

Abstract
Replication fork arrest is a source of genome re arrangements, and the recombinogenic properties of blocked forks are likely to depend on the cause of blockage. Here we study the fate of replication forks blocked at natural replication arrest sites. For this purpose, Escherichia coli replication terminator sequences Ter were placed at ectopic positions on the bacterial chromosome. The resulting strain requires recombinational repair for viability, but replication forks blocked at Ter are not broken. Linear DNA molecules are formed upon arrival of a second round of replication forks that copy the DNA strands of the first blocked forks to the end. A model that accounts for the requirement for homologous recombination for viability in spite of the lack of chromosome breakage is proposed. This work shows that natural and accidental replication arrests sites are processed differently.
Keywords: double-strand break repair/Escherichia coli/homologous recombination
Introduction
Genome instability is observed in numerous somatic and hereditary disorders and therefore plays a crucial role in human diseases (Luo et al., 2000; Khanna and Jackson, 2001). In addition, genome rearrangements are an important motor of species evolution (Spratt et al., 2001). DNA double-strand breaks (DSBs) are considered as one of the main sources of genetic rearrangements (Jasin, 2000; van Gent et al., 2001). Deletions, translocations or inversions often result from the joining of non-homologous sequences, catalyzed by the enzymatic complexes that process DNA DSBs. The properties of these enzymatic complexes are the subject of extensive research (reviewed in Paques and Haber, 1999; Aguilera et al., 2000; Petrini, 2000; Cromie et al., 2001). In contrast, the causes of spontaneous DSB or occurrence of double-stranded ends have remained more elusive. Failure to repair DNA lesions accurately or in a timely fashion is a recognized source of DNA double-stranded ends. Recently, DNA replication blockage has also emerged as playing an important role in genome instability. Natural replication arrest sites have been found in many organisms, several of them associated with local high recombination frequency (reviewed in Hyrien, 2000; Rothstein et al., 2000). Accidental replication arrests, such as those caused by a deficiency in a replication protein or by replication inhibitors, also induce chromosome instability (Bierne et al., 1997b; Saveson and Lovett, 1997; Chen and Kolodner, 1999; Myung et al., 2001; Saintigny et al., 2001; reviewed in Flores-Rozas and Kolodner, 2000). Natural and accidental replication pauses are therefore a potential source of genome instability in all organisms, from bacteria to mammals (reviewed in Haber, 1999; Schar, 2001; Sonoda et al., 2001).
A double-stranded end can be formed by replication when a replication fork runs into a single-strand interruption in the template DNA, a reaction termed collapse (Kuzminov, 1995, 2001). In Escherichia coli, several observations argue that a defect in a replication protein can lead to a specific reaction termed replication fork reversal (Seigneur et al., 1998; Flores et al., 2001; reviewed in Michel, 2000; Michel et al., 2001). According to the replication fork reversal model, when a replication fork is stalled, the newly synthesized strands unwind from the template strands and pair, allowing the parental strands to re-anneal. The four-way junction is recognized by RuvAB, a complex that catalyzes branch migration of Holliday junction recombination intermediates, and can be resolved by RuvC, resulting in chromosome breakage. However, annealing of the newly synthesized strands also produces a DNA double-stranded end, a target of RecBCD, the recombination complex that specifically repairs DSBs in E.coli. The RecBCD complex can either degrade the linear portion of DNA via its exonuclease V activity or initiate RecA-dependent homologous recombination, which results in the re-integration of the double-stranded end into the chromosome. Action of RecBCD prior to RuvABC prevents chromosome breakage.
Paradoxically, in spite of the potential deleterious consequences of arresting replication forks, replication arrest occurs naturally at specific replication arrest sites. In E.coli, bi-directional chromosome replication initiates at a specific origin (oriC), and the two replication forks progress at a similar speed to the terminus region, which is approximately opposite oriC. The terminus region contains several specific sites, Ter, which, upon binding of a protein named Tus, block replication in a polar manner (reviewed in Hill, 1996; Bussiere and Bastia, 1999). Ter sites are oriented in the terminus region to form a replication trap in which a fork coming from one direction is forced to wait for the arrival of the fork coming from the other direction (Figure 1A). The Tus–Ter complex acts as an anti-helicase, blocking the progression of the replisome helicase DnaB. The polarity of replication arrest results from the existence of specific interactions between DnaB and Tus proteins (Mulugu et al., 2001).

Fig. 1. Schematic representation of Lac-Phe-Ter chromosome. (A) Chromosome on which replication is blocked at Terphe and Terlac. (B) Cleavage of both leading and lagging strands. (C) Cleavage of two strands of the same polarity (leading or lagging strands), at both Ter sites. (D) DNA replication initiated at oriC and progressing to Terphe and Terlac. Natural terminators are symbolized by gray indented rectangles, and Terphe and Terlac by white indented rectangles. The chromosome origins (oriC) are shown as small circles. Each line represents double-stranded DNA. Cleavage sites are indicated by black arrows.
The rnh mutation allows replication initiation from non-oriC sequences on the E.coli chromosome and, in an rnh mutant, initiation of replication from a region close to natural Ter sites stimulates homologous recombination in the vicinity of Ter, in a Tus-dependent way. This finding suggested that replication forks blocked at Ter provided an entrance for the RecBCD enzyme into duplex DNA (Horiuchi et al., 1994). Escherichia coli strains carrying an additional Ter site were used to characterize further the hyper-recombination phenotype associated with replication arrest. In one case, a Ter site was inserted in the lacZ gene, blocking the clockwise replication fork halfway (lacZ-Ter strain; Horiuchi and Fujimura, 1995). In another study, two Ter sites were inserted back to back in the terminus region of the chromosome, blocking both forks and preventing the replication of a 2 kb spacer (invTer strain; Sharma and Hill, 1995). Expression of Tus in lacZ-Ter and invTer strains slightly compromised cell viability and led to SOS induction. Interestingly, lacZ-Ter and invTer strains required the homologous recombination proteins RecA and RecBC for viability. Furthermore, measures of recombination frequencies indicated an increased level of RecBC-dependent homologous recombination in the vicinity of the lacZ-Ter site, similar to the recombinational hot spot previously observed at Ter sites in their natural position (Horiuchi et al., 1994; Horiuchi and Fujimura, 1995). Since RecBCD specifically initiates DSB repair by homologous recombination in E.coli, these data led to the conclusion that arresting replication forks by additional Ter sites leads to the formation of DNA double-stranded ends, possibly by direct breakage of blocked forks. Additional support for this notion came from the observation that Ter-blocked forks can promote illegitimate recombination by a process that involves linear DNA intermediates (Bierne et al., 1997a).
In the present work, we investigated at the molecular level the consequences of prolonged replication blockage at a natural replication arrest site. For this purpose, we used a strain carrying two additional replication terminators, so that both replication forks are arrested midway between the origin and terminus (Figure 1A). The presence of these two terminators allowed us to test directly whether DSBs occur at Ter-blocked forks. We find that the DNA at blocked forks is not broken, nor reversed. Ter-blocked forks remain stable until a second round of replication reaches Ter and generates linear DNA by copying the first leading and lagging strands arrested at Ter to the end. We propose that homologous recombination, which is essential for viability in our strain, is required for the re-integration of this linear DNA into the intact chromosome and may play a role in the removal of Tus protein.
