

Abstract
Fibroblast growth factor-1 (FGF-1) has both extra- and intracellular functions. To identify intracellular binding partners for FGF-1, we isolated proteins from U2OS human osteosarcoma cells interacting specifically with FGF-1. One of the isolated proteins was identified as protein kinase CK2 (CK2). We here provide evidence that FGF-1 binds to both the catalytic α-subunit and to the regulatory β-subunit of CK2. The interaction between FGF-1 and CK2α and β was characterized by surface plasmon resonance, giving KD values of 0.4 ± 0.3 and 1.2 ± 0.2 µM, respectively. By using a novel assay for intracellular protein interaction, FGF-1 and CK2α are shown to interact in vivo. In vitro, FGF-1 and FGF-2 are phosphorylated by CK2, and the presence of FGF-1 or FGF-2 was found to enhance the autophosphorylation of CK2β. A correlation between the mitogenic potential of FGF-1 mutants and their ability to bind to CK2α was observed. The possible involvement of CK2 in the FGF-induced stimulation of DNA synthesis is discussed.
Keywords: autophosphorylation/FGF-1/mitogenicity/protein kinase CK2
Introduction
Fibroblast growth factor-1 (FGF-1) is a member of the large FGF family of growth factors, consisting of 30 members in humans (Lander et al., 2001). It is involved in various cellular processes such as stimulation of DNA synthesis and cell proliferation, as well as differentiation and cell migration (Burgess and Maciag, 1989; Basilico and Moscatelli, 1992; Wiedlocha et al., 1994; Szebenyi and Fallon, 1999).
FGF-1 binds at the cell surface to any of four closely related high affinity FGF receptors (FGFR1–4) and induces a phosphorylation cascade that results in downstream signalling by phospholipase Cγ, activation of the mitogen-acivated protein (MAP) kinase cascade and expression of immediate early genes such as c-jun and c-fos (Mason, 1994; Szebenyi and Fallon, 1999). FGF-1 also binds to a Golgi-localized cysteine-rich receptor without known biological function and to cell surface heparan sulfate proteoglycans (HSPGs) (Szebenyi and Fallon, 1999). Binding to surface HSPGs leads to a high local concentration of FGF-1 at the cell surface, which facilitates dimerization and activation of the high affinity receptors (Spivak-Kroizman et al., 1994).
FGF-1, like FGF-2 and a few other members of the FGF family, is synthesized as an intracellular protein lacking a signal sequence for secretion (Burgess and Maciag, 1989; Szebenyi and Fallon, 1999). In response to stress situations such as heat shock, serum starvation or HIV-TAT transformation of cells, it is released by a mechanism bypassing the classical export route taken by most secreted proteins via the endoplasmic reticulum (ER) and the Golgi apparatus (Jackson et al., 1992; Opalenik et al., 1995; Shin et al., 1996).
During the last decade, an increasing amount of evidence has suggested that FGF-1 is present in the nucleus and that nuclear translocation of the growth factor is required for the induction of DNA synthesis (Imamura et al., 1990; Wiedlocha et al., 1994, 1996). Both signalling through FGF receptors and nuclear localization of FGF-1 may be required for the stimulation of cell proliferation (Wiedlocha et al., 1996). Like FGF-1, FGF-2 has also been found in the nucleus. Here FGF-2 can activate rDNA transcription, and its nuclear accumulation is associated with cellular proliferation (Bouche et al., 1987; Amalric et al., 1994).
A mutant FGF-1 where Lys132 has been changed to glutamic acid has greatly reduced mitogenic activity (Burgess et al., 1990). In spite of this, the mutant retains the ability to bind to heparin, it has a normal receptor-binding activity and it is capable of stimulating receptor tyrosine kinase activity and proto-oncogene expression (Burgess et al., 1990, 1991). In addition, both wild-type and mutant FGF-1 are found in the nucleus following transfection of NIH 3T3 cells with the relevant construct, even though only wild-type FGF-1 induces a transformed phenotype (Burgess et al., 1991). The nuclear translocation of this mutant was studied using an approach where the wild-type or the K132E mutant of FGF-1 was fused to diphtheria toxin A fragment (Klingenberg et al., 1998). The proteins were reconstituted with diphtheria toxin B fragment and translocated to the cytosol via the diphtheria toxin pathway in U2OSDR1 cells, which lack FGF receptors. Even though both proteins were translocated to the cytosol and subsequently found in the nuclear fraction, only wild-type FGF-1 stimulated DNA synthesis.
Taken together, all these findings point to both an extra- and an intracellular role for FGF-1. While the extracellular action of FGF-1 is understood in some detail, the intracellular trafficking of FGF-1, as well as its mode of action, remains largely unexplained. We reasoned that in order to exert its intracellular role, FGF-1 must interact with intracellular proteins that are involved with the trafficking and functioning of FGF-1. FGF-1 intracellular binding protein (FIBP) and mortalin are two such proteins that have been identified previously (Kolpakova et al., 1998; Mizukoshi et al., 1999). Also, the proteins synaptotagmin and S100A13, which are involved in the release of FGF-1 from cells, form complexes with FGF-1 (Carreira et al., 1998; Tarantini et al., 1998).
Here we report binding of mitogenic FGF-1, but not of a non-mitogenic mutant, to protein kinase CK2, a pleiotropic, constituitively active serine/threonine kinase (Guerra et al., 1999). Protein kinase CK2 is known to be required for viability in yeast, for cell cycle progression of mammalian cells and it is found to be elevated in proliferating tissues (Pinna and Meggio, 1997; Lebrin et al., 2001). In addition, CK2 plays a prominent role in the early phases of mitogenic signal transduction and in subsequent signal processing (Pyerin, 1994; Tawfic et al., 2001).
Results
Identification of proteins selectively binding to FGF-1
To identify new intracellular proteins binding to FGF-1, we used a co-precipitation approach. A fusion protein of maltose-binding protein and FGF-1 (MBP–FGF-1) was bound covalently to Sepharose beads and used to adsorb proteins from a lysate of U2OS cells labelled with [35S]methionine/cysteine. MBP and MBP–interferon-γ (IFN-γ) were used as negative controls. MBP fusion proteins were used since both MBP–FGF-1 and MBP–FGF-2 have been found in control experiments to be as potent as wild-type FGF-1 and FGF-2 in binding to and activating high affinity receptors (results not shown). Much less unspecific binding was seen when using GST and GST–FGF-1 in the co-precipitation experiments. However, GST–FGF-1 was not able to compete with wild-type FGF-1 for binding to high affinity receptors and was therefore assumed not to be biologically active (results not shown).
Several proteins were found to bind to MBP–FGF-1 (Figure 1A, lanes 2, 3 and 5) that did not bind to the controls (Figure 1A, lanes 1 and 4). Furthermore, these protein bands were essentially absent when the incubation was performed in the presence of excess free FGF-1 (Figure 1B, compare lanes 3 and 4). In proteins adsorbed with MBP–IFN-γ as a bait, no difference was seen between the absence and presence of free FGF-1 (Figure 1B, compare lanes 1 and 2).

