

Abstract
Efficient expression of most bacteriophage λ early genes depends upon the formation of an antiterminating transcription complex to overcome transcription terminators in the early operons, pL and pR. Forma tion of this complex requires the phage-encoded protein N, the first gene product expressed from the pL operon. The N leader RNA contains, in this order: the NUTL site, an RNase III-sensitive hairpin and the N ribosome-binding site. N bound to NUTL RNA is part of both the antitermination complex and an autoregulatory complex that represses the translation of the N gene. In this study, we show that cleavage of the N leader by RNase III does not inhibit antitermination but prevents N-mediated translation repression of N gene expression. In fact, by preventing N autoregulation, RNase III activates N gene translation at least 200-fold. N-mediated translation repression is extremely sensitive to growth rate, reflecting the growth rate regulation of RNase III expression itself. Given N protein’s critical role in λ development, the level of RNase III activity therefore serves as an important sensor of physiological conditions for the bacteriophage.
Keywords: antitermination/bacteriophage λ/growth rate regulation/RNase III/translation repression
Introduction
Coliphage λ uses N-mediated antitermination to regulate the expression of early phage functions (Friedman and Gottesman, 1983; Das, 1992; Friedman and Court, 1995; Weisberg and Gottesman, 1999). Transcription of the early operons of λ, pL and pR is interrupted by multiple transcription terminators. The expression of the early phage function N from pL facilitates the assembly of terminator-resistant transcription complexes. N and Escherichia coli Nus factors interact at a site in the pL and pR transcripts called NUT to convert RNA polymerase to this antiterminating form.
The NUT site of the pL transcript is located in the leader of the N gene from +34 to +64 (Figure 1). In addition to antitermination, two other processes act through the N leader, affecting the expression of N. A large stem–loop structure at +76 to +208 in the N leader is a substrate for the double-strand-specific ribonuclease RNase III (Figure 1) (Lozeron et al., 1976, 1977; Steege et al., 1987; Court, 1993). This hairpin, which ends immediately upstream of the N Shine–Dalgarno sequence, interferes with translation of the N gene (Kameyama et al., 1991). RNase III cleavage removes the inhibitory structure and stimulates N gene expression. The translation of N is also affected by an autoregulatory mechanism in which N bound at the NUTL site represses the expression of its own gene (Wilson et al., 1997).

Fig. 1. Structure of the N leader. Nucleotides are numbered from +1 of the pL transcript. Shown are NUTL, the RNase III-sensitive hairpin with cleavage sites (arrows) and the Shine–Dalgarno sequence (SD) and initiation codon of the N ribosome-binding site.
In a previous paper, we proposed that the structure of the N leader reflects the temporal order of the three regulatory processes acting through the N leader (Wilson et al., 1997). Once NUTL is transcribed, the antitermination complex is assembled. Upon completion of the RNase III-sensitive hairpin, this site in the N leader can be cleaved by RNase III. Once the N ribosome-binding site is transcribed, N bound at the NUTL site can cause translation repression of N gene expression. Cleavage by RNase III eliminates the RNA connection between NUTL at the 5′ end of the transcript and the transcribing antitermination complex, as well as between NUTL and the N ribosome-binding site and structural gene. RNase III cleavage is expected to occur after assembly of the antitermination complex but prior to N-mediated translation repression. In this study, we investigate the relationship between RNase III cleavage of the N leader and the two N-mediated functions, antitermination and, in greater detail, autoregulation. Our observations suggest that N expression is controlled by an appealing mechanism in which one regulatory pathway counteracts another in a highly temporally sensitive manner.
Results
Studying the effect of RNase III on N-mediated antitermination and translation repression using a double reporter system
To study antitermination and the regulation of N gene expression, we have constructed a gene fusion on the E.coli chromosome that permits simultaneous measurement of N-mediated translation repression and transcription antitermination (Figure 2) (Wilson et al., 1997). This pL-nutL-N-lacZ-galK fusion contains the pL promoter, the N leader, the first 33 codons of the N structural gene fused to the ninth codon of lacZ and, far downstream, the gal operon which includes the galK gene. The effect of N and RNase III (the latter encoded by the E.coli rnc gene) on the expression of the N gene can be studied by monitoring β-galactosidase (β-gal) expression from N-lacZ. There are multiple transcription terminators between pL and galK which virtually eliminate galK expression unless an antitermination complex forms at pL. Therefore, N-mediated antitermination can be studied by measuring levels of galactokinase, the galK gene product. In our constructs, the pL promoter is under the control of the temperature-sensitive repressor, CI857.

Fig. 2. The pL-nutL-N-lacZ-galK double reporter fusion. The expression of the gal operon is under the control of pL and is blocked under N– conditions by transcription terminators (T) including one in an IS2 element in the gal leader. N-mediated antitermination allows gal expression in N+ cells. The left attachment site of the λ prophage carrying pL-N-lacZ is designated as att.
In all experiments below using fusion-containing strains, cells growing exponentially at 30°C were shifted to 42°C to inactivate CI857 and induce expression from pL. All data are enzyme activities in samples collected after 60 min of heat induction. In these experiments, N without its regulatory sequences was constitutively expressed in trans from pLAC on a derivative of the medium copy-number plasmid pGB2 (Wilson et al., 1997).
The data in Table I show a multitude of regulatory effects, three of which have been reported previously. First, N-mediated antitermination through transcription terminators significantly enhances galK expression (compare galK units of N– with N+, Table IA) (Adhya et al., 1974). Secondly, RNase III cleavage of the inhibitory hairpin immediately adjacent to the N ribosome-binding site (Figure 1) stimulates N-lacZ translation (compare β-gal units of Table IA with B under N– conditions) (Kameyama et al., 1991). Thirdly, N almost completely represses the translation of N-lacZ in rnc– cells through an autoregulatory mechanism (compare β-gal units of N– with N+, Table IA) (Wilson et al., 1997).
