

Abstract
In this paper, we studied the fate of endocytosed glycosylphosphatidyl inositol anchored proteins (GPI- APs) in mammalian cells, using aerolysin, a bacterial toxin that binds to the GPI anchor, as a probe. We find that GPI-APs are transported down the endocytic pathway to reducing late endosomes in BHK cells, using biochemical, morphological and functional approaches. We also find that this transport correlates with the association to raft-like membranes and thus that lipid rafts are present in late endosomes (in addition to the Golgi and the plasma membrane). In marked contrast, endocytosed GPI-APs reach the recycling endosome in CHO cells and this transport correlates with a decreased raft association. GPI-APs are, however, diverted from the recycling endosome and routed to late endosomes in CHO cells, when their raft association is increased by clustering seven or less GPI-APs with an aerolysin mutant. We conclude that the different endocytic routes followed by GPI-APs in different cell types depend on the residence time of GPI-APs in lipid rafts, and hence that raft partitioning regulates GPI-APs sorting in the endocytic pathway.
Keywords: aerolysin/GPI/late endosome/raft/recycling endosome
Introduction
A variety of cell surface eukaryotic proteins are not transmembrane but anchored into the outer leaflet of the lipid bilayer via a glycosylphosphatidyl inositol (GPI) moiety (Kinoshita et al., 1997). These lipid-anchored proteins can have very different roles: some act as receptors for circulating ligands, others as adhesion molecules or enzymes. In polarized epithelial cells, these proteins are mainly found at the apical plasma membrane due to selective sorting in the trans-Golgi network (Rodriguez-Boulan and Powell, 1992). The prevalent model is that GPI-anchored proteins (GPI-APs) laterally associate into liquid-ordered, cholesterol- and sphingo lipid-rich domains, or rafts, which are then targeted to their proper plasma membrane destination (for a review, see Brown and London, 1998).
Endocytosis of GPI-APs appears to occur via a clathrin-independent pathway (Parton et al., 1994; Skretting et al., 1999; Nichols et al., 2001; Sabharanjak et al., 2002). The intracellular route subsequently followed by these proteins is still a matter of debate. In Chinese hamster ovary (CHO) cells, GPI-APs were found to be transported to the GEEC (GPI-AP enriched endosomal compartment) (Sabharanjak et al., 2002), which might be a sub-compartment of early endosomes, and then to peri-centriolar recycling endosomes before returning to the plasma membrane (Mayor et al., 1998). In HeLa cells, indirect evidence, obtained with exogenously expressed GPI-APs, suggests that they are transported to early endosomes and then further down the endocytic pathway (Rijnboutt et al., 1996; Skretting et al., 1999). Finally, in COS-7 cells, a direct route for GPI-APs between the plasma membrane and the Golgi (Nichols et al., 2001), possibly via caveolin-1 positive endosomal compartments (Nichols, 2002), has been proposed. However, these observations have recently been challenged, both in COS-7 and CHO cells (Sabharanjak et al., 2002).
In this paper, we have studied the endocytic transport of GPI-APs by making use of the bacterial toxin aerolysin, which is a monovalent probe for the glycan core of these lipid-based proteins (Abrami et al., 2000). We found that internalized GPI-APs are transported to the early endosome both in CHO and baby hamster kidney (BHK) cells, but that their subsequent fate differs depending on their association with lipid domains.
Results
Aerolysin
The bacterial toxin aerolysin is produced as a precursor that requires proteolytic removal of a C-terminal peptide to become active (Figure 1A; Abrami et al., 2000). Once bound to the cell surface, the mature toxin undergoes circular polymerization into an amphipathic heptameric complex, which spontaneously inserts into a lipid bilayer and forms a pore. GPI-APs serve as cell surface receptors, as demonstrated for example by observations that PI-PLC treatment protects cells against the toxin (Abrami et al., 1998) and that generation of aerolysin-resistant cell lines leads to GPI-deficient mutant cells (Abrami et al., 2001). Importantly, aerolysin does not bind to protein free GPIs (Abrami et al., 2002). Since aerolysin binds GPI-APs with high affinity both on cells and in vitro, using a toxin overlay assay (Abrami et al., 1998), we used the toxin as a monovalent probe to study GPI-APs. We also used a double mutant of aerolysin (referred to here as ASSP) in which residues 202 and 445 are changed to cysteines, creating a disulfide bridge between the C-terminal propeptide and the mature toxin (Figure 1A; van der Goot et al., 1994). Like the wild-type toxin, ASSP binds specifically to GPI-APs (Abrami et al., 1998), but remains inactive, even after C-terminal proteolysis, unless it becomes reduced.

Fig. 1. Mapping endogenous GPI-APs using aerolysin. (A) Activation of the wild-type precursor is triggered by proteolytic removal of a C-terminal peptide. The resulting mature toxin is able to oligomerize into a heptameric ring that forms a pore in the target membrane. The ASSP aerolysin mutant is inactive, even after C-terminal proteolysis, due to an engineered disulfide bridge that links the propeptide to the mature toxin (van der Goot et al., 1994). Disulfide reduction is required for ASSP to oligomerize and form pores. (B) BHK GPI-APs (indicated by arrows) were purified from 35S-labeled cells (lane labeled GPI-APs; see Materials and methods). The control lane corresponds to non-GPI-APs released from the Triton X-114 detergent phase. Contaminants are labeled with asterisks. In parallel, an aerolysin overlay assay was performed on BHK DRMs (30 µg of protein). (C–F) 2D maps of endogenous GPI-APs were generated by performing aerolysin overlays on DRMs of BHK (C), wild-type (E) and GPI-deficient CHO (F) cells (50 µg of protein per gel). (D) The protein profile revealed by silver stain of a Triton X-114 detergent phase prepared from BHK DRMs (150 µg of protein). The most abundant BHK GPI-APs are labeled in (C) and (D).
Mapping GPI-APs using aerolysin
Our first goal was to map the GPI-APs expressed in BHK cells. After GPI-AP purification from metabolically labeled cells (see Materials and methods), we identified three polypeptides with apparent mol. wts of 55, 80 and 130 kDa (Figure 1B). Since GPI-APs at the cell surface are enriched in lipid rafts, which have the characteristic property of being insoluble in Triton X-100 at 4°C (Brown and London, 1998), we also prepared detergent-resistant membranes (DRMs) (Figure 1B). Using the toxin overlay assay, we identified three bands in DRMs with the same apparent mol. wts, as well as an additional ∼30 kDa GPI-AP (see below), indicating that this assay is more sensitive for detecting GPI-APs. A high-resolution map was then obtained after toxin overlay of two-dimensional (2D) gels, which revealed nine different, highly N-glycosylated GPI-APs (Figure 1C). GPI-AP1–3 were clearly visible by silver staining of the gels (Figure 1D), but GPI-AP4, corresponding to Thy-1, required N-deglycosylation to be detected (Fivaz et al., 2000). GPI-AP1–3 protein spots, originating from N-deglycosyl ated or non-deglycosylated DRMs, were identified by nanoelectrospray tandem mass spectrometry as N-CAM120, semaphorin 7 and CD14, respectively (see Supplementary table I available at The EMBO Journal Online).
