

Abstract
Background
Research using the model system Xenopus laevis has provided critical insights into the mechanisms of early vertebrate development and cell biology. Large scale sequencing efforts have provided an increasingly important resource for researchers. To provide full advantage of the available sequence, we have analyzed 350,468 Xenopus laevis Expressed Sequence Tags (ESTs) both to identify full length protein encoding sequences and to develop a unique database system to support comparative approaches between X. laevis and other model systems.
Description
Using a suffix array based clustering approach, we have identified 25,971 clusters and 40,877 singleton sequences. Generation of a consensus sequence for each cluster resulted in 31,353 tentative contig and 4,801 singleton sequences. Using both BLASTX and FASTY comparison to five model organisms and the NR protein database, more than 15,000 sequences are predicted to encode full length proteins and these have been matched to publicly available IMAGE clones when available. Each sequence has been compared to the KOG database and ~67% of the sequences have been assigned a putative functional category. Based on sequence homology to mouse and human, putative GO annotations have been determined.
Conclusion
The results of the analysis have been stored in a publicly available database XenDB http://bibiserv.techfak.uni-bielefeld.de/xendb/. A unique capability of the database is the ability to batch upload cross species queries to identify potential Xenopus homologues and their associated full length clones. Examples are provided including mapping of microarray results and application of 'in silico' analysis. The ability to quickly translate the results of various species into 'Xenopus-centric' information should greatly enhance comparative embryological approaches.
Supplementary material can be found at http://bibiserv.techfak.uni-bielefeld.de/xendb/.
Background
Following the publication of the first automated cDNA sequencing study in 1991 demonstrating the utility of large scale random clone cDNA sequencing approaches [1], there has been a rapid and accelerating growth of such Expressed Sequence Tags (EST). The initial study of 600 partial human sequences has grown to more than 20.0 × 106 while more than 30 organisms have more than 100,000 sequences. To make sense of the resulting sequence, a variety of bioinformatic approaches have been developed to identify protein coding sequences and domains [2-4] and generate 'unigene' sets based on agglomerative clustering methods [5,6]. Clustering EST sequences is a widely used method for analyzing the transcriptome of a genome. Especially for organisms whose genome is not (yet) sequenced, the EST data is a valuable source of information. While enormously useful, most current analysis tools result in the loss of significant biological information such as alternatively spliced transcripts and polymorphisms [7-18]. Alternative splicing in particular plays important roles during both development and in the mature organism [7-15]. Moreover, most EST based approaches appear to overestimate the number of unique sequences compared to gene predictions based on whole genome sequencing efforts [19-22].
There are different approaches for EST clustering; the most commonly used being (1) each cluster represents a distinct gene, alternative transcripts of the same gene are grouped together into the same cluster. UniGene is one approach that uses this gene-based strategy [23-27]. (2) Alternative transcripts are represented by distinct clusters. Using genome assembly tools like CAP3 [28] or Phrap [29,30] results in such a clustering, as these tools cannot (and are not designed to) handle the kinds of differences in the EST sequences. (3) STACK [6] groups ESTs based on their tissue source first, and clusters are then generated for each tissue separately. Our approach first generates gene-oriented clusters and then attempts to generate separate contigs which potentially correspond to alternative transcripts.
The underlying principle for each of these approaches is a pairwise comparison of all sequences to identify common subsequences of a given length and identity that is subsequently used to group sequences into clusters. The types of pairwise comparisons result in a runtime that is quadratic in the number of sequences to be compared. To achieve better running times, most tools try to identify promising pairs of sequences by applying word-based algorithms, which consider the frequency of common words in each pair of sequences [31]. In any case these approaches have to compare all possible pairs of sequences, resulting in a running time that grows quadratically with the number of sequences. We have implemented a pipeline for rapid processing and clustering of EST data, based on enhanced suffix arrays [32-34]. Compared to other methods it reduces the running time tremendously. While we focus on generating gene-based clusters, we also assembled each cluster separately using CAP3 to generate consensus sequences for further analyses. Liang et al. evaluated Phrap, CAP3, TA-EST and TIGR Assembler and found in their analysis that CAP3 consistently out-performed the other programs [35]. We therefore chose CAP3 for cluster assembly.
All sequence and clustering information obtained with our approach was stored in a relational database system. To allow for extensive queries, GenBank annotations were incorporated including the library source, tissue type, cell type and developmental stage. Results of all sequence analyses performed on the consensus sequences were stored in the database. This way, comparative queries could be answered to identify e.g. full length clones, sequences unique to X. laevis, or shared between Xenopus and another organism. The comparative query also allows the identification of the set of Xenopus sequences most related to a set from another organism. Thus, the XenDB database is designed to address a critical issue facing many researchers: the comparison of genomic studies in one organism and their application to studies in another model organism. This task is faced by many laboratories attempting to extract the information gained in human, mouse, fly and worm microarray and library sequencing studies which often consist of large tables of genes.
While other databases such as UniGene [36] or TIGR Gene Indices [37] also provide collections of clustered ESTs, the unique batch functionality of mapping results from other organisms to Xenopus laevis and retrieving their potential full length clones was not available before. Moreover, our implementation is specifically designed and focused on relating Xenopus sequence data to the major model organisms. Thus, one can search for the Xenopus homologue directly using the human or mouse protein.
Construction and Content
Sequence sources and cleanup
350,468 Sequences were downloaded from GenBank release 138 and stored in a relational database using the open source ORDBMS PostgreSQL. The following divisions were included: Vertebrate Sequences (VRT, 5,506 sequences), EST (344,747 sequences) and High Throughput cDNA (HTC, 215 sequences). 228,496 sequences were annotated as 5' ESTs and 116,122 as 3' ESTs. 245,415 different cDNA clones were represented in the data set, out of which 92,463 had both 5' and 3' sequences. Entries annotated as being genomic sequences were excluded from the analysis. To enhance the usability and search capabilities of the database, complete GenBank entries were incorporated. Annotations including but not limited to library source, tissue type, cell type and developmental stage were extracted directly from GenBank entries (feature: source, qualifiers: clone_lib, tissue_type, cell_type and dev_stage). Unfortunately, the sequences are not very well annotated in GenBank. 34% of the sequences do not have a tissue type assigned and 36% have no developmental stage information. Distributions of tissue types, developmental stages and clone libraries are shown in supplemental files [see additional files 2, 3 and 4 respectively].
197,888 ESTs (57.4% of the EST sequences) had information about high quality start or end of sequencing reads. This information was used to trim sequences according to high quality regions to insure best sequence quality. Vector sequence was downloaded from GenBank and VectorDB [38] and the sequence masked using the program Vmatch [39] developed by Stefan Kurtz. Vmatch is based on a novel sequence index (enhanced suffix arrays, [32-34]), allowing for the rapid identification of similarities in large sequence sets. ESTs were trimmed to eliminate vector sequence located at either the 5' or 3' end (6678 ESTs, 1.9% of total sequence set). In some cases, additional non vector sequence preceded or followed known vector sequence. If such non-vector sequence was less than 20 bases long, it was trimmed from the EST together with the vector sequence. ESTs that had vector sequences left after trimming were discarded completely. Repetitive elements were obtained from Repbase [40] and GenBank and masked using RepeatMasker [41]. In addition, if hits against ribosomal RNA and mitochondrial sequences were found in the downloaded sequence set, the corresponding sequences were removed. The availability of complete mitochondrial genomic and ribosomal sequences makes the inclusion of these sequences unnecessary while masking was performed to minimize possible clustering errors arising from these common sequences. Sequences that had less than 100 consecutive bases left after cleanup were discarded completely (21,039 sequences, 6.0%). The resulting sequence set consisted of 317,242 sequences (90.5%) with an average length of 536 bases (see Table 1).
Table 1.
Summary of Xenopus EST cleanup and clustering.
| Total number of ESTs and cDNAs | 350,468 |
| Number of distinct clones | 245,415 |
| Number of good sequences | 317,242 |
| Average trimmed EST length (bp) | 536 |
| Number of 3' EST sequences | 116,122 |
| Number of 5' EST sequences | 228,496 |
| Clones with 5' and 3' sequences | 92,463 |
| Number of clusters | 25,971 |
| Number of singletons | 40,877 |
| Number of CAP3 contigs | 31,353 |
| Number of CAP3 singletons | 4,801 |
| Average CAP3 contig length (bp) | 1,045 |
| Max. cluster size (no. of ESTs) | 6,332 |
| Average cluster size (no. of ESTs) | 10.6 |
| Cluster sizes: | # EST |
| 4,097 – 8,192 | 1 |
| 2,049 – 4,096 | 1 |
| 1,025 – 2,048 | 2 |
| 513 – 1,024 | 15 |
| 257 – 512 | 35 |
| 129 – 256 | 116 |
| 65 – 128 | 414 |
| 33 – 64 | 973 |
| 17 – 32 | 1,755 |
| 9 – 16 | 2,974 |
| 5 – 8 | 4,571 |
| 3 – 4 | 6,444 |
| 2 | 8,670 |
Clustering and assembly of tentative contig sequences
The cleaned X. laevis EST sequence set was grouped into gene specific clusters using Vmatch. Vmatch preprocesses the EST sequences into an index structure: an enhanced suffix array. This data structure has been shown to be as powerful as suffix trees, with the advantage of a reduced space requirement and reduced processing time. Further on, enhanced suffix arrays have been shown to be superior to other matching tools for a variety of applications [33,42,43]. For a detailed introduction of enhanced suffix arrays see Abouelhoda et al. [34]. Briefly, the index efficiently represents all substrings of the sequences and allows the solution of matching tasks, in time independent of the size of the index (unlike BLAST). Vmatch was chosen for the following reasons: (1) At first, there was no clustering tool available which could handle large data sets efficiently, and which was documented well enough to allow a detailed replication and evaluation of existing clusters. (2) Second, Vmatch identifies similarities between sequences rapidly, and it provides additional options to cluster a set of sequences based on these matches. Furthermore, the Vmatch output provides information about how the clusters were derived. Due to the efficiency of Vmatch, we were able to perform the clustering for a wide variety of parameters on the complete sequence set (see below). This allowed us to study the effect of the parameter choice on the clustering. Moreover, in the future, the efficiency will allow us to more frequently update the data set. A longer term goal of the project is to generate a data set that maintains the different alleles in this pseudotetraploid animal as separate entries. The clustering approach has been integrated into an analysis pipeline which can be applied to other organisms that often receive less attention from the bioinformatics community.
The database sequences were clustered according to the matches found in a self comparison of the index. Initially each database sequence is put into its own cluster. Then all pairs of matches are generated and each pair is evaluated to possibly form single linkage clusters. To identify matching sequences, Vmatch first computes all maximal exact matches of a given minimal length (seeds) between all sequences. These seeds are extended in both directions allowing for matches, mismatches, insertions, and deletions using the X-Drop alignment strategy as described previously. This greedy alignment strategy was developed for comparing highly similar DNA sequences that differ only by sequencing errors, or by equivalent errors from other sources [44].
In an attempt to objectively define appropriate clustering criteria, we took advantage of the speed of the Vmatch clustering approach to systematically vary the relevant parameters (overlap length, % identity, seedlength and X-drop value). It was hypothesized that the 'correct' parameters would be revealed as an abrupt change in the curve on the resulting graph. An example of such an analysis showing the effect of varying the overlap length and % identity is presented in supplemental materials [see additional file 1]. Here a number of conclusions become apparent. First, at this level of resolution (~30 independent clusterings), a distinct point indicating the 'correct' parameter does not become readily apparent. Second, the collapse of the cluster set to few clusters containing every larger numbers of individual sequences serves as a reminder that all sequences (regardless of species) can be considered part of a single cluster. Finally, as the length overlap decreased, we observed the formation of 'superclusters' containing >10,000 sequences clearly derived from multiple gene families. These problem of 'superclusters' diminished at an overlap length of ~135 (data not shown, and not apparent in additional file 1). These clusters appear to be due to the presence of undefined repetitive elements, chimeric sequences and possibly transposed elements. Studies on the nature of the clustered sequences and the effects of parameter variation are ongoing.