Fig. 1. Identification of proteins binding selectively to FGF-1. (A) U2OS cells were incubated overnight (16 h) with [35S]cysteine/methionine, then washed and lysed. The cell lysates were rotated for 1 h at 4°C with either MBP (lane 1), MBP–FGF-1 (lane 2) or MBP–IFN-γ (lane 4) covalently bound to Sepharose beads (∼20 µg of protein). The lysates were then transferred to another Eppendorf tube and subjected to a second round of precipitation for 1 h at 4°C with Sepharose-bound MBP–FGF-1. The Sepharose beads were washed, the proteins eluted with SDS sample buffer and the eluates were subjected to SDS–PAGE [12% (w/v) gel] and fluorography. An asterisk marks proteins that bound to MBP–FGF-1 after the lysate had first been incubated with MBP, and two asterisks mark proteins that bound to MBP–FGF-1 after the lysate had first been incubated with MBP–IFN-γ. Arrows point to proteins that bind FGF-1 specifically. Also shown are the positions of molecular weight standards. (B) The same conditions as in (A), but the rotations with fusion proteins were performed in the absence (lanes 2 and 4) or presence (lanes 1 and 3) of excess free FGF-1 (500 µg). Arrows mark proteins that bound FGF-1 specifically and which were competed out by excess FGF-1.
The experiment was scaled up and two bands with apparent molecular weights of ∼28 and 35 kDa (arrows in Figure 1A and B), which appeared to be specific for FGF-1 binding, and which were present in large enough quantities for mass spectrometric analysis, were excised from the dried Coomassie Blue-stained gel. Both proteins were digested with trypsin and analysed by matrix-assisted laser desorption/ionization-time of flight-mass spectrometry (MALDI-TOF-MS). The molecular masses obtained from the analyses were used to search protein databases using the ProFound, MS-Fit and Mascot search engines. The p28 protein was identified as protein kinase CK2β (CK2β) and will be discussed here. p35 is discussed in a separate paper (Skjerpen et al., 2002).
FGF-1 and CK2 interact
Protein kinase CK2 is a pleiotropic, constitutively active, heterotetrameric 130 kDa kinase consisting of two α- and two β-subunits (Allende and Allende, 1995; Guerra et al., 1999). To study the interaction between CK2 and FGF-1, equal amounts of U2OS cell lysate were incubated with increasing amounts of MBP–FGF-1. The bound proteins were analysed by western blotting with an antibody against the β-subunit of CK2. As shown in Figure 2, endogenous CK2 binds extensively to FGF-1. The signal of bound CK2 increased with the amount of MBP–FGF-1 added, and reached saturation at 10 µg of MBP–FGF-1.
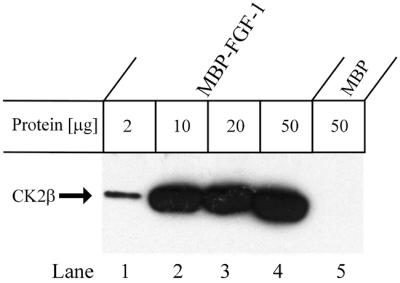
Fig. 2. Ability of CK2β to bind to FGF-1. MBP–FGF-1 or MBP was pre-bound to Sepharose beads via an anti-MBP antibody (Kolpakova et al., 1998). U2OS cells were lysed and lysate was incubated for 1 h at 4°C with the indicated amount of immobilized MBP–FGF-1 or MBP. The bound proteins were separated by SDS–PAGE, transferred to a PVDF membrane and blotted with anti-CK2β antibodies. The arrow marks the position of CK2β.
Although CK2 from most organisms has been found predominantly as a heterotetramer of α2β2, αα′β2 or α′2β2 stoichiometry, free subunits have also been detected (Heller-Harrison and Czech, 1991; Allende and Allende, 1995). To test whether FGF-1 binds CK2 as a complex of both α- and β-subunits, or whether it binds to the β-subunit alone, we incubated MBP–FGF-1 with cell lysate and, after separation by SDS–PAGE, immunoblotting was used to detect the presence of CK2β and then CK2α. Both the β- (upper panel) and α- (lower panel) subunits were shown to be present in the material binding to FGF-1 (Figure 3A). In agreement with this, a protein with an apparent molecular mass of ∼43 kDa that could not be identified by mass spectrometry can be seen in Figure 1B. This protein could represent CK2α.

Fig. 3. Binding of FGF-1 to both CK2α and β. (A) U2OS cells were lysed and incubated for 1 h at 4°C with the indicated amounts of MBP–FGF-1 or MBP immobilized on Sepharose beads. The Sepharose beads were washed and the bound proteins were eluted with SDS sample buffer and separated by SDS–PAGE. The proteins were then transferred to a PVDF membrane and the membrane was probed with an anti-CK2β antibody (upper panel). Then the membrane was stripped and reprobed with an antibody against CK2α (lower panel). (B) MBP–FGF-1 (lanes 1–12) or MBP (lanes 13–15) bound to protein A–Sepharose beads was incubated for 2 h at 4°C with the indicated volumes of in vitro translated CK2β (lanes 2–4 and 13), CK2α (lanes 6–9 and 14) or CK2α′ (lanes 10–12 and 15). The beads were washed and the bound proteins were subjected to SDS–PAGE and fluorography. For comparison, 1 µl of the in vitro translated proteins used in the precipitations was run in separate lanes (1, 5 and 9). (C) GST–CK2α (lanes 1, 4 and 7), GST–CK2β (lanes 2, 5 and 8) or GST (lanes 3, 6 and 9) bound to glutathione–Sepharose was incubated with MBP–FGF-1 (lanes 1–3), MBP–FGF-2 (lanes 4–6) or MBP (lanes 7–9) for 2 h at 4°C. After binding, the samples were washed and analysed by western blotting with an antibody against MBP.
FGF-2 previously has been found to bind only to the β-subunit of CK2 (Bonnet et al., 1996). To test whether this is also the case for FGF-1, MBP–FGF-1–Sepharose was incubated with increasing amounts of in vitro translated CK2 α, α′ or β. FGF-1 was able to precipitate the α-, α′- and β-subunits, although binding to the α-subunit is apparently strongest (Figure 3B). FGF-1 does not seem to discriminate between the α- or the α′-subunits since both bound strongly to FGF-1.
As a further test of whether the binding between FGF-1 and CK2α and β is direct, and as a test as to whether CK2α as such can bind to FGF-2, we conducted a GST pull-down experiment with purified proteins. GST fusion proteins of CK2α and β, or GST alone, were bound to glutathione– Sepharose and then incubated with MBP fusions of FGF-1 or FGF-2, or MBP alone. Both FGF-1 and FGF-2 interacted directly with CK2α and CK2β (Figure 3C), although the latter interaction was clearly much weaker.
We then investigated whether FGF-1 binds to CK2α and CK2β as a complex of all three proteins or whether FGF-1 binding abrogates the αβ interaction in CK2. Excess CK2β was able to compete out the binding of CK2α to FGF-1 (Figure 4A). Excess free FGF-1 was also able to disrupt the binding between the α- and β-subunits (Figure 4B). This suggests that binding of FGF-1 is preferentially to free α- or β-subunits.