A 2D map of CHO GPI-APs was also generated (Figure 1E). Western blotting showed that the main GPI-AP is N-CAM (data not shown, but see overlays in Figure 1C and E for comparison), and that these cells also contain four less abundant GPI-APs. The high specificity of the aerolysin overlay assay is illustrated by the total absence of signal when performing the assay on DRMs from GPI-deficient CHO cells (Figure 1F; Abrami et al., 2001). These observations show that the GPI-AP content of BHK and CHO cells differs, and indicate that aerolysin binds selectively to all GPI-APs expressed in BHK cells, in agreement with the fact that the glycan core, common to all mammalian GPI anchors (Kinoshita et al., 1997), is the major determinant for aerolysin binding (Diep et al., 1998).
An Alexa-546-labeled fluorescent aerolysin derivative was next used to localize GPI-APs on fixed cells. On non-permeabilized BHK and CHO cells, the staining was uniform at the surface (Supplementary figure S1), as is generally the case for GPI-APs (Maxfield and Mayor, 1997). No staining was observed when binding of Alexa–aerolysin was competed away with an excess of unlabeled toxin (data not shown) or when CHO GPI-deficient cells were analyzed (Supplementary figure S1). On permeabilized cells, an additional intracellular staining pattern was observed (Supplementary figure S1) that was different in CHO and in BHK cells, suggesting that GPI-APs accumulate in different intracellular compartments in the two cell types.
Endocytosed GPI-APs are transported to late endosomes in BHK cells
Endocytosis of Alexa–ASSP was followed as a function of time in BHK cells. After a brief time period, the labeled mutant toxin showed a punctate pattern and co-localized with FITC–dextran internalized for 5 min, but not with FITC–transferrin (Figure 2). Then, the toxin also co-localized with the rab5 effector EEA1 (Supplementary figure S2), although the toxin distribution was somewhat broader, consistent with the highly restricted localization of EEA1 (Zerial and McBride, 2001). These data thus indicate that GPI-APs had reached the early endosome. Then, Alexa–ASSP gradually moved towards more perinuclear structures containing the late endosomal lipid lysobisphosphatidic acid (LBPA) (Kobayashi et al., 1998). Complete overlap between Alexa–ASSP and LBPA labeling was reached after 3–4 h (Figure 2).

Fig. 2. Endocytosis of GPI-APs in BHK cells. BHK cells were incubated with the GPI-AP-specific probe Alexa–ASSP (a-ASSP, 10 nM) for 1 h at 4°C, rinsed, and further incubated at 37°C for various times. Early endosomes were labeled by 5-min internalized FITC–dextran, recycling endosomes by 15-min internalized FITC–transferrin and late endosomes by immunolabeling against LBPA. Arrows indicate examples of co-localization. Scale bar, 10 µm. Note that cell surface labeling with transferrin was low due to the fact that 70% of the receptor is intracellular on these cells and that the cell surface receptors are diluted over a large surface.
When cells were exposed to non-labeled ASSP for several hours, they remained viable, but underwent severe vacuolation (Figure 3A) (note that Alexa labeling alters the pore-forming activity even of the wild-type toxin). Consistent with the localization of internalized Alexa– ASSP in late endosomes, ASSP-induced vacuoles were positive for the late endosomal/lysosomal glycoprotein lamp-1 (Aniento et al., 1993; Figure 2B) and for the small GTPase rab7, another late endosomal marker (Chavrier et al., 1991), tagged with green fluorescent protein (data not shown).

Fig. 3. ASSP is activated in late endosomes in BHK cells. (A) BHK cells were incubated or not with ASSP (10 nM, 37°C). After 6 h, large cytoplasmic vacuoles were observed by phase-contrast microscopy in ASSP-treated cells but not in control cells. Scale bar, 10 µm. (B) The ASSP-induced vacuoles were immunostained with an anti-lamp-1 antibody. (C) BHK cells were treated with trypsin-nicked ASSP (10 nM) or left untreated (control cells) and then harvested after various incubation times at 37°C. Total cell extracts were analyzed by SDS–PAGE under non-reducing conditions (30 µg per lane) followed by western blotting using an anti-aerolysin antibody. The migration patterns of pure trypsin-nicked ASSP in the absence and presence of DTT are shown as markers of the non-reduced (NR) and reduced (R) forms. Note that the aerolysin heptamer, which can only be formed after disulfide reduction of ASSP, is resistant to SDS and therefore migrates at the top of the gel. The asterisk indicates the presence of a cellular protein cross-reacting with the anti-aerolysin antibody. (D) BHK cells were incubated with trypsin-nicked ASSP (10 nM) for 1 h at 4°C or 24 h at 37°C, and fractionated on a sucrose step gradient to separate late endosomes from other membranes. The three membrane fractions of the gradient were analyzed by non-reducing SDS–PAGE and assayed for the presence of ASSP and rab7 by western blotting. (E) BHK cells were incubated with trypsin-nicked biotin–ASSP (10 nM) for 24 h at 37°C, and fractionated on a sucrose step gradient to separate late endosomes from other membranes. Two forms of biotinylated ASSP were used: biotin–ASSP (lane 1) and the cleavable counterpart biotin-SS–ASSP (lane 2). The late endosomal fractions of the gradients were analyzed by non-reducing SDS–PAGE, transferred onto a nitrocellulose membrane, and assayed for the presence of biotin–ASSP using streptavidin–HRP. Note that biotinylation inhibits oligomerization and thereby allows degradation of the toxin.
The ASSP-induced late endosomal vacuolation raised the possibility that the mutant toxin became active. This not only requires proteolysis, which can be performed with trypsin prior to the addition to cells (as was done for the present biochemical experiments), but also release of the C-terminal peptide, which can only occur if the engineered disulfide bond is reduced (Figure 1A). We therefore analyzed, by SDS–PAGE, whether peptide release occurred in cells that had internalized trypsin-nicked ASSP. A faster migrating species, corresponding to reduced ASSP, indeed appeared when the toxin was internalized, but not when ASSP was bound to the cell surface (4°C) (Figure 3C). Concomitantly, the SDS-resistant heptameric pore-forming complex was formed (Figure 3C).
To investigate whether disulfide reduction of ASSP occurred in late endosomes, we purified this organelle using a well-characterized subcellular fractionation procedure (Aniento et al., 1993). The endocytosed mutant toxin co-fractionated with the small GTPase rab7 in the low buoyancy late endosomal fraction 3 containing the bulk of cellular LBPA (Kobayashi et al., 1998), and was essentially in a reduced and oligomerized state (Figure 3D). As a control, the distribution of the toxin was analyzed in cells that had been treated with ASSP at 4°C. The surface-bound toxin was in a non-reduced state and was highly enriched in fraction 2, as expected since this fraction contains plasma membrane rafts (van der Goot, 1997).
To exclude the possibility that loss of the toxin propeptide was due to complete proteolytic breakdown and not to disulfide reduction, these experiments were repeated using ASSP that had been biotinylated using a spacer arm with (biotin-SS–ASSP) or without (biotin– ASSP) a disulfide bond. The reduced and oligomerized biotinylated toxin could be revealed using strepta vidin–HRP when using biotin–ASSP (Figure 3E, lane 1) but not when using biotin-SS–ASSP (Figure 3E, lane 2), indicating that in the latter case biotin molecules had been lost by disulfide reduction. To our knowledge, this is the first report that late endosomes, or a sub-compartment of late endosomes, exhibit a reducing activity (see discussion in Supplementary data). Our morphological, biochemical and function observations thus indicate that endocytosed GPI-APs are transported to reducing late endosomes in BHK cells.