For the current data set, we tried to select parameters which mimic the parameters that were probably used for generating the UniGene clusters. Unfortunately, the algorithm used for constructing the UniGene clusters is not sufficiently documented to allow complete reproduction. We selected parameters designed to produce a stringent clustering of the available sequences. For the described data set, sequences were clustered when a pairwise match of at least 150 nucleotides and 98% identity was found (seedlength = 33, X-Drop = 3). The construction of the enhanced suffix array took 33 minutes on a SUN UltraSparc III (900 MHz) CPU. Clustering took another 17 minutes. This resulted in 25,971 clusters containing 276,365 sequences (87.11% of the input set) and 40,877 singletons (12.89%). The average cluster size was 10.6 (std. dev 51.8) sequences. The distribution of cluster sizes is shown in Table 1. 22,834 clusters were composed of ESTs only, 61 clusters of mRNA sequences (VRT and HTC divisions) only and 3,076 clusters of both mRNAs and ESTs. Among the singletons are 4262 sequences which contain less than 150 nt (after sequence cleanup described above) and would therefore be incapable of being joined in a cluster. Less than 25% of these sequences have a significant match against NR database and less than 2% of the sequences match full length cDNA criteria described below.
Next, a consensus sequence was generated for each cluster using CAP3 [28]. The aim of this approach was to both refine the number of clusters and to improve the overall sequence quality. This latter aim simplifies the design of oligonucleotide probes. The 25,971 clusters produced 31,353 tentative contig (TC) sequences (avg. length: 1,045 bp, std. dev: 729 bp) and 4,801 singlets (avg. length: 664 bp, std. dev: 424 bp). The longest TC was 13,130 bp (DNA-dependent protein kinase catalytic subunit, accession: [Genbank:AB016434]), while the smallest TC was 154 bases long. Here, it became obvious that CAP3 is a genome assembly program not designed to assemble EST clusters containing potential splice variants: CAP3 assembly subsequently split a fraction of the clusters into separate contigs and singletons. On average, a cluster was split into 1.2 (std. dev 3.0) TCs and 1.8 (std. dev 11.3) singlets by CAP3. As illustrated in Table 1, the average length of the sequences increased from 536 bp (average for input ESTs) to 1,045 bp (average for CAP3 contig sequences) which was lower than the average length for previously characterized Xenopus full length sequences (sequences selected as full length by XGC had an average length of 2,115 bp).
There are many genes whose transcript is significant longer than 2× the current state of the art sequencing run of ~1000 bp. This means that 5' and 3' sequences derived from a >2 kb transcript are unable to be joined without sequence from incomplete cDNA clones which provide a source of nested deletions. Sequences from both ends can be linked by annotation, and this has been done by a variety of clustering approaches including NCBI UniGene which uses a double linkage rule. Non-overlapping 5' and 3' ESTs are assigned to the same cluster if clone IDs are found that link at least two 5' ends from one cluster with at least two 3' ends from another cluster and the two clusters are merged. We have examined the effect of double linkage joining using the clone annotation. In this analysis, 17,588 clusters were stable and the total number of clusters was reduced from 25,971 to 21,249. Most of the joined clusters (3,122) were created from two clusters while three clusters were combined 456 times. While the number of clusters is decreased by this joining, our overall analysis is not affected. Potential full length clones selected as part of the P5P group (see below) are also unaffected by annotation linkage. We provide the identity of clusters 'linked by annotation' as part of the XenDB output.
Sequence analysis
We have performed a variety of sequence comparisons at the protein level including translation analysis. The sequences of cluster TCs and all singletons were subject to extensive BLASTX [45] and FASTY [46] homology searches vs. the non-redundant protein database (NR) from NCBI and the proteomes of five major model organisms using the high throughput analysis pipeline of the Genlight system [47] Proteome sets for H. sapiens, M. musculus and R. norvegicus were obtained from the International Protein Index [48,49]. The IPI provides a top-level guide to the main databases: Swiss-Prot, TrEMBL, RefSeq and Ensembl. It curates minimally redundant yet maximally complete sets of the indexed organisms. C. elegans and D. melanogaster protein sequences were retrieved from the UniProt database [50]. UniProt proteome sets are solely derived from Swiss-Prot and TrEMBL entries. Additionally, all available protein sequences for X. laevis and X. tropicalis were extracted from GenBank. additional file 5 provides an overview of the downloaded data sets. Performing separate comparisons allows a search for matching sequences based on the identity of any gene known from each species as well as query for genes which have matches in some but not all databases. We believe that this will aid in the discovery and analysis of conserved and unique genes. In addition to these databases, we have included BLASTX searches in the KOG database and have used the results to functionally classify the Xenopus sequences. All sequences resulting from the clustering and assembly processes were compared to these protein sets using BLASTX with an E-value cutoff of 1.0e-6. ESTs are often of low sequence quality, and sequencing errors can still exist in the assembled TC sequences. Therefore, all analyses against the protein databases were also done using FASTY (E-value cutoff: 1.0e-6) a version of FASTA that compares a DNA sequence to a protein sequence database, translates the DNA sequence in three forward (or reverse) frames and allows (in contrast to BLASTX) for frame shifts, maximizing the length of the resulting alignments.
Identification of chimeric sequences
A significant issue in EST clustering methods is the presence of chimeric sequence which inappropriately joins unrelated genes into a single cluster. While the number of chimeric sequences is estimated at less than 1% [51,52], their presence has disproportionate effects on the clustering outcome. To identify potential chimeric sequences, we analyzed the FASTY hits in the protein NR database and applied the following simple procedure: Matches of at least 100 bp in length were mapped back to the TC sequences to identify the regions that are covered by a match. If two matches overlap, the region will be extended accordingly. If after the mapping two clearly separated regions remain, the TC is flagged as potential chimera (see Figure 3).
Figure 3.
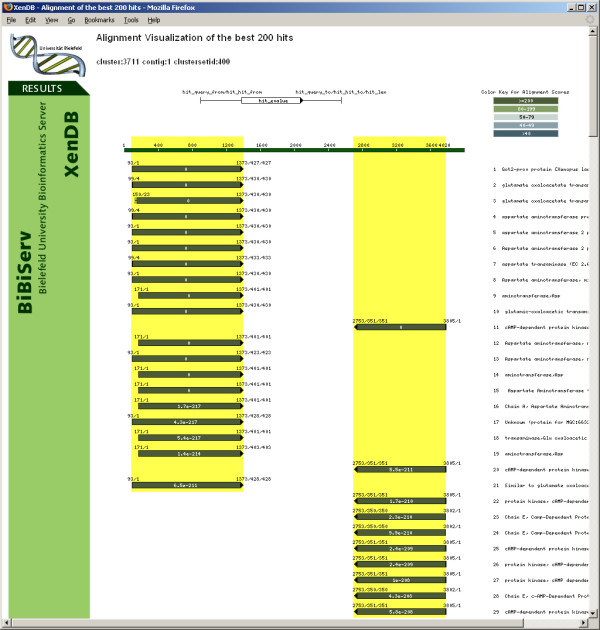
Identification of chimeric TCs: Matches of at least 100 bp in length were mapped back to the TC sequences to identify the regions that are covered by a match (yellow boxes). If two matches overlap, the region will be extended accordingly. If after the mapping two clearly separated regions remain as shown here, the TC is flagged as potential chimera.
Examination of the identified chimeric sequences reveals three major classes. In the first, two distinct FASTY hits can be identified which do not overlap and are in opposite orientation. In the second, the second identified FASTY hit matches retroviral or transposable element related sequences. This suggests the possibility that these may reflect real transcripts in which a mobile element has been inserted into the genome. A close evaluation of such sequences may provide some insights into the evolutionary history of various populations of Xenopus. The final class of potential chimeric sequences identified contains short predicted or hypothetical proteins. This class may in fact not be chimeric at all but may reflect errors in protein coding prediction methods.
The described procedure identified 113 potential chimeric TCs (0.3% of the 33,034 sequences with matches against the protein NR database), which are flagged in the database as such. We do not eliminate these potential chimeras, as they don't significantly affect the results of the sequence analyses done later on, which are mainly based on the best hit only. In fact, the analysis underestimates the number of full length sequences, as some chimeras cover two full length protein matches. A complete identification of chimeric sequences is practically impossible without a comparison to the underlying genome sequence. And even then, polycistronic transcripts which may exist cannot be separated from chimeras perfectly [53].
Definitions
In the subsequent analyses we were interested in three kinds of information: (1) Full Length Orf containing COntigs (FLOCOs), (2) Full Length Insert containing CLones (FLICLs), and (3) Predicted 5' (P5P) sequences. The result of the clustering and CAP3 analysis generates a set of tentative contig sequences (TC). FLOCOs are defined as TC sequences that have an (almost) full length hit against a known protein. These sequences are especially useful for gene identification. Full length insert containing clones, FLICLs, were predicted. Such clones are distinguished by sequence homologies corresponding to the amino terminal part of a protein but are not restricted at the carboxy-terminus. These sequences are derived from clones which are predicted to carry a full length insert (see below), though the full length sequence has not been determined, usually because of single pass EST sequencing from the 5' end. Finally, we identified sequences that we call P5P for which sequence similarity did not extend through the amino-terminal end of the protein but whose length was sufficient to include a full length coding sequence of a similarly sized protein.
Identification of Full Length Orf containing COntigs (FLOCOs)
We were especially interested in full length hits of the TC sequences vs. known proteins. For this purpose, BLASTX and FASTY hits were categorized into four classes, representing the quality of the full length matches (see Figure 1): (1) Matches cover 100% of the sequence of a known protein. Additionally, the matched protein sequence has to begin with the conserved methionine and has to end at a conserved STOP codon. (2) Matches covering 100% of the sequence of a known protein. Additionally, the matched protein sequence has to include the initial methionine. (3) Matches capable of covering 100% of the matched protein sequence with no additional constraints. (4) Matches that cover the protein over almost its full length, allowing the match to start or end maximal ten amino acids after/before the start or end of the protein.
Figure 1.

Full length clone selection (top) and TC categories (bottom). ESTs derived from different clones were clustered and assembled. The CAP3 contig was compared to protein databases using BLASTX and FASTY and hits categorized in 4 categories. Class 1 hits had to match the whole protein sequence and start with an ATG in the TC and M in the protein and the hit had to end at a STOP codon. Class 2 hits had to match the whole protein sequence, start with an ATG in the TC and M in the protein. Class 3 had to match the full protein sequence (without further restrictions), class 4 had to cover the protein over almost its full length, allowing the match to start or end maximal 10 ten amino acids after/before the start or end of the protein. Predicted 5' TCs (P5P) had to have enough sequence to fill up the missing 5' end of the protein sequence. Clone selection: Clone A and B were discarded because of missing IMAGE id. Clone 54321 does not span 5' end of protein match. Clone 21345 was selected as most 5' clone fulfilling the requirements.
Table 2 shows the number of identified FLOCOs using BLASTX. 3,942 TCs were Class 1 hits in the non-redundant protein database. As the stringency of the full length definition was relaxed, the number of TCs characterized as full length increases to 5,050 (Class 2), 7,792 (Class 3) and 12,389 (Class 4) TCs respectively. As EST sequences have many sequencing errors, and even the assembly of clusters can not correct all of these, FASTY comparisons were done for the same data set (Table 3). This way, the length of the resulting alignments could be maximized. A comparison of Table 2 and Table 3 shows the effect of frame shift corrections obtained by FASTY. The number of TCs having Class 1 hits could be increased to 5,139 while the less stringent categories increased similarly by an average of 20%. The effect of frameshift correction can clearly be seen in Figure 2. Table 4 and Table 5 show the average lengths of TCs for each of the four categories. Here, the average length of the TCs is 2,210 bp for Class 1 TCs having FASTY matches against X. laevis, corresponding very well to already known Xenopus proteins. Overall, the average length decreases with lower quality categories as expected, especially for Class 4, where the alignment can miss 20 amino acids on both ends of the matching protein. The only exceptions are Drosophila and C. elegans, where the average length increases for Class 4 sequences.
Table 2.
Number of X. laevis TCs with full length BLASTX hits in the non-redundant protein database (NCBI), five model organisms, and available X. laevis and X. tropicalis proteins, determined by BLASTX. Lower quality categories include sequences from higher, more stringent categories.
| Class | Protein NR | Human | Mouse | Rat | Fruitfly | C. elegans | X. laevis | X. tropicalis |
| 1 | 3,942 | 1,760 | 1,765 | 1,455 | 219 | 140 | 2,918 | 495 |
| 2 | 5,050 | 2,067 | 2,076 | 1,736 | 311 | 233 | 3,104 | 541 |
| 3 | 7,792 | 2,647 | 2,919 | 2,592 | 392 | 283 | 3,898 | 590 |
| 4 | 12,389 | 5,587 | 5,841 | 3,078 | 2,071 | 1,856 | 5,024 | 1,033 |
| P5P | 15,870 | 13,942 | 14,179 | 13,113 | 8,425 | 8,117 | 9,227 | 4,334 |
Table 3.