Results
A strain carrying two additional replication terminators requires homologous recombination for viability
In order to study the fate of E.coli replication forks arrested at natural termination sites, the Lac-Phe-Ter strain was constructed. This strain carries two additional TerB sites, one inserted in the lacZ gene (Terlac site) and one in the pheA gene (Terphe site) (Figure 1A, see Materials and methods for construction). To be able to control replication arrest, a Δtus mutant was used and a plasmid carrying an inducible ara–tus fusion was introduced at the last step of strain constructions (pBADtus; Sharma and Hill, 1995). In the final strains, Tus is expressed from the plasmid in the presence of arabinose, promoting replication arrest at Ter sites, and is repressed in the presence of glucose, rendering Ter sites inactive. The accumulation of stalled forks at Terlac and Terphe sites was verified by Southern hybridization. Ter-specific replication intermediates appeared 30 min after Tus induction and in 1 h reached a level of ∼50% of total DNA, which did not change significantly during at least another 2 h of growth in the presence of arabinose (not shown). In spite of the accumulation of replication intermediates, Tus induction only slightly compromised the viability of the Lac-Phe-Ter strain (Figure 2, wild type). The Lac-Phe-Ter strain formed colonies on arabinose and glucose plates with equal efficiencies; however, as previously observed for strains carrying ectopic Ter sites (Sharma and Hill, 1995), colonies were smaller on arabinose compared with glucose medium.

Fig. 2. recA, recB and ruvABC mutations are lethal in the Lac-Phe-Ter strain, dif deletion is not. At time 0, a culture growing in LB supplemented with glucose was centrifuged, resuspended in LB medium and split into two parts to which either glucose or arabinose was added. At each indicated time, an aliquot was withdrawn and appropriate dilutions were plated on LB medium supplemented with glucose. Open symbols, growth in glucose; closed symbols, growth in arabinose. Circles, Lac-Phe-Ter strain; squares, Lac-Phe-Ter dif; rectangles, Lac-Phe-Ter recA; diamonds, Lac-Phe-Ter recBC; triangles, Lac-Phe-Ter ruvABC.
It was reported previously that replication-blocked strains require at least two homologous recombination functions for viability: RecA, the main homologous recombination protein; and RecBC, which specifically binds to DNA double-stranded ends and is essential for the recombinational repair of DSBs (Horiuchi and Fujimura, 1995; Sharma and Hill, 1995). As expected, recA or recB inactivation caused a decrease in cell viability upon Tus induction (Figure 2). The lethality of the Lac-Phe-Ter strain in combination with the recB mutation was not due to the inactivation of the exonuclease V function of RecBCD, as a recD mutation did not affect Lac-Phe-Ter strain viability (not shown); these observations suggest a requirement for the repair of DSBs by homologous recombination. This hypothesis is strengthened by the need for RuvABC, the proteins that catalyze resolution of Holliday junctions, an intermediate in homologous recombination (Figure 2, ruvABC; similar results were obtained with the ruvC mutation, not shown). Further more, in contrast to wild-type and recD Lac-Phe-Ter strains, recA, recB, ruvABC and ruvC Lac-Phe-Ter strains did not form colonies on Luria broth (LB) agar plates or on minimal medium containing arabinose (not shown).
Chromosome recombination can lead to dimerization. Chromosome dimers prevent cell division and hence are lethal unless they are resolved into monomers by the action of the XerCD resolvase at the dif site (reviewed in Barre et al., 2001). Inactivation of Xer or deletion of dif did not cause a significant loss of viability of the Lac-Phe-Ter strain in the presence of arabinose (Figure 2), suggesting that dimer resolution is not required. Further more, dif (or xerC) Lac-Phe-Ter colonies appeared with equal efficiency on arabinose- and glucose-containing plates and were smaller on arabinose. Colony size was reduced on arabinose plates due to Tus induction similarly for dif and wild-type Lac-Phe-Ter strains (see Materials and methods). Altogether, these results confirm previous observations that blocking E.coli replication with additional Ter sites creates a need for DSB recombinational repair and show that such repair generally does not lead to the formation of chromosome dimers.
DNA double-stranded ends are formed at Ter sites
To determine whether the double-stranded ends acted upon by RecBCD are close to the replication arrest site, we treated the DNA of Lac-Phe-Ter recB cells, imbedded in agarose plugs, with NotI restriction enzyme, and analyzed it by Southern hybridization, using a recN PCR fragment as a probe (recN is located 14 kb upstream of Terphe; Figure 3). Only the expected linear NotI restriction fragment encompassing Ter hybridized with the recN probe was found when cells were grown in glucose, i.e. when Ter was inactive (Figure 3, lane 3). Two additional fragments were observed upon Tus induction (Figure 3, lane 4): a slightly more retarded band corresponding to Y-structure molecules with stalled forks; and a fast migrating band of ∼35 kb, the size of which corresponds to the distance between the upstream NotI site and Terphe. This experiment suggests that a DNA double-stranded end is formed at Terphe in replication blockage conditions.

Fig. 3. Tus induction leads to the appearance of a linear fragment upstream of Terphe only. Lanes 1 and 2, ethidium bromide staining of a pulsed-field gel of a NotI-digested Lac-Phe-Ter recB chromosome. Lanes 3 and 4, Southern blot of the same gel using recN DNA as a probe. Lanes 5 and 6, Southern blot of the same gel using recO DNA as a probe. Lanes 1, 3 and show 5 growth in glucose (G), and lanes 2, 4 and 6 show growth in arabinose (A). Sizes of the AB1157 chromosome NotI fragments are indicated on the left (Perkins et al., 1993). Because of replication arrest at Terphe and Terlac, the intensity of DNA fragments located downstream of these Ter sites is decreased in arabinose compared with glucose. It can be noted that the smallest Not band that hybridizes with recN is smeary, probably due to the in vivo erosion of the DNA end by various single-stranded exonucleases, such as SbcB, RecJ or ExoVII. The position of the gel origin (O), linear NotI fragments encompassing Terphe (L), replication intermediate molecules formed upon fork blockage at Terphe (Y) and the expected positions of migration of linear fragments from the upstream NotI to Terphe (NotI–Ter) and from Terphe to the downstream NotI site (Ter–NotI) are indicated. A schematic representation of the NotI fragment encompassing the Terphe site is shown. The positions of recN and recO genes used as probes and of Terphe are indicated. The indented rectangle represents a Tus-bound Terphe site. Chromosome replication proceeds from left to right. The distances from Terphe to upstream and downstream NotI sites are indicated (34.7 and 222.9 kb, respectively).
A recO probe located 38 kb downstream of Terphe was used to test the presence of a linear fragment corresponding to the sequence downstream of Ter. No linear molecule corresponding to the distance between Terphe and the downstream NotI site (220 kb) was detected, whereas, as expected, the recO probe hybridized with the linear NotI fragment encompassing Ter and with the Y-structure DNA molecules (Figure 3, lanes 5 and 6). This result suggests that the formation of the NotI–Terphe 35 kb linear fragment, upstream of Ter, is not accompanied by the formation of a detectable Terphe–NotI fragment, downstream of Ter.