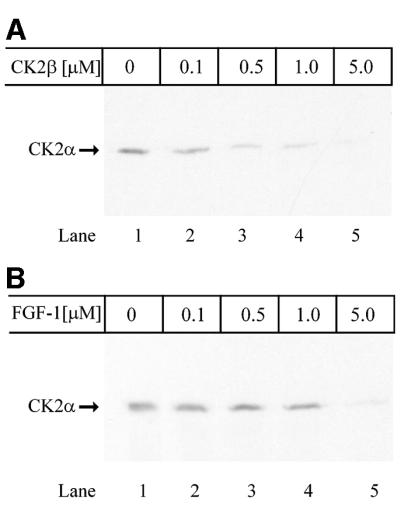
Fig. 4. Binding of FGF-1 to free subunits of CK2. (A) MBP–FGF-1 covalently bound to CNBr-activated Sepharose beads was incubated for 90 min with in vitro translated [35S]methionine-labelled CK2α in the presence of increasing amounts of CK2β as indicated. The beads were washed and the bound proteins were subjected to SDS–PAGE and fluorography. (B) The same conditions as described in (A), but MBP–CK2β was bound to CNBr-activated Sepharose and incubated with in vitro translated [35S]methionine-labelled CK2α in the presence of increasing concentrations of FGF-1.
Characterization of the interaction between FGF-1 and CK2
To characterize the interaction between FGF-1 and CK2α and β, we first tested the salt sensitivity of the binding. [35S]methionine-labelled, in vitro translated CK2 α- or β-subunits were incubated with MBP–FGF-1–Sepharose and subsequently washed with different salt concentrations. The binding was highly salt sensitive. A salt concentration of 0.3 M NaCl reduced the interaction with both the α- and β-subunits (Figure 5), and 0.5 M NaCl essentially eliminated the binding.

Fig. 5. Sensitivity to salt of the binding between FGF-1 and CK2α and β. MBP–FGF-1 covalently bound to CNBr-activated Sepharose beads was incubated in a 1:1 mixture of lysis buffer and PBS for 90 min at 4°C with in vitro translated [35S]methionine-labelled CK2α (lanes 1–4) or CK2β (lanes 5–8). The samples were washed in the same buffer (lanes 1 and 5) or the same buffer supplemented with either 0.3 (lanes 2 and 6), 0.5 (lanes 3 and 7) or 1.0 M NaCl (lanes 4 and 8). The bound proteins were then eluted with SDS sample buffer and analysed by SDS–PAGE and fluorography.
To quantify the kinetics of the interaction between FGF-1 and CK2α and β, association and dissociation kinetics were measured using surface plasmon resonance. GST–CK2α or β was immobilized onto a sensor surface using anti-GST antibodies, and sensorgrams were recorded upon injection of different concentrations of FGF-1. The interaction between both CK2α (Figure 6A) and β (Figure 6B) and FGF-1 showed characteristics of a typical 1:1 binding (Langmuir isotherm kinetics). From the association and dissociation curves, we determined the equilibrium constant (KD) for the binding of FGF-1 to CK2α and CK2β to be 0.4 ± 0.3 and 1.2 ± 0.2 µM, respectively. While the dissociation constants of the two interactions were very similar, the association constant for the binding between FGF-1 and CK2α was about three times as high as that for the binding to CK2β.

Fig. 6. Kinetics of binding between FGF-1 and CK2α or CK2β as measured by surface plasmon resonance. FGF-1 was injected over a sensorchip loaded with either GST–CK2α (A) or GST–CK2β (B). Sensorgrams obtained at the indicated concentrations from one representative series are shown. The calculated association (Ka), dissociation (Kd) and equilibrium (KD) constants (± SDs) based on four separate series are given.
Co-localization of FGF-1 and CK2 in transfected cells
CK2 is found widely distributed throughout the cell (Allende and Allende, 1998; Faust and Montenarh, 2000). Similarly, FGF-1 is found to be present in both the cytosol and nucleus (Wiedlocha et al., 1995). A dotted co-localization between FGF-1 and CK2α and between FGF-1 and CK2β was seen in the cytosol of transiently transfected U2OS cells. In the nucleus, FGF-1 co-localized with both CK2α and β (Figure 7).

Fig. 7. Co-localization of FGF-1 and CK2 in U2OS cells transfected with GFP–FGF-1. U2OS cells grown on coverslips were transfected with pEGFP-FGF-1 and pRc/CMV-CK2α-HA or pEGFP-FGF-1 and pcDNA3-CK2β, fixed in 3% paraformaldehyde, permeabilized and labelled with either a polyclonal antibody against CK2α or a monoclonal antibody against CK2β. The cells were subsequently stained with anti-rabbit-Cy3 (CK2α) or anti-mouse–lissamine–rhodamine (CK2β) secondary antibodies and analysed by confocal microscopy.
Due to the ubiquitous expression of both proteins, it is difficult to decide whether they really interact in the living cell. To test this, we developed a novel co-localization system where we targeted FGF-1 to the peroxisomes, which are distinctly visible under the microscope, and studied whether CK2α would partly co-localize. It has been demonstrated that proteins containing signals for targeting to peroxisomes can be imported directly into this organelle without prior unfolding (Walton et al., 1995). Other studies have shown that oligomeric complexes of >400 kDa and where only one protein contains the peroxisomal targeting signal can be imported (McNew and Goodman, 1994; Titorenko et al., 2002).
We constructed a mutant FGF-1 with a C-terminal type 1 signal (SerLysLeu) for targeting to peroxisomes (FGF-1pts). When expressed in cells, this construct was found in peroxisomes (Figure 8A, B and D) as determined by co-staining with anti-catalase, a marker for peroxisomes. CFP-CK2α was then co-transfected with myc-FGF-1 with or without the peroxisomal targeting signal. In the cells co-transfected with CK2α and FGF-1pts, >80% of the peroxisomes containing FGF-1 also contained CK2α (Figure 8C, E and M). By merging the confocal images of CFP-CK2α and myc-FGF-1pts nine pixels askew (∼1.5 times the peroxisomal size), we found that no more than 8% of this targeting was due to random overlap with cytosolic proteins (Figure 8F and M). Hardly any CK2α was found in peroxisomes in cells transfected with only CFP-CK2α (results not shown) or in cells co-transfected with CFP-CK2α and myc-FGF-1 lacking the peroxisome targeting signal (Figure 8L). The data indicate that when expressed together, FGF-1 and CK2α bind to each other in the living cell.

Fig. 8. FGF-1 interacts with CK2α in vivo. HeLa cells were transfected with either pcDNA3-myc-FGF-1 containing a signal for targeting to peroxisomes (myc-FGF-1pts) and pECFP-CK2α (A–F) or pcDNA3-myc-FGF-1 and pECFP-CK2α (G–L). After depletion of the cytosol with digitonin, the cells were fixed, permeabilized with Triton X-100 and double stained with rabbit anti-catalase and mouse anti-c-Myc antibodies followed by staining with anti-rabbit Cy5 (catalase) and anti-mouse lissamine–rhodamine (c-Myc). (M) Quantitation of peroxisomes containing both FGF-1pts and CK2α. The number of peroxisomes that stained for both FGF-1pts and CK2α was counted in a number of cells (20) both by accurately merging the pictures taken of FGF-1pts and CK2α and by merging them nine pixels askew. The number of peroxisomes containing both FGF-1pts and CK2α in the image merged askew is taken as the amount of unspecific co-localization.