GPI-APs are present in late endosomes of BHK cells at steady state
We next analyzed whether GPI-APs were present in BHK late endosomes at steady state. After fixation and permeabilization, Figure 4A shows that LBPA-positive structures were double-labeled with Alexa–aerolysin. The strong perinuclear labeling observed with Alexa– aerolysin, however, partly corresponds to the Golgi apparatus, as witnessed by double labeling for the Golgi enzyme mannosidase II.

Fig. 4. Steady-state distribution of GPI-APS in BHK cells. (A) BHK cells were fixed/permeabilized and double labeled with 7.5 nM Alexa–aerolysin (a-aero) and anti-LBPA or anti-mannosidase II (ManII) antibodies. In the upper panel, examples of co-localization are shown with arrows. Scale bar, 10 µm. (B) BHK cells were fixed in paraformaldehyde/glutaraldehyde and processed for frozen sectioning. Ultrathin sections were incubated with biotinylated aerolysin followed by 10 nm anti-biotin–gold. Specific labeling is associated with multi-vesicular late endocytic structures (L) and with the Golgi complex (G; arrows show gold particle in the Golgi); N, nucleus. Scale bar, 200 nm.
The steady-state distribution GPI-APs was further analyzed by electron microscopy after directly labeling cryosections with biotinylated aerolysin followed by anti-biotin–gold. As shown in Figure 4B, multi-vesicular late endosomes, as well as the Golgi apparatus, were clearly labeled with the toxin. These data demonstrate that aerolysin is a powerful tool for immunoelectron microscopy detection of GPI-APs, and confirm the presence of GPI-APs in late endosomes and in the Golgi complex of BHK cells.
ASSP–GPI-AP complexes are in DRMs in BHK late endosomes
We previously showed that aerolysin that had been bound to the cell surface in vivo is recovered in DRMs after fractionation (Abrami and van der Goot, 1999), as expected from the presence of GPI-APs in DRMs (Figure 1B). We thus investigated whether ASSP– GPI-AP complexes that had reached late endosomes still co-purified with DRMs. ASSP in late endosomes, i.e. reduced and oligomerized, indeed floated to the top of the gradient (Figure 5A). Importantly, monomeric reduced ASSP was also associated with DRMs, confirming that the toxin was still associated with GPI-APs. Monomeric aerolysin is readily soluble and would have remained at the bottom of the tube together with the other soluble proteins had it not been GPI bound. We consistently found that aerolysin–GPI-AP interactions are resistant to acid washes (pH 2) (data not shown), and thus uncoupling is unlikely to occur at the acidic pH of late endosomes (pH 5–5.5).

Fig. 5. DRM domains in late endosomes of BHK cells. (A) DRMs were prepared from BHK cells treated for 24 h with trypsin-nicked ASSP (10 nM), by flotation on an OptiPrep gradient using a Beckman SW60 rotor. Fractions of 200 µl were collected. SDS–PAGE was performed under non-reducing conditions. The gel was loaded by yield. (B and C) DRMs were prepared from purified late endosomes by flotation on an OptiPrep gradient using a TLS-55 Beckman rotor and 400 µl samples were collected. (B) The total cholesterol content of each fraction of the gradient was determined by 1D TLC and quantified using the ScanAnalysis software. Cholesterol contents were expressed as a percentage of the total cholesterol content of the gradient. Values correspond to the mean of four experiments. Error bars represent the standard deviation. (C) The total content of each fraction was loaded on a 15% SDS gel. The distributions of lamp-1, flotillin-1 and rab7 were analyzed by western blotting and that of GPI-APs by aerolysin overlay. Note that we are here analyzing the late endosomal lamp-1 population and not lysosomal lamp-1, which is found in heavy organelles and not low buoyancy fractions. (D) Subcellular fractionation of BHK cells. BHK cells were fractionated on a sucrose density gradient to separate late endosomes for other cellular membranes. Three membrane fractions were collected at the sucrose interfaces, and probed for caveolin-1 and flotillin-1 by western blotting (10 µg of proteins per lane). Fraction 3 is highly enriched in late endosomes (Kobayashi et al., 1998). Fraction 2 contains plasma membrane rafts (van der Goot, 1997) and early endosomes (Aniento et al., 1996). (E) BHK cells were fixed/permeabilized and double labeled for flotillin-1 and LBPA. The anti-flotillin-1 antibody also stained the plasma membrane; however, brief exposure times were chosen so as to highlight the intracellular staining.
BHK late endosomes contain raft-like membrane domains
The above observations suggest that late endosomes contain DRMs. To address this issue more directly, purified late endosomes were treated with detergent at 4°C. DRMs could be isolated and were enriched in cholesterol (Figure 5B), and in the four major GPI-APs of BHK cells (Figure 5C). A significant fraction of the GPI-APs was detergent soluble. This could reflect the fact that a fraction of GPI-APs are not raft-associated in late endosomes or, more likely, results from the higher detergent:protein ratio used in this experiment for technical reasons, when compared with that used on whole cells.
Only a subset of late endosomal proteins specifically associated with these DRMs (<15% of total late endosome protein; data not shown), and these lacked the geranyl-geranylated small GTPase rab7 and the transmembrane protein lamp-1 (Figure 5C), but contained the four major BHK GPI-APs. Moreover, we found that flotillin-1, which was documented as an integral membrane protein of plasma membrane rafts (Bickel et al., 1997), co-fraction ated with late endosomes, in contrast to caveolin-1 (Figure 5D), co-localized with the late endosomal lipid LBPA (Figure 5E) and was highly enriched in late endosomal DRMs (Figure 5C).
Endocytosed GPI-APs are transported to recycling endosomes in CHO cells
Since internalized GPI-APs were reported to reach recycling endosomes in CHO cells (Mayor et al., 1998; Sabharanjak et al., 2002) in apparent contradiction with our findings in BHK cells, we extended our studies to CHO cells. Much as in BHK cells, endocytosed Alexa– ASSP showed a punctate distribution after 15 min and co-localized with 5-min internalized FITC dextran (Figure 6A) and with EEA1 (Supplementary figure S2). However, internalized Alexa–ASSP was also found in a juxtanuclear dot-like compartment containing internalized FITC–transferrin (Figure 6A). Quantitative image analysis showed that ∼30% of the total cell bound Alexa–ASSP reached the transferrin-positive compartment after 60 min (Figure 6B). Thus, as previously observed by others and in contrast to what we observe for BHK cells, endocytosed GPI-APs accumulate in CHO recycling endosomes. Functional evidence that endocytosed GPI-APs reach different organelles in BHK and CHO cells came with the observation that CHO cells do not undergo vacuolation when exposed to ASSP, even after prolonged incubation (Figure 6C). In agreement, ASSP remained in a non-reduced, and hence a non-heptameric, non-channel forming state (Figure 6D). A slight reduction could be observed with time, suggesting that a minor fraction of the toxin did reach a reducing degradative compartment, too little, however, to allow oligomerization, and was degraded (see arrow in Figure 6D). This presumably reflects the minor pathway of GPI-AP turnover in late endosomes and lysosomes.