Replication arrest at Ter induces the appearance of an ∼2 Mb linear chromosome fragment
Since the viability of the Lac-Phe-Ter strain depends on recombination enzymes that act at DNA double-stranded ends, the possibility that linearization of chromosomes occurs in Lac-Phe-Ter recB cells was analyzed by pulsed-field gel electrophoresis (PFGE). In PFGE, circular chromosomes remain trapped in the wells, whereas linear DNA migrates according to size. Intact cellular DNA of different strains was run on pulsed-field gels. As expected, no linear DNA was detected in RecB+ cells regardless of Tus induction (not shown), consistent with the notion that RecBCD degrades or recombines linear molecules in vivo. In the Phe-Lac-Ter recB strain, a band migrating as 4–6 Mb linear controls was present in Tus-repressed conditions (Figure 4, lane 3). Linear DNA of this size is also observed in recB single mutants and presumably results from spontaneous chromosome breakage (Seigneur et al., 1998; Figure 4, lane 2). Tus induction led to a decrease in the 4–6 Mb DNA and the appearance of a specific ∼2 Mb linear fragment, a size corresponding to the distance between the two additional Ter sites (Figure 4, lane 4). The ∼2 Mb linear DNA was not observed in strains that carry only one additional Ter site (not shown).
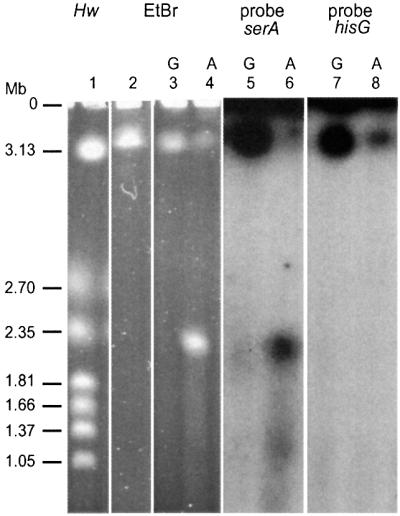
Fig. 4. Tus induction leads to the formation of 2 Mb linear molecules that hybridize with the oriC half of the chromosome only. Lane 1, Hanselina wingei (Hw) chromosome size marker. Lanes 2–4, ethidium bromide staining of a pulsed-field gel of intact recB (lane 2) or Lac-Phe-Ter recB (lanes 3–4) chromosome. Lanes 5 and 6, Southern blot of lanes 3 and 4 using the serA gene DNA as a probe (oriC-proximal probe). Lanes 7 and 8, Southern blot of the same lanes using the hisG gene DNA as a probe (terminal region probe). Lanes 3, 5 and 7 show growth in glucose, and lanes 2, 4, 6 and 8 show growth in arabinose. The position of the gel origin (O) is indicated. Schizosaccharomyces pombe chromosomes were also used routinely as size markers, indicating that linear chromosomes of 3–6 Mb migrate in pulsed-fields gels in the conditions used. It should be noted that for unknown reasons, the apparent intensities of bands in pulsed-field gels performed with entire E.coli chromosomes may be misleading, which is the reason why precise quantification of the amount of DNA in different regions of the gels was performed by counting [3H]thymidine-labeled DNA.
The ∼2 Mb linear fragment that appears upon Tus induction could correspond to the origin containing half of the chromosome, or the terminus half, or a mixture of both. To distinguish between these possibilities, pulsed-field gels were analyzed by Southern hybridization with probes located in either half of the chromosome. The oriC-proximal probe (serA gene) hybridized with the ∼2 Mb fragment, whereas the terminal region probe (hisG gene) did not (Figure 4, lanes 6 and 8). As expected, both probes hybridized with the intact DNA trapped in the wells and with the >3.5 Mb linear DNA. Lack of hybridization of the hisG probe suggests that sequences downstream of Ter are not present within the ∼2 Mb DNA band, which is consistent with the lack of detection of sequences downstream of Terphe in the hybridization experiment of NotI restriction fragments (Figure 3).
Specific cleavage of either only the lagging strand templates or only the leading strand templates at both Terlac and Terphe blocked forks would result in the formation of linear molecules larger than a chromosome unit size, 4.6 Mb (Figure 1C). The size of the linear fragment (∼2 Mb, Figure 4) argues against such strand-specific cleavage. Random cleavage of leading and lagging strand templates would generate an oriC-carrying half and a terminus-carrying half of the chromosome, each of ∼2.3 Mb (Figure 1B), which is difficult to reconcile with recombinational repair of broken chromosomes, with the lack of detection of a double-stranded end downstream of Terphe (Figure 3) and with the absence of hybridization of the Ter-induced linear DNA with a terminus-proximal probe (Figure 4). The appearance of linear molecules of ∼2 Mb that hybridize only with the oriC half of the chromosome suggests a model in which Ter-blocked forks are stable, and double-stranded ends are formed when a second round of replication copies the first leading and lagging strands to the end, a reaction hereafter named collapse (Figure 1D). To test this hypothesis, pulsed-field gels with labeled DNA were analyzed.
Replication arrest at Ter leads to collapse of subsequent replication forks
To quantify the effects of Tus, chromosomes were labeled in vivo with [3H]thymidine. After PFGE, each gel lane was cut into slices and the amount of tritiated DNA in each slice was measured. In a first series of experiments, [3H]thymidine was present prior to and during Tus induction. The total amounts of DNA entering gels (30 ± 6%) and the gel profiles were similar in recB cells carrying no or one additional Ter site only, regardless of the presence of Tus (Figure 4; Table I, first line). In the Lac-Phe-Ter recB strain, calculation of the percentage of migrating DNA in nine independent experiments showed that Tus induction did not significantly modify the proportion of DNA entering gels (30 ± 5% and 35 ± 6% of the total radioactivity in glucose and arabinose, respectively). In contrast, as expected from the observation of ethidium bromide-stained gels (Figure 4), the profile of linear DNA was clearly affected by Tus induction (Figure 5A). In Tus-repressed conditions, a large fraction of the linear DNA formed a sharp peak migrating as molecules >3.5 Mb (peak 1, Figure 5A). In Tus-induced conditions, most linear DNA migrated as molecules of ∼2 Mb, forming a peak followed by a shoulder (this 8–10 slice region of the gel is named peak 2). The percentage of linear DNA in both peak 1 and peak 2 was calculated from nine independent experiments (Table I, line 2). Upon Tus induction, the amount of DNA in peak 1 decreased significantly (from 46.6 to 22.5%), whereas the amount of linear DNA in peak 2 increased significantly (from 31.1 to 56.6%).
