

Abstract
The atypical protein kinase C isoform, ζPKC, has been implicated in the control of extracellular signal-regulated kinase (ERK) and nuclear factor (NF)-κB pathways. Recent evidence from ζPKC knock-out mice demonstrates that this kinase is important for NF-κB transcriptional activity but not for ERK activation in embryonic fibroblasts. The lack of ζPKC produces in mice a number of alterations in the development of secondary lymphoid tissues that could be accounted for, at least in part, by defects in B-cell function. Here, we present evidence that the loss of ζPKC selectively impairs signaling through the B-cell receptor, resulting in inhibition of cell proliferation and survival, as well as defects in the activation of ERK and the transcription of NF-κB-dependent genes. Furthermore, ζPKC–/– mice are unable to mount an optimal T-cell-dependent immune response. Collectively, these results genetically establish a critical role for ζPKC in B-cell function in vitro and in vivo.
Keywords: B-cell signaling/protein kinase C/ζ isoform
Introduction
Nuclear factor (NF)-κB is a transcription factor formed by complexes of different family members: p65 (RelA), c-Rel, RelB, NF-κB1 (p50) and NF-κB2 (p52) (Thanos and Maniatis, 1995; Verma et al., 1995; Baldwin, 1996). Several findings demonstrate that they play critical roles in a number of biological functions, including innate and adaptative immune responses in mammals and Drosophila (Israel, 2000; Karin and Ben-Neriah, 2000; Silverman and Maniatis, 2