Fig. 6. GPI-APs in CHO cells are delivered to the recycling endosome. CHO were incubated with aerolysin–ASSP (10 nM) for 1 h at 4°C followed by various times at 37°C and processed for fluorescence microscopy. (A) Alexa–ASSP was allowed to internalize for 15 min. During the last 5 min, FITC–dextran (upper panels) was internalized to label early endosomes. Arrows indicate examples of co-localization. In a parallel experiment, ASSP was internalized for 15 min in the presence of FITC–transferrin (lower panels) in order to label the recycling endosome (indicated by an arrowhead in the upper panel and an arrow in the lower). (B) Quantification of Alexa–ASSP uptake. More than 20 cells from two independent experiments were analyzed for each condition, and standard deviations of the mean were calculated. (C) CHO cells were treated with trypsin-nicked ASSP (10 nM) for 24 h and imaged by phase-contrast microscopy. (D) Total cell extracts of wild-type and GPI-deficient CHO cells treated or not with trypsin-nicked ASSP (10 nM) were analyzed by non-reducing SDS–PAGE and western blotting using anti-aerolysin antibodies (30 µg/lane). The arrow indicates an aerolysin degradation product. The asterisk indicates the presence of a protein cross-reacting with the anti-aerolysin antibody. (E) CHO cells were fixed 3% paraformaldehyde and permeabilized with 0.1% saponin. A triple-labeling experiment was performed using Alexa–aerolysin (7.5 nM), filipin (specific probe for cholesterol) and antibodies against the transferrin receptor (Tf-R). Scale bar, 10 µm. (F) CHO cells were fixed in paraformaldehyde/glutaraldehyde and processed for frozen sectioning. Ultrathin sections were incubated with the biotinylated aerolysin probe followed by 10 nm anti-biotin–gold. Specific labeling (arrows) is associated with the Golgi complex (G; upper panel) and with tubulo-vesicular profiles in close proximity to the centriole (C; lower panel), presumed to be reycling endosomes. Scale bars, 200 nm.
Labeling of GPI-APs with Alexa–aerolysin on fixed/permeabilized cells (Figure 6E) showed that GPI-APs are present at steady state in the cholesterol-rich transferrin receptor containing recycling endosomes. Electron microscopy analysis, performed using biotinylated aerolysin, confirmed this localization, as witnessed by staining of tubulo-vesicular profiles close to the centriole, and showed that GPI-APs were also present in the Golgi (Figure 6F), again in agreement with Sabharanjak et al. (2002).
Differential sorting does not depend on the nature of expressed GPI-APs
Since CHO and BHK cells express, in part, different sets of GPI-APs, a trivial explanation for the differential routing of Alexa–ASSP in the two cell types could be that sorting is determined by the nature of the expressed GPI-APs. To address this issue, we followed endocytosis of the same GPI-AP, CD14, in the two cell types using antibodies. Human CD14 was ectopically expressed in CHO, since these cells do not express CD14, and in BHK cells (which express CD14), for comparison. Then, FITC-conjugated anti-human CD14 monoclonal antibody was bound to cell surface CD14 at 4°C, and cells were incubated for 1 h at 37°C. The internalized antibody-tagged CD14 accumulated in transferrin-positive recycling endosomes of CHO cells (Figure 7A), but not of BHK cells (Figure 7B). To follow the subsequent fate of CD14 in BHK cells, endogenous CD14 was tagged at the cell surface with a polyclonal anti-CD14 antibody (to allow double labeling with a monoclonal antibody). After 4 h or more, the internalized antibody co-localized with LBPA in late endosomes (Figure 7B). The fact that internalized CD14 followed different routes in the two cell types, as did ASSP, suggests that sorting depends on the cell type and not on the nature of the GPI-AP. Also, transport of CD14 was not altered by the antibody, since the behavior was the same as that observed with ASSP. Differential sorting of CD14 in the two cell types could be due to differences in the interacting partners. Alternatively, the sorting mechanism, perhaps lipid based, may be differentially tuned in different cells.

Fig. 7. Differential endocytic fate of CD14 in CHO and BHK cells. CHO (A) or BHK (B) cells, transiently transfected with human CD14, were incubated with FITC-conjugated monoclonal antibodies against human CD14 (1 µg/ml) at 4°C, shifted to 37°C for 60 min and incubated with rhodamine-transferrin during the last 30 min. To better resolve intracellular staining of CD14, the surface-bound anti-CD14 monoclonal antibody was removed by a brief acid wash. BHK cells were also incubated with polyclonal anti-CD14 antibodies (2 µg/ml), revealing endogenous CD14, for 15 h (37°C). Cells were fixed/permeabilized and a double immunostaining was performed against CD14 (secondary antibody only) and LBPA.
Detergent solubility of internalized ASSP–GPI-AP complexes in CHO cells
We next analyzed the association of internalized ASSP with rafts in CHO cells. Since ASSP does not undergo disulfide reduction in CHO cells (Figure 6D), internalized ASSP could not be distinguished from extracellular ASSP by SDS–PAGE. Moreover, surface-bound ASSP could not be reduced by the membrane-impermeant reducing agent sodium 2-mercaptoethanesulfonate (MesNa), since the engineered disulfide bond is very resistant [full reduction in vitro requires 15 min incubation at 37°C with 50 mM dithiothreitol (DTT)]. We therefore made use of biotin-SS–ASSP. MesNa treatment removed 95% of the biotin from surface-bound ASSP (4°C), as quantified using [125I]streptavidin (Figure 8B), indicating that this method was suitable to distinguish internalized from extracellular ASSP. When biotin-SS–ASSP was internalized for 45 min, ∼30% became MesNa resistant (Figure 8A and B), in good agreement with the internalization studies of Alexa–ASSP (Figure 6B).

Fig. 8. Detergent solubility of intracellular GPI-APs in CHO cells. CHO cells were incubated with biotin-SS–ASSP (10 nM) at 4°C, shifted or not at 37°C for 45 min, then treated or not with 30 mM MesNa (15 min at 4°C) and PNSs were prepared. Thirty micrograms of protein were analyzed by SDS–PAGE (10% gel), blotted on nitrocellulose and the membrane was probed with [125I]streptavidin. (A) A representative experiment and (B) quantification by phosphoimager analysis (n = 4; bars represent standard deviation of the mean). The percentage of MesNa-resistant biotin-SS–ASSP was derived from the ratio of signal intensities obtained with and without MesNa treatment and is a direct measure of biotin-SS–ASSP internalization. (C and D) Cells were incubated with biotin-SS–ASSP (10 nM) at 4°C, shifted to 37°C for 45 min, treated or not with 30 mM MesNa (15 min at 4°C) and then extracted in 2% cold Triton for 1 h. DRMs were isolated on a sucrose density gradient using a Beckman SW60 rotor. Fractions of 700 µl were collected, precipitated, analyzed on a 12.5% gel and probed with streptavidin–HRP (C) or [125I]streptavidin (D) for quantification. In (D) the percentage of detergent-resistant biotin-SS–ASSP (expressed as the ratio of the signal in the top two fractions over the sum of the signals in all fractions) was plotted (n = 4; bars represent standard deviation of the mean).
DRMs were prepared from CHO cells that had been incubated with biotin-SS–ASSP for 45 min at 37°C to allow transport to the RE (recycling endosome), and then treated or not with MesNa. In the absence of MesNa treatment, i.e. when monitoring plasma membrane (70%) plus intracellular (30%) biotin-SS–ASSP, the toxin could be detected in the DRM fractions at the top of the gradient (Figure 8C and D), as expected from the known association of GPI-APs with plasma membrane rafts. Interestingly, upon MesNa treatment of cells, the relative amount of biotin-SS–ASSP detectable in DRMs was drastically reduced (taking into account the fact that the overall amount of toxin detected by streptavidin was ∼3-fold lower than in the absence of MesNa). These experiments show that ASSP–GPI-AP complexes are more sensitive to solubilization by Triton X-100 in recycling endosomes than at the plasma membrane, strongly suggesting that GPI-APs are associated to different membrane domains in these two compartments of CHO cells.