Table I. Proportion of linear DNA in peak 1 and in peak 2 in different strains and in different growth conditions.
| Relevant genotype | I | II |
|||||
|---|---|---|---|---|---|---|---|
| |
Tus repressed (glucose) |
Tus induced (arabinose) |
|||||
| [3H]thymidine | [3H]thymidine | Peak 1 | Peak 2 | [3H]thymidine | Peak 1 | Peak 2 | |
| recB (Terlac or Terphe only) | + | + | 45.5 ± 3.4 (4) | 30.4 ± 2.7 (4) | + | 42.9 ± 3 (4) | 33.6 ± 3.3 (4) |
| Lac-Phe-Ter recB | + | + | 46.6 ± 6.2 (9) | 31.3 ± 9.1 (9) | + | 22.5 ± 2.7 (9) | 56.6 ± 7.6 (9) |
| Lac-Phe-Ter recB | – | + | 53.6 ± 9.4 (4) | 26.5 ± 7.4 (4) | + | 22.3 ± 2.3 (4) | 51.2 ± 4.3 (4) |
| Lac-Phe-Ter recB | + | – | 38.7 ± 2.9 (3) | 32.1 ± 5.3 (3) | – | 33.5 ± 7.6 (3) | 36.1 ± 7.7 (3) |
| Lac-Phe-Ter recB ruvABC | + | + | 35.1 ± 4.9 (4) | 32.7 ± 5.9 (4) | + | 15.1 ± 2.6 (4) | 56.5 ± 6.6 (4) |
Cultures were grown for 3 h in Tus-repressed conditions (glucose) and then divided into two parts in which Tus was either still repressed (glucose) or induced (arabinose). I, labeling conditions during the first 3 h of growth in Tus-repressed conditions. II, labeling and Tus conditions during the following 3 h of growth. The percentage of labeled DNA in each peak was calculated relative to the total amount of labeled linear DNA. The borders of peak 1 and peak 2 were defined in each gel from a Lac-Phe-Ter recB sample, labeled throughout the experiment and Tus induced during the last 3 h. Numbers in parentheses indicate the number of independent experiments. First line: different strains carrying only one Ter site were used; similar results were obtained and the average is presented.

Fig. 5. Peak 2 is formed by newly synthesized DNA. The proportion of labeled DNA in each gel slice relative to the total amount of linear DNA migrating in a pulsed-field gel is shown. The gel origin is not shown. The positions of peak 1 and peak 2 are indicated. (A) Cells were grown for 3 h in [3H]thymidine glucose medium, washed and then shifted for 3 h to either [3H]thymidine glucose medium (dashed line, crosses) or to [3H]thymidine arabinose medium (full lines, squares). Peak 1 and peak 2 encompass all slices in which [3H]thymidine is significantly more abundant in glucose and in arabinose, respectively. (B) Formation of peak 2 during Tus induction. Cells were grown for 3 h in cold glucose medium, washed and then shifted to either glucose or arabinose tritiated medium. Dashed line with crosses, shift to [3H]thymidine glucose medium and incubation for 3 h. Full lines, shift to [3H]thymidine arabinose medium and incubation for: open squares, 1 h; gray squares, 2 h; filled squares, 3 h. (C) Peak 2 is not labeled when [3H]thymidine is present only prior to Tus induction. Cells were grown for 3 h in [3H]thymidine glucose medium, washed and then shifted to glucose or arabinose cold medium. Dashed line with crosses: shift to glucose cold LB and incubation for 3 h; full line with black squares: shift to arabinose cold LB and incubation for 3 h.
According to the ‘collapse’ model, the ∼2 Mb linear molecules (peak 2) are synthesized after Tus induction; consequently, they should be labeled if the tritium is added only in the presence of arabinose. Cells were grown in glucose cold medium and shifted to medium containing either [3H]thymidine and arabinose, to induce Tus, or [3H]thymidine and glucose. DNA was analyzed at different times after the shift (Figure 5B). Peak 2 appeared 1 h after Tus induction and increased with time in the presence of arabinose, reaching 51.2% of the total linear DNA in 3 h (Table I, line 3). Addition of [3H]thymidine 1 h after arabinose also led to the appearance of peak 2 (not shown). Comparison of the amounts of labeled DNA in peak 1 and in peak 2, in cells labeled throughout the experiments (Table I, line 2) and in cells labeled only in the presence of arabinose (Table I, line 3), confirmed that most of the labeled DNA in peak 2 was synthesized after Tus induction.
In a third series of experiments, the DNA was labeled in glucose, i.e. in conditions where Tus is inactive, and then shifted to cold arabinose-containing medium. The Tus-induced ∼2 Mb linear band was observed by ethidium bromide staining (not shown), but was not labeled (Figure 5C). The average amount of labeled DNA in peak 1 and in the gel slices corresponding to peak 2 was calculated from three independent experiments (Table I, line 4). Indeed, when tritium was present only during the first 3 h of growth in glucose, Tus induction did not lead to a significant modification of the amount of labeled DNA in peak 2. This result indicates that the chromosomes labeled in glucose did not generate linear DNA, and hence are not broken upon Tus binding.
Tus-dependent linear DNA is RuvABC independent
We previously reported that in recB cells, replication forks arrested because of the inactivation of a replication protein are broken by the action of the RuvABC complex on a Holliday junction formed by fork reversal (Seigneur et al., 1998; Flores et al., 2001). The present study suggests that replication arrest at Ter does not cause breakage of the fork. A Lac-Phe-Ter recB ruvABC strain was used to verify that the formation of Tus-induced linear DNA is indeed independent of RuvABC (Table I, line 5). The amount of DNA in the Tus-dependent peak (peak 2) was not affected by the ruvABC mutation. Only the Tus-independent peak (peak 1) was slightly less abundant in the absence of the Ruv proteins, suggesting that part of spontaneous DSBs are RuvABC dependent, as previously observed (Seigneur et al., 1998). The lack of effects of the ruvABC mutation on the formation of Tus-dependent linear DNA shows that replication forks arrested at Ter sites are not converted into Holliday junction, in contrast to forks arrested by inactivation of a replication protein.
Discussion
In this work, we analyzed the fate of bacterial chromosomes when additional Ter sites block both replication forks halfway. The presence of additional terminators prevented the removal of the Tus block of the arrested fork by the fork approaching from the opposite direction. We confirm previous observations that blocking replication renders homologous recombination essential for viability and observed that blocking replication yields, as expected, linear molecules. However, we found no evidence for fork breakage, and rather observe that linear DNA molecules are generated by re-initiation of DNA replication on chromosomes that carry blocked forks and progression of these second rounds of replication to the end of the DNA strands at the blocked forks. Homologous recombination is therefore not required to repair broken forks, but rather to re-incorporate linear DNA formed by ensuing rounds of replication.
Replication forks arrested because of a replication mutation are acted upon by recombination proteins and undergo replication fork reversal (see Introduction). Two observations argue against replication fork reversal at Ter sites. First, DBSs at reversed forks are dependent on the presence of functional RuvABC proteins, whereas in the present work inactivating RuvABC did not modify the proportion of Tus-dependent linear DNA (Table I). Secondly, in replication mutants that undergo replication fork reversal, RuvABC-dependent breakage generates mainly a peak of linear molecules migrating as 4–6 Mb (Seigneur et al., 2000). The specific formation of such large linear chromosomes may be accounted for by the mode of action of the RuvABC complex. Since the DNA strands cleaved by RuvC are determined by the direction of migration of the RuvAB complex (van Gool et al., 1999; Cromie and Leach, 2000; Michel et al., 2000), breakage of both replication forks by RuvABC often may be symmetrical and therefore generate mainly linear molecules larger than a chromosome unit size (as in Figure 1C). In contrast, the Tus-induced linear fragments observed here are only ∼2 Mb. In addition, the size of Tus-induced linear DNA also rules out a model in which either leading or lagging strands would be cleaved specifically at both forks by the action of a protein other than RuvABC (Figure 1C).