Number of X. laevis TCs with full length FASTY hits in the non-redundant protein database (NCBI), five model organisms, and available X. laevis and X. tropicalis proteins, determined by FASTY. Lower quality categories include sequences from higher, more stringent categories.
| Class | Protein NR | Human | Mouse | Rat | Fruitfly | C. elegans | X. laevis | X. tropicalis |
| 1 | 5,139 | 2,347 | 2,337 | 1,930 | 268 | 190 | 3,862 | 660 |
| 2 | 6,243 | 2,692 | 2,671 | 2,248 | 383 | 296 | 4,119 | 721 |
| 3 | 9,576 | 3,528 | 3,774 | 3,374 | 473 | 357 | 4,967 | 796 |
| 4 | 14,094 | 6,467 | 6,701 | 6,341 | 2,249 | 1,918 | 5,701 | 1,241 |
| P5P | 15,651 | 13,578 | 13,954 | 13,085 | 8,108 | 7,746 | 9,055 | 4,159 |
Figure 2.

Comparison of a BLASTX alignment with corresponding full length FASTY alignment, as generated by the Genlight system. Blue boxes in (a) indicate open reading frames, green boxes start and red boxes stop codons, respectively. The assembled TC sequence has a frameshift at position 1150 from frame 1 to 3, generating two distinct HSPs in the BLASTX alignment (b). FASTY clearly corrects this frameshift and generates a full length alignment (c).
Table 4.
Average length of X. laevis TCs for different BLASTX full length TC categories.
| Class | Protein NR | Human | Mouse | Rat | Fruitfly | C. elegans | X. laevis | X. tropicalis |
| 1 | 1984 | 1835 | 1805 | 1788 | 1620 | 1541 | 2171 | 1743 |
| 2 | 1831 | 1806 | 1776 | 1775 | 1541 | 1391 | 2120 | 1697 |
| 3 | 1630 | 1813 | 1775 | 1834 | 1560 | 1429 | 1981 | 1693 |
| 4 | 1393 | 1680 | 1675 | 496 | 1638 | 1640 | 1879 | 1660 |
Table 5.
Average length of X. laevis TCs for different FLASTY full length TC categories.
| Class | Protein NR | Human | Mouse | Rat | Fruitfly | C. elegans | X. laevis | X. tropicalis |
| 1 | 2007 | 1888 | 1859 | 1843 | 1659 | 1575 | 2210 | 1807 |
| 2 | 1837 | 1856 | 1821 | 1819 | 1563 | 1440 | 2152 | 1774 |
| 3 | 1553 | 1790 | 1772 | 1804 | 1569 | 1441 | 2019 | 1768 |
| 4 | 1329 | 1683 | 1673 | 1664 | 1611 | 1563 | 1910 | 1703 |
Comparing the numbers of full length sequences in Table 2 and Table 3, the matches in human, mouse, rat and X. laevis are in general agreement (2619 full length sequences for Class 1 on average). What is striking is the deviation of both the number of full length TCs as well as the average length of TCs having matches against Drosophila and C. elegans: only 268 and 190 full length sequences with average lengths of 1659 and 1575 bp for Drosophila and C. elegans in Class 1, respectively. Only within the Class 4 category there are 2,249 and 1,918 TCs with average lengths of 1,611 bp and 1,563 bp, respectively. A possible explanation for this difference is the divergence of the vertebrate species from these invertebrate model systems.
Selection of putative Full Length Insert containing CLones (FLICLs)
Often, biologists are interested in identifying a full length clone for further study and this desire has been met by the establishment of a number of the Gene Collections (the Mammalian Gene Collection [54], the Xenopus Gene Collection [55] and the Zebrafish Gene Collection [56]). We have extended our analysis described above to select potential full length insert containing clones (FLICLs) that are available through the IMAGE consortium and provide a simple yet powerful search tool to rapidly match homologous genes of interest to their Xenopus counterparts. The Gene Collections are an NIH initiative that supports the production of cDNA libraries, clones and 5'/3' sequences to provide a set of full-length (ORF) sequences and cDNA clones of expressed genes for a variety of model systems.
Since the average length of the characterized full length vertebrate protein is 1,400 bases and the average sequence length of a TC is 1,045 bases, many sequences which are full length will not be detected by the previous approach and will contain sequence gaps of approximately 350 bases. To identify additional clones that potentially carry a full length insert, we queried the database for sequence matches which were sufficiently long to include the start methionine but which did not have sufficient homology to be detected by the previous methods Thus, a sequence with a query start position (Startq) which is greater than the subject start site (Starts) is potentially a full length open reading frame (hereafter referred to as P5P, predicted 5 prime). Clearly, the value of such a prediction decreases as the values of Startq increases and the predictive value increases with lower values of Starts. Full length clones predicted by this method are subject to 3' truncations due to mispriming in poly(A) rich regions rather than at the polyA tail. Such regions would be characterized by the presence of the amino acid lysine (codons AAA, AAG) or asparagine (codons AAU, AAC).
Best FASTY hits were extracted for TCs from all four full length categories as well as the P5P categories as described above. For TCs matching these categories, the most 5' EST contributing to the CAP3 contig sequence was selected. In addition, the selected clone had to span the amino-terminal end of the FASTY protein match. Finally, to ensure the ready availability of the clones and therefore the utility of the analysis, the selected clone had to be available through the IMAGE consortium. See Figure 1 for an illustration of 5' clone selection. The P5P criteria selected 15,651 potential full length insert containing clones out of which 10,500 are distinct IMAGE clones, which represents an additional 1,557 sequences compared to Class 4. Two examples of such predicted protein coding sequences are presented in Figure 4. We have mapped these clones to 7,782 distinct clusters. To assess the quality of the FL prediction method, we compared our set to the IMAGE clone set selected by the Xenopus Gene Collection (XGC, [55]) for full length sequencing. As of April 2004 the XGC had selected 10,482 IMAGE clones for sequencing. Our analysis selected 3,152 IMAGE clones that were identical to clones selected by the XGC. Of the remaining 7,348 clones from our set, 4,866 selected IMAGE clones were found in an identical cluster as 4,465 XGC selected clones (note that some of these clones are in the same cluster). In addition, 1,154 XGC clones did not have sequence available to be included in our analysis. The remaining 1,711 IMAGE clones selected for sequencing by XGC are not found in our predicted set while 2,482 clones were unique to our set. In an effort to examine why the 1,711 sequences selected for sequencing were not identified as full length, we compared the startq and starts values as described above. Using the P5P prediction criteria described above, we identify 107 XGC selected IMAGE clones that we predict are not full length but have an alternative clone which we predict is full length. Though final confirmation of the results requires additional sequencing, our method appears to be successful at identifying full length sequences and distinguishing non-full length sequences identified by an independent method. The FL clones are labeled in the XenDB web interface (see below), allowing a rapid identification of potential FL clones for a gene of interest.
Figure 4.

Two examples of TCs derived from clones predicted to have a full length insert (P5P). The start positions in the hit suggest that the unmatched amino-terminal protein sequence is not well conserved between X. laevis and the matched organisms, here rabbit (top) and human (bottom), but the open reading frames (blue boxes) indicate that the clones the sequences were derived from do actually contain a full length insert. (Screenshots of the results were generated by the Genlight system.)
Due to the large number of sequences, we are unable to examine each sequence individually. Since the analysis depends on the overall degree of conservation among the sequences, such an approach will not be as successful on weakly conserved genes. In general, it seems likely that decreasing e-values correspond to higher quality predictions. On a global basis, the results need to be carefully considered, as an independent assessment of the distribution of conservation among the ensemble of sequences is not available.
Gene Ontology prediction and Functional Classification
The Gene Ontology (GO) project [57] is an ongoing international collaborative effort to generate consistent descriptions of gene products using a set of three controlled vocabularies or ontologies: biological processes, cellular components, and molecular functions. The GO vocabulary allows consistent searching of databases using uniform queries. The availability of such vocabularies can be critical to the interpretation of high through put approaches such as microarrays. Based on FASTY homologies with both mouse and human sequence, we have mapped GO annotations to the Xenopus sequences. Of the 30,683 TCs with matches to mouse (29,971) or human IPI sequences (29,963), 19,721 TCs have been assigned putative GO annotations. Among the 10,500 potential full length ORF containing IMAGE clones, 6,886 have been assigned GO annotations.
The non-redundant X. laevis data set was then classified based on their homology to known proteins from the KOG [58] database (BLASTX 1.0e-5 E-value cutoff, best hit selection). KOGS are euKaryotic clusters of Orthologous Groups. KOG includes proteins from 7 eukaryotic genomes: C. elegans, D. melanogaster, H. sapiens, A. thaliana, S. cerevisiae, S. pombe, E. cuniculi.17,624 sequences (67.3%) had a hit against the KOG database and could be assigned a functional category.
Identification of conserved genes not found in major model organisms
To identify additional genes within the dataset that are not found by comparison to protein sets of the major model organisms and to assess the extent of diverged or non conserved sequences, open reading frames of 600 nucleotides or longer were selected from the clustered data set for analysis. 219 sequences that did not have any hit in the previous analyses were identified (188 TCs representing 178 clusters and 31 singlets). We further restricted the number of sequences by re-running the BLASTX and FASTY analysis with E-value cutoffs of 0.01. 111 sequences (91 TCs representing 87 clusters consisting of an average of 6 ESTs per cluster and 19 singlets) without any significant similarity in protein databases could be identified and these were examined by TBLASTN against the human, mouse and 'others' EST databases (22.7 million sequences total). Signal peptides were identified by SignalP [59] as well as transmembrane domains by TMHMM [60,61]. Results are presented in Table 6. The analysis identified 46 sequences with similarity to other organisms (E<0.01) with 11 sequences matching chicken (Gallus gallus), 10 sequences matching zebrafish (Danio rerio) and 6 sequences matching the rainbow trout (Oncorhynchus mykiss). Three of the sequences matched human sequences with less significance than the cutoff used above (i.e. 1.0e-6). Among the sequences with highly significant BLAST hits were two matches to the eastern tiger salamander (Ambystoma tigrinum tigrinum) and one to the rainbow trout (Oncorhynchus mykiss). A surprising match was to barley (Hordeum vulgare, E = 9.0e-35) which was the only plant represented among these hits. The remaining 65 sequences did not have significant homology to existing public database sequences. For 7 sequences both signal peptide cleavage sites and transmembrane domains could be identified. Another 15 sequences had either a signal peptide cleavage site or a transmembrane domain. These 22 sequences are potentially novel membrane proteins.
Table 6.
Xenopus Long Open Reading Frames (>= 600 nt) without homology to major model organism protein sequences. ORF sequences were compared to all available EST data using TBLASTN. The 46 sequences shown here have homologies to ESTs from other organisms (E < 0.01). For each TC, the number of ESTs in the TC and the accession, SignalP and TMHMM results, and description and E-value of the best hit is shown. Additionally (not shown here), both signal peptides and transmembrane domains could be predicted in: clSignal peptides only in: cl4857_sin8, cl11312_sin2, cl11866_ctg2, cl14117_ctg1, cl16548_ctg1, cl19372_ctg2; Transmembrane domains only in: cl3994_ctg1, vimsin144578, cl18799_ctg1, cl18978_ctg1, cl18978_ctg2, cl25690_ctg1, cl23256_ctg1.