FGF phosphorylation and autophosphorylation by CK2
Earlier studies have shown that protein kinase C (PKC) is the major kinase responsible for phosphorylating FGF-1, while FGF-2 is phosphorylated by both PKA and PKC (Feige and Baird, 1989; Klingenberg et al., 1999). However, FGF-1 also possesses two putative CK2 phosphorylation sites. In addition, both FGF-1 and FGF-2 have been shown previously to be weakly phosphorylated by CK2 (Feige and Baird, 1989). To examine the phosphorylation of both FGF-1 and FGF-2 and to explore whether the addition of either growth factor would have an effect on the autophosphorylation of CK2, we performed in vitro phosphorylation experiments. Both FGF-1 and FGF-2 were weakly phosphorylated by CK2 (Figure 9). More importantly, low concentrations of FGF-1 or FGF-2 were found to stimulate the autophosphorylation of CK2. This stimulatory effect was not observed with the non-mitogenic FGF-1(K132E) mutant.

Fig. 9. Phosphorylation of FGF-1 and FGF-2 by CK2 and the ability of the growth factors to stimulate autophosphorylation of CK2β. Purified rat liver CK2 was incubated for 20 min at 30°C either alone or with the indicated quantity of FGF-1 (lanes 2–4), FGF-2 (lanes 5–8) or FGF-1(K132E) (lane 9). The samples were then incubated for 2 h with either heparin–Sepharose (lanes 1–7 and 9) or anti-FGF-2 coupled to protein A–Sepharose (lane 8). The bound proteins were eluted with sample buffer, analysed by SDS–PAGE and exposed to a phosphoimager. It should be noted that CK2 binds to heparin–Sepharose independently of FGF.
Correlation between mitogenic activity and interaction of different FGF-1 mutants with CK2α
CK2 is known to have a prominent role in the early phases of mitogenic signal transduction and subsequent signal processing (Tawfic et al., 2001). We therefore tested the ability of CK2 to bind to a number of FGF-1 mutants with different mitogenic activities. Specific mutations in an exposed loop of FGF-1, which contains a phosphorylation site for PKC, led to reduced abilities of the mutants to stimulate DNA synthesis when added externally to cells (Klingenberg et al., 1999). We tested whether there was a correlation between mitogenicity and ability to bind to CK2. The mitogenicity of several of the mutants made up in a reticulocyte lysate has been assessed previously (Klingenberg et al., 1999). However, since we were working with MBP fusions made in bacteria, we wanted to test the mitogenicity of these proteins. The results (Figure 10A) were roughly similar to those of the in vitro translated proteins. Some differences could, however, be observed. Generally, the differences in the mitogenic potentials were smaller than observed previously. The FGF-1 mutants S130E, K132R and S113A all had roughly the same mitogenic activity as wild-type FGF-1 or FGF-2. The other mutants had a decreased (S130E, S130A/K132E or K132A) or essentially no (K132E) mitogenic potential.

Fig. 10. Correlation between mitogenic potential and binding to CK2α. (A) NIH 3T3 cells were serum starved for 24 h before 10 U/ml heparin and different amounts of FGF-1, mutants of FGF-1, FGF-2 or the corresponding amount of MBP as in the fusion protein constructs were added. The cells were incubated for another 24 h, the last 6 h in the presence of [3H]thymidine. The cell-associated, TCA-precipitable radioactivity was then measured. The data shown are from a representative experiment out of eight. (B) MBP–FGF-2 (lane 1), MBP–FGF-1 (lane 2) or MBP fused with the indicated FGF-1 mutants (lanes 3–10) were incubated for 2 h at 4°C with GST–CK2α bound to glutathione–Sepharose. The beads were washed and the bound proteins were analysed by western blotting with an antibody against MBP. Indicated above each lane is the mitogenicity of the corresponding mutant as a percentage of the mitogenicity of wild-type FGF-1, which is estimated from at least three independent DNA stimulation experiments. The amount of growth factor necessary to induce half-maximal stimulation of DNA synthesis was determined. The mitogenicity is taken as the reciprocal of the amount of growth factor needed and normalized to 100 for the mitogenic activity of wild-type FGF-1. (C) U2OS cells were lysed and incubated for 1 h at 4°C with Sepharose-bound MBP–FGF-1 or with the indicated mutant. The beads were washed, and adsorbed proteins were eluted with SDS sample buffer and separated by SDS–PAGE. The proteins were then transferred to a PVDF membrane and the membrane was probed with an anti-CK2β antibody. The mitogenicity was calculated as in (B). (D) NIH 3T3 cells were starved for 26 h and then 10 U/ml heparin and 5 ng/ml FGF-1, mutants of FGF-1, FGF-2 or MBP were added. The cells were stimulated for 10 min, washed, lysed with SDS sample buffer and analysed by western blotting with antibodies against phosphorylated p42/p44 MAP kinase (upper panel) and total p42/p44 MAP kinase (lower panel). The data shown are from a representative experiment.
A mutant FGF-2 with strongly reduced mitogenic potential (S117A) previously has been shown not to bind CK2 (Bailly et al., 2000). The FGF-1 mutant S113A corresponds to FGF-2(S117A). This mutant was constructed and tested for both its mitogenicity and its ability to interact with CK2. The mitogenicity and binding were similar to those observed with wild-type FGF-1.
A correlation between the ability of MBP–FGF-1 mutants to bind to GST–CK2α and their mitogenic activities could be observed (Figure 10B). In particular, the non-mitogenic K132E mutant showed no binding to CK2α.
A less clear correlation was found between the mitogenic potential of the different FGF-1 mutants and their ability to bind endogenous CK2 in a cell lysate (measured with an antibody against the β-subunit, Figure 10C). The only mutant that bound CK2 as strongly as wild-type FGF-1 was K132R. This mutant was found to have about two-thirds of the mitogenic activity of wild-type FGF-1. Also in this experiment, no binding to the non-mitogenic K132E mutant could be detected. By exposing the membrane for a longer time, one could also detect weak binding of the S130E mutant, which had mitogenic activity similar to wild-type FGF-1 (data not shown).
To test the possibility that the difference in the mitogenic effect of the different FGF-1 mutants was due to different abilities to activate FGF receptors rather than related to their intracellular activity, we tested their ability to induce phosphorylation of MAP kinase in cells. Activation of MAP kinase occurs after binding of growth factor to high affinity FGF-receptors and subsequent receptor autophosphorylation. We studied the phosphorylation of p42/p44 MAP kinase after stimulation with either 2, 5 (Figure 10D) or 20 ng/ml growth factor. In no case was there any difference between the amount of phosphorylated and therefore active MAP kinase. This shows that the proteins used are biologically active and they support the view that the observed difference in mitogenic potential is due to altered interactions with an intracellular component.
Discussion
In this report, we present evidence that both the α- and the β-subunits of CK2 bind to FGF-1. Furthermore, whereas CK2 exhibited good binding to wild-type FGF-1, there was almost no binding to the non-mitogenic mutant FGF-1(K132E). A correlation was also observed between the mitogenic activity of a number of FGF-1 mutants and their ability to bind to the α-subunit of CK2.
During the last decade, >160 different proteins have been reported to bind to the regulatory β-subunit of CK2. One of these is FGF-2 (Bonnet et al., 1996; Pinna and Meggio, 1997). Fewer proteins have been found to interact with the catalytic α-subunit, but they include bovine prion protein, protein phosphatase 2A and heterogenous nuclear ribonucleoprotein A2 (Heriche et al., 1997; Pancetti et al., 1999; Meggio et al., 2000). In addition, counting both in vitro and in vivo studies, >160 proteins have been found to be phosphorylated by CK2 (Pinna and Meggio, 1997).