Table I. The effect of RNase III on N-mediated antitermination and translation repression.
| rnc allele | β-galactosidase (β-gal) units |
galactokinase (galK) units |
|||
|---|---|---|---|---|---|
| N– | N+ | N– | N+ | ||
| (A) pL-nutL-RIIIshp+-N-lacZ-galK | rnc– (rnc14) | 838 | <10 | <1 | 159 |
| (B) pL-nutL-RIIIshp+-N-lacZ-galK | rnc+ | 2080 | 3945 | <1 | 310 |
| (C) pL-nutL-RIIIshp+-N-lacZ-galK | rnc70 | 908 | <10 | ND | ND |
| (D) pL-nutL-RIIIshpΔ-N-lacZ-galK | rnc– (rnc14) | 3906 | 141 | ND | ND |
| (E) pL-nutL-RIIIshpΔ-N-lacZ-galK | rnc+ | 4357 | 20 | ND | ND |
| (F) pL-nutL-RIIIshp+-N-lacZ-galK | rnc40 (1×) | 1053 | 24 | ND | ND |
| (G) pL-nutL-RIIIshp+-N-lacZ-galK | rnc40 (2×) | ND | 70 | ND | ND |
| (H) pL-nutL-RIIIshp+-N-lacZ-galK | rnc40 (≥3×) | ND | 154 | ND | ND |
Shown are enzyme activities in exponentially growing cells after 60 min of heat induction with zero time values subtracted. Data are an average of at least two experiments. Variability between averaged values for β-galactosidase assays was <10% and for galactokinase assays was <29%. N– and N+ cells carry pGB2 or pZH124 (plac-N+), respectively. RIIIshp refers to the RNase III-sensitive hairpin. ND indicates values that were not determined.
New to this study are the effects of RNase III cleavage of the N leader on N-mediated antitermination and translation repression. RNase III did not inhibit antitermination (compare galK units of Table IA and B under N+ conditions); in fact, galK expression consistently was enhanced 2-fold in rnc+ cells. However, RNase III did prevent N-mediated translation repression (compare β-gal units of Table IA and B under N+ conditions).
The rnc70 allele encodes a mutant RNase III that binds but does not cleave its substrate (Inada et al., 1989; Dasgupta et al., 1998). N repressed N-lacZ translation in an rnc70 background (Table IC), indicating that binding by RNase III is not sufficient to prevent N autoregulation. To determine whether this interference with N autoregulation is caused by RNase III cleaving the N leader or some other RNase III-sensitive site, we analyzed N-mediated translation repression using a construct with the RNase III-sensitive hairpin deleted in the N leader (Wilson et al., 1997). This deletion precisely deletes the entire RNase III-sensitive stem–loop structure shown in Figure 1 from +76 through +208. In these deletion strains, N repressed N-lacZ expression in both rnc– and rnc+ cells, demonstrating that it is specifically cleavage of the N leader that prevents N-mediated translation repression (Table ID and E). Surprisingly, when the RNase III-sensitive hairpin was deleted, N-mediated translation repression was more effective in rnc+ than rnc– cells (repressing to 20 versus 141 U, respectively). This difference may be a consequence of RNase III affecting translation through its role in rRNA processing (Dunn and Studier, 1973; Nikolaev et al., 1973) or influencing the expression of some other component of the N autoregulatory complex.
A weak transcription terminator in the N leader
We attribute the 2-fold increase in N-lacZ expression caused by deletion of the RNase III-sensitive hairpin (compare β-gal units of Table IB with D and E under N– conditions) to the deletion of a transcription terminator in the N leader (Adhya et al., 1977). The activating effect of antitermination through this terminator can be observed when RNase III is present and preventing translation repression (compare β-gal units of Table IB under N– and N+ conditions). The precise location of the terminator within the RNase III-sensitive stem–loop structure has not been determined.
The effect of RNase III on pL-N+ expression
In the experiments presented in Table I, we studied the effect of RNase III on N-mediated translation repression using an N-lacZ fusion as the reporter and N protein constitutively expressed in trans from a multicopy plasmid as the repressing molecule. In the normal context of the prophage, N expressed from pL should repress its own expression in rnc– cells. Using immunoblotting, we investigated the effect of RNase III on N protein being expressed and regulated in this more natural context from an N+ prophage containing the entire pL operon. In these strains, as in our N-lacZ fusion strains, all of the genes expressed from pR, as well as the right attachment site, have been deleted. Cultures growing exponentially at 30°C were shifted to 42°C to induce expression from pL. We observed a delay in detectable levels of N expressed in an rnc– (rnc14) strain relative to an rnc+ strain (Figure 3). This delay can be a consequence of one or both of the roles of the intact RNase III-sensitive hairpin on N translation: either as a structural component of the N autoregulatory complex or as a steric inhibitor of ribosome entry in an N-independent manner. The nutL44 mutation eliminates N binding to NUT and, consequently, N-mediated translation repression (Salstrom and Szybalski, 1978; Wilson et al., 1997), but should have no effect on the N-independent inhibition by the RNase III-sensitive hairpin. The 2.5- to 3-fold difference between N levels at 9 min after induction in rnc+ versus rnc– nutL+ prophage-containing strains was reduced at least 50% by the nutL44 mutation (data not shown). Thus, at least some of the RNase III-mediated effect on N expression is due to N autoregulation.