| Contig/ORF | #ESTs | Accession | SignalP | TM | Description (best hit) | E-value |
| cl9703_ctg1_1 | 53 | CN060851 | Ambystoma tigrinum tigrinum | 5.00E-112 | ||
| cl15798_ctg1_1 | 4 | CN061938 | Ambystoma tigrinum tigrinum | 6.00E-53 | ||
| cl9914_ctg1_1 | 11 | BX864357 | Oncorhynchus mykiss | 4.00E-45 | ||
| vmsin143901_1 | 1 | CK600275 | Rattus norvegicus | 4.00E-43 | ||
| cl10823_ctg1_1 | 3 | CA471690 | Danio rerio | 5.00E-39 | ||
| cl2563_ctg2_1 | 10 | AV913994 | Hordeum vulgare subsp. Vulgare | 9.00E-35 | ||
| cl1723_ctg1_1 | 12 | BU129000 | Gallus gallus | 2.00E-34 | ||
| vmsin213651_1 | 1 | CD218114 | Gallus gallus | 7.00E-30 | ||
| cl15560_ctg1_1 | 3 | CK871392 | Danio rerio | 2.00E-24 | ||
| cl10197_ctg1_1 | 3 | CA975598 | Danio rerio | 2.00E-22 | ||
| cl11603_ctg1_1 | 4 | AJ456928 | Gallus gallus | 7.00E-22 | ||
| cl2506_ctg1_1 | 7 | BJ494402 | Oryzias latipes | 3.00E-19 | ||
| vmsin144578_1 | 1 | BU241764 | Gallus gallus | 1.00E-17 | ||
| cl24411_ctg1_1 | 2 | BW379961 | Ciona intestinalis | 2.00E-16 | ||
| cl25096_ctg1_1 | 2 | BX269216 | Gallus gallus | 5.00E-16 | ||
| vmsin117573_1 | 1 | BU114361 | Gallus gallus | 3.00E-14 | ||
| vmsin141365_1 | 1 | BI385350 | Amphioxus Branchiostoma fl. | 3.00E-14 | ||
| vmsin275700_1 | 1 | CN024469 | Danio rerio | 4.00E-14 | ||
| cl5895_ctg1_1 | 12 | BX870166 | Oncorhynchus mykiss | 6.00E-14 | ||
| cl18998_ctg1_1 | 2 | BW156550 | Ciona intestinalis | 8.00E-13 | ||
| cl5042_ctg1_1 | 14 | CN316430 | Danio rerio | 3.00E-12 | ||
| cl9402_ctg2_1 | 2 | AJ448952 | Gallus gallus | 2.00E-11 | ||
| cl19097_ctg1_1 | 4 | CN023422 | yes | Danio rerio | 2.00E-09 | |
| cl4943_ctg1_1 | 4 | AJ450094 | Gallus gallus | 5.00E-09 | ||
| cl19576_ctg1_1 | 2 | BX862425 | Oncorhynchus mykiss | 4.00E-08 | ||
| vmsin9176_1 | 1 | CO051215 | Leucoraja erinacea | 1.00E-07 | ||
| cl5371_ctg1_1 | 9 | CD295994 | Strongylocentrotus purpuratus | 5.00E-07 | ||
| cl10375_ctg1_1 | 9 | BU133150 | yes | Gallus gallus | 7.00E-07 | |
| cl3127_ctg2_1 | 19 | CF577195 | Saccharum sp. | 2.00E-05 | ||
| vmsin5140_1 | 1 | BM265659 | Danio rerio | 6.00E-05 | ||
| cl15473_ctg1_1 | 8 | CN502421 | Danio rerio | 7.00E-05 | ||
| cl3097_ctg2_1 | 30 | CA374396 | Oncorhynchus mykiss | 8.00E-05 | ||
| cl9923_ctg1_1 | 2 | DAA01768 | Lytechinus variegatus | 1.00E-04 | ||
| cl15340_ctg1_1 | 14 | CN180033 | yes | 1 | Danio rerio | 3.00E-04 |
| cl11246_ctg1_1 | 6 | BX302229 | Oncorhynchus mykiss | 5.00E-04 | ||
| cl4857_ctg3_1 | 7 | BF718744 | Homo sapiens | 6.00E-04 | ||
| cl18267_ctg1_1 | 2 | AAS58046 | Babesia bovis | 0.001 | ||
| cl5917_ctg1_1 | 6 | CN004343 | Canis familiaris | 0.002 | ||
| cl9934_ctg1_1 | 4 | CD740019 | yes | 2 | Gallus gallus | 0.002 |
| cl3233_ctg1_3 | 12 | CN506386 | yes | 5 | Danio rerio | 0.003 |
| cl22258_ctg1_1 | 2 | BM485921 | Gallus gallus | 0.004 | ||
| cl14723_ctg1_1 | 3 | BF037758 | Homo sapiens | 0.005 | ||
| cl5206_ctg1_1 | 9 | BG166355 | Homo sapiens | 0.005 | ||
| cl5199_ctg2_1 | 8 | BM627372 | yes | 1 | Anopheles gambiae | 0.006 |
| vmsin18077_1 | 1 | BX877871 | yes | 1 | Oncorhynchus mykiss | 0.007 |
| cl5686_ctg1_1 | 2 | BG783827 | Strongylocentrotus purpuratus | 0.008 |
Utility
User interface
The results of the analyses described above have been incorporated into an SQL database amenable to complex queries. The database can be accessed through a user friendly web based interface (XenDB). XenDB allows individual and batch queries using Xenopus accession, GI, and XenDB, UniGene and TIGR cluster IDs. In addition, the user can query the Xenopus sequence hits using any protein accession/GI number both singly and in batch mode. This allows a rapid identification of Xenopus TCs and their corresponding clones with hits to given protein sequences. The output of various queries displays the matching Xenopus cluster(s) and links to a web page as presented in Figure 5. For each cluster, links to the best hit for a number of model organisms are provided as well as links to the assembly result, consensus sequence generated by CAP3, and visual alignments of all FASTY results. GenBank accession numbers for each EST in the cluster and whether the corresponding clone has been identified as full length are provided. Additionally, for each TC the COG and KOG classification, as well as the GO terms are available.
Figure 5.
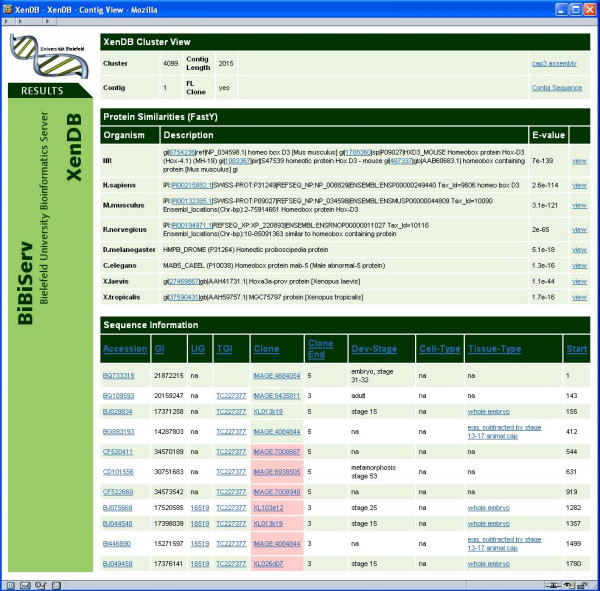
Cluster view of the XenDB Web interface. Best FASTY hits to NR protein database, five model organisms and Xenopus proteins are shown on top. Gene Ontologies (GO) are based on best human and mouse IPI hits, functional categories on hits to COG and KOG databases. Below, additional information for each EST in the cluster is shown, such as accession, UniGene and TGI id, clone, cell and tissue type. Clones predicted not to be full length are colored red. Links to CAP3 assembly and TC sequence are provided.
The analysis and database system provides a very powerful tool which will enable the Xenopus community to take advantage of a number of technical and experimental advances. We have selected a couple of examples to illustrate possible types of queries. In considering the results, it is important to bear in mind that these examples can be combined to further refine the sequence set. In the first example, we sought to identify all the genes of a known type or class. In the second example, we wished to identify the set of Xenopus sequences which best matched a set of genes from another species identified using the CGAP database administered by the National Cancer Institute (NCI) [62,63]. A final example demonstrates the ability of the system to translate results identified by microarray technologies, or other related high throughput technologies, to identify likely Xenopus homologues.
Homeobox gene identification
Homeobox containing proteins are a very important group of transcriptional regulators that play key roles in developmental processes. They can be divided into a 'complex' and a 'dispersed' super class representing the homeotic genes and the large number of homeodomain containing proteins dispersed (and diverged) within the genome [64]. The homeotic (Hox) genes play key roles in the anterior-posterior patterning of both vertebrate and invertebrate embryos and in Xenopus are often used as markers of anterior-posterior development. [65-67]. The vertebrate homeotic genes are organized into four clusters arranged in the same order in which they are expressed in the anterior-posterior axis [64]. Of the 39 vertebrate Hox genes, we have identified 28 homologs in Xenopus laevis, while 19 are present in the protein database (Table 7). For those sequences not identified, we sought to determine whether they had been identified in the genome of Xenopus tropicalis. To do so, we used TBLASTX, provided as a tool on the Xenopus tropicalis website [68] to search for the missing sequences. Strong matches were identified for all of the remaining Hox genes except HoxD12. Using the BLASTN tool on the genome site, we confirmed that the gene order was conserved within each scaffold (data not shown). Interestingly, we were unable to identify HoxD12 within the predicted region though both HosxD11 and HoxD13 were recognized.
Table 7.
Homeobox genes in X. laevis: for each HOX gene the corresponding cluster and TC is shown, as well as the most 5' clone in the assembly and the protein accession number, if available. When X. laevis genes were not identified, an identifier corresponding X. tropicalis sequence is provided.
| IPI Accession | Description | Xenopus cluster/contig | FASTY e-value | BLASTX e-value | FL Clone | Protein Accession |
| IPI00027694 | HOX-A1. | cluster:4123 contig:1 | 4.0e-85 | 1.99E-99 | 5536792 | AAH44984 |
| IPI00012049 | HOX-A2. | cluster:7495 contig:1 | 4.1e-130 | 7.64E-145 | 3556495 | AAG30508 |
| IPI00012050 | HOX-A3. | cluster:10945 contig:1 | 6.5e-91 | 2.89E-111 | 4683538 | AAH41731 |
| IPI00020926 | HOX-A4. | fgenesh.C_1023000005 | ||||
| IPI00302291 | HOX-A5. | cluster:25739 contig:1 | 6.9e-44 | 1.27E-38 | ||
| IPI00010742 | HOX-A6. | fgenesh.C_1023000003 | ||||
| IPI00010743 | HOX-A7. | cluster:3210 contig:1 | 5.8e-40 | 1.17E-64 | XL071e19 | AAA49753 |
| IPI00010744 | HOX-A9. | vm_singlet:264323 | 1.2e-33 | 3.48E-29 | ||
| IPI00010731 | HOX-A10. | fgenesh.C_1487000003 | ||||
| IPI00010754 | HOX-A11. | cluster:6499 contig:1 | 7.2e-42 | Was C11 | XL088b06 | |
| IPI00305850 | HOX-A13. | vm_singlet:174355 | 3.8e-57 | 1.22E-97 | ||
| IPI00294724 | HOX-B1. | fgenesh.C_2225000001 | ||||
| IPI00027261 | HOX-B2. | fgenesh.C_2225000002 | ||||
| IPI00027259 | HOX-B3. | fgenesh.C_2225000003 | ||||
| IPI00014540 | HOX-B4. | cluster:22503 contig:1 | 1.2e-27 | |||
| IPI00012514 | HOX-B5. | vm_singlet:57425 | 8.5e-35 | 3.92E-59 | ||
| IPI00015075 | HOX-B6. | cluster:2339 contig:1 | 6.2e-42 | 2.52E-72 | XL098k02 | |
| IPI00172584 | HOX-B7. | cluster:1985 singlet:1 | 2.6e-65 | 8.16E-77 | 4201615 | P04476 |
| IPI00014536 | HOX-B8. | cluster:16406 contig:1 | 2.8e-28 | 9.90E-43 | ||
| IPI00014539 | HOX-B9. | cluster:8543 contig:1 | 4.0e-30 | 1.05E-50 | XL069k06 | P31272 |
| IPI00030703 | HOX-B10. | cluster:24736 contig:1 | 5.6e-48 | 6.95E-74 | ||
| IPI00295561 | HOX-C4. | fgenesh.C_202000010 | ||||
| IPI00022893 | HOX-C5. | vm_singlet:33065 | 1.5e-41 | 6.14E-32 | ||
| IPI00015921 | HOX-C6. | cluster:9871 singlet:1 | 4.2e-93 | 3.16E-109 | 4202432 | P02832 |
| IPI00010756 | HOX-C8. | cluster:11257 contig:1 | 5.2e-95 | 9.74E-118 | XL045l21 | AAB71818 |
| IPI00010757 | HOX-C9. | fgenesh.C_202000007 | ||||
| IPI00020947 | HOX-C10. | cluster:3243 contig:1 | 1.3e-51 | 1.63E-127 | 4970594 | AAO25534 |
| IPI00011610 | HOX-C11. | fgenesh.C_202000005 | ||||
| IPI00010758 | HOX-C12. | vm_singlet:240042 | 2.4e-46 | 2.75E-22 | ||
| IPI00010759 | HOX-C13. | cluster:21388 contig:1 | 2.0e-80 | 5.86E-89 | XL064e01 | |
| IPI00001551 | HOX-D1. | cluster:9419 contig:1 | 2.5e-50 | 1.68E-65 | 3475513 | AAA49745 |
| IPI00215882 | HOX-D3. | cluster:4099 contig:1 | 2.6e-114 | 3.48E-121 | 4684054 | |
| IPI00012390 | HOX-D4. | cluster:21685 contig:1 | 7.1e-67 | 7.99E-83 | 5571854 | AAQ95789 |
| IPI00008481 | HOX-D8. | cluster:11793 contig:1 | 5.8e-62 | 2.08E-74 | 5543040 | AAH60408 |
| IPI00292734 | HOX-D9. | cluster:13847 contig:1 | 6.5e-38 | 5.28E-55 | XL045k22 | CAC44973 |
| IPI00292735 | HOX-D10. | cluster:6503 contig:1 | 3.8e-135 | 3.97E-143 | 4032032 | CAC44974 |
| IPI00305856 | HOX-D11. | fgenesh.C_1333000003 | ||||
| IPI00018803 | HOX-D12. | missing | ||||
| IPI00018806 | HOX-D13. | cluster:13386 contig:1 | 1.7e-93 | 2.17E-112 | 3399571 | AAO25535 |
Homologue identification from the Cancer Genome Anatomy Project (CGAP)
A second example takes advantage of the CGAP database [69] administered by the National Cancer Institute (NCI). This database and resource incorporates a large number of interconnected modules aimed at gene expression in cancer. Among the modules are a Serial Analysis of Gene Expression (SAGE) database [70,71]. The SAGE approach counts polyadenylated transcripts by sequencing a short 14 bp tag at the genes 3'end and is a quantitative method to examine gene expression [70]. Another module is the Digital Gene Expression Displayer (DGED) which distinguishes statistical differences in gene expression between two pools of libraries [72]. Each method generates tables of genes based on a wide variety of selection criteria. As would be expected, the source for the vast majority of the available data comes from either human or mouse thus demanding a tool to cross match the results in Xenopus.