We demonstrate using several techniques that FGF-1 binds to both the enzymatically active α-subunit and to the regulatory β-subunit of CK2. The binding to the α-subunit is clearly the strongest interaction in the case of both FGF-1 and the closely related FGF-2. It was reported earlier that FGF-2 binds only to the β-subunit of Drosophila melanogaster CK2 (Bonnet et al., 1996). Our experiments showing that both FGF-1 and FGF-2 bind to the α-subunit is in apparent disagreement with their findings (Bonnet et al., 1996). The use of human CK2 as opposed to CK2 from D.melanogaster cannot be the reason for the observed discrepancy, since we obtained the same results with CK2α and β from D.melanogaster (our unpublished data). The finding that FGF-1 and FGF-2 bind to both the α- and the β-subunit of CK2 is unusual, but not unique. Heterogenous nuclear ribonucleoprotein A2, a protein involved in pre-mRNA interaction, processing and protection, also binds to both subunits of CK2 (Pancetti et al., 1999).
CK2 binds to wild-type FGF-1 and to the K132R mutant with about the same affinity, whereas mutants in which this lysine had been exchanged for a neutral or a negatively charged residue showed little or no binding to CK2. Exchanging Ser130 for either alanine or glutamic acid leads to a loss of affinity for CK2. This indicates that the amino acids in this exposed loop are important for the binding to CK2. These two amino acids are also part of a phosphorylation site for PKC, although phosphorylation does not seem to play a role in the binding (our unpublished results).
A number of mutations in the same exposed loop of FGF-1 affect, to varying extents, the ability of the growth factor to stimulate DNA synthesis and to induce cell multiplication (Klingenberg et al., 1999). Here we find that while the correlation between mitogenic activity of the different FGF-1 mutants and binding to CK2 holoenzyme is not clear, there is in most cases a correlation between mitogenicity and binding to CK2α. A perfect correlation can, perhaps, not be expected as we are comparing interactions in a simple bimolecular system with the more complicated interactions in cells, which may involve several proteins.
It was suggested previously that altered interactions of FGF-1 with a cellular component could be the reason for the differences in mitogenic activity (Klingenberg et al., 1999). Our study suggests that CK2α could be the missing component. The results demonstrate that although the different mutants of FGF-1 exhibit different mitogenic potential, their ability to stimulate p42/p44 MAP kinase is similar. MAP kinase is a downstream target of FGF-1 after signalling through their high affinity receptors. The differences in mitogenic potential, but not the ability to signal through MAP kinase, indicates further that the differences between the described point mutants are in their intracellular action. Our findings are in agreement with observations made with the FGF-2(S117A) mutant (Bailly et al., 2000). This mutant, which supports differentiation, but has a reduced mitogenic potential, also does not interact with CK2.
FGF-1, but not the non-mitogenic mutant FGF-1(K132E), has been previously shown to bind to the cellular protein FIBP (Kolpakova et al., 1998). Together with the results obtained in this study, the hypothesis that the residues located around Lys132 are important for binding to other proteins is strengthened.
The equilibrium binding constant (KD) for the interaction between FGF-1 and CK2α was determined by surface plasmon resonance to be 0.4 µM, while the KD for the binding to CK2β was found to be 1.2 µM. While both these interactions are relatively weak, they are clearly in the biologically relevant range and, in the case of the α-subunit, can be compared with the KD of interaction between FGF-1 and FIBP (200 nM; our unpublished results) while the interaction with the β-subunit is similar in strength to the binding of Scr homology 2 (SH2) domains to phosphotyrosine residues (Ladbury et al., 1995). The amount of FGF-1 found in the cytoplasm and nucleus of the cell after incubation overnight with 5 ng/ml FGF-1 was estimated to be 70 000 molecules (Wiedlocha et al., 1995). This amount corresponds roughly to 0.1 µM, assuming a cell size of 10–12 l for NIH 3T3 mouse fibroblast. Recent experiments carried out in our laboratory have even indicated that the number may be 2–3 times higher (our unpublished results). These experiments clearly argue in favour of the determined equilibrium binding constants being in the biologically relevant range. Both binding constants are in the range expected for a transient association in a trafficking or an activation process.
The probability that CK2 and FGF-1 interact in vivo is strengthened further by the results obtained in the peroxisomal targeting assay. The finding that CK2α is transported to peroxisomes after binding to a peroxisomally targeted FGF-1 shows that not only do FGF-1 and CK2α interact in vivo, but the interaction is sufficiently strong for FGF-1 to redirect CK2α.
Protein kinase CK2, like FGF-1, is found both in the cytosol and in the nucleus, and it was reported recently that CK2 is widely distributed throughout the cell (Pinna and Meggio, 1997; Faust and Montenarh, 2000). It is unlikely that the interaction between FGF-1 and CK2 redistributes CK2 between the cytosol and nucleus. Thus, stimulation of U2OS cells with FGF-1, followed by fractionation, revealed no apparent translocation of protein from the cytosol to the nucleus (our unpublished results). FGF-1 could instead change the substrate specificity of CK2 and thereby induce phosphorylation of a different set of proteins.
The finding that both FGF-1 and FGF-2 have the ability to stimulate autophosphorylation of CK2β suggests that FGF-1 can change the accessibility of the autophosphorylation site in the N-terminal region of CK2 (Ser2 and Ser3), towards the CK2α catalytic region (Lin et al., 1994). The stimulation of autophosphorylation also seems to depend upon a physical interaction between the proteins. Thus, the mutant FGF-1(K132E), which has a strongly reduced capacity to interact with CK2 and to stimulate DNA synthesis, was not able to stimulate autophosphorylation of CK2. Autophosphorylation of CK2β has been shown to correlate with a modest decrease in activity (Lin et al., 1994). In fact, when we phosphorylated p53 in vitro with CK2 in the presence of increasing concentrations of FGF-1, we observed a decrease in phos phorylation of p53 that correlated with increased autophosphorylation of CK2β (our unpublished results). The decrease in stimulation of autophosphorylation observed with increasing concentrations of FGF-1 or FGF-2 could be a consequence of competition for the catalytic α-subunit. Whether or not the interaction of FGF-1 with CK2, and/or autophosphorylation of CK2β, is a necessary step in the signalling from FGF-1 towards stimulation of DNA synthesis still remains to be elucidated.
The results provided in this work demonstrate that FGF-1 interacts with both subunits of CK2 and that there is a correlation between binding of a series of different FGF-1 mutants to CK2α and mitogenic potential of the growth factor when added externally to cells. This might implicate CK2 in the FGF-1-induced stimulation of DNA synthesis.
Materials and methods
Cells and transfections
U2OS cells were propagated in Dulbecco’s modified Eagle’s medium (DMEM) with 10% (v/v) fetal calf serum (FCS) in a 5% CO2 atmosphere at 37°C. Transient expression of green fluorescent protein (GFP)–FGF-1, myc-FGF-1, CK2α-HA and CK2β was achieved by transiently transfecting U2OS cells with the appropriate plasmids (pEGFP-FGF-1, pcDNA3-myc-FGF-1, pRc/CMV-HA-CK2α and pcDNA3-CK2β) using the Fugene-6 (Boehringer Mannheim) transfection agent according to the manufacturer’s description. Cells were used for experiments 20–24 h after transfection.