Theoretically, the 2 Mb Tus-dependent fragments could be produced either by direct breakage of the arrested replication fork at both leading and lagging strands or by ‘collapse’ of ensuing rounds of replication. Random breakage of both leading and lagging strands at blocked forks (Figure 1B) is unlikely for two reasons: (i) it would lead to linear DNA that cannot be repaired by homologous recombination, whereas the viability of the Lac-Phe-Ter strain depends on recombinational repair (Figure 2); and (ii) it should produce both the oriC and terminus halves of the chromosome, which was not observed here (Figures 3 and 4). DSB formation of oriC-containing fragments of 2 Mb could only result from coordinated cleavage of the leading strand template at one fork and the lagging strand template at the other fork. This model is unlikely since a Tus-induced linear molecule should then carry labeled nucleotides when cells are propagated in tritiated medium prior to Tus induction only, which was not observed (Figure 5C). It is of course difficult to exclude entirely the possibility that there may be some fork breakage. However, if such breakage does occur in vivo, then it is too weak to be detected by the methods we have employed. Direct evidence that ∼2 Mb linear fragments are formed by DNA synthesis on chromosomes carrying blocked forks is provided by the observation that these fragments are labeled when [3H]thymidine is added at the same time, or after the addition of arabinose (i.e. Tus induction). This result directly supports the collapse model. Altogether, the present work shows that replication arrest at additional Ter sites does not cause either fork reversal or fork breakage, and rather causes replication fork collapse. The absence of fork reversal or fork cleavage suggests that Ter blocked forks may be protected, either by Tus or by components of the replisome.
Previous studies showed the occurrence of increased RecBCD-dependent recombination in the vicinity of Ter sites (Horiuchi et al., 1994; Horiuchi and Fujimura, 1995), consistent with our detection of a DNA double-stranded end adjacent to Ter (Figure 3). In another study, strains carrying back to back replication terminators in the terminus region of the chromosome were used (InvTer strain; Sharma and Hill, 1995). Kinetic measurements of SOS induction were performed in synchronized cultures, and SOS was found to be induced not by blockage of the first forks arriving at Ter but at later times, consistent with SOS induction upon arrival of a second round of replication. Assuming that the SOS-inducing signal in such strains is a DNA double-stranded end, the authors proposed that double-stranded end formation requires a second round of replication and results either from breakage caused by piling up of successive forks or from replication collapse (Sharma and Hill, 1995). The term ‘collapse’ was proposed originally to describe a reaction in which a replication fork runs off when encountering a single-stranded interruption in one of the template strands (Kuzminov, 1995). Collapse indeed was directly observed upon encounter of a replication fork with a nick introduced by a phage protein in a plasmid molecule (Kuzminov, 2001). This observation suggests that the replication machinery does not ‘sense’ the status of the downstream DNA and may indeed run into interrupted DNA without stopping. Our results directly support the collapse model, in which subsequent rounds of replication forks do not stop behind blocked forks and rather copy interrupted DNA strands to the ends.
A viable cell can only be formed if Tus is removed from the Terphe and Terlac sites, allowing completion of chromosome replication. The requirement for RecA and RecBC for viability in spite of the absence of DNA breakage indicates that the Tus–Ter complex does not dissociate spontaneously. Two classes of models can be proposed, depending on whether Tus is removed by a second round of replication that reaches Ter or later on. Let us first consider that arrival of the second round of replication allows Tus removal from blocked forks (Figure 6A). This may occur, for example, if one (or several) replication protein of the first fork remains associated with Tus and removal of the stalled replication complex is facilitated by the lack of direct interaction between Tus and the DnaB helicase of the second replication fork machinery. After Tus removal, replication would restart by assembly of replication proteins at the fork formed by collapse (Figure 6A2). The observation that homologous recombination is essential for viability implies that re-incorporation of the double-stranded ends formed by collapse is required for the formation of a viable cell (Figure 6A3). Re-incorporation of the linear DNA into the circular chromosome by homologous recombination would allow the assembly of new replication forks at recombination intermediates (Kogoma, 1996; Liu et al., 1999). During recombination, sequences belonging to different forks are exchanged (Figure 6A4). Presumably, productive recombination will occur only when Holliday junctions migrate away from the chromosome ends, since migration in the other direction would disrupt the recombination intermediate. The existence of a bias in the strands cleaved during Holliday junction resolution, imposed by the direction of branch migration (van Gool et al., 1999; Cromie and Leach, 2000; Michel et al., 2000), implies that strands of opposite polarity will be used for the resolution of clockwise and counterclockwise replication forks (Figure 6A4 and B). Consequently, predominantly chromosome dimers will form (Figure 6B). According to this model, formation of viable cells requires chromosome dimer resolution. This is at odds with our observation that inactivation of XerCD or dif does not cause lethality of the Phe-Lac-Ter strain (Figure 2).

Fig. 6. Replication collapse model for the formation of Ter-induced linear DNA. In the first step, a clockwise (1C) and a counterclockwise (1CC) replication fork initiated at the chromosome origin (o) are blocked by Tus bound at Terphe and Terlac, respectively (indented rectangle). In the second step, a second round of replication has started from the origin (2C and 2CC). Black lines, template DNA; blue lines, strands of the first round of replication; red lines, strands of the second round of replication; yellow ovals, progressing replisomes. (A) Only the clockwise migrating forks are shown in this part of the model. (1) The second round of replication dislodges Tus. (2) The replisome is reassembled at the Tus-free LacTer site. (3) The linear DNA recombines with the homologous chromosome. A replisome is assembled at the recombination intermediates. (4) Resolution of Holliday junctions; the strands favored by migration of Holliday junctions toward the origin are exchanged. (B) Only the top chromatid with both clockwise and counterclockwise replication forks is shown here. Branch migration toward the origin, hence in two opposite directions, of Holliday junctions formed at clockwise and counterclockwise forks implies exchange of different strands at both junctions. (5) The first clockwise and counterclockwise forks reach the terminus. (6) The second replication forks reach the terminus region; a dimer molecule is formed. A chromosome dimer is also formed from the bottom chromatid (not shown), resulting in a cell that contains two dimeric chromosomes. (C) Only the clockwise migrating forks are shown here. (1) The second round of replication does not dislodge Tus and is blocked at Ter; linear DNA is formed by copy of the first strands to the end (collapse). (2) The linear DNA recombines with the homologous chromosome and a replisome is assembled at the recombination intermediates. (3) Holliday junctions are resolved; the exchanged strands are determined by the direction of branch migration, toward the origin. The forks reassembled at the recombination intermediates reach Ter. These forks dislodge Tus, and linear DNA is formed as the result of a second collapse reaction. (4) The linear DNA molecules recombine with the homologous chromosome and a replisome is assembled at the recombination intermediates. (5) Holliday junctions are resolved by RuvABC. Since productive branch migration is again directed toward the origin, the exchanged strands are the same as in the first homologous recombination reaction. (D) Only the top chromatid with both clockwise and counterclockwise replication forks is shown here. (6) The first clockwise and counterclockwise forks reach the terminus. (7) The second replication forks reach the terminus. Two monomers are formed from each chromatid, four per cell.