For this particular example, we selected a tissue based query (DGED) derived from SAGE data in which we sought a set of genes that might include potential markers for glial or astrocyte fates. For this query, we selected all brain, cortex, cerebellum and spinal cord libraries excluding any libraries derived from cell lines. This yielded 58 potential libraries. From this we selected any library labeled as a glioblastoma for pool A and libraries labeled astrocytoma for pool B while excluding the remaining libraries (which included medulloblastomas, ependymomas, etc.). We did not distinguish between cancer grades. This limited the total number of libraries to six glioblastoma and nine astrocytoma libraries containing 487,197 and 863,610 SAGE tags each, respectively. Submission of the query resulted in the identification of 395 tags with a 2× expression factor and a 0.05 significance factor (default CGAP query values). These 395 tags represented 308 different sequences (180 were >2 fold higher in glioblastoma and 128 were >2 fold higher in astrocytoma) which corresponded to 278 proteins in the public database (115 glioblastoma, 163 astrocytoma) and were matched using the batch GenBank accession module available online in XenDB to 100 and 142 Xenopus sequences, respectively. (In the interests of space we have not included the extended table but provide the saved DGED query [see additional file 6] and the two text files [see additional files 7 and 8] that can be uploaded to the XenDB database). The results table includes links to the matching cluster and TC, the e-value and rank and whether a full length clone has been identified. The contig web link leads to additional information including the consensus analysis, the top FASTY hits to five model organisms and links to the Xenopus EST sequences in the TC (Figure 5). Among the genes identified are vimentin (15×, P = 0.01) and sox10 (7.6×, P = 0.03), genes previously established as markers of glial and oligodendrocyte fate respectively [73-75] as well as genes downstream of the Notch signalling pathway, known to be important for glia formation [76]. Thus the system developed and presented here allows 'in silico' based tools established for the study and analysis of other organisms, particularly human and mouse, to be easily and rapidly applied to the Xenopus model system.
Homologues of Drosophila eye development genes
In the final example, we take advantage of the database to perform a comparative analysis of microarray expression data. In many instances, the outcome of an array type experiment is a variety of tables listing regulated genes and the associated expression changes. Currently, there are few published Xenopus array studies available [77-85] while there exist extensive databases of expression for a variety of model organisms. The NCBI maintains a common database, the Gene Expression Omnibus [86] which contains data from over 15,000 samples including 337 Human, 92 mouse and 12 Drosophila experiments (average 25 samples/experiment). Based on an ongoing interest in eye development, we selected a recent paper by Michaut and co-workers in the Gehring lab which examined gene expression changes induced by ectopic expression of the eyeless gene (ey/Pax-6) in Drosophila imaginal disks [87]. The development of the eye is evolutionarily conserved among both vertebrates and invertebrates [88,89]. Many important insights into eye development have come from studies in Drosophila which has defined a genetic cascade of evolutionarily conserved regulatory factors [90]. One such factor is Pax-6/eyeless which is capable of inducing ectopic eyes on both flies [91] and vertebrates [92]. In the Michaut study, 371 eye-induced genes are detected using two different oligonucleotide based array platforms (Affymetrix and Hoffmann-LaRoche) and 73 are discussed in detail within the text (Michaut et al., Table 1, 2). To identify likely homologues of these genes in Xenopus, GenBank accession numbers were obtained from the NCBI Gene Expression Omnibus ([93], accession # GSE271) and used to query the XenDB database to identify 47 potential homologues of the Drosophila Pax6/ey regulated genes and included 32 predicted full length sequences (Table 8). As these sequences are available from commercial sources, they can be readily obtained and tested using the various experimental approaches available to Xenopus such as gain of function studies by microinjection.
Table 8.
Xenopus matches to Pax6/ey Regulated Genes identified by Michaut et al.
| # | Cluster | Ctg | FL clone | Protein Accession | Description | E-value | DM Rank | All Rank |
| 1 | 21344 | 1 | YES | AAA19592 | Lola protein short isoform | 3.90E-10 | 42 | 508 |
| 2 | 21344 | 1 | YES | AAA19593 | Lola protein long isoform | 7.00E-10 | 67 | 553 |
| 3 | 22774 | NO | AAA21879 | atonal protein | 1.20E-17 | 3 | 21 | |
| 4 | 5646 | 1 | YES | AAA28528 | fasciclin II | 4.30E-28 | 5 | 61 |
| 5 | 3838 | 1 | NO | AAA28723 | eyes absent | 1.80E-118 | 1 | 49 |
| 6 | 10868 | 1 | YES | AAB61239 | bunched gene product | 2.00E-21 | 6 | 39 |
| 7 | BJ063320 | NO | AAC46506 | Dachshund | 1.60E-16 | 8 | 44 | |
| 8 | 10334 | 1 | YES | AAC47196 | Lozenge | 2.90E-56 | 4 | 77 |
| 9 | 7019 | 1 | YES | AAD38602 | scratch | 4.40E-35 | 15 | 83 |
| 10 | 4763 | 2 | YES | AAD38642 | BcDNA.GH11415 | 2.90E-146 | 3 | 15 |
| 11 | 16925 | 1 | YES | AAD38646 | BcDNA.GH11973 | 8.20E-14 | 1 | 14 |
| 12 | 18882 | 1 | NO | AAD52845 | Pebble | 7.40E-62 | 2 | 14 |
| 13 | 3666 | 1 | YES | AAF24476 | Sticky ch1 | 1.70E-11 | 3 | 56 |
| 14 | 7799 | 2 | YES | AAF48990 | CG12238-PA | 1.70E-22 | 3 | 64 |
| 15 | 19264 | 1 | YES | AAF55415 | CG5407-PA | 6.30E-198 | 3 | 10 |
| 16 | 5529 | 1 | YES | AAF57639 | CG15093-PA | 2.20E-45 | 1 | 24 |
| 17 | CD327522 | NO | AAK06753 | roughoid/rhomboid-3 | 1.10E-29 | 8 | 26 | |
| 18 | 22774 | NO | AAK14073 | DNA-binding transcription factor | 8.60E-10 | 11 | 158 | |
| 19 | 1415 | 445 | YES | AAL86442 | slamdance | 5.50E-70 | 26 | 194 |
| 20 | BU911996 | NO | AAN74533 | transcription factor fruitless | 7.90E-10 | 28 | 459 | |
| 21 | CD329851 | NO | BAA78210 | white protein | 2.20E-36 | 17 | 54 | |
| 22 | 21321 | 1 | YES | CAA33450 | glass protein | 2.20E-45 | 21 | 1739 |
| 23 | 2426 | 1 | YES | CAA38746 | neurotactin | 2.40E-24 | 103 | 706 |
| 24 | 9209 | 1 | YES | CAA52934 | Drosophila cyclin E type I | 2.50E-56 | 2 | 25 |
| 25 | 18485 | 1 | NO | CAA76941 | UNC-13 protein | 2.70E-165 | 1 | 14 |
| 26 | 17438 | 1 | NO | NP_523928 | CG7525-PA | 8.70E-24 | 101 | 1508 |
| 27 | 570 | 1 | YES | NP_524354 | CG4236-PA | 0 | 1 | 17 |
| 28 | BI349728 | NO | NP_573095 | CG9170-PA | 2.70E-17 | 1 | 7 | |
| 29 | 1761 | 1 | NO | NP_609033 | CG9536-PA | 1.20E-21 | 1 | 6 |
| 30 | 12008 | 1 | YES | NP_609545 | CG14946-PA | 9.10E-25 | 8 | 63 |
| 31 | 440 | 2 | YES | NP_610108 | CG8663-PA | 5.70E-17 | 5 | 90 |
| 32 | 9019 | 1 | YES | NP_611013 | CG11798-PA | 1.40E-07 | 156 | 2411 |
| 33 | 10147 | 2 | YES | NP_648269 | CG5653-PA | 1.90E-16 | 5 | 48 |
| 34 | 3752 | 1 | YES | NP_649919 | CG9427-PA | 4.10E-13 | 1 | 32 |
| 35 | 20081 | 1 | YES | NP_725617 | CG5522-PF | 7.10E-49 | 1 | 18 |
| 36 | 2636 | 2 | YES | NP_729075 | CG10625- | 1.70E-28 | 16 | 1185 |
| 37 | 8386 | YES | O18381 | Eyeless protein | 3.90E-70 | 7 | 75 | |
| 38 | 11614 | 1 | YES | P00528 | Tyrosine-protein kinase Src64B | 4.30E-152 | 3 | 150 |
| 39 | 4073 | 1 | NO | P10181 | Homeobox protein rough | 3.10E-14 | 13 | 165 |
| 40 | 919 | NO | P20483 | String protein (Cdc25-like | 3.30E-40 | 3 | 43 | |
| 41 | 1777 | 1 | YES | P36872 | Twins protein (PR55) | 0 | 2 | 41 |
| 42 | 9517 | 1 | YES | P48554 | Ras-related protein Rac2 | 1.00E-109 | 1 | 22 |
| 43 | 7661 | 1 | YES | Q01070 | E(spl) mgamma | 5.50E-19 | 5 | 52 |
| 44 | 7661 | 1 | YES | Q01071 | E(spl) mdelta | 1.10E-15 | 7 | 63 |
| 45 | 4146 | 2 | YES | Q23989 | Villin-like protein quail | 6.30E-23 | 9 | 138 |
| 46 | 10061 | 1 | YES | Q27324 | Derailed protein | 1.20E-45 | 23 | 400 |
| 47 | 14903 | 1 | YES | Q27350 | Sine oculis protein | 3.90E-87 | 1 | 20 |
| Sequences without significant homology | ||||||||
| 48 | O77459 | transcription factor Ken | 60 | NP_651346 | CG11849-PA | |||
| 49 | AAF46666 | CG10527-PA | 61 | Q23997 | Chitinase-like protein DS47 precursor | |||
| 50 | NP_728586 | CG9134-PA | 62 | AAD09748 | Gasp precursor | |||
| 51 | NP_609450 | CG17124-PA | 63 | AAF63503 | SP2523 | |||
| 52 | CG140595 | Zea mays genomic | 64 | AAF47412 | CG13897-PA | |||
| 53 | NP_570064 | CG10803-PA | 65 | AAL27368 | zinc finger C2H2 protein sequoia | |||
| 54 | NP_650785 | CG5835-PA | 66 | NP_730444 | CG32209: CG32209-PB | |||
| 55 | AAF51847 | CG11370-PA | 67 | NP_723827 | CG18507-PA | |||
| 56 | AAG46059 | SKELETOR | 68 | NP_611728 | CG13532-PA | |||
| 57 | AAN61340 | BcDNA:GH10711 | 69 | NP_651343 | CG13651-PA | |||
| 58 | NP_729183 | CG10121-PA | 70 | NP_995997 | CG12605-PA | |||
| 59 | AAO39528 | RE22242p | 71 | NP_610067 | CG9335-PA | |||
Discussion
Comparative approaches to important biological problems have resulted in enormous progress in the past decades. The advent of genomic and proteomic approaches has led to a torrent of data in many organisms and has demanded increasingly sophisticated bioinformatic approaches to organize and manage the information. We have developed an integrated information resource with a user-friendly interface powered by an automated clustering pipeline which will allow researchers to take advantage of the wealth of knowledge available in the public domain.