Plasmids
The MBP fusions of FGF-2, FGF-1 and the FGF-1 mutants have been described previously (Klingenberg et al., 1999), except for the MBP–FGF-1(S113A) mutant, which was made using PCR-directed mutagenesis with the plasmid MBP-FGF-1 as template. pEGFP-FGF-1 was made by inserting the cDNA for FGF-1 into the multiple cloning site of the vector pEGFP (Clontech). pcDNA3-myc-FGF-1 was constructed by inserting the cDNA for FGF-1 into the multiple cloning site of the vector pcDNA3 (Invitrogen), and the myc tag was added to the N-terminus of FGF-1 by PCR. pcDNA3-myc-FGF-1pts was then constructed by adding the three amino acids SGG and then the type 1 peroxisomal targeting signal (PTS1) SKL to the C-terminus of FGF-1 in pcDNA3-myc-FGF-1 using PCR. CK2α and CK2α′ in the plasmids pRc/CMV-CK2α-HA and pRc/CMV-HA-CK2α′, respectively, were gifts from Dr Litchfield, Manitoba Institute of Cell Biology, Winnipeg, Canada (Penner et al., 1997). CK2α was subcloned from the pRc/CMV-HA- CK2α vector into the ApaI site of pGEX-5X-3. This construct was named pGEX-CK2α. CK2α was also subcloned into the EcoRI–SalI site of pECFP to make pECFP-CK2α using PCR. CK2β was subcloned from the expression vector pCMVES-CK2β (a gift from Dr C.Götz, University of the Saarland, Germany) (Gotz et al., 2000), into the BamHI–SalI sites of pGEX-5X-3 or pECFP using PCR.
Expression and purification of MBP and GST fusion proteins and recombinant FGF-1, and preparation of Sepharose beads containing MBP fusion proteins
Expression and purification of MBP and GST fusion proteins and recombinant FGF-1 was carried out as described previously. The preparation of Sepharose beads containing MBP fusion proteins has been described by Skjerpen et al. (2002).
Affinity adsorption and purification of proteins binding to FGF-1
Subconfluent U2OS cells were labelled overnight with [35S]methionine/cysteine, washed with phosphate-buffered saline (PBS) and lysed on ice for 20 min in lysis buffer (100 mM NaCl, 10 mM Na2HPO4 pH 7.2, 1% Triton X-100, 1 mM EDTA) with 10 mM dithiothreitol (DTT) and complete protease inhibitor cocktail. The cells were collected with a cell scraper and centrifuged at 3020 g for 10 min at 4°C. The supernatant was diluted 1:1 with PBS and incubated for 2 h at 4°C with Sepharose without additional bound protein. The precipitation mixture was centrifuged at 3020 g for 5 min at 4°C and the supernatant was incubated for another 2 h at 4°C with Sepharose-bound MBP–IFN-γ (control). After another centrifugation, the supernatant was incubated with Sepharose-bound MBP–FGF-1 for 2.5 h at 4°C. The beads were then washed four times with 1:1 lysis buffer:PBS, and the bound proteins were eluted with 2 M NaCl in PBS on ice for 15 min. Proteins were precipitated with 5% trichloroacetic acid (TCA) on ice for 1 h and the pellet was extracted three times with ether. The proteins were analysed by SDS–PAGE [12% (w/v)] followed by staining with Coomassie Blue-G, and the dried gel was subjected to autoradiography. Defined bands were excised from the gel and subjected to in-gel trypsin treatment followed by either MALDI-TOF-MS alone or MALDI-TOF-MS and internal sequencing. The protein sequence data were obtained at the Rockefeller University Protein/DNA Technology Center (New York) (Fernandez et al., 1994, 1998).
In vitro transcription and translation
[35S]methionine-labelled HA-CK2α, HA-CK2α′ and CK2β were produced in a rabbit reticulocyte lysate system (Promega). Plasmid DNA (pRc/CMV or pcDNA3 with the appropriate inserts) was linearized downstream of the coding sequence, and transcription and translation were performed as described previously (Skjerpen et al., 2002).
Co-precipitation, western blotting and in vitro binding assay with radioactively labelled proteins
Co-precipitation of proteins from U2OS cells and subsequent western blotting, or co-precipitation with radiolabelled in vitro translated proteins were performed as described by Skjerpen et al. (2002). The antibodies used for western blotting were rabbit anti-CK2α (Upstate Biotechnology) and mouse anti-CK2β (Transduction Laboratories).
GST pull-down assay
A 2 µg aliquot of GST fusion protein was bound to 20 µl of glutathione–Sepharose for 2 h at 4°C, washed and incubated in a 1:1 mixture of PBS and lysis buffer for 2 h with 1 µg of MBP fusion protein at 4°C. The samples were washed, eluted with sample buffer and analysed by SDS–PAGE and western blotting with a rabbit anti-MBP primary antibody (NEB).
Surface plasmon resonance
The KD for the binding between FGF-1 and CK2α or CK2β was determined using a BIAcore X (BIAcore AB, Uppsala, Sweden) at 25°C. CK2α and β were expressed and purified as GST fusion proteins and coupled to a carboxymethyl (CM) 5 sensorchip (BIAcore) using the GST Kit for fusion capture (BIAcore). The assay was performed as described by Skjerpen et al. (2002).
Peroxisomal targeting of the FGF-1–CK2α complex by immunofluorescence microscopy
HeLa cells were seeded on sterile coverslips and co-transfected with either pcDNA3-myc-FGF-1pts and pECFP-CK2α or pcDNA3-myc- FGF-1 and pECFP-CK2α. At 24 h post-transfection, the cells were washed in PBS and permeabilized for 10 min with 40 µg/ml digitonin in PBS. The cells were washed twice in PBS to deplete the cell of cytosolic material and fixed for 60 min with 3% paraformaldehyde in PBS. Autofluorescence was quenched by incubating for 10 min in 50 mM NH4Cl in PBS and the peroxisomal membrane was permeabilized by incubating for 5 min with 0.1% Triton X-100 in PBS. Subsequently, the cells were washed in PBS and blocked for 20 min with 5% FCS in PBS. The cells were then incubated with primary (anti-c-Myc and anti-catalase) and secondary (anti-mouse rhodamine–lissamine and anti-rabbit Cy5) antibodies before they were mounted in Mowiol. Immunofluores cence images were taken using a Leica (Wezlar, Germany) confocal microscope and they were processed using Adobe Photoshop 5.0 (Adobe, Mountain View, CA).
Phosphorylation assay
The assay was carried out in a phosphorylation buffer consisting of 10 mM MgCl2, 50 mM MOPS pH 7.0, 150 mM NaCl and 20 µCi/ml [γ-33P]ATP. FGF-1, FGF-2 or FGF-1(K132E) (0–1.0 µg) in phosphorylation buffer without [γ-33P]ATP was added and the reaction was started by adding 0.03 U of purified rat liver CK2. The reactions were carried out in a total volume of 15 µl. The mixtures were incubated for 20 min at 30°C. The reaction was terminated by placing the samples on ice and adding heparin–Sepharose or anti-FGF-2 antibody (Santa Cruz Biotechnology). The samples were incubated for 2 h at 4°C and the proteins were eluted by addition of sample buffer. All the samples were analysed by SDS–PAGE followed by exposure of the gel to a phosphorimager.