GPI-AP clustering inhibits transport to CHO recycling endosomes
We next analyzed whether clustering of GPI-APs, which increases raft association (Harder et al., 1998), would affect their transport. GPI-APs were first labeled with Alexa–ASSP at 4°C and clustering was induced by the sequential addition at 4°C of a polyclonal anti-aerolysin antibody followed by a secondary antibody. Upon 30 min internalization at 37°C of the clustered complexes, the toxin no longer reached the perinuclear recycling endosomes (Figure 9A). We were concerned that antibody sandwiches might lead to the formation of large clusters, which could in itself act as a degradation signal. To generate small, well-controlled clusters, we used the Y221G aerolysin mutant. This mutant forms heptameric complexes, thus leading to clustering of a maximum of seven GPI-APs, but is inactive (Nelson et al., 1999) because the heptamers do not insert into the membrane (Y.Tsitrin, C.J.Morton, C.El Bez, P.Paumard, M.-C.Velluz, M.Adrian, M.Dubochet, M.W.Parker, S.Lanzavecchia and F.G.van der Goot, submitted). When trypsin-nicked Y221G was added to CHO cells, oligomerization occurred even at 4°C (Figure 9B), and led to increased DRM association compared with monomeric ASSP (Figure 9C). Upon internalization, Y221G did not reach recycling endosomes, in contrast to ASSP, but was transported to LBPA-positive late endosomes (Figure 9D). In marked contrast, Maxfield and co-workers showed that cross-linking of 10 transferrin receptors using multivalent transferring (Tf10) did not prevent recycling (Marsh et al., 1995), supporting the notion that targeting of the transferrin receptor and GPI-APs to CHO recycling endosomes occurs by different mechanisms.
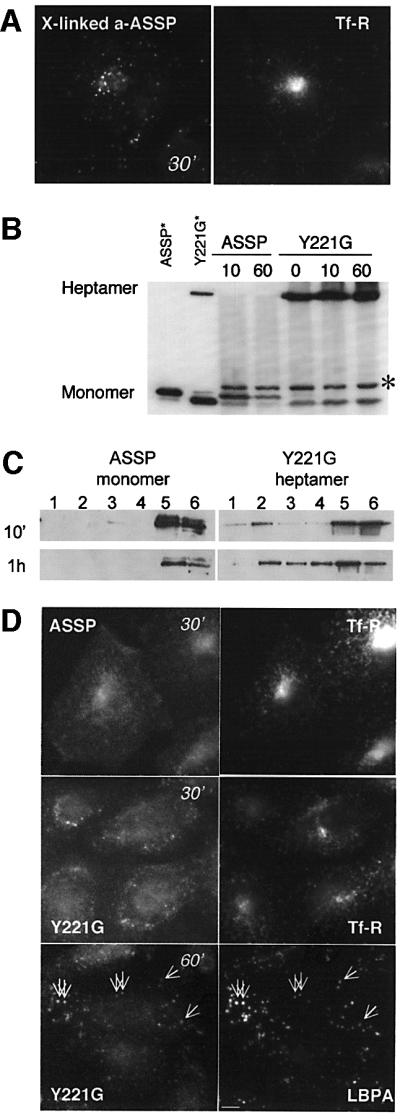
Fig. 9. Clustering of GPI-APs inhibits trafficking to the recycling endosome of CHO cells. (A) CHO cells were sequential incubated at 4°C with Alexa–ASSP (10 nM) for 1 h, anti-aerolysin polyclonal antibody (1 h) and anti-chicken antibody (1 h) and then warmed to 37°C for 30 min in the presence of FITC–transferrin. (B–D) CHO cells were incubated at 4°C with trypsin-nicked ASSP or Y221G (10 nM) and then warmed up at 37°C for various times. Cells were lysed in 1% Triton X-100, analyzed by SDS–PAGE and western blotting for the presence of the various forms of the toxin (non-reducing 10% gel, 40 µg of protein per lane except for the standards) (B) or fractionated on a sucrose density gradient to isolate DRMs (C). The asterisk in (B) indicates the presence of a protein cross-reacting with the anti-aerolysin antibody. In (C), DRMs were prepared by a short centrifugation on sucrose gradients in order to visualize the difference in behavior between ASSP and Y221G. (D) ASSP and Y221G were detected with anti-aerolysin antibodies followed by an anti-chicken secondary antibody. We used anti-aerolysin antibodies since Alexa labeling of Y221G inhibited oligomerization. The anti-aerolysin antibody gives a background staining in particular in the nucleus. Cells were incubated for the last 30 min with FITC–transferrin or labeled with an anti-LBPA antibody. Scale bar, 10 µm.
Discussion
Here we have analyzed the endocytic trafficking routes of endogenous GPI-APs. Based on biochemical, morphological and functional data, we report that the intracellular fate of GPI-APs in the endocytic pathway is cell type dependent. In CHO cells, GPI-APs were found to accumulate in the recycling endosome, whereas in BHK cells, they were transported to late endosomes. These two pathways are not due to permanent cell line genetic instability, since both were found in different primary cells: whereas GPI-APs were transported to the recycling endosome in rat astrocytes (Supplementary figure S3), they were routed to late endosomes in primary human skin fibroblasts (Supplementary figure S4). The fact that the endocytic route of GPI-APs is cell type specific could explain the apparent discrepancies found in the literature (Fransson et al., 1995; Mayor et al., 1998; Skretting et al., 1999; Nichols et al., 2001).
In neither CHO nor BHK cells was uptake of GPI-APs affected by the overexpression of a dominant-negative mutant of the clathrin-interacting protein Eps15 (data not shown), in agreement with previous observations showing that GPI-APs enter via a clathrin-independent pathway (Skretting et al., 1999; Nichols et al., 2001; Sabharanjak et al., 2002). However, in both cell types, GPI-APs were transported within 15 min to early endosomes. How is subsequent differential sorting achieved? Our results strongly suggest that sorting does not depend on the ectodomain of GPI-APs, since the same GPI-AP (CD14) is differentially transported in the two cell types, although the possibility that sorting is mediated by the ectodomain through interaction with different binding partners in different cell types cannot be ruled out. Since GPI-APs lack a cytoplasmic signal that could be involved in sorting, it is attractive to speculate that sorting could be at least partly lipid based.
Association of GPI-APs with DRMs in BHK late endosomes
In BHK cells, we found that internalized GPI-APs remain associated with DRMs, as they reach late endosomes, suggesting that GPI-APs interact with raft-like domains all along the degradative pathway. This is consistent with the observation that lipid analogues with long and saturated fatty acyl chains, which partition in liquid-ordered membranes, are preferentially targeted to late endosomes/lysosomes (Kok et al., 1991; Sandhoff and Klein, 1994; Mukherjee et al., 1999), in contrast to short unsaturated lipids, which are recycled (see below).
Our results also led to the interesting finding that raft-like domains exist in late endosomes. These late endosomal rafts were enriched in cholesterol, GPI-APs and flotillin-1, but devoid of rab7 and lamp-1. It has previously been proposed that the degradative pathway has reduced raft levels due to the NPC-1-dependent clearance of LDL-derived cholesterol from late endosomes (Lusa et al., 2001). The present findings, however, illustrate that, under physiological conditions, rafts are present in late endosomes or a late endosome population. Their level in this compartment is probably tightly regulated. Accumulation of rafts, above a certain threshold, as possibly occurs in sphingolipid storage diseases (Puri et al., 1999), could alter the properties of the compartment and, in particular, lead to a jam in lipid and protein trafficking (Simons and Gruenberg, 2000).