The alternative model supposes that the second round of replication forks reaching Ter does not dislodge Tus (Figure 6C). This would occur if, for example, the replication proteins have disassembled from the blocked fork, so that the second replication forks directly encounter Tus. As in the previous model, re-incorporation of the collapsed double-stranded ends leads to exchanges of chromosome strands and to replication re-initiation at the recombination intermediates (Figure 6C2). However, since the Tus protein is still bound, the replication forks initiated at recombination intermediates will again collapse at Ter (Figure 6C3). Interestingly, these third replication forks that reach the Tus–Ter complex have been initiated at a recombination intermediate, no more than a few tens of kilobases upstream of Tus, in contrast to the previous replication forks that came from the replication origin. These forks reach Ter soon after the previous ones, eventually before disassembly of the replication machinery, which would facilitate Tus removal by preventing direct Tus–DnaB interactions. Alternatively, initiation of replication from a recombination intermediate may enable forks to remove Tus, by modifying the proteins associated with the replisome (Figure 6C3). Re-incorporation of the second collapse products would cause a second round of exchanges between chromosomes (Figure 6C4). The same strands are likely to be used for Holliday junction resolution in the first and in the second recombination reactions, since productive junctions migrate away from the interrupted forks, and, consequently, viable chromosomes carrying patched regions of exchanges will be generated (Figure 6C5). In contrast to model A, model C ends up with chromosome monomers because it involves two successive recombination events per half chromosome (Figure 6D). This model, where Tus is not removed by replication initiated at the origin but rather by replication initiated at recombination intermediates, is therefore compatible with the absence of a requirement for dimer resolution. The collapse model accounts for the paradoxical observation that replication blockage at Ter renders double-stranded end recombinational repair essential for viability in spite of the fact that it does not cause a significant increase in the amount of in vivo linear DNA, which remains at ∼30–35% of total DNA. According to the model shown in Figure 6C and D, RecBC-dependent recombination is required to remove Tus.
In conclusion, the present study stresses several important notions. First, it illustrates how DNA double-stranded ends can be formed upon replication arrest by a purely replicative process, without single- or double-strand DNA cleavage. As pointed out in the Introduction, these double-stranded ends may be used as substrates for homologous or non-homologous recombination, and lead to chromosome rearrangements. Replication arrest-induced genomic instability may therefore occur in the absence of actual DNA damage. Secondly, in the absence of DNA breakage, the requirement for homologous recombination in Ter-blocked strains cannot derive from a need for DSB repair, suggesting that it may result from the capability of recombination-dependent replication to remove Tus. This implies that replication forks initiated at recombination intermediates, or forks that have traveled only a short distance, are able to remove Tus, in contrast to replication forks initiated at a replication origin, which have traveled megabases of DNA. Although alternative models are not excluded, our results raise the possibility that replication forks that have been initiated at a recombination intermediate have properties different from those that issued from the chromosomal origin. Recombination-dependent replication has also been described in eukaryotes, named break-induced replication (BIR; reviewed in Haber, 1999, 2000), and possible differences between break-induced and ori-initiated replication in yeast have been proposed (Paques et al., 1998). Finally, this work raises the important notion that ‘accidental arrests’ and ‘natural arrests’ may have different consequences. Ter sites have been selected during evolution as a bona fide replication arrest site in E.coli and, consequently, forks blocked at Ter do not suffer either replication fork reversal or replication fork breakage. Even when Ter is inserted at a random position in the chromosome, Ter-blocked forks remain stable. This stability is fundamentally different from the processing of forks that are arrested because of a defect in a replication protein and suggests that replication fork reversal occurs only at non-programmed replication arrests.
Materials and methods
Strain construction
Strain JJC907 containing TerB in lacZ was constructed as follows. First, the EcoRI–DraI fragment of plasmid pSKS107 (Shapira et al., 1983) containing the lacZ gene was inserted between EcoRI and HindIII (blunted) sites of plasmid pGB2ts (Clerget, 1991). The EcoRV site of the resulting plasmid, pGBtslacZ, was used to insert the blunted PstI–NheI fragment of plasmid pBRToriC (Bierne et al., 1997a) containing the E.coli TerB site, leading to the plasmid pGBtslac::Ter. TerB was inserted in the chromosome lacZ gene of JJC256 (ΔTus::kan; Sharma and Hill, 1995) using the plasmid pGBtslac::Ter and the thermosensitive property of pGB2ts replication as follows. JJC256 cells containing pGBtslac::Ter plasmid were propagated at 42°C in the presence of spectinomycin (Sp; plasmid-borne resistance marker). Cells in which the plasmid has integrated into the chromosome by simple crossing-over between chromosomal and plasmid lacZ sequences were obtained. These cells were grown for ∼3 generations at 30°C, then at 42°C up to saturation, plated at 42°C and Sp-sensitive β-galactosidase-negative colonies were screened. The presence of the Terlac cassette and the absence of the vector moiety in chromosomes were verified by Southern hybridization.
The strain JJC1818 that contains TerB inserted in the pheA gene was constructed as follows. Part of the pheA gene was amplified from the E.coli chromosome using oligonucleotides 5′-CGGTCGCCATTAACAACG and 5′-CGCACTCAGCACGCATCG and was cloned in the EcoRV site of pBR322. The blunted BssHII site present in the pheA moiety of the resulting plasmid pBRphe was used to clone the blunted SalI–PstI fragment of the plasmid pBRToriC, which contains the TerB site and the spectinomycin resistance gene (SpR). For insertion of the Terphe cassette in the phe gene of E.coli chromosomes, the PheTer::SpR fragment was amplified from pBRphe::ter using the above oligonucleotides and was used to transform JJC256 (ΔTus::kan) competent cells containing the plasmid pKD46 as described (Datsenko and Wanner, 2000).
The strain Lac-Phe-Ter (JJC1819), which contains two ectopic ter sites, was constructed by P1 transduction of the PheTer::SpR cassette in strain JJC907 with selection for Sp resistance. The structure of the Terphe and Terlac regions was verified by Southern hybridization.