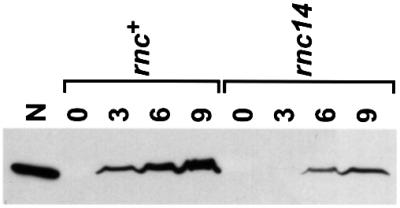
Fig. 3. Western blot analysis of N expression in rnc+ and rnc14 (rnc–) pL-N+ strains. Numbers indicate time in minutes of sample collection following heat induction of pL-N expression. The first lane contains 20 ng of pure N protein.
The effect of intermediate levels of RNase III on N-mediated translation repression
N binding to NUT, RNase III cleavage of the N leader and translation of N are tightly coupled events. It takes a minimum of 4 s to transcribe from the end of the nut site, through the RNase III-sensitive site to the N ribosome-binding site (Figure 1) (Gotta et al., 1991). The distance between the end of NUT and the end of the RNase III-sensitive hairpin allows sufficient time for the antitermination complex to form prior to cleavage (Barik et al., 1987). However, the end of the RNase III-sensitive hairpin and the N ribosome-binding site are so close that we envisage that RNase III must process quickly to prevent N-mediated translation repression of N gene expression.
In a preliminary experiment to determine how levels of RNase III affect N-mediated translation repression, we constructed a derivative of the pL-N-lacZ strain with the rnc40 allele. The rnc40 mutation is a mini-Tn10 insertion in the leader region of the rnc operon (Takiff et al., 1989). This mutation blocks the normal rnc promoter and RNase III-mediated regulation of the operon. In this mutant, the rnc operon is expressed from a tetracycline-dependent promoter within the mini-Tn10. The level of transcription from this tetracycline-dependent promoter is predicted, based on lacZ operon fusion data, to be 10% of that from the rnc promoter (Takiff et al., 1992). This level of transcription is sufficient to supply enough of the essential protein Era, which is expressed as part of the rnc operon, for cell viability and to permit a level of 30S rRNA processing by RNase III that is intermediate to that seen in rnc+ and rnc– cells (data not shown). Much to our surprise, N-mediated translation repression at the level of RNase III activity expressed from this allele was almost as strong as that in rnc– cells (Table IF). We constructed additional derivatives of the pL-N-lacZ strain with two and at least three copies of the rnc40 allele (see Materials and methods). Although our western blotting technique was not sensitive enough to measure RNase III levels in these strains directly (Figure 5), we expected the amount of RNase III to increase incrementally as the copy number of the rnc40 allele increased. The level of N-mediated translation repression in these strains remained strong at these presumably higher levels of RNase III activity (Table IG and H). These results suggest that below a certain unexpectedly high threshold, RNase III does not prevent N autoregulation efficiently. The intracellular levels of RNase III in this experiment were manipulated artificially and could not be measured precisely, but our results encouraged us to search for physiological conditions in which the level of RNase III activity was sufficiently reduced for N-mediated translation repression to operate in rnc+ cells.
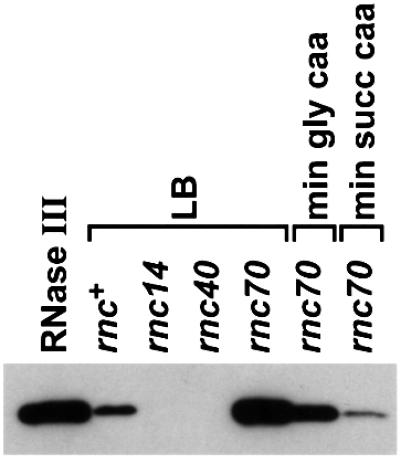
Fig. 5. Western blot analysis of rnc expression. A 20 ng aliquot of pure RNase III is run relative to 20 µl of rnc+, rnc40 and rnc14 cell lysates grown on LB and 10 µl of rnc70 cell lysates grown on the media indicated.
The effect of growth medium on N-mediated translation repression
rRNA precursor is an important substrate for RNase III (Dunn and Studier, 1973; Nikolaev et al., 1973), and the synthesis of rRNA decreases significantly with decreasing growth rate (Bremer and Dennis, 1996). In a previous study, we observed that the expression of rnc-lacZ protein fusions decreased as cells grow more slowly, suggesting growth rate regulation of rnc translation (Britton et al., 1998). The N autoregulation experiments reported up to this point were done using cells grown in rich LB medium in which RNase III activity is high. Therefore, we next tested whether N autoregulation occurs in cells growing slowly in poor media where RNase III activity should be reduced.
The growth rate of the pL-nutL-N-lacZ-galK fusion-containing strain was manipulated over a 4-fold range by growing cells in liquid minimal media supplemented with increasingly poor carbon sources (Figure 4). The control experiments show first that, when cells lacked functional genes for both RNase III and N (rnc– N–, Figure 4), N-lacZ expression was only slightly affected by growth medium and, secondly, that when these rnc– cells expressed N, N-mediated translation repression was complete in all media (rnc– N+, Figure 4). As was already shown in Table I, N-lacZ expression was high in rnc+ N+ cells growing rapidly in LB medium because there was no N-mediated translation repression (Figure 4). However, N-mediated repression increased significantly with decreasing growth rate, suggesting a reduction of RNase III activity as the cells grew more slowly (rnc+ N+, Figure 4). At a doubling time of ∼240 min, N-mediated translation repression of N gene expression was virtually complete in rnc+ cells.

Fig. 4. The effect of growth rate on N-mediated translation repression. Media from right to left are LB, M63 glucose plus casamino acids, M63 glycerol plus casamino acids, M63 glucose, M63 glycerol, and M63 succinate plus casamino acids. Shown are β-galactosidase activities in samples collected after 60 min of heat induction with the zero time values subtracted. To simplify comparison of the β-galactosidase activity of each strain in each medium, all data are plotted relative to rnc+ doubling times. The rnc– strains grow between 21 and 8% more slowly than rnc+ strains depending on the medium. N+ cells carry pZH124 and N– cells carry pGB2. Data shown are an average of five experiments with <23% variability between averaged values.