GPI-anchored proteins in the recycling endosome of CHO cells
Since GPI-AP trafficking in CHO cells was different, we compared the biochemical properties of GPI-APs in recycling endosomes and at the plasma membrane, and found that intracellular GPI-APs were more sensitive to detergent extraction than plasma membrane GPI-APs. This finding seemed a priori surprising, since some raft components (sphingomyelin, cholesterol and caveolin-1) co-fractionate with the transferrin receptor in Madin– Darby canine kidney cells (Gagescu et al., 2000). However, raft association was not analyzed in this study. Also, cholesterol (Mayor et al., 1998) and sphingomyelin (Chatterjee et al., 2001) play a role in the recycling kinetics of the GPI-anchored folate receptor in CHO cells, pointing to a role of lipid rafts, but these lipids may well act in early endosomes or in the GEEC. Importantly, Mukherjee et al. (1999) found, also in CHO cells, that lipid analogues with short or unsaturated fatty acyl chains, with a preference for more fluid domains, were efficiently transported to recycling endosomes. These observations support our findings that fluid membrane domains are preferentially transported to recycling endosomes. This may contribute to explaining why the transferrin receptor, which is typically not raft associated and seems to lack recycling signals, can recycle efficiently, perhaps along a default pathway.
Conclusion
Transport of BHK GPI-APs to late endosomes correlates with association to raft-like membranes (DRMs), whereas transport of CHO GPI-APs to recycling endosomes is accompanied by decreased raft association. Based on these data, we propose the following model for differential sorting of GPI-APs along the endocytic pathway. Jacobson and co-workers have shown that GPI-APs such as Thy-1 associate with rafts only transiently (Sheets et al., 1997). They found that raft partitioning, or what we here call raft residence time τ, can be modulated by subtle changes in lipid composition (Dietrich et al., 2001). Therefore, τ is likely to vary between cell types and/or according to the physiological/growth conditions or cell activation state. Our hypothesis is that τ is shorter in CHO than in BHK cells for GPI-APs. How would this affect trafficking? The early endosome is composed of tubular and vesicular elements (Mukherjee and Maxfield, 2000), the former being involved in transport to recycling endosomes and the later in transport down the degradative pathway. As reviewed recently (Mukherjee and Maxfield, 2000), fluid domain-preferring lipids exit the early endosome rapidly to be recycled back to the plasma membrane (half-time <10 min), whereas lipids preferring more rigid domains are slowly trafficked to late endosomes. If GPI-APs have a long raft residence time, they will be transported to late endosomes. On the other hand, if τ is small, the time spent outside of rafts will allow rapid exit from early endosomes, and efficient recycling. Note that the effect of τ on sorting proposed here could also affect the initial entry route of GPI-APs into the cell. One prediction of this model is that if the raft association of GPI-APs is increased in CHO cells by some means, then transport to the recycling endosome should be inhibited. This indeed is the case. We found that clustering of seven or less GPI-APs using the Y221G aerolysin mutant enhanced raft association, impaired transport to recycling endosomes and caused targeting to late endosomes. In conclusion, we proposed that trafficking of GPI-APs is controlled by their residence time in rafts.
Differential sorting may be of physiological importance for the intracellular delivery of GPI-AP ligands as well as perhaps the infectivity of the GPI-anchored PrP prion protein. Indeed, certain cell types cannot be infected by prion despite the presence of PrPC (Raeber et al., 1999), and this might be due to the fact that PrPc is not transported to late endosomes/lysosomes where conversion is thought to occur (Taraboulos et al., 1990).
Finally, we would like to speculate that this sorting mechanism may apply to other lipid rafts components, and may be particularly relevant to signaling proteins, whose residence time in rafts may be modulated by external stimuli. As ligand activation of receptors often results in increased partition in rafts, our model predicts that activated signaling platforms at the plasma membrane may be efficiently turned off by preferential endocytic targeting to the degradative pathway.
Materials and methods
Cell lines and reagents
BHK, CHO and GPI-deficient CHO cells were grown as described previously (Abrami et al., 1998, 2001). FITC–dextran (10 kDa) and FITC- or rhodamine-conjugated transferrins were purchased from Molecular Probes. PI-PLC was kindly provided by M.Low. Wild-type aerolysin was purified and radiolabeled as described previously (Abrami et al., 1998). The ASSP aerolysin double mutant G202C-I445C (van der Goot et al., 1994) was kindly provided by T.Buckley. His-tagged versions of the ASSP and Y221G mutants, produced in Escherichia coli and purified on a nickel column (Novagen), were also used. Nicked ASSP was obtained by trypsin treatment (100:1 ratio w/w) for 10 min at room temperature, before addition of a 10-fold excess of trypsin inhibitor. Wild-type and ASSP were conjugated with Alexa-546 (Molecular Probes), NHS-biotin or NHS-S-S-Biotin (Pierce) according to the manufacturers’ instructions. Transient cDNA transfections were carried out using the Fugene transfection reagent (Roche). The human CD14 cDNA was cloned into a pRc/RSV mammalian expression vector (Invitrogen).
Isolation of DRMs
For 2D SDS–PAGE analysis, DRMs were prepared from BHK and CHO cells and N-deglycosylation of DRM proteins was performed as described previously (Fivaz et al., 2000). Otherwise, DRMs were prepared using either OptiPrep or sucrose gradients. In the former case, cells were treated for 30 min at 4°C in 200 µl of lysis buffer (25 mM Tris–HCl pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100) in the presence of Complete, a cocktail of protease inhibitors (Roche), adjusted to 40% OptiPrep (Nycodenz), overlaid with 30 and 0% OptiPrep cushions, and centrifuged for 4 h at 40 000 r.p.m. (4°C) in a Beckman SW60 rotor. In the latter case, cells were extracted in lysis buffer containing 2% Triton X-100 (4°C) for 60 min and DRMs were separated on a sucrose step gradient (cushions of 40, 35 and 15%) by 7 h centrifugation at 40 000 r.p.m. (4°C) using a SW60 rotor or 2 h centrifugation at 55 000 r.p.m. (4°C) using a TLS.55 rotor. Fractions were collected and precipitated with 6% trichloroacetic acid in the presence of sodium deoxycholate as a carrier.
To prepare DRMs from late endosomes, this organelle was first purified by subcellular fractionation of BHK cells (van der Goot, 1997). Late endosomes (0.2–0.4 mg/ml) were then diluted four times, sedimented by centrifugation (TLS.55 Beckman rotor, 30 min, 55 r.p.m.) and resuspended in 200 µl of lysis buffer containing 1% Triton X-100. After 20 min of incubation at 4°C, fractions were separated on an OptiPrep gradient as described above.
Purification of GPI-APs
BHK cells were metabolically labeled with [35S]methionine. DRMs were then prepared as described above, dialyzed against 10 mM Tris pH 7.4, 150 mM NaCl and submitted to two rounds of Triton X-114 phase separation (Bordier, 1981; Fivaz et al., 2000). The detergent phase was treated with PI-PLC (1 U/ml) for 2 h at 37°C. After another round of Triton X-114 phase separation, GPI-APs were specifically recovered in the aqueous phase. As a negative control, the experiment was carried out as above, except that PI-PLC treatment was performed prior to the first Triton X-114 phase separation.