Measurements of DNA synthesis and MAP kinase activation
The measurement of 3H incorporation in response to stimulation with growth factor was performed as described by Klingenberg et al. (1998). To test for MAP kinase activation, NIH 3T3 cells were starved in DMEM with 0.5% FCS for 24 h. The medium was replaced with fresh DMEM/0.5% FCS and the cells grown for 2 h. Heparin (10 U/ml) was added and the cells were stimulated for 10 min with 2, 5 or 20 ng/ml growth factor. After washing with PBS, the cells were lysed with SDS sample buffer, sonicated (5 s) and analysed by western blotting. The antibodies used were mouse anti-phospho-MAP kinase (P42/p44) (NEB) and rabbit anti-MAP kinase (p42/p44) (Cell Signaling Technology).
Acknowledgments
Acknowledgements
The expert help of Dr David J.Gillooly with the BIAcore measurements and with helpful comments on the manuscript, and the skilful work with the cell cultures by Mette Sværen is gratefully acknowledged. C.S.S. and J.W. are fellows of Norwegian Cancer Society. This work was supported by the Novo Nordisk Foundation, The Norwegian Research Council for Science and Humanities, Blix Legat, Rachel and Otto Kr Bruun’s Legat and by the Jahre Foundation.
References
- Allende J.E. and Allende,C.C. (1995) Protein kinases. 4. Protein kinase CK2: an enzyme with multiple substrates and a puzzling regulation. FASEB J., 9, 313–323. [DOI] [PubMed] [Google Scholar]
- Allende C.C. and Allende,J.E. (1998) Promiscuous subunit interactions: a possible mechanism for the regulation of protein kinase CK2. J. Cell. Biochem. Suppl., 30–31, 129–136. [PubMed] [Google Scholar]
- Amalric F., Bouche,G., Bonnet,H., Brethenou,P., Roman,A.M., Truchet,I. and Quarto,N. (1994) Fibroblast growth factor-2 (FGF-2) in the nucleus: translocation process and targets. Biochem. Pharmacol., 47, 111–115. [DOI] [PubMed] [Google Scholar]
- Bailly K., Soulet,F., Leroy,D., Amalric,F. and Bouche,G. (2000) Uncoupling of cell proliferation and differentiation activities of basic fibroblast growth factor. FASEB J., 14, 333–344. [PubMed] [Google Scholar]
- Basilico C. and Moscatelli,D. (1992) The FGF family of growth factors and oncogenes. Adv. Cancer Res., 59, 115–165. [DOI] [PubMed] [Google Scholar]
- Bonnet H., Filhol,O., Truchet,I., Brethenou,P., Cochet,C., Amalric,F. and Bouche,G. (1996) Fibroblast growth factor-2 binds to the regulatory β subunit of CK2 and directly stimulates CK2 activity toward nucleolin. J. Biol. Chem., 271, 24781–24787. [DOI] [PubMed] [Google Scholar]
- Bouche G., Gas,N., Prats,H., Baldin,V., Tauber,J.P., Teissie,J. and Amalric,F. (1987) Basic fibroblast growth factor enters the nucleolus and stimulates the transcription of ribosomal genes in ABAE cells undergoing G0–G1 transition. Proc. Natl Acad. Sci. USA, 84, 6770–6774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess W.H. and Maciag,T. (1989) The heparin-binding (fibroblast) growth factor family of proteins. Annu. Rev. Biochem., 58, 575–606. [DOI] [PubMed] [Google Scholar]
- Burgess W.H., Shaheen,A.M., Ravera,M., Jaye,M., Donohue,P.J. and Winkles,J.A. (1990) Possible dissociation of the heparin-binding and mitogenic activities of heparin-binding (acidic fibroblast) growth factor-1 from its receptor-binding activities by site-directed mutagenesis of a single lysine residue. J. Cell Biol., 111, 2129–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess W.H., Shaheen,A.M., Hampton,B., Donohue,P.J. and Winkles,J.A. (1991) Structure–function studies of heparin-binding (acidic fibroblast) growth factor-1 using site-directed mutagenesis. J. Cell. Biochem., 45, 131–138. [DOI] [PubMed] [Google Scholar]
- Carreira C.M., LaVallee,T.M., Tarantini,F., Jackson,A., Lathrop,J.T., Hampton,B., Burgess,W.H. and Maciag,T. (1998) S100A13 is involved in the regulation of fibroblast growth factor-1 and p40 synoptagmin-1 release in vitro. J. Biol. Chem., 273, 22224–22231. [DOI] [PubMed] [Google Scholar]
- Faust M. and Montenarh,M. (2000) Subcellular localization of protein kinase CK2. A key to its function? Cell Tissue Res., 301, 329–340. [DOI] [PubMed] [Google Scholar]
- Feige J.J. and Baird,A. (1989) Basic fibroblast growth factor is a substrate for protein phosphorylation and is phosphorylated by capillary endothelial cells in culture. Proc. Natl Acad. Sci. USA, 86, 3174–3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez J., Andrews,L. and Mische,S.M. (1994) An improved procedure for enzymatic digestion of polyvinylidene difluoride-bound proteins for internal sequence analysis. Anal. Biochem., 218, 112–117. [DOI] [PubMed] [Google Scholar]
- Fernandez J., Gharahdaghi,F. and Mische,S.M. (1998) Routine identification of proteins from sodium dodecyl sulfate– polyacrylamide gel electrophoresis (SDS–PAGE) gels or polyvinyl difluoride membranes using matrix assisted laser desorption/ionization-time of flight-mass spectrometry (MALDI-TOF-MS). Electrophoresis, 19, 1036–1045. [DOI] [PubMed] [Google Scholar]
- Gotz C., Kartarius,S., Scholtes,P. and Montenarh,M. (2000) Binding domain for p21(WAF1) on the polypeptide chain of the protein kinase CK2 β-subunit. Biochem. Biophys. Res. Commun., 268, 882–885. [DOI] [PubMed] [Google Scholar]
- Guerra B., Boldyreff,B., Sarno,S., Cesaro,L., Issinger,O.G. and Pinna,L.A. (1999) CK2: a protein kinase in need of control. Pharmacol. Ther., 82, 303–313. [DOI] [PubMed] [Google Scholar]
- Heller-Harrison R.A. and Czech,M.P. (1991) Enhanced casein kinase II activity in COS-1 cells upon overexpression of either its catalytic or noncatalytic subunit. J. Biol. Chem., 266, 14435–14439. [PubMed] [Google Scholar]
- Heriche J.K., Lebrin,F., Rabilloud,T., Leroy,D., Chambaz,E.M. and Goldberg,Y. (1997) Regulation of protein phosphatase 2A by direct interaction with casein kinase 2α. Science, 276, 952–955. [DOI] [PubMed] [Google Scholar]
- Imamura T. et al. (1990) Recovery of mitogenic activity of a growth factor mutant with a nuclear translocation sequence. Science, 249, 1567–1570. [DOI] [PubMed] [Google Scholar]