The repressive effect of poor growth medium on N-lacZ expression in rnc+ N– cells also suggests a decrease in RNase III activity (Figure 4). This 2-fold decrease of N-lacZ expression in the absence of N in the slowest growing cells relative to the fastest growing cells tested is consistent with a loss of RNase III activation of N-lacZ translation through cleavage of the inhibitory hairpin (Kameyama et al., 1991). In fact, in the poorest growth medium, the level of N-lacZ expression in rnc+ N– cells equaled that in rnc– N– cells, as if the former are rnc– in terms of N gene expression.
The effect of growth medium on RNase III levels
We used immunoblotting to monitor directly RNase III protein in LB and minimal medium supplemented with casamino acids plus either glycerol or succinate. In these media, we observed essentially no, intermediate or complete N-mediated translation repression of N-lacZ expression in rnc+ cells, respectively (Figure 4). The sensitivity of our immunodetection assay allowed us to detect RNase III in rnc+ strains grown on LB (Figure 5) but not those grown on the minimal media tested (data not shown). While this result is consistent with growth rate regulation of rnc expression, we preferred to detect RNase III protein in all media tested. To increase rnc expression overall and thus the sensitivity of our assay, we used a strain carrying the rnc70 allele. The synthesis of RNase III protein is higher in this background because the mutant protein cannot cleave the rnc leader RNA and inhibit rnc expression (Dasgupta et al., 1998). The rnc70 allele increases rnc expression 5- to 6-fold or 5- to 12-fold, as determined using rnc-lacZ fusions (Dasgupta et al., 1998) or immunoblotting (Figure 5), respectively.
We retested the effect of growth rate on rnc expression using the rnc70 strain (Figure 5). The level of RNase III protein was reduced 4- to 7-fold in cells grown on minimal glycerol casamino acids and 7- to 14-fold in cells grown on minimal succinate casamino acids relative to those grown on LB. These data corroborate the conclusions from experiments using rnc-lacZ fusions that rnc expression is growth rate regulated. More important to this study, we observe that as RNase III levels decreased, N autoregulation of N-lacZ expression increased in rnc+ cells, consistent with RNase III blocking N-mediated translation repression in a growth rate-dependent manner. The rnc+ cells grown on minimal succinate casamino acids or rnc40 strains grown on LB appear to have similar levels of RNase III (∼10% of the level in rnc+ cells grown on LB) and virtually complete repression of N-lacZ expression by N.
Discussion
The RNA leader of the N gene contains sites for assembly of an antitermination complex, RNase III cleavage and autoregulatory repression of N gene translation. The order in which these regulatory sites in the N leader are transcribed appears to determine the sequence in which the mechanisms acting at those sites can operate (Figure 1). RNase III cleavage of a hairpin downstream of NUTL does not interfere with antitermination. These results suggest that the antitermination complex assembles quickly on RNA polymerase after transcription of the nutL site (Barik et al., 1987) and that the RNase III-induced cleavage does not disrupt this complex once assembled (compare Figure 6A and B). Because it has been demonstrated that NUT sites do not act in trans to promote antitermination (Devito and Das, 1992), it is unlikely that assembly occurs after RNase III cleaves to release NUT RNA from the transcript.

Fig. 6. Model of the antitermination/autoregulation complex transcribing the N gene before (A) or after (B) cleavage at the RNase III-sensitive hairpin (RIIIshp). The model suggests that the contact inhibitory to translation is between the N Shine–Dalgarno sequence (SD) and N protein in the complex, but is not meant to depict precisely the nature of this contact.
In contrast, RNase III cleavage of this hairpin, lying immediately upstream of the N ribosome-binding site, interferes with the ability of N bound at NUTL to repress N gene translation. This has been shown using both pL-N-lacZ fusions with N protein constitutively expressed in trans (Table I) and a pL-N+ prophage with the N expressed in this context regulating its own expression (Figure 3). In our working model of N autoregulation, translation repression of N gene expression occurs within the antitermination complex (Figure 6A; D.Yu, H.Wilson, J.G.Zhou and D.Court, manuscript in preparation). We propose that RNase III cleavage enhances N translation by preventing incorporation of the N transcript into this inhibitory complex (compare Figure 6A and B). This transcript remains unoccluded and is free to be translated. If the transcript is uncleaved either because of a mutant enzyme encoded by rnc14 or rnc70, for example, or a mutant substrate as in the N-lacZ fusion strain with the RNase III-sensitive hairpin deleted, the N ribosome-binding site is incorporated into the antitermination/autoregulation complex and repression is complete. Our work leads us to envisage an intriguing mechanism in which at least one function of a protein/RNA machine is regulated by RNase cleavage.
The RNase III-sensitive hairpin has multiple roles in the N leader. First, since N-mediated translation repression acts only on a ribosome-binding site that is positioned at the base of the RNase III-sensitive hairpin, this structure apparently holds the N gene in the proper position for repression by N bound at NUTL (Wilson et al., 1997). Secondly, even in the absence of N protein and the antitermination complex, the stable hairpin structure inhibits translation of N 2-fold (Kameyama et al., 1991). Lastly, cleavage of this hairpin eliminates both of these repressive effects, resulting in a >400-fold regulation of N gene expression by RNase III under N+ conditions. The modest 2-fold effect of N-independent RNase III activation of N gene translation is presumed to be most important very early in infection when N has not yet been synthesized. As N levels increase, RNase III can exert a stronger effect on N translation by preventing N autoregulation (compare β-gal units of Table IA and B under N– with N+ conditions) and allowing an optimal steady-state level of N. While N-mediated translation repression amplifies the effect of RNase III on N gene expression, we propose that the critical regulatory element in N autoregulation is not N itself, but RNase III.