Endocytosis of ASSP, Y221G, anti-CD14 antibodies, transferrin and dextran
CHO or BHK cells were incubated with Alexa-546-labeled ASSP (10 nM) for 1 h at 4°C in IM (GMEM medium buffered with 10 mM HEPES pH 7.4), rinsed and incubated in cell culture medium at 37°C (in 5% CO2) for the indicated times. Alternatively, cells were continuously incubated with Alexa–ASSP. Cells transiently expressing human CD14 were incubated for 1 h at 4°C with FITC-conjugated anti-human CD14 monoclonal antibody (1 µg/ml) in IM, followed by various times of incubation at 37°C. When indicated, surface-bound FITC-conjugated anti-hCD14 was removed by a brief acid wash (4 min at 15°C in 100 mM citrate pH 2, 140 mM NaCl, 1 mM CaCl2, 1 mM MgCl2) and the remaining FITC surface fluorescence further quenched for 30 min at 4°C using an anti-FITC antibody. To label the recycling pathway, cells were incubated with FITC–transferrin (50 µg/ml) for 15 or 30 min at 37°C. To label early endosomes, cells were incubated with FITC–dextran (1 mg/ml) for 5 min at 37°C. Cells were then fixed with 3% paraformaldehyde, permeabilized or not with 0.1% saponin, and processed for immuno cytochemistry.
Analysis of intracellular ASSP in CHO cells
CHO cells were incubated with ASSP, labeled with biotin containing a thiol-cleavable spacer (biotin-SS–ASSP) (10 nM in IM) for 1h at 4°C, rinsed and shifted or not to 37°C for 45 min, put back on ice, and incubated for 15 min in buffer containing 50 mM Tris–HCl pH 8.3, 150 mM NaCl with or without 30 mM MesNa to remove biotin from cell surface biotin-SS–ASSP. Cells were then washed and excess MesNa was quenched with iodoacetamide (0.5 M) for 5 min. Post-nuclear supernatant (PNS) or DRMs were prepared as described above and samples were analyzed by SDS–PAGE under non-reducing conditions. Biotin-SS–ASSP was revealed on blots using streptavidin–HRP or [125I]streptavidin for quantification.
Epifluorescence microscopy and image analysis
Epifluorescence microscopy was carried out using an inverted Axiovert 135 TV Zeiss microscope equipped with a cooled-CCD camera (Princeton Instruments), driven by the IP laboratory imaging system software. To quantify the amount of Alexa–ASSP that reached the RE in CHO cells, outlines of the RE (identified by Alexa–ASSP and FITC–transferrin co-localization) and the entire cell (identified by plasma membrane staining of Alexa–ASSP) were traced out manually and the total fluorescence intensity for each surface was determined. RE fluorescence was corrected for plasma membrane background by subtracting the fluorescence intensity of a region, of identical surface, of the cell away from the RE. The percentage of Alexa–ASSP in the RE was determined by calculating the ratio of (corrected RE)/(entire cell surface) fluorescence intensities.
Analytical techniques
One-dimensional SDS–PAGE was carried out on 9–15% acrylamide gradient gels unless stated otherwise. 2D gel electrophoresis and sequencing by nano spray mass spectrometry were performed as described previously (Fivaz et al., 2000). Aerolysin overlays were performed as described previously (Abrami et al., 1998). HRP-conjugated streptavidin or secondary antibodies were revealed using the chemiluminescence reagent from Pierce. [125I]streptavidin was quantified using a Bio-Rad phosphoimager (Molecular Imager FX) driven by the Quantify One (v. 4.2.1) software.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We would like to thank F.Lafont and S.Clarkson for providing us with rat astrocytes and human skin fibroblasts, respectively; M.Low and T.Buckley for providing us with PI-PLC and the ASSP mutant, respectively; and G.Emery, V.Cavalli, J.Gruenberg, J.Fauré and F.Lafont for critical reading of the manuscript. This work was supported by grants from the Swiss National Science Foundation to F.G.v.d.G., and the American Diabetes Association and the National Institutes of Health (5P30 DK56341-02) to P.E.B.
References
- Abrami L. and van der Goot,F.G. (1999) Plasma membrane micro domains act as concentration platforms to facilitate intoxication by aerolysin. J. Cell Biol., 147, 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrami L., Fivaz,M., Glauser,P.E., Parton,R.G. and van der Goot,F.G. (1998) A pore-forming toxin interact with a GPI-anchored protein and causes vacuolation of the endoplasmic reticulum. J. Cell Biol., 140, 525–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrami L., Fivaz,M. and van der Goot,F.G. (2000) Adventures of a pore-forming toxin at the target cell surface. Trends Microbiol., 8, 168–172. [DOI] [PubMed] [Google Scholar]
- Abrami L., Fivaz,M., Kobayashi,T., Kinoshita,T., Parton,R.G. and van der Goot,F.G. (2001) Cross-talk between caveolae and glycosylphosphatidylinositol-rich domains. J. Biol. Chem., 276, 30729–30736. [DOI] [PubMed] [Google Scholar]
- Abrami L., Velluz,M.C., Hong,Y., Ohishi,K., Mehlert,A., Ferguson,M., Kinoshita,T. and Gisou van der Goot,F. (2002) The glycan core of GPI-anchored proteins modulates aerolysin binding but is not sufficient: the polypeptide moiety is required for the toxin-receptor interaction. FEBS Lett., 512, 249–254. [DOI] [PubMed] [Google Scholar]
- Aniento F., Emans,N., Griffiths,G. and Gruenberg,J. (1993) Cytoplasmic dynein-dependent vesicular transport from early to late endosomes. J. Cell Biol., 123, 1373–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aniento F., Gu,F., Parton,R.G. and Gruenberg,J. (1996) An endosomal beta COP is involved in the pH-dependent formation of transport vesicles destined for late endosomes. J. Cell Biol., 133, 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickel P.E., Scherer,P.E., Schnitzer,J.E, Oh,P., Lisanti,M.P. and Lodish,H.F. (1997) Flotillin and epidermal surface antigen define a new family of caveolae-associated integral membrane proteins. J. Biol. Chem., 272, 13793–13802. [DOI] [PubMed] [Google Scholar]
- Bordier C. (1981) Phase separation of integral membrane proteins in Triton X-114 solution. J. Biol. Chem., 256, 1604–1607. [PubMed] [Google Scholar]
- Brown D.A. and London,E. (1998) Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol., 14, 111–136. [DOI] [PubMed] [Google Scholar]
- Chatterjee S., Smith,E.R., Hanada,K., Stevens,V.L. and Mayor,S. (2001) GPI anchoring leads to sphingolipid-dependent retention of endocytosed proteins in the recycling endosomal compartment. EMBO J., 20, 1583–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavrier P., Gorvel,J.P., Stelzer,E., Simons,K., Gruenberg,J. and Zerial,M. (1991) Hypervariable C-terminal domain of rab proteins acts as a targeting signal. Nature, 353, 769–772. [DOI] [PubMed] [Google Scholar]
- Diep D.B., Nelson,K.L., Raja,S.M., Pleshak,E.N. and Buckley,J.T. (1998) Glycosylphosphatidylinositol anchors of membrane glyco proteins are binding determinants for the channel-forming toxin Aerolysin. J. Biol. Chem., 273, 2355–2360. [DOI] [PubMed] [Google Scholar]