Comparison to human and mouse
Human and mouse are the best studied vertebrate organisms at the molecular level. In addition to the well publicized genome projects, both have extensive EST collections. This has led to the prediction and characterization of 44,775+ human sequences and 36,182 mouse sequences [94]. As vertebrate development is well conserved, it is important to assess the extent to which the Xenopus EST project has identified the known vertebrate genes. At the same time, one would like to identify any genes that are unique to Xenopus. Most gene prediction programs rely on homology thus eliminating this approach to unique gene identification. Sequences without significant homology could arise from incomplete sequencing that does not extend into the coding region. Results of the human genome project suggest that this would not be the case for a majority of the sequences analyzed in this report. The average 5' UTR in humans is 240 bp and the 3' UTR is 400 bp [95]. Sequencing reactions with current technologies yield readable sequence of 700 bases on average. Therefore, at least some subset of sequences would yield their protein sequence to analysis. An alternative origin of non-homologous sequences would be unspliced or improperly spliced transcripts. This possibility is also minimized by the utilization of polyA tails for RNA selection and reverse transcription priming using oligo(dT). A final, obvious and expensive approach is to select non-homologous sequences for full length double stranded sequencing. Sequence without errors more easily yields the desired open reading frame in even the simplest bioinformatic programs.
Sequences without hits
A class of sequences includes those without significant BLAST hits. In our analysis we have used a cutoff e-value of 10e-6. This of course is necessarily arbitrary, since as mentioned above it is not known what the exact level of similarity is between any given sequence pair. Based on this value, we remain with 43,753 sequences that neither have a BLASTX nor a FASTY hit to a known model organism sequence. The lack of similarity could be due to significant divergence of the sequence, the lack of an appropriate homologue in the public dataset, sequencing errors inherent in EST data or due to the presence of non-coding, presumably regulatory sequences, in the EST clone set. These unmatched sequences mirror the situation in the UniGene set for both mouse and human with greater than 3 and 4 × 106 EST sequences in 76,000 and 106,000 clusters respectively while fewer than 25,000 coding sequences have been recognized [21,94,96]. The source of these discrepancies are currently unclear, but may arise from non coding RNA (ncRNA)[97], micro RNA precursors [98], incompletely or unspliced transcripts [99]. In particular, ncRNAs are a likely source for a large fraction of the discrepancy based on estimates of a 10-fold greater number of non-coding transcription units than protein coding genes [100]. It has been estimated that >95% of transcription is non-coding [101]. Much of the analysis and identification of ncRNA relies on the availability of genomic sequence which is currently unavailable for X. laevis and incomplete for X. tropicalis, the highly homologous diploid species.
Completeness of Xenopus EST set
We have compared all the Xenopus sequences to the human and mouse protein sets to identify conserved proteins. An obvious question is how complete is the Xenopus EST set and what percentage of genes have been identified assuming that the vast majority of protein coding sequences have been evolutionarily conserved. Of the ~40,000 sequences in the IPI databases, 9,225 human and 7,664 mouse sequences do not have a strong match (E < 1.0e-6). Thus, there is a considerable effort remaining to develop a complete Xenopus protein coding set. In the course of our analysis we note the high degree of similarity between the allotetraploid laevis and diploid tropicalis Xenopus species which depended on the length of the matching sequence. For sequences covering >= 95% of the query, there was an average of 94% identity while the average identity dropped to 91% and 88% as the coverage dropped to 90 and 80% respectively. This conservation may allow sequences from both species to be combined to generate a more complete set.
It is well known that the outcome of clustering methods on a large scale depends on the variety of involved parameters. A systematic comparison between UniGene or TIGR Gene Indices and our results turns out to be extremely difficult, mainly because the underlying sequence sets differ as well due to different sequence cleanup and masking approaches. To maximize the utility and usability of our analysis, we have incorporated UniGene and TGI information into our dataset and provide simple tools for identifying the related UniGene and TGI identifier.
Future prospects
Both the clustering and consensus generation approaches are very rapid: 50 minutes for clustering on a single 900 MHz SPARC-CPU and a few hours for assembly on a cluster of 20 heterogeneous SPARC-based machines with 450 to 900 MHz. We therefore have achieved the design goal of being able to frequently update this aspect of the analysis. The subsequent comparative sequence analysis requires significantly greater resources and time (several weeks on same cluster of heterogeneous workstations). The analysis described above is performed by various PERL based scripts developed during the course of our analysis which will allow updates and application to other model systems. We are currently working on a tool to compare clusters over time which will allow the sequence analysis described below to be performed on the restricted set of modified/new clusters rather than to the entire ensemble. The effect of CAP3 consensus generation is that a given cluster can be split into several separate TC sequences, usually due to low sequence quality or differences in the UTR regions of the sequences. The UTR end splitting is likely due to the differences between the in-paralogs in this allotetraploid species. We believe that such information will be of value to those researchers interested in a variety of evolutionary questions, examples of which will be discussed below. The difference in ploidy makes Xenopus laevis distinct from all of the other organisms for which similar analysis have been performed.
As with all ongoing high throughput sequencing efforts, certain aspects of the results change in proportion to the total number of sequences. As noted above, a complete gene set for Xenopus will require additional sequencing. The generation of tetra, octo and dodecaploid species of Xenopus between 80 and 10 million years ago [102] offers opportunities in the field of evolutionary biology. For example, comparisons of 3' UTR regions between in-paralogs of Xenopus laevis and their counterpart diploid tropical species may improve statistical models of molecular evolution. At the genome level, the potential availability of genome data from the polyploid species may provide insight into questions of chromosome segregation and silencing. The selection of Xenopus as a model organism by the NIH http://www.nih.gov/science/models/ and the establishment of the Trans-NIH Xenopus Initiative [103] have directly led to the support of EST and genome sequencing efforts. Among the priorities identified is the establishment and funding of a Xenopus Database [104] which will integrate sequence, expression and other Xenopus data. We hope to be able to update the results described here on a regular basis and contribute to the community effort.
Conclusion
One of the primary goals of the effort was to provide a resource of gene-oriented EST clusters and transcript oriented TCs, enriched with various information from heterogeneous sources, that would be of value to the biology community and the Xenopus community in particular. Using the XenDB system, the biologist can identify sequences of interest using simple gene name queries, accessions, or gene ontologies. The identified sequences have been mapped to public resources like NCBI's UniGene and TIGR Gene Indices and a consensus sequence prepared. In addition, we have identified publicly available IMAGE clones that maximizes the 5' sequence to provide a full length construct when possible. These clones are available from IMAGE consortium providers.
Availability and requirements
Sequence availability, XenDB database and results display
The database and associated files are freely accessible through the XenDB website: http://bibiserv.techfak.uni-bielefeld.de/xendb/. The GenBank accession numbers and FASTA formatted files of the masked and clipped input sequences, as well as the TC sequences and results of the example applications (see below) can be downloaded. Additionally, the list of full length clones is available to researchers interested in performing genome-wide studies. Programs, scripts and database dumps are available from the authors upon request. The XenDB database should be cited with the present publication as a reference.
List of abbreviations used
EST: Expressed Sequence Tag, ORDBMS: Object Relational Database Managemant System, TC: tentative contig sequence, KOG: clusters of euKaryotic Orthologous Groups, GO: Gene Ontology, VRT: Vertebrate Sequences, HTC: High Throughput cDNA, XGC: Xenopus Gene Collection, MGC: Mammalian Gene Collection, ZGC: Zebrafish Gene Collection, FL: Full Length, IPI: International Protein Index, CGAP: Cancer Genome Anatomy Project, DGED: Differential Gene Expression Database, SAGE: Serial Analysis of Gene Expression, ncRNA: non-coding RNA, TGI: TIGR Gene Index
Authors' contributions
A.S. developed and implemented the Vmatch based clustering pipeline. M.B. contributed his high throughput sequence analysis system Genlight. A.S. and M.B. developed the XenDB database schema, performed the post clustering data analyses and contributed to the manuscript. A.H.B. provided supervision and guidance on the development of the project design goals and the interpretation of analysis output with regard to biological significance. R.G. provided supervision and guidance on the development of the clustering pipeline and provided essential infrastructure. C.R.A. provided advice and guidance on the development of the clustering pipeline, the incorporation of analysis into the database and performed and interpreted the various queries presented and wrote a significant portion of the manuscript.
Supplementary Material
Table S1, Distribution of EST sequences in the analysis based on the annotated tissue source for the preparation of the library. (NOTE: annotations are imported directly from GenBank entries and are dependent on the original annotation.)
Table S2: The 20 most abundant developmental stage annotations in the X. laevis data set as annotated in GenBank: Distribution of EST sequences in the analysis based on the annotated developmental stage of the source library. (NOTE: annotations are imported directly from GenBank entries and are dependent on the original annotation.)
Table S3: The 30 most abundant Clone Libraries in the X. laevis data set as determined by the GenBank annotation. (NOTE: annotations are imported directly from GenBank entries and are dependent on the original annotation.)
Figure S1, Effect of Parameter Variation on EST Clustering: Masked and trimmed EST sequences were clustered using the Vmatch algorithm using different overlap length and percentage identity values. The total number of clusters (blue) and the number of singletons (red) are plotted against the minimal overlap length. Values were plotted at different percentage identities (squares 98%, stars 96%, circles 94%).
Table S4: Sizes of protein sets used for sequence analysis of clustered sequences.
file containing the SAGE database query used in the glioblastoma and astrocytoma analysis.
File containing protein accession numbers of SAGE glioblastoma genes for upload to XenDb system
File containing protein accession numbers of SAGE astrocytoma genes for upload to XenDb system
Acknowledgments
Acknowledgements
The authors thank Jan Reinkensmeier for his help in setting up the XenDB Web pages, Alin Vonika, Trent Clarke and Stefan Kurtz for comments on the manuscript. The FSU School of Computation Science and Information Technology and FSU Supercomputing Facility provided computing resources. CRA was supported by an FSU Research Foundation Program Enhancement Grant.
Contributor Information
Alexander Sczyrba, Email: asczyrba@techfak.uni-bielefeld.de.
Michael Beckstette, Email: mbeckste@TechFak.Uni-Bielefeld.de.
Ali H Brivanlou, Email: brvnlou@mail.rockefeller.edu.
Robert Giegerich, Email: robert@TechFak.Uni-Bielefeld.de.
Curtis R Altmann, Email: curtis.altmann@med.fsu.edu.