- Jackson A., Friedman,S., Zhan,X., Engleka,K.A., Forough,R. and Maciag,T. (1992) Heat shock induces the release of fibroblast growth factor 1 from NIH 3T3 cells. Proc. Natl Acad. Sci. USA, 89, 10691–10695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg O., Widlocha,A., Rapak,A., Munoz,R., Falnes,P. and Olsnes,S. (1998) Inability of the acidic fibroblast growth factor mutant K132E to stimulate DNA synthesis after translocation into cells. J. Biol. Chem., 273, 11164–11172. [DOI] [PubMed] [Google Scholar]
- Klingenberg O., Wiedlocha,A. and Olsnes,S. (1999) Effects of mutations of a phosphorylation site in an exposed loop in acidic fibroblast growth factor. J. Biol. Chem., 274, 18081–18086. [DOI] [PubMed] [Google Scholar]
- Kolpakova E., Wiedlocha,A., Stenmark,H., Klingenberg,O., Falnes,P.O. and Olsnes,S. (1998) Cloning of an intracellular protein that binds selectively to mitogenic acidic fibroblast growth factor. Biochem. J., 336, 213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladbury J.E., Lemmon,M.A., Zhou,M., Green,J., Botfield,M.C. and Schlessinger,J. (1995) Measurement of the binding of tyrosyl phosphopeptides to SH2 domains: a reappraisal. Proc. Natl Acad. Sci. USA, 92, 3199–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander E.S. et al. (2001) Initial sequencing and analysis of the human genome. Nature, 409, 860–921. [DOI] [PubMed] [Google Scholar]
- Lebrin F., Chambaz,E.M. and Bianchini,L. (2001) A role for protein kinase CK2 in cell proliferation: evidence using a kinase-inactive mutant of CK2 catalytic subunit α. Oncogene, 20, 2010–2022. [DOI] [PubMed] [Google Scholar]
- Lin W.J., Sheu,G.T. and Traugh,J.A. (1994) Effects of autophos phorylation on casein kinase II activity: evidence from mutations in the β subunit. Biochemistry, 33, 6998–7004. [DOI] [PubMed] [Google Scholar]
- Mason I.J. (1994) The ins and outs of fibroblast growth factors. Cell, 78, 547–552. [DOI] [PubMed] [Google Scholar]
- McNew J.A. and Goodman,J.M. (1994) An oligomeric protein is imported into peroxisomes in vivo. J. Cell Biol., 127, 1245–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meggio F., Negro,A., Sarno,S., Ruzzene,M., Bertoli,A., Sorgato,M.C. and Pinna,L.A. (2000) Bovine prion protein as a modulator of protein kinase CK2. Biochem. J., 352, 191–196. [PMC free article] [PubMed] [Google Scholar]
- Mizukoshi E., Suzuki,M., Loupatov,A., Uruno,T., Hayashi,H., Misono,T., Kaul,S.C., Wadhwa,R. and Imamura,T. (1999) Fibroblast growth factor-1 interacts with the glucose-regulated protein GRP75/mortalin. Biochem. J., 343, 461–466. [PMC free article] [PubMed] [Google Scholar]
- Opalenik S.R., Shin,J.T., Wehby,J.N., Mahesh,V.K. and Thompson,J.A. (1995) The HIV-1 TAT protein induces the expression and extracellular appearance of acidic fibroblast growth factor. J. Biol. Chem., 270, 17457–17467. [DOI] [PubMed] [Google Scholar]
- Pancetti F., Bosser,R., Krehan,A., Pyerin,W., Itarte,E. and Bachs,O. (1999) Heterogeneous nuclear ribonucleoprotein A2 interacts with protein kinase CK2. Biochem. Biophys. Res. Commun., 260, 17–22. [DOI] [PubMed] [Google Scholar]
- Penner C.G., Wang,Z. and Litchfield,D.W. (1997) Expression and localization of epitope-tagged protein kinase CK2. J. Cell. Biochem., 64, 525–537. [PubMed] [Google Scholar]
- Pinna L.A. and Meggio,F. (1997) Protein kinase CK2 (‘casein kinase-2’) and its implication in cell division and proliferation. Prog. Cell Cycle Res., 3, 77–97. [DOI] [PubMed] [Google Scholar]
- Pyerin W. (1994) Human casein kinase II: structures, genes, expression and requirement in cell growth stimulation. Adv. Enzyme Regul., 34, 225–246. [DOI] [PubMed] [Google Scholar]
- Shin J.T., Opalenik,S.R., Wehby,J.N., Mahesh,V.K., Jackson,A., Tarantini,F., Maciag,T. and Thompson,J.A. (1996) Serum-starvation induces the extracellular appearance of FGF-1. Biochim. Biophys. Acta, 1312, 27–38. [DOI] [PubMed] [Google Scholar]
- Skjerpen C.S., Wesche,J. and Olsnes,S. (2002) Identification of ribosome-binding protein p34 as an intracellular protein that binds acidic fibroblast growth factor. J. Biol. Chem., 277, 23864–23871. [DOI] [PubMed] [Google Scholar]
- Spivak-Kroizman T. et al. (1994) Heparin induced oligomerization of FGF molecules is responsible for FGF receptor dimerization, activation and cell proliferation. Cell, 79, 1015–1024. [DOI] [PubMed] [Google Scholar]
- Szebenyi G. and Fallon,J.F. (1999) Fibroblast growth factors as multifunctional signaling factors. Int. Rev. Cytol., 185, 45–106. [DOI] [PubMed] [Google Scholar]
- Tarantini F., LaVallee,T., Jackson,A., Gamble,S., Carreira,C.M., Garfinkel,S., Burgess,W.H. and Maciag,T. (1998) The extravesicular domain of synoptagmin-1 is released with the latent fibroblast growth factor-1 homodimer in response to heat shock. J. Biol. Chem., 273, 22209–22216. [DOI] [PubMed] [Google Scholar]
- Tawfic S., Yu,S., Wang,H., Faust,R., Davis,A. and Ahmed,K. (2001) Protein kinase CK2 signal in neoplasia. Histol. Histopathol., 16, 573–582. [DOI] [PubMed] [Google Scholar]
- Titorenko V.I., Nicaud,J.M., Wang,H., Chan,H. and Rachubinski,R.A. (2002) Acyl-CoA oxidase is imported as a heteropentameric, cofactor-containing complex into peroxisomes of Yarrowia lipolytica. J. Cell Biol., 156, 481–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton P.A., Hill,P.E. and Subramani,S. (1995) Import of stably folded proteins into peroxisomes. Mol. Biol. Cell, 6, 675–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedlocha A., Falnes,P.Ø., Madshus,I.H., Sandvig,K. and Olsnes,S. (1994) Dual mode of signal transduction by externally added acidic fibroblast growth factor. Cell, 76, 1039–1051. [DOI] [PubMed] [Google Scholar]
- Wiedlocha A., Falnes,P.O., Rapak,A., Klingenberg,O., Munoz,R. and Olsnes,S. (1995) Translocation of cytosol of exogenous, CAAX-tagged acidic fibroblast growth factor. J. Biol. Chem., 270, 30680–30685. [DOI] [PubMed] [Google Scholar]
- Wiedlocha A., Falnes,P.O., Rapak,A., Munoz,R., Klingenberg,O. and Olsnes,S. (1996) Stimulation of proliferation of a human osteosarcoma cell line by exogenous acidic fibroblast growth factor requires both activation of receptor tyrosine kinase and growth factor internalization. Mol. Cell. Biol., 16, 270–280. [DOI] [PMC free article] [PubMed] [Google Scholar]