Mandeville,R., Maekawa,T. and Ishii,S. (1999) ATF-2 is a common nuclear target of Smad and TAK1 pathways in transforming growth factor-β signaling. J. Biol. Chem., 274, 27161–27167. [DOI] [PubMed] [Google Scholar]
- Selvakumaran M., Lin,H.K., Sjin,R.T., Reed,J.C., Liebermann,D.A. and Hoffman,B. (1994) The novel primary response gene MyD118 and the proto-oncogenes myb, myc and bcl-2 modulate transforming growth factor β1-induced apoptosis of myeloid leukemia cells. Mol. Cell. Biol., 14, 2352–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoane J., Pouponnot,C., Staller,P., Schader,M., Eilers,M. and Massagué,J. (2001) TGFβ influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat. Cell Biol., 3, 400–408. [DOI] [PubMed] [Google Scholar]
- Shin I., Bakin,A.V., Rodeck,U., Brunet,A. and Arteaga,C.L. (2001) Transforming growth factor β enhances epithelial cell survival via Akt-dependent regulation of FKHRL1. Mol. Biol. Cell, 12, 3328–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Dijke P., Miyazono,K. and Heldin,C.H. (2000) Signaling inputs converge on nuclear effectors in TGF-β signaling. Trends Biochem. Sci., 25, 64–70. [DOI] [PubMed] [Google Scholar]
- Thiery J.P. and Chopin,D. (1999) Epithelial cell plasticity in development and tumor progression. Cancer Metastasis Rev., 18, 31–42. [DOI] [PubMed] [Google Scholar]
- Wakefield L.M. and Roberts,A.B. (2002) TGF-β signaling: positive and negative effects on tumorigenesis. Curr. Opin. Genet. Dev., 12, 22–29. [DOI] [PubMed] [Google Scholar]
- Whitman M. (1998) Smads and early developmental signaling by the TGF-β superfamily. Genes Dev., 12, 2445–2462. [DOI] [PubMed] [Google Scholar]
- Wieser R., Wrana,J.L. and Massagué,J. (1995) GS domain mutations that constitutively activate TβR-I, the downstream signaling component in the TGF-β receptor complex. EMBO J., 14, 2199–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrana J.L. (2000) Regulation of Smad activity. Cell, 100, 189–192. [DOI] [PubMed] [Google Scholar]
- Wrana J.L., Attisano,L., Cárcamo,J., Zentella,A., Doody,J., Laiho,M., Wang,X.F. and Massagué,J. (1992) TGF β signals through a heteromeric protein kinase receptor complex. Cell, 71, 1003–1014. [DOI] [PubMed] [Google Scholar]
- Yagi K., Furuhashi,M., Aoki,H., Goto,D., Kuwano,H., Sugamura,K., Miyazono,K. and Kato,M. (2002) c-myc is a downstream target of the Smad pathway. J. Biol. Chem., 277, 854–861. [DOI] [PubMed] [Google Scholar]
- Zawel L., Dai,J.L., Buckhaults,P., Zhou,S., Kinzler,K.W., Vogelstein,B. and Kern,S.E. (1998) Human Smad3 and Smad4 are sequence-specific transcription activators. Mol. Cell, 1, 611–617. [DOI] [PubMed] [Google Scholar]
- Zhang Y. and Derynck,R. (1999) Regulation of Smad signalling by protein associations and signalling crosstalk. Trends Cell Biol., 9, 274–279. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Feng,X.H., Wu,R.Y. and Derynck,R. (1996) Receptor-associated Mad homologues synergize as effectors of the TGF-β response. Nature, 383, 168–172. [DOI] [PubMed] [Google Scholar]