Derivatives of Lac-Phe-Ter strains were constructed by P1 transduction using the alleles Δ(recA-srl)::Tn10 (JJC1821), recB268::Tn10 (JJC1820), ruvABC::Cm (JJC1822), ruvC::cm (JJC1823), recD1901::Tn10 (JJC1825) [donor strains are described in Seigneur et al. (1998)], Δdif::cm (JJC1826), ΔxerC::cm (JJC1827) (donor strains are PK3477 and PK3384 respectively, described in Kuempel et al., 1996). The mutations were P1 transduced using antibiotic resistance for selection, and the phenotype of the resulting strains was verified. recA, recBC and ruv strains were verified by measurement of UV sensitivity; the exoV– phenotype of recB and recD strains was verified by plating of gpII– phage T4; the dif and xerC phenotypes were verified by the presence of ∼15–20% filamented cells in exponential cultures. The plasmid pBADTus (Sharma and Hill, 1995) in which the tus gene is under control of the arabinose promoter was used as the inducible donor of Tus protein. To determine the effect of dimer resolution inactivation on Lac-Phe-Ter viability, Lac-Phe-Ter and dif Lac-Phe-Ter colonies grown for 24 h at 37°C on arabinose- or glucose-containing plates were resuspended in salt medium, diluted appropriately and plated on glucose-containing medium. Lac-Phe-Ter and dif Lac-Phe-Ter colonies formed on glucose contained 2.4 × 107 ± 4.4 × 106 and 9.3 × 106 ± 2 × 106 c.f.u., respectively, corresponding to 23–24 generations in 24 h. Lac-Phe-Ter and dif Lac-Phe-Ter colonies formed on arabinose contained 8.4 × 104 ± 9 × 103 and 3 × 104 ± 1.1 × 104 c.f.u., respectively, corresponding to 15–16 generations. Longer incubation at 37°C allowed wild-type and dif colonies to reach normal size on arabinose medium. In order to compare the amount of Ter-specific replication intermediates in different strains and at different times after Tus induction, Southern hybridizations of EcoRV-cleaved chromosomes with Phe or Lac probes were performed. EcoRV cleavage at sites that flank Ter leads to the formation of Y molecules when replication is blocked. The ratio of Y to linear molecules was quantified by phosphoimager. Recombination mutations did not modify significantly the amount of Y-form replication intermediates, and hence the efficiency of replication arrest in the Lac-Phe-Ter strain (data not shown).
Growth conditions
LB was used for cell growth and Tus induction. In cells containing pBADTus, 1% glucose was used for Tus repression and 0.2% arabinose for induction. Antibiotics were added to solid and liquid media at the following concentrations (µg/ml): ampicillin, 50; chloramphenicol, 20; kanamycin, 20; and spectinomycin, 60. For growth curves, overnight cultures in LB glucose were diluted 1000-fold in LB glucose, grown for 2 h at 37°C, washed and resuspended in LB supplemented with either 1% glucose or 0.2% arabinose. Aliquots of the cultures were withdrawn every hour, and appropriate dilutions were plated on LB plates containing 1% glucose and incubated for 24 h at 37°C.
Cellular DNA labeling
DNA labeling in vivo was performed in LB. For DNA labeling prior to Tus induction, overnight cultures were diluted 100-fold in LB supplemented with glucose (1%), deoxyadenosine (100 µg/ml) and [3H]thymidine (83.8 Ci/mmol; 5 µl/ml) and grown at 37°C for 3 h. To induce Tus, cells were washed twice in M63 minimal medium and diluted to OD 0.05 in cold LB. Cultures were then divided into two parts to which glucose (1%) or arabinose (0.2%) were added, and incubation continued for a further 3 h at 37°C. Induction was performed in the presence of either [3H]thymidine and deoxyadenosine for DNA labeling or cold thymidine (100 µg/ml).
Pulsed-field gel electrophoresis
Plug preparation and treatment, and PFGE of total DNA were as previously described (Seigneur et al., 1998). Digestion of agarose-embedded DNA by NotI endonuclease (New England Biolabs) was performed according to the supplier’s protocol. NotI-treated plugs were used for PFGE in 1% agarose gel (SeaKem GTG; FMC BioProducts), 0.5% TBE buffer, with the following parameters: switch time 5–30 min; 6 V/cm; included angle 120°; total run time 17 h.
DNA hybridization
The fragments of hisG, serA, recO and recN genes were used as DNA probes in Southern hybridization. They were amplified from the JJC256 chromosome using the following oligonucleotides: 5′-GTTATCTCGACCAGAAAGGC (hisG1); 5′-ACAGATTCCGTACCAAACGC (hisG2); 5′-GAAGCCAATGCTAAAGCG (serA1); 5′-GATGTTGACGCCCTGCTC (serA2); 5′-CGATATGGTCAATGTTGACGCC (recO1); 5′-GAGTAACTCCGATGGAAGGCTG (recO2); 5′-CGACTATGTTGGCAC AACTGACC (recN1); and 5′-GTTTACTCTGACCGTGAAGCAGG (recN2).
Acknowledgments
Acknowledgements
We are very grateful to Danielle Canceill and Philippe Noirot for helpful reading of the manuscript. This work is supported in part by the Programme de Recherche Fondamentale en Microbiologie, Maladies Infectieuses et Parasitaire. B.M. is on the CNRS staff.
References
- Aguilera A., Chavez,S. and Malagon,F. (2000) Mitotic recombination in yeast: elements controlling its incidence. Yeast, 16, 731–754. [DOI] [PubMed] [Google Scholar]
- Barre F.X., Soballe,B., Michel,B., Aroyo,M., Robertson,M. and Sherratt,D. (2001) Circles: the replication–recombination– chromosome segregation connection. Proc. Natl Acad. Sci. USA, 98, 8189–8195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierne H., Ehrlich,S.D. and Michel,B. (1997a) Deletions at stalled replication forks occur by two different pathways. EMBO J., 16, 3332–3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierne H., Vilette,D., Ehrlich,S.D. and Michel,B. (1997b) Isolation of a dnaE mutation which enhances RecA-independent homologous recombination in the Escherichia coli chromosome. Mol. Microbiol., 24, 1225–1234. [DOI] [PubMed] [Google Scholar]
- Bussiere D.E. and Bastia,D. (1999) Termination of DNA replication of bacterial and plasmid chromosomes. Mol. Microbiol., 31, 1611–1618. [DOI] [PubMed] [Google Scholar]
- Chen C. and Kolodner,R.D. (1999) Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat. Genet., 23, 81–85. [DOI] [PubMed] [Google Scholar]
- Clerget M. (1991) Site-specific recombination promoted by a short DNA segment of plasmid R1 and by a homologous segment in the terminus region of the Escherichia coli chromosome. New Biol., 3, 780–788. [PubMed] [Google Scholar]
- Cromie G.A. and Leach,D.R.F. (2000) Control of crossing-over. Mol. Cell, 6, 815–826. [DOI] [PubMed] [Google Scholar]
- Cromie G.A., Connelly,J.C. and Leach,D.R.F. (2001) Recombination at double-strand breaks and DNA ends: conserved mechanisms from phage to humans. Mol. Cell, 8, 1163–1174. [DOI] [PubMed] [Google Scholar]
- Datsenko K.A. and Wanner,B.L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl Acad. Sci. USA, 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores M.J., Bierne,H., Ehrlich,S.D. and Michel,B. (2001) Impairment of lagging strand synthesis triggers the formation of a RuvABC substrate at replication forks. EMBO J., 20, 619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Rozas H. and Kolodner,R.D. (2000) Links between replication, recombination and genome instability in eukaryotes. Trends Biochem. Sci., 25, 196–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber J.E. (1999) DNA recombination: the replication connection. Trends Biochem. Sci., 24, 271–275. [DOI] [PubMed] [Google Scholar]
- Haber J.E. (2000) Recombination: a frank view of exchanges and vice versa. Curr. Opin. Cell Biol., 12, 286–292. [DOI] [PubMed] [Google Scholar]
- Hill T.M. (1996) Features of the chromosomal terminus region. In Neidhardt,F.C. et al. (eds), Escherichia coli and Salmonella thyphimurium: Cellular and Molecular Biology. ASM Press, Washington, DC, pp. 1602–1614.