References
- Adams MD, Kelley JM, Gocayne JD, Dubnick M, Polymeropoulos MH, Xiao H, Merril CR, Wu A, Olde B, Moreno RF, . Complementary DNA sequencing: expressed sequence tags and human genome project. Science. 1991;252:1651–1656. doi: 10.1126/science.2047873. [DOI] [PubMed] [Google Scholar]
- Zhang MQ. Computational prediction of eukaryotic protein-coding genes. Nat Rev Genet. 2002;3:698–709. doi: 10.1038/nrg890. [DOI] [PubMed] [Google Scholar]
- Henderson J, Salzberg S, Fasman KH. Finding genes in DNA with a Hidden Markov Model. J Comput Biol. 1997;4:127–141. doi: 10.1089/cmb.1997.4.127. [DOI] [PubMed] [Google Scholar]
- Besemer J, Borodovsky M. Heuristic approach to deriving models for gene finding. Nucleic Acids Res. 1999;27:3911–3920. doi: 10.1093/nar/27.19.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontius JU, Wagner L, Schuler GD. The NCBI Handbook. Bethesda, MD, National Center for Biotechnology Information; 2003. UniGene: a unified view of the transcriptome; pp. 21-1–21-12. [Google Scholar]
- Christoffels A, van Gelder A, Greyling G, Miller R, Hide T, Hide W. STACK: Sequence Tag Alignment and Consensus Knowledgebase. Nucleic Acids Res. 2001;29:234–238. doi: 10.1093/nar/29.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov AA, Fickett JW, Gelfand MS. Frequent alternative splicing of human genes. Genome Res. 1999;9:1288–1293. doi: 10.1101/gr.9.12.1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladd AN, Cooper TA. Finding signals that regulate alternative splicing in the post-genomic era. Genome Biol. 2002;3:reviews0008. doi: 10.1186/gb-2002-3-11-reviews0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscombe D, Pan JQ, Gray AC. Functional diversity in neuronal voltage-gated calcium channels by alternative splicing of Ca(v)alpha1. Mol Neurobiol. 2002;26:21–44. doi: 10.1385/MN:26:1:021. [DOI] [PubMed] [Google Scholar]
- Stamm S. Signals and their transduction pathways regulating alternative splicing: a new dimension of the human genome. Hum Mol Genet. 2002;11:2409–2416. doi: 10.1093/hmg/11.20.2409. [DOI] [PubMed] [Google Scholar]
- Venables JP. Alternative splicing in the testes. Curr Opin Genet Dev. 2002;12:615–619. doi: 10.1016/S0959-437X(02)00347-7. [DOI] [PubMed] [Google Scholar]
- Roberts GC, Smith CW. Alternative splicing: combinatorial output from the genome. Curr Opin Chem Biol. 2002;6:375–383. doi: 10.1016/S1367-5931(02)00320-4. [DOI] [PubMed] [Google Scholar]
- Oklu R, Hesketh R. The latent transforming growth factor beta binding protein (LTBP) family. Biochem J. 2000;352 Pt 3:601–610. doi: 10.1042/0264-6021:3520601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarone G, Hirsch E, Brancaccio M, De Acetis M, Barberis L, Balzac F, Retta SF, Botta C, Altruda F, Silengo L, Retta F. Integrin function and regulation in development. Int J Dev Biol. 2000;44:725–731. [PubMed] [Google Scholar]
- Klint P, Claesson-Welsh L. Signal transduction by fibroblast growth factor receptors. Front Biosci. 1999;4:D165–D177. doi: 10.2741/klint. [DOI] [PubMed] [Google Scholar]
- Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Muller WE, Wetter T, Suhai S. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004;14:1147–1159. doi: 10.1101/gr.1917404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kota R, Rudd S, Facius A, Kolesov G, Thiel T, Zhang H, Stein N, Mayer K, Graner A. Snipping polymorphisms from large EST collections in barley (Hordeum vulgare L.) Mol Genet Genomics. 2003;270:24–33. doi: 10.1007/s00438-003-0891-6. [DOI] [PubMed] [Google Scholar]
- Useche FJ, Gao G, Harafey M, Rafalski A. High-throughput identification, database storage and analysis of SNPs in EST sequences. Genome Inform Ser Workshop Genome Inform. 2001;12:194-203.:194–203. [PubMed] [Google Scholar]
- Nekrutenko A. Reconciling the numbers: ESTs versus protein-coding genes. Mol Biol Evol. 2004;21:1278–1282. doi: 10.1093/molbev/msh125. [DOI] [PubMed] [Google Scholar]
- Wang JP, Lindsay BG, Leebens-Mack J, Cui L, Wall K, Miller WC, DePamphilis CW. EST clustering error evaluation and correction. Bioinformatics. 2004;20:2973–84. doi: 10.1093/bioinformatics/bth342. [DOI] [PubMed] [Google Scholar]
- Genome-Consortium Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- Ewing B, Green P. Analysis of expressed sequence tags indicates 35,000 human genes. Nat Genet. 2000;25:232–234. doi: 10.1038/76115. [DOI] [PubMed] [Google Scholar]
- Wheeler DL, Church DM, Edgar R, Federhen S, Helmberg W, Madden TL, Pontius JU, Schuler GD, Schriml LM, Sequeira E, Suzek TO, Tatusova TA, Wagner L. Database resources of the National Center for Biotechnology Information: update. Nucleic Acids Res. 2004;32 :D35–D40. doi: 10.1093/nar/gkh073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DL, Church DM, Federhen S, Lash AE, Madden TL, Pontius JU, Schuler GD, Schriml LM, Sequeira E, Tatusova TA, Wagner L. Database resources of the National Center for Biotechnology. Nucleic Acids Res. 2003;31:28–33. doi: 10.1093/nar/gkg033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler GD. Pieces of the puzzle: expressed sequence tags and the catalog of human genes. J Mol Med. 1997;75:694–698. doi: 10.1007/s001090050155. [DOI] [PubMed] [Google Scholar]
- Schuler GD, Boguski MS, Stewart EA, Stein LD, Gyapay G, Rice K, White RE, Rodriguez-Tome P, Aggarwal A, Bajorek E, Bentolila S, Birren BB, Butler A, Castle AB, Chiannilkulchai N, Chu A, Clee C, Cowles S, Day PJ, Dibling T, Drouot N, Dunham I, Duprat S, East C, Hudson TJ, . A gene map of the human genome. Science. 1996;274:540–546. doi: 10.1126/science.274.5287.540. [DOI] [PubMed] [Google Scholar]
- Boguski MS, Schuler GD. ESTablishing a human transcript map. Nat Genet. 1995;10:369–371. doi: 10.1038/ng0895-369. [DOI] [PubMed] [Google Scholar]
- Huang X, Madan A. CAP3: A DNA sequence assembly program. Genome Res. 1999;9:868–877. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Phrap sequence assember website. Laboratory of Phil Green, HHMI Genome Sciences Department, University of Washington; 2005. http://www.phrap.org/ [Google Scholar]
- Burke J, Davison D, Hide W. d2_cluster: a validated method for clustering EST and full-length cDNAsequences. Genome Res. 1999;9:1135–1142. doi: 10.1101/gr.9.11.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abouelhoda MI, Ohlebusch E, Kurtz S. Proceeding of the Ninth International Symposium on String Processing and Information Retieval. 2476. Springer Verlag; 2002. Optimal exact string matching based on suffix arrays; pp. 31–43. (Lecture Notes in Computer Science). [Google Scholar]
- Abouelhoda MI, Kurtz S, Ohlebusch E. Proceedings of the Second Workshop on Algorithms in Bioinformatics. 2452. Springer Verlag; 2002. The Enhanced Suffix Array and its Applications to Genome Analysis; pp. 449–463. (Lecture Notes in Computer Science). [Google Scholar]
- Abouelhoda MI, Kurtz S, Ohlebusch E. Replacing Suffix Trees with Enhanced Suffix Arrays. Journal of Discrete Algorithms. 2004;2:53–86. doi: 10.1016/S1570-8667(03)00065-0. [DOI] [Google Scholar]
- Liang F, Holt I, Pertea G, Karamycheva S, Salzberg SL, Quackenbush J. An optimized protocol for analysis of EST sequences. Nucleic Acids Res. 2000;28:3657–3665. doi: 10.1093/nar/28.18.3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Church DM, DiCuccio M, Edgar R, Federhen S, Helmberg W, Kenton DL, Khovayko O, Lipman DJ, Madden TL, Maglott DR, Ostell J, Pontius JU, Pruitt KD, Schuler GD, Schriml LM, Sequeira E, Sherry ST, Sirotkin K, Starchenko G, Suzek TO, Tatusov R, Tatusova TA, Wagner L, Yaschenko E. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2005;33:D39–D45. doi: 10.1093/nar/gki062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quackenbush J, Cho J, Lee D, Liang F, Holt I, Karamycheva S, Parvizi B, Pertea G, Sultana R, White J. The TIGR Gene Indices: analysis of gene transcript sequences in highly sampled eukaryotic species. Nucleic Acids Res. 2001;29:159–164. doi: 10.1093/nar/29.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vector Database Website. 2005. http://seq.yeastgenome.org/vectordb/
- The Vmatch large scale sequence analysis software website. 2005. http://www.vmatch.de/
- Jurka J. Repbase update: a database and an electronic journal of repetitive elements. Trends Genet. 2000;16:418–420. doi: 10.1016/S0168-9525(00)02093-X. [DOI] [PubMed] [Google Scholar]
- Smit A, Green P. Repeat Masker Website and Server. 2005. http://www.repeatmasker.org/
- Beckstette M, Strothmann D, Homann R, Giegerich R, Kurtz S. PoSSuMsearch: Fast and Sensitive Matching of Position Specific Scoring Matrices Using Enhanced Suffix Arrays. In Proceedings of the German Conference on Bioinformatics (GCB 2004); GI Lecture Notes in Informatics, 53:53-64. [Google Scholar]
- Kruger J, Sczyrba A, Kurtz S, Giegerich R. e2g: an interactive web-based server for efficiently mapping large EST and cDNA sets to genomic sequences. Nucleic Acids Res. 2004;32:W301–W304. doi: 10.1093/nar/gkh586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000;7:203–214. doi: 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson WR, Wood T, Zhang Z, Miller W. Comparison of DNA sequences with protein sequences. Genomics. 1997;46:24–36. doi: 10.1006/geno.1997.4995. [DOI] [PubMed] [Google Scholar]
- Beckstette M, Mailänder JT, Marhöfer RJ, Sczyrba A, Ohlebusch E, Giegerich R, Selzer PM. Journal of Integrative Bioinformatics. Vol. 8. Magdeburg, IMBio, Informationsmanagement in der Biotechnologie e.V.; 2004. Genlight: Interactive high-throughput sequence analysis and comparative genomics; pp. 79–94. (Yearbook Bioinformatics 2004). Hofestädt R. [Google Scholar]
- European Bioinformatics Institute International Protein Index Website. 2005. http://www.ebi.ac.uk/IPI
- Kersey PJ, Duarte J, Williams A, Karavidopoulou Y, Birney E, Apweiler R. The International Protein Index: an integrated database for proteomics experiments. Proteomics. 2004;4:1985–1988. doi: 10.1002/pmic.200300721. [DOI] [PubMed] [Google Scholar]
- Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O'Donovan C, Redaschi N, Yeh LS. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 2004;32 (Database issue):D115–D119. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aaronson JS, Eckman B, Blevins RA, Borkowski JA, Myerson J, Imran S, Elliston KO. Toward the development of a gene index to the human genome: an assessment of the nature of high-throughput EST sequence data. Genome Res. 1996;6:829–845. doi: 10.1101/gr.6.9.829. [DOI] [PubMed] [Google Scholar]
- Hillier LD, Lennon G, Becker M, Bonaldo MF, Chiapelli B, Chissoe S, Dietrich N, DuBuque T, Favello A, Gish W, Hawkins M, Hultman M, Kucaba T, Lacy M, Le M, Le N, Mardis E, Moore B, Morris M, Parsons J, Prange C, Rifkin L, Rohlfing T, Schellenberg K, Marra M, . Generation and analysis of 280,000 human expressed sequence tags. Genome Res. 1996;6:807–828. doi: 10.1101/gr.6.9.807. [DOI] [PubMed] [Google Scholar]
- Komar AA, Hatzoglou M. Internal ribosome entry sites in cellular mRNAs: The mystery of their existence. J Biol Chem. 2005 doi: 10.1074/jbc.R400041200. [DOI] [PubMed] [Google Scholar]
- The Mammalian Gene Collection. 2005. http://mgc.nci.nih.gov/
- The Xenopus Gene Collection. 2005. http://xgc.nci.nih.gov/
- The Zebrafish Gene Collection. 2005. http://zgc.nci.nih.gov/
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Fedorova ND, Jackson JD, Jacobs AR, Krylov DM, Makarova KS, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Rogozin IB, Smirnov S, Sorokin AV, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004;5:R7. doi: 10.1186/gb-2004-5-2-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Sonnhammer EL, von Heijne G, Krogh A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol. 1998;6:175–182. [PubMed] [Google Scholar]
- Lash AE, Tolstoshev CM, Wagner L, Schuler GD, Strausberg RL, Riggins GJ, Altschul SF. SAGEmap: a public gene expression resource. Genome Res. 2000;10:1051–1060. doi: 10.1101/gr.10.7.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strausberg RL, Buetow KH, Greenhut SF, Grouse LH, Schaefer CF. The cancer genome anatomy project: online resources to reveal the molecular signatures of cancer. Cancer Invest. 2002;20:1038–1050. doi: 10.1081/CNV-120005922. [DOI] [PubMed] [Google Scholar]