- Dietrich C., Volovyk,Z.N., Levi,M., Thompson,N.L. and Jacobson,K. (2001) Partitioning of Thy-1, GM1 and cross-linked phospholipid analogs into lipid rafts reconstituted in supported model membrane monolayers. Proc. Natl Acad. Sci. USA, 98, 10642–10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fivaz M., Vilbois,F., Pasquali,C. and van der Goot,F.G. (2000) Analysis of glycosyl phosphatidylinositol-anchored proteins by two-dimensional gel electrophoresis. Electrophoresis, 21, 3351–3356. [DOI] [PubMed] [Google Scholar]
- Fransson L.A., Edgren,G., Havsmark,B. and Schmidtchen,A. (1995) Recycling of a glycosylphosphatidylinositol-anchored heparan sulphate proteoglycan (glypican) in skin fibroblasts. Glycobiology, 5, 407–415. [DOI] [PubMed] [Google Scholar]
- Gagescu R., Demaurex,N., Parton,R.G., Hunziker,W., Huber,L.A. and Gruenberg,J. (2000) The recycling endosome of Madin–Darby canine kidney cells is a mildly acidic compartment rich in raft components. Mol. Biol. Cell, 11, 2775–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder T., Scheiffele,P., Verkade,P. and Simons,K. (1998) Lipid domain structure of the plasma membrane revealed by patching of membrane components. J. Cell Biol., 141, 929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita T., Ohishi,K. and Takeda,J. (1997) GPI-anchor synthesis in mammalian cells: genes, their products and a deficiency. J. Biochem. (Tokyo), 122, 251–257. [DOI] [PubMed] [Google Scholar]
- Kobayashi T., Stang,E., Fang,K.S., de Moerloose,P., Parton,R.G. and Gruenberg,J. (1998) A lipid associated with the antiphospholipid syndrome regulates endosome structure and function. Nature, 392, 193–197. [DOI] [PubMed] [Google Scholar]
- Kok J.W., Babia,T. and Hoekstra,D. (1991) Sorting of sphingolipids in the endocytic pathway of HT29 cells. J. Cell Biol., 114, 231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusa S., Blom,T.S., Eskelinen,E.L., Kuismanen,E., Mansson,J.E., Simons,K. and Ikonen,E. (2001) Depletion of rafts in late endocytic membranes is controlled by NPC1-dependent recycling of cholesterol to the plasma membrane. J. Cell Sci., 114, 1893–1900. [DOI] [PubMed] [Google Scholar]
- Marsh E.W., Leopold,P.L., Jones,N.L. and Maxfield,F.R. (1995) Oligomerized transferrin receptors are selectively retained by a lumenal sorting signal in a long-lived endocytic recycling compartment. J. Cell Biol., 129, 1509–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxfield F.R. and Mayor,S. (1997) Cell surface dynamics of GPI-anchored proteins. Adv. Exp. Med. Biol., 419, 355–364. [DOI] [PubMed] [Google Scholar]
- Mayor S., Sabharanjak,S. and Maxfield,F.R. (1998) Cholesterol-dependent retention of GPI-anchored proteins in endosomes. EMBO J., 17, 4626–4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S. and Maxfield,F.R. (2000) Role of membrane organization and membrane domains in endocytic lipid trafficking. Traffic, 1, 203–211. [DOI] [PubMed] [Google Scholar]
- Mukherjee S., Soe,T.T. and Maxfield,F.R. (1999) Endocytic sorting of lipid analogues differing solely in the chemistry of their hydrophobic tails. J. Cell Biol., 144, 1271–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson K.L., Brodsky,R.A. and Buckley,J.T. (1999) Channels formed by subnanomalar concentrations of the toxin aerolysin trigger apoptosis of T lymphomas. Cell. Microbiol., 1, 69–74. [DOI] [PubMed] [Google Scholar]
- Nichols B.J. (2002) A distinct class of endosome mediates clathrin-independent endocytosis to the Golgi complex. Nat. Cell Biol., 4, 374–378. [DOI] [PubMed] [Google Scholar]
- Nichols B.J., Kenworthy,A.K., Polishchuk,R.S., Lodge,R., Roberts,T.H., Hirschberg,K., Phair,R.D. and Lippincott-Schwartz,J. (2001) Rapid cycling of lipid raft markers between the cell surface and Golgi complex. J. Cell Biol., 153, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton R.G., Joggerst,B. and Simons,K. (1994) Regulated internalization of caveolae. J. Cell Biol., 127, 1199–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri V., Watanabe,R., Dominguez,M., Sun,X., Wheatley,C.L, Marks,D.L. and Pagano,R.E. (1999) Cholesterol modulates membrane traffic along the endocytic pathway in sphingolipid-storage diseases. Nat. Cell Biol., 1, 386–388. [DOI] [PubMed] [Google Scholar]
- Raeber A.J., Sailer,A., Hegyi,I., Klein,M.A., Rulicke,T., Fischer,M, Brandner,S., Aguzzi,A. and Weissmann,C. (1999) Ectopic expression of prion protein (PrP) in T lymphocytes or hepatocytes of PrP knockout mice is insufficient to sustain prion replication. Proc. Natl Acad. Sci. USA, 96, 3987–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijnboutt S., Jansen,G., Posthuma,G., Hynes,J.B., Schornagel,J.H. and Strous,G.J. (1996) Endocytosis of GPI-linked membrane folate receptor-α. J. Cell Biol., 132, 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Boulan E. and Powell,S.K. (1992) Polarity of epithelial and neuronal cells. Annu. Rev. Cell. Biol., 8, 395–427. [DOI] [PubMed] [Google Scholar]
- Sabharanjak S., Sharma,P., Parton,R.G. and Mayor,S. (2002) GPI-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated, clathrin-independent pinocytic pathway. Dev. Cell, 2, 411–423. [DOI] [PubMed] [Google Scholar]
- Sandhoff K. and Klein,A. (1994) Intracellular trafficking of glyco sphingolipids: role of sphingolipid activator proteins in the topology of endocytosis and lysosomal digestion. FEBS Lett., 346, 103–107. [DOI] [PubMed] [Google Scholar]
- Sheets E.D., Lee,G.M., Simson,R. and Jacobson,K. (1997) Transient confinement of a glycosylphosphatidylinositol-anchored protein in the plasma membrane. Biochemistry, 36, 12449–12458. [DOI] [PubMed] [Google Scholar]
- Simons K. and Gruenberg,J. (2000) Jamming the endosomal system: lipid rafts and lysosomal storage diseases. Trends Cell Biol., 10, 459–462. [DOI] [PubMed] [Google Scholar]
- Skretting G., Torgersen,M.L., van Deurs,B. and Sandvig,K. (1999) Endocytic mechanisms responsible for uptake of GPI-linked diphtheria toxin receptor. J. Cell Sci., 112, 3899–3909. [DOI] [PubMed] [Google Scholar]
- Taraboulos A., Serban,D. and Prusiner,S.B. (1990) Scrapie prion proteins accumulate in the cytoplasm of persistently infected cultured cells. J. Cell Biol., 110, 2117–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Goot F.G. (1997) Separation of early steps in endocytic membrane transport. Electrophoresis, 18, 2689–2693. [DOI] [PubMed] [Google Scholar]
- van der Goot F.G., Hardie,K.R., Parker,M.W. and Buckley,J.T. (1994) The C-terminal peptide produced upon proteolytic activation of the cytolytic toxin aerolysin is not involved in channel formation. J. Biol. Chem., 269, 30496–30501. [PubMed] [Google Scholar]
- Zerial M. and McBride,H. (2001) Rab proteins as membrane organizers. Nat. Rev. Mol. Cell. Biol., 2, 107–117. [DOI] [PubMed] [Google Scholar]