- Horiuchi T. and Fujimura,Y. (1995) Recombinational rescue of the stalled DNA replication fork: a model based on analysis of an Escherichia coli strain with a chromosome region difficult to replicate. J. Bacteriol., 177, 783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi T., Fujimura,Y., Nishitani,H., Kobayashi,T. and Hidara,M. (1994) The DNA replication fork blocked at the Ter site may be an entrance for the RecBCD enzyme in to duplex DNA. J. Bacteriol., 176, 4656–4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyrien O. (2000) Mechanisms and consequences of replication fork arrest. Biochimie, 82, 5–17. [DOI] [PubMed] [Google Scholar]
- Jasin M. (2000) Chromosome breaks and genomic instability. Cancer Invest., 18, 78–86. [DOI] [PubMed] [Google Scholar]
- Khanna K.K. and Jackson,S.P. (2001) DNA double-strand breaks: signaling, repair and the cancer connection. Nat. Genet., 27, 247–254. [DOI] [PubMed] [Google Scholar]
- Kogoma T. (1996) Recombination by replication. Cell, 85, 625–627. [DOI] [PubMed] [Google Scholar]
- Kuempel P., Hogaard,A., Nielsen,M., Nagappan,O. and Tecklenburg,M. (1996) Use of a transposon (Tndif) to obtain suppressing and nonsuppressing insertions of the dif resolvase site of Escherichia coli. Genes Dev., 10, 1162–1171. [DOI] [PubMed] [Google Scholar]
- Kuzminov A. (1995) Collapse and repair of replication forks in Escherichia coli. Mol. Microbiol., 16, 373–384. [DOI] [PubMed] [Google Scholar]
- Kuzminov A. (2001) Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc. Natl Acad. Sci. USA, 98, 8241–8246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.I., Xu,L.W., Sandler,S.J. and Marians,K.J. (1999) Replication fork assembly at recombination intermediates is required for bacterial growth. Proc. Natl Acad. Sci. USA, 96, 3552–3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo G.B., Santoro,I.M., McDaniel,L.D., Nishijima,I., Mills,M., Youssoufian,H., Vogel,H., Schultz,R.A. and Bradley,A. (2000) Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nat. Genet., 26, 424–429. [DOI] [PubMed] [Google Scholar]
- Michel B. (2000) Replication fork arrest and DNA recombination. Trends Biochem. Sci., 25, 173–178. [DOI] [PubMed] [Google Scholar]
- Michel B., Recchia,G.D., PenelColin,M., Ehrlich,S.D. and Sherratt,D.J. (2000) Resolution of Holliday junctions by RuvABC prevents dimer formation in rep mutants and UV-irradiated cells. Mol. Microbiol., 37, 180–191. [DOI] [PubMed] [Google Scholar]
- Michel B., Flores,M.J., Viguera,E., Grompone,G., Seigneur,M. and Bidnenko,V. (2001) Rescue of arrested replication forks by homologous recombination. Proc. Natl Acad. Sci. USA, 98, 8181–8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulugu S., Potnis,A., Shamsuzzaman, Taylor,J., Alexander,K. and Bastia,D. (2001) Mechanism of termination of DNA replication of Escherichia coli involves helicase–contrahelicase interaction. Proc. Natl Acad. Sci. USA, 98, 9569–9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung K., Chen,C. and Kolodner,R.D. (2001) Multiple pathways cooperate in the suppression of genome instability in Saccharomyces cerevisiae. Nature, 411, 1073–1076. [DOI] [PubMed] [Google Scholar]
- Paques F. and Haber,J.E. (1999) Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev., 63, 349–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paques F., Leung,W.Y. and Haber,J.E. (1998) Expansions and contractions in a tandem repeat induced by double-strand break repair. Mol. Cell. Biol., 18, 2045–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins J.D., DonHeath,J., Sharma,B.R. and Weinstock,G.M. (1993) XbaI and BlnI genomic cleavage maps of Escherichia coli K-12 strain MG1655 and comparative analysis of other strains. J. Mol. Biol., 232, 419–445. [DOI] [PubMed] [Google Scholar]
- Petrini J.H.J. (2000) The Mre11 complex and ATM: collaborating to navigate S phase. Curr. Opin. Cell Biol., 12, 293–296. [DOI] [PubMed] [Google Scholar]
- Rothstein R., Michel,B. and Gangloff,S. (2000) Replication fork pausing and recombination or ‘gimme a break’. Genes Dev., 14, 1–10. [PubMed] [Google Scholar]
- Saintigny Y., Delacote,F., Vares,G., Petitot,F., Lambert,S., Averbeck,D. and Lopez,B.S. (2001) Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J., 20, 3861–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saveson C.J. and Lovett,S.T. (1997) Enhanced deletion formation by aberrant DNA replication in Escherichia coli. Genetics, 146, 457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schar P. (2001) Spontaneous DNA damage, genome instability and cancer—when DNA replication escapes control. Cell, 104, 329–332. [DOI] [PubMed] [Google Scholar]
- Seigneur M., Bidnenko,V., Ehrlich,S.D. and Michel,B. (1998) RuvAB acts at arrested replication forks. Cell, 95, 419–430. [DOI] [PubMed] [Google Scholar]
- Seigneur M., Ehrlich,S.D. and Michel,B. (2000) RuvABC-dependent double-strand breaks in dnaBts mutants require RecA. Mol. Microbiol., 38, 565–574. [DOI] [PubMed] [Google Scholar]
- Shapira S.K., Chou,J., Richaud,F.V. and Casadaban,M.J. (1983) New versatile plasmid vectors for expression of hybrid proteins coded by a cloned gene fused to lacZ gene sequences encoding an enzymatically active carboxy-terminal portion of β-galactosidase. Gene, 25, 71–82. [DOI] [PubMed] [Google Scholar]
- Sharma B. and Hill,T.M. (1995) Insertion of inverted Ter sites into the terminus region of the Escherichia coli chromosome delays completion of DNA replication and disrupts the cell cycle. Mol. Microbiol., 18, 45–61. [DOI] [PubMed] [Google Scholar]
- Sonoda E., Takata,M., Yamashita,Y.M., Morrison,C. and Takeda,S. (2001) Homologous DNA recombination in vertebrate cells. Proc. Natl Acad. Sci. USA, 98, 8388–8394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spratt B.G., Hanage,W.P. and Feil,E.J. (2001) The relative contribution of recombination and point mutation to the diversification of bacterial clones. Curr. Opin. Microbiol., 4, 602–606. [DOI] [PubMed] [Google Scholar]
- van Gent D.C., Hoeijmakers,J.H. and Kanaar,R. (2001) Chromosomal stability and the DNA double-stranded break connection. Nat. Rev. Genet., 2, 196–206. [DOI] [PubMed] [Google Scholar]
- van Gool A.J., Hajibagheri,N.M., Stasiak,A. and West,S.C. (1999) Assembly of the Escherichia coli RuvABC resolvasome directs the orientation of Holliday junction resolution. Genes Dev., 13, 1861–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]