- Gehring WJ, Affolter M, Burglin T. Homeodomain proteins. Annu Rev Biochem. 1994;63:487–526. doi: 10.1146/annurev.bi.63.070194.002415. [DOI] [PubMed] [Google Scholar]
- Cox WG, Hemmati-Brivanlou A. Caudalization of neural fate by tissue recombination and bFGF. development. 1995;121:4349–4358. doi: 10.1242/dev.121.12.4349. [DOI] [PubMed] [Google Scholar]
- Wright CV, Morita EA, Wilkin DJ, De Robertis EM. The Xenopus XIHbox 6 homeo protein, a marker of posterior neural induction, is expressed in proliferating neurons. Development. 1990;109:225–234. doi: 10.1242/dev.109.1.225. [DOI] [PubMed] [Google Scholar]
- Isaacs HV, Pownall ME, Slack JM. Regulation of Hox gene expression and posterior development by the Xenopus caudal homologue Xcad3. EMBO J. 1998;17:3413–3427. doi: 10.1093/emboj/17.12.3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JGI Xenopustropicalis Web Site . 2005. http://genome.jgi-psf.org/Xentr3/Xentr3.home.html
- Cancer Genome Anatomy Project. 2005. http://cgap.nci.nih.gov/
- Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial analysis of gene expression. Science. 1995;270:484–487. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- Boon K, Osorio EC, Greenhut SF, Schaefer CF, Shoemaker J, Polyak K, Morin PJ, Buetow KH, Strausberg RL, De Souza SJ, Riggins GJ. An anatomy of normal and malignant gene expression. Proc Natl Acad Sci U S A. 2002;99:11287–11292. doi: 10.1073/pnas.152324199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal A, Lash AE, Altschul SF, Velculescu V, Zhang L, McLendon RE, Marra MA, Prange C, Morin PJ, Polyak K, Papadopoulos N, Vogelstein B, Kinzler KW, Strausberg RL, Riggins GJ. A public database for gene expression in human cancers. Cancer Res. 1999;59:5403–5407. [PubMed] [Google Scholar]
- Kuhlbrodt K, Herbarth B, Sock E, Hermans-Borgmeyer I, Wegner M. Sox10, a novel transcriptional modulator in glial cells. J Neurosci. 1998;18:237–250. doi: 10.1523/JNEUROSCI.18-01-00237.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M. Intermediate filament proteins define different glial subpopulations. J Neurosci Res. 2001;63:284–289. doi: 10.1002/1097-4547(20010201)63:3<284::AID-JNR1022>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Colman DR. Glial-defined rhombomere boundaries in developing Xenopus hindbrain. J Comp Neurol. 2000;424:47–57. doi: 10.1002/1096-9861(20000814)424:1<47::AID-CNE4>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Gaiano N, Fishell G. The role of notch in promoting glial and neural stem cell fates. Annu Rev Neurosci. 2002;25:471–490. doi: 10.1146/annurev.neuro.25.030702.130823. [DOI] [PubMed] [Google Scholar]
- Konig R, Baldessari D, Pollet N, Niehrs C, Eils R. Reliability of gene expression ratios for cDNA microarrays in multiconditional experiments with a reference design. Nucleic Acids Res. 2004;32:e29. doi: 10.1093/nar/gnh027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump D, Werry K, Veldhoen N, Van Aggelen G, Helbing CC. Exposure to the herbicide acetochlor alters thyroid hormone-dependent gene expression and metamorphosis in Xenopus Laevis. Environ Health Perspect. 2002;110:1199–1205. doi: 10.1289/ehp.021101199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Sanjuan I, Bell E, Altmann CR, Vonica A, Brivanlou AH. Gene profiling during neural induction in Xenopus laevis: regulation of BMP signaling by post-transcriptional mechanisms and TAB3, a novel TAK1-binding protein. development. 2002;129:5529–5540. doi: 10.1242/dev.00097. [DOI] [PubMed] [Google Scholar]
- Tran PH, Peiffer DA, Shin Y, Meek LM, Brody JP, Cho KW. Microarray optimizations: increasing spot accuracy and automated identification of true microarray signals. Nucleic Acids Res. 2002;30:e54. doi: 10.1093/nar/gnf053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altmann CR, Bell E, Sczyrba A, Pun J, Bekiranov S, Gaasterland T, Brivanlou AH. Microarray-based analysis of early development in Xenopus laevis. Dev Biol. 2001;236:64–75. doi: 10.1006/dbio.2001.0298. [DOI] [PubMed] [Google Scholar]
- Arima K, Shiotsugu J, Niu R, Khandpur R, Martinez M, Shin Y, Koide T, Cho KW, Kitayama A, Ueno N, Chandraratna RA, Blumberg B. Global analysis of RAR-responsive genes in the Xenopus neurula using cDNA microarrays. Dev Dyn. 2005;232:414–431. doi: 10.1002/dvdy.20231. [DOI] [PubMed] [Google Scholar]
- Peiffer DA, von Bubnoff A, Shin Y, Kitayama A, Mochii M, Ueno N, Cho KW. A Xenopus DNA microarray approach to identify novel direct BMP target genes involved in early embryonic development. Dev Dyn. 2005;232:445–456. doi: 10.1002/dvdy.20230. [DOI] [PubMed] [Google Scholar]
- Shin Y, Kitayama A, Koide T, Peiffer DA, Mochii M, Liao A, Ueno N, Cho KW. Identification of neural genes using Xenopus DNA microarrays. Dev Dyn. 2005;232:432–444. doi: 10.1002/dvdy.20229. [DOI] [PubMed] [Google Scholar]
- Chung HA, Hyodo-Miura J, Kitayama A, Terasaka C, Nagamune T, Ueno N. Screening of FGF target genes in Xenopus by microarray: temporal dissection of the signalling pathway using a chemical inhibitor. Genes Cells. 2004;9:749–761. doi: 10.1111/j.1356-9597.2004.00761.x. [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaut L, Flister S, Neeb M, White KP, Certa U, Gehring WJ. Analysis of the eye developmental pathway in Drosophila using DNA microarrays. Proc Natl Acad Sci U S A. 2003;100:4024–4029. doi: 10.1073/pnas.0630561100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser T, Walton DS, Maas RL. Genomic structure, evolutionary conservation and aniridia mutations in the human PAX6 gene. Nat Genet. 1992;2:232–239. doi: 10.1038/ng1192-232. [DOI] [PubMed] [Google Scholar]
- Gehring WJ, Ikeo K. Pax 6: mastering eye morphogenesis and eye evolution. Trends Genet. 1999;15:371–377. doi: 10.1016/S0168-9525(99)01776-X. [DOI] [PubMed] [Google Scholar]
- Gehring WJ. The genetic control of eye development and its implications for the evolution of the various eye-types. Int J Dev Biol. 2002;46:65–73. [PubMed] [Google Scholar]
- Halder G, Callaerts P, Gehring WJ. Induction of ectopic eyes by targeted expression of the eyeless gene in Drosophila [see comments] Science. 1995;267:1788–1792. doi: 10.1126/science.7892602. [DOI] [PubMed] [Google Scholar]
- Chow RL, Altmann CR, Lang RA, Hemmati-Brivanlou A. Pax6 induces ectopic eyes in a vertebrate. development. 1999;126:4213–4222. doi: 10.1242/dev.126.19.4213. [DOI] [PubMed] [Google Scholar]
- The NCBI Gene Expression Omnibus. 2005. http://www.ncbi.nlm.nih.gov/geo/
- Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, Baertsch R, Bailey J, Barlow K, Beck S, Berry E, Birren B, Bloom T, Bork P, Botcherby M, Bray N, Brent MR, Brown DG, Brown SD, Bult C, Burton J, Butler J, Campbell RD, Carninci P, Cawley S, Chiaromonte F, Chinwalla AT, Church DM, Clamp M, Clee C, Collins FS, Cook LL, Copley RR, Coulson A, Couronne O, Cuff J, Curwen V, Cutts T, Daly M, David R, Davies J, Delehaunty KD, Deri J, Dermitzakis ET, Dewey C, Dickens NJ, Diekhans M, Dodge S, Dubchak I, Dunn DM, Eddy SR, Elnitski L, Emes RD, Eswara P, Eyras E, Felsenfeld A, Fewell GA, Flicek P, Foley K, Frankel WN, Fulton LA, Fulton RS, Furey TS, Gage D, Gibbs RA, Glusman G, Gnerre S, Goldman N, Goodstadt L, Grafham D, Graves TA, Green ED, Gregory S, Guigo R, Guyer M, Hardison RC, Haussler D, Hayashizaki Y, Hillier LW, Hinrichs A, Hlavina W, Holzer T, Hsu F, Hua A, Hubbard T, Hunt A, Jackson I, Jaffe DB, Johnson LS, Jones M, Jones TA, Joy A, Kamal M, Karlsson EK, Karolchik D, Kasprzyk A, Kawai J, Keibler E, Kells C, Kent WJ, Kirby A, Kolbe DL, Korf I, Kucherlapati RS, Kulbokas EJ, Kulp D, Landers T, Leger JP, Leonard S, Letunic I, Levine R, Li J, Li M, Lloyd C, Lucas S, Ma B, Maglott DR, Mardis ER, Matthews L, Mauceli E, Mayer JH, McCarthy M, McCombie WR, McLaren S, McLay K, McPherson JD, Meldrim J, Meredith B, Mesirov JP, Miller W, Miner TL, Mongin E, Montgomery KT, Morgan M, Mott R, Mullikin JC, Muzny DM, Nash WE, Nelson JO, Nhan MN, Nicol R, Ning Z, Nusbaum C, O'Connor MJ, Okazaki Y, Oliver K, Overton-Larty E, Pachter L, Parra G, Pepin KH, Peterson J, Pevzner P, Plumb R, Pohl CS, Poliakov A, Ponce TC, Ponting CP, Potter S, Quail M, Reymond A, Roe BA, Roskin KM, Rubin EM, Rust AG, Santos R, Sapojnikov V, Schultz B, Schultz J, Schwartz MS, Schwartz S, Scott C, Seaman S, Searle S, Sharpe T, Sheridan A, Shownkeen R, Sims S, Singer JB, Slater G, Smit A, Smith DR, Spencer B, Stabenau A, Stange-Thomann N, Sugnet C, Suyama M, Tesler G, Thompson J, Torrents D, Trevaskis E, Tromp J, Ucla C, Ureta-Vidal A, Vinson JP, Von Niederhausern AC, Wade CM, Wall M, Weber RJ, Weiss RB, Wendl MC, West AP, Wetterstrand K, Wheeler R, Whelan S, Wierzbowski J, Willey D, Williams S, Wilson RK, Winter E, Worley KC, Wyman D, Yang S, Yang SP, Zdobnov EM, Zody MC, Lander ES. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Morey C, Avner P. Employment opportunities for non-coding RNAs. FEBS Lett. 2004;567:27–34. doi: 10.1016/j.febslet.2004.03.117. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Gupta S, Zink D, Korn B, Vingron M, Haas SA. Genome wide identification and classification of alternative splicing based on EST data. Bioinformatics. 2004;20:2579–2585. doi: 10.1093/bioinformatics/bth288. [DOI] [PubMed] [Google Scholar]
- Yelin R, Dahary D, Sorek R, Levanon EY, Goldstein O, Shoshan A, Diber A, Biton S, Tamir Y, Khosravi R, Nemzer S, Pinner E, Walach S, Bernstein J, Savitsky K, Rotman G. Widespread occurrence of antisense transcription in the human genome. Nat Biotechnol. 2003;21:379–386. doi: 10.1038/nbt808. [DOI] [PubMed] [Google Scholar]
- Mattick JS. Non-coding RNAs: the architects of eukaryotic complexity. EMBO Rep. 2001;2:986–991. doi: 10.1093/embo-reports/kve230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sammut B, Marcuz A, Pasquier LD. The fate of duplicated major histocompatibility complex class Ia genes in a dodecaploid amphibian, Xenopus ruwenzoriensis. Eur J Immunol. 2002;32:1593–1604. doi: 10.1002/1521-4141(200206)32:6<1593::AID-IMMU1593>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Trans-NIH Xenopus Initiative Website. 2005. http://www.nih.gov/science/models/Xenopus/
- Xenbase Xenopus Web Resource Website. 2005. http://xenbase.org
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, Distribution of EST sequences in the analysis based on the annotated tissue source for the preparation of the library. (NOTE: annotations are imported directly from GenBank entries and are dependent on the original annotation.)
Table S2: The 20 most abundant developmental stage annotations in the X. laevis data set as annotated in GenBank: Distribution of EST sequences in the analysis based on the annotated developmental stage of the source library. (NOTE: annotations are imported directly from GenBank entries and are dependent on the original annotation.)
Table S3: The 30 most abundant Clone Libraries in the X. laevis data set as determined by the GenBank annotation. (NOTE: annotations are imported directly from GenBank entries and are dependent on the original annotation.)
Figure S1, Effect of Parameter Variation on EST Clustering: Masked and trimmed EST sequences were clustered using the Vmatch algorithm using different overlap length and percentage identity values. The total number of clusters (blue) and the number of singletons (red) are plotted against the minimal overlap length. Values were plotted at different percentage identities (squares 98%, stars 96%, circles 94%).
Table S4: Sizes of protein sets used for sequence analysis of clustered sequences.
file containing the SAGE database query used in the glioblastoma and astrocytoma analysis.
File containing protein accession numbers of SAGE glioblastoma genes for upload to XenDb system
File containing protein accession numbers of SAGE astrocytoma genes for upload to XenDb system