The seemingly unfortunate observation that in rich medium there is no N-mediated translation repression of N gene expression in rnc+ cells gave us the opportunity to observe how growth rate-associated changes in RNase III levels affect N gene expression. The rapid progression of events during phage development makes the timing of regulatory steps critical. Because of the proximity of the RNase III-sensitive hairpin to the N ribosome-binding site (Figure 1), the opportunity to cleave and activate only immediately precedes the opportunity for N bound at NUTL to repress N gene translation. The consequence is a system that is exquisitely sensitive to the level of RNase III. Our results with the rnc40 allele suggest that a threshold level of activity must be reached before RNase III cleavage of the N leader is competitive with the N repression complex (Table I). Figure 4 shows that we have identified growth conditions in which N expression is autoregulated presumably because intracellular RNase III levels are not high enough to block formation of the autoregulation complex completely. Therefore, the level of RNase III activity can serve as an important sensor of intracellular conditions for λ and, through its effect on the expression of the λ antitermination protein N, can influence the expression of virtually all other λ genes. Thus, this study reinforces the view that RNase III can act as an important global regulator in E.coli (Gitelman and Apirion, 1980; Britton et al., 1998).
The relationship between the level of RNase III activity and the extent of N autoregulation may be complex and not simply a matter of total intracellular RNase III activity. The N-mediated translation repression complex, once formed, may protect the hairpin from RNase III cleavage; alternatively, cleavage subsequent to the formation of the complex may not free the transcript to be translated. More importantly, host- and phage-specified RNase III-sensitive sites in E.coli have a hierarchy of sensitivity, and presumably are competing with the N leader for a limited amount of RNase III. The most important competitor is probably the rRNA precursor, the synthesis of which is changing with growth rate, as is the synthesis of RNase III and the level of N-mediated translation repression. The ribosome itself should also be considered as a possible effector of our growth rate-dependent translation repression mechanism (Comer et al., 1996). Ribosome concentration, which changes coordinately with growth rate, may influence how effectively the ribosome competes with the N repression complex for the cleaved transcript.
Upon infection, λ may replicate either lytically, producing many phage particles and destroying the host, or lysogenically, with the phage DNA passively maintained in the host chromosome. The typical λ plaque contains phage in both modes of development. The plaque itself is, of course, a consequence of the lytic killing of cells and spread of progeny phage. The plaque appears turbid because it contains growing lysogenic cells which are immune to further lytic infection.
RNase III regulates the expression of a number of λ functions that are all competing to determine the final developmental outcome. The gene products of cII, cIII and int are important for the establishment of lysogeny. RNase III cleaves a sense–antisense hybrid of the cII message, thus promoting degradation of the cII transcript (Krinke and Wulff, 1987, 1990). Binding by RNase III is proposed to favor a secondary structure in the cIII transcript that enhances the translation of this gene (Altuvia and Oppenheim, 1986; Altuvia et al., 1987, 1989). In addition, depending on the structure at the 3′ end of the int transcript, RNase III either cleaves to promote degradation or may bind without cleaving to stabilize this message (Gottesman et al., 1982; Court, 1993). Interestingly, RNase III cleavage at a site downstream of N in the N-antiterminated pL transcript destabilizes the N region of this transcript (Lozeron et al., 1976, 1977; Wilder and Lozeron, 1979; Anevski and Lozeron, 1981). This could represent another mechanism whereby N and RNase III regulate N expression, but clearly not the dominant mechanism since N levels are higher in rnc+ than rnc– cells (Figure 3). Finally, RNase III cleaves the transcript of lytic late genes necessary for virus assembly (Daniels et al., 1988).
Does RNase III also influence the lysis/lysogeny decision through its effect on N autoregulation? λ forms clear plaques on cells constitutively overexpressing N from a plasmid, demonstrating that inappropriate N expression can affect this choice. λ also makes clear plaques on rnc– cells in which N levels are reduced by autoregulation (Figure 3). Suppressors of this clear plaque phenotype on rnc– strains increase cIII translation by stabilizing the hairpin structure in the cIII leader that RNase III binding putatively favors (Altuvia and Oppenheim, 1986; Altuvia et al., 1987, 1989). There fore, λ lytic growth presumably is favored on rnc– cells because the loss of RNase III action on the cIII transcript is overriding all the other effects including that of N autoregulation. All the RNase III-sensitive sites important in λ development are also competing for RNase III in poor growth medium, which is a physiologically different condition from that which exists in an rnc– cell. Low intracellular levels of RNase III will have different biological effects from those of no RNase III. For example, the efficiency of lysogeny has been shown to be greater in poor relative to rich medium at low phage-to-cell ratios (<1 phage per cell) (Kourilsky and Knapp, 1974). This is in contrast to the decreased efficiency of lysogeny seen on rnc– cells relative to rnc+ cells. To understand the role of N autoregulation in λ development, we need to separate it from all this complexity. Therefore, we currently are designing a mutant phage that will allow us to study N-mediated translation repression independently of the pleiotropic RNase III- and growth rate-mediated effects.
Materials and methods
Bacterial strains and plasmids
The construction of the pL-nutL-N-lacZ-galK double reporter strains is described in a previous study (Wilson et al., 1997). To construct the pL-N+ strains used in Figure 3, a cI857 λ lysogen of WJW349 [W3110 Δ(argF-lacZ)U169 gal490*(IS2) pglΔ8] was made Δ(cro-bioA)<>TetR by homologous recombination between a PCR product and the chromosome (Yu et al., 2000). This strain was made rnc– by transducing to chloramphenicolR (at 10 µg/ml) using P1 phage grown on an rnc<>cat derivative of DY330 [W3110 Δ(argF-lacZ)U169 gal490*(IS2) pglΔ8 λ cI857 Δ(cro-bioA] (this laboratory). Other mutant rnc strains were made by transducing to tetracycline resistance (at 12.5 µg/ml) using P1 phage grown on HT115 (W3110 rnc14::ΔTn10), HT120 (W3110 rnc40:: ΔTn10) (Takiff et al., 1989) or SDF217 (C600 rnc70 TD1-17::ΔTn10) (this laboratory; Dasgupta et al., 1998) and confirming that λ forms clear plaques on the chosen isolate. TD1-17::ΔTn10 alone has no effect on expression of the fusion (data not shown).
A λ phage carrying the rnc40 allele as part of the rnc operon (TD1 λimm21 rnc40::ΔTn10) (Takiff et al., 1989) was used to lysogenize the N-lacZ fusion strain already containing rnc40 at the natural position in the E.coli chromosome. This attΔ phage integrates by homologous recombination into the rnc operon. Strains with one or more copies of the rnc40 phage were distinguished by the ‘ter’ test (Mousset and Thomas, 1968). The strain designated as having one copy of rnc40 in Table I contains the rnc40 allele brought in by P1 transduction. The strain designated as having two copies of rnc40 contains the original rnc40 allele plus one copy of the rnc40 phage. The strain designated as having three or more copies of rnc40 contains the original rnc40 allele plus at least two copies of the rnc40 phage as determined by the ter test. The copy number of the rnc40 phage in this last strain is predicted based on an incremental increase in β-galactosidase activity above that of the strain carrying two copies of the rnc40 allele. For comparison, multiple lysogens of the rnc40 phage expressing even higher levels of β-galactosidase activity were isolated and are hypothesized to carry three and more copies of the phage (data not shown).
Medium copy-number plasmids pGB2 and pZH124 have been described previously (Churchward et al., 1984; Wilson et al., 1997).
Enzyme assays
The cells used for enzyme assays reported in Table I were grown as described previously (Wilson et al., 1997) except that the tetracycline-dependent rnc40 strains were grown in LB medium supplemented with tetracycline at 12.5 µg/ml. Bacteria for β-galactosidase assays shown in Figure 4 were grown as reported previously (Wilson et al., 1997) except overnight cultures were diluted in growth curve media to a starting OD600 of 0.004 and the overnight medium for the five minimal growth curve media was M63 plus 1 µg/ml thiamine, 1 µg/ml biotin, 0.4% glucose, 0.05% casamino acids and 100 µg/ml spectinomycin. Prior to dilution into the minimal growth curve medium, the cells were collected by centrifugation, washed in 5 ml of M63 and resuspended in M63 to an approximate OD600 of 2.0. In addition to LB plus 100 µg/ml spectinomycin, the growth curve media in this experiment were M63 plus 1 µg/ml thiamine, 1 µg/ml biotin, 100 µg/ml spectinomycin and either 0.4% glucose and 0.05% casamino acids, 0.4% glycerol and 0.05% casamino acids, 0.4% glucose, 0.4% glycerol, or 0.4% succinate and 0.05% casamino acids.
Prior to harvesting cells for enzyme assays, cultures growing exponentially at 30°C were shifted to 42°C for 60 min to induce expression of pL. β-galactosidase and galactokinase assays were performed as described previously (Miller, 1972; McKenney et al., 1981).
Western blot analysis
To immunoblot for N, overnight cultures were grown at 30°C, diluted 1:400 in 50 ml of fresh LB broth and grown with shaking at 30°C to an OD600 of 0.6. A 2 ml aliquot of culture was taken as the zero time sample. A 10 ml aliquot of the remaining culture was dispensed to each of three 50 ml flasks. These cultures were shifted to 42°C with aeration (200 r.p.m.), and one flask per culture was chilled in an ice–water bath at the indicated times after initiating heat induction. Samples were kept on ice until all time points were collected. Cells in 2 ml of each culture were harvested by centrifugation (10 min at 18 000 g, 4°C), resuspended in 50 µl of treatment buffer (0.0625 M Tris–HCl pH 6.8, 2% SDS, 10% glycerol, 5% 2-mercaptoethanol, 0.05% Bromphenol Blue) and heated at 100°C for 4 min. To eliminate viscosity, samples were disrupted with a sonic oscillator tip (Branson Sonifier cell disrupter 200, 10 pulses at output setting of 1.5) and cellular debris was removed by centrifugation (10 min at 18 000 g, 4°C). Samples (0.24 OD600 units) were separated on an 18% NOVEX pre-cast Tris-glycine gel and proteins were transferred to a nitrocellulose membrane essentially according to the manufacturer’s protocols (Invitrogen, Carlsbad, CA). N protein was detected using affinity-purified anti-N polyclonal primary antibody from rabbit and then goat anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibody (Promega, Madison, WI) essentially as described (Feather-Henigan et al., 2000) using 0.2% Tween-20 and 5% non-fat dry milk in the Tris-buffered saline blocking buffer. The N-antibody complex was visualized using SuperSignal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL) essentially according to the manufacturer’s instructions.
Cell lysate preparation and immunoblotting for RNase III were performed essentially as described for N except cells were grown as described for Figure 4 without the 42°C heat induction, 6 ml of each culture were harvested at OD600 = 0.2, samples were separated on a 12% NOVEX Tris-glycine gel, transferred to a PVDF membrane and protein was detected using anti-RNase III monoclonal antibody from mouse and anti-mouse HRP-conjugated antibody from goat.
Two approaches were taken to verify that samples within an experiment contained roughly equivalent amounts of total protein. First, blots were stained for total protein using GelCode Blue Stain Reagent (Pierce) according to the manufacturer’s instructions. Secondly, rnc70 strains were grown on the media shown in Figure 5 and prepared as described for immunoblotting except that a modified treatment buffer lacking 2-mercaptoethanol and Bromphenol Blue was used. The protein concentration in these samples was determined using the MicroBCA Protein Assay (Pierce). The results were consistent with those reported in Bremer and Dennis (1996). The total protein per OD is 1.4-fold greater in cultures grown on minimal glycerol plus casamino acids and 1.2-fold greater in cultures grown on minimal succinate plus casamino acids than in cultures grown on LB.
Fold differences between N or RNase III levels detected using immunoblotting were approximated by scanning the developed membranes with a Typhon 8600 Variable Mode Imager and analyzing the resulting data using ImageQuant (Molecular Dynamics, Piscataway, NJ).
Acknowledgments
Acknowledgements
The authors gratefully acknowledge David Friedman for critical reading of the manuscript, Teresa Baker for construction of the DY330 rnc<>cat strain, Jack Greenblatt and Joyce Li for affinity-purified anti-N antibody and purified N protein, Su-Min Chen for anti-RNase III antibody, James Sawitzke and Lynn Thomason for assistance with various computer programs, Mikhail Kashlev’s laboratory for the use of their Typhon 8600 Variable Mode Imager, and Peter Johnson and Jon Shuman for considerable advice on western blotting techniques.
References
- Adhya S., Gottesman,M. and De Crombrugghe,B. (1974) Release of polarity in Escherichia coli by gene N of phage λ: termination and antitermination of transcription. Proc. Natl Acad. Sci. USA, 71, 2534–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhya S., Gottesman,M. and Court,D. (1977) Independence of gene N and tof functions of bacteriophage λ. J. Mol. Biol., 112, 657–660. [DOI] [PubMed] [Google Scholar]
- Altuvia S. and Oppenheim,A.B. (1986) Translational regulatory signals within the coding region of the bacteriophage λ cIII gene. J. Bacteriol., 167, 415–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altuvia S., Locker-Giladi,H., Koby,S., Ben-Nun,O. and Oppenheim,A.B. (1987) RNase III stimulates the translation of the cIII gene of bacteriophage λ. Proc. Natl Acad. Sci. USA, 84, 6511–6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altuvia S., Kornitzer,D., Teff,D. and Oppenheim,A.B. (1989) Alterna tive mRNA structures of the cIII gene of bacteriophage λ determine the rate of its translation initiation. J. Mol. Biol., 210, 265–280. [DOI] [PubMed] [Google Scholar]
- Anevski P.J. and Lozeron,H.A. (1981) Multiple pathways of RNA processing and decay for the major leftward N-independent RNA transcript of coliphage λ. Virology, 113, 39–53. [DOI] [PubMed] [Google Scholar]
- Barik S., Ghosh,B., Whalen,W., Lazinski,D. and Das,A. (1987) An antitermination protein engages the elongating transcription apparatus at a promoter-proximal recognition site. Cell, 50, 885–899. [DOI] [PubMed] [Google Scholar]
- Bremer H. and Dennis,P.P. (1996) Modulation of chemical composition and other parameters of the cell by growth rate. In Neidhardt,F.C. et al. (eds), Escherichia coli and Salmonella: Cellular and Molecular Biology. Vol. 2, American Society for Microbiology Press, Washington, DC, pp. 1553–1569.
- Britton R.A., Powell,B.S., Dasgupta,S., Sun,Q., Margolin,W., Lupski,J.R. and Court,D.L. (1998) Cell cycle arrest in Era GTPase mutants: a potential growth rate-regulated checkpoint in Escherichia coli. Mol. Microbiol., 27, 739–750. [DOI] [PubMed] [Google Scholar]
- Churchward G., Belin,D. and Nagamine,Y. (1984) A pSC101-derived plasmid which shows no sequence homology to other commonly used cloning vectors. Gene, 31, 165–171. [DOI] [PubMed] [Google Scholar]
- Comer M.M., Dondon,J., Graffe,M., Yarchuk,O. and Springer,M. (1996) Growth rate-dependent control, feedback regulation and steady-state mRNA levels of the threonyl-tRNA synthetase gene of Escherichia coli. J. Mol. Biol., 261, 108–124. [DOI] [PubMed] [Google Scholar]
- Court D.L. (1993) RNA processing and degradation by RNase III. In Belasco,J. and Brawerman,G. (eds), Control of mRNA Stability. Academic Press, New York, NY, pp. 71–116.
- Daniels D.L., Subbarao,M.N., Blattner,F.R. and Lozeron,H.A. (1988) Q-mediated late gene transcription of bacteriophage λ: RNA start point and RNase III processing sites in vivo. Virology, 167, 568–577. [PubMed] [Google Scholar]
- Das A. (1992) How the phage λ N gene product suppresses transcription termination: communication of RNA polymerase with regulatory proteins mediated by signals in nascent RNA. J. Bacteriol, 174, 6711–6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta S., Fernandez,L., Kameyama,L., Inada,T., Nakamura,Y., Pappas,A. and Court,D.L. (1998) Genetic uncoupling of the dsRNA-binding and RNA cleavage activities of the Escherichia coli endoribonuclease RNase III—the effect of dsRNA binding on gene expression. Mol. Microbiol., 28, 629–640. [DOI] [PubMed] [Google Scholar]
- Devito J. and Das,A. (1992) Directing an operon-specific antiterminator to the target polymerase by an artificial tethering mechanism. J. Cell. Biochem., Suppl., 16E, 177. [Google Scholar]
- Dunn J.J. and Studier,F.W. (1973) T7 early RNAs and Escherichia coli ribosomal RNAs are cut from large precursor RNAs in vivo by ribonuclease III. Proc. Natl Acad. Sci. USA, 70, 3296–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feather-Henigan K., Lipton,B., Hines,K. and Brotcke,T. (2000) Western Blotting with Chemiluminescent Substrates. Pierce Chemical Company, Rockford, IL.
- Friedman D.I. and Court,D.L. (1995) Transcription antitermination: the λ paradigm updated. Mol. Microbiol., 18, 191–200. [DOI] [PubMed] [Google Scholar]
- Friedman D.I. and Gottesman,M. (1983) Lytic mode of λ development. In Hendrix,R.W., Roberts,J.W., Stahl,F.W. and Weisberg,R.A. (eds), Lambda II. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 21–51.
- Gitelman D.R. and Apirion,D. (1980) The synthesis of some proteins is affected in RNA processing mutants of Escherichia coli. Biochem. Biophys. Res. Comm., 96, 1063–1070. [DOI] [PubMed] [Google Scholar]
- Gotta S.L., Miller,O.L. and French,S.L. (1991) rRNA transcription rate in Escherichia coli. J. Bacteriol., 173, 6647–6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman M., Oppenheim,A. and Court,D. (1982) Retroregulation: control of gene expression from sites distal to the gene. Cell, 29, 727–728. [DOI] [PubMed] [Google Scholar]
- Inada T., Kawakami,K., Chen,S.M., Takiff,H.E., Court,D.L. and Nakamura,Y. (1989) Temperature-sensitive lethal mutant of era, a G protein in Escherichia coli. J. Bacteriol., 171, 5017–5024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameyama L., Fernandez,L., Court,D.L. and Guarneros,G. (1991) RNase III activation of bacteriophage λ N synthesis. Mol. Microbiol., 5, 2953–2963. [DOI] [PubMed] [Google Scholar]
- Kourilsky P. and Knapp,A. (1974) Lysogenization by bacteriophage λ. III. Multiplicity dependent phenomena occurring upon infection by λ. Biochimie, 56, 1517–1523. [PubMed] [Google Scholar]
- Krinke L. and Wulff,D.L. (1987) OOP RNA, produced from multicopy plasmids, inhibits λ cII gene expression through an RNase III-dependent mechanism. Genes Dev., 1, 1005–1013. [DOI] [PubMed] [Google Scholar]
- Krinke L. and Wulff,D.L. (1990) RNase III-dependent hydrolysis of λ cII-O gene mRNA mediated by λ OOP antisense RNA. Genes Dev., 4, 2223–2233. [DOI] [PubMed] [Google Scholar]
- Lozeron H.A., Dahlberg,J.E. and Szybalski,W. (1976) Processing of the major leftward mRNA of coliphage λ. Virology, 71, 262–277. [DOI] [PubMed] [Google Scholar]
- Lozeron H.A., Anevski,P.J. and Apirion,D. (1977) Antitermination and absence of processing of the leftward transcript of coliphage λ in the RNAase III-deficient host. J. Mol. Biol., 109, 359–365. [DOI] [PubMed] [Google Scholar]
- McKenney K., Shimatake,H., Court,D., Schmeissner,U., Brady,C. and Rosenberg,M. (1981) A system to study promoter and termination signals recognized by Escherichia coli RNA polymerase. In Chirikjian,J.G. and Papas,T.S. (eds), Gene Amplification and Analysis Vol. II. Structural Analysis of Nucleic Acids. Elsevier/North-Holland, New York, NY, pp. 383–415. [PubMed]
- Miller J.H. (1972) Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Mousset S. and Thomas,R. (1968) Dilysogenic excision: an accessory expression of the termination function? Cold Spring Harbor Symp. Quant. Biol., 33, 749–754. [DOI] [PubMed] [Google Scholar]
- Nikolaev N., Silengo,L. and Schlessinger,D. (1973) Synthesis of a large precursor to ribosomal RNA in a mutant of Escherichia coli. Proc. Natl Acad. Sci. USA, 70, 3361–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salstrom J.S. and Szybalski,W. (1978) Coliphage λnutL–: a unique class of mutants defective in the site of gene N product utilization for antitermination of leftward transcription. J. Mol. Biol., 124, 195–221. [DOI] [PubMed] [Google Scholar]
- Steege D.A., Cone,K.C., Queen,C. and Rosenberg,M. (1987) Bacterio phage λ N gene leader RNA. RNA processing and translational initiation signals. J. Biol. Chem., 262, 17651–17658. [PubMed] [Google Scholar]
- Takiff H.E., Chen,S.M. and Court,D.L. (1989) Genetic analysis of the rnc operon of Escherichia coli. J. Bacteriol., 171, 2581–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takiff H.E., Baker,T., Copeland,T., Chen,S.M. and Court,D.L. (1992) Locating essential Escherichia coli genes by using mini-Tn10 transposons: the pdxJ operon. J. Bacteriol., 174, 1544–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg R.A. and Gottesman,M.E. (1999) Processive antitermination. J. Bacteriol., 181, 359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilder D.A. and Lozeron,H.A. (1979) Differential modes of processing and decay for the major N-dependent RNA transcript of coliphage λ. Virology, 99, 241–256. [DOI] [PubMed] [Google Scholar]
- Wilson H.R., Kameyama,L., Zhou,J.G., Guarneros,G. and Court,D.L. (1997) Translational repression by a transcriptional elongation factor. Genes Dev., 11, 2204–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D., Ellis,H.M., Lee,E.C., Jenkins,N.A., Copeland,N.G. and Court,D.L. (2000) An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl Acad. Sci. USA, 97, 5978–5983. [DOI] [PMC free article] [PubMed] [Google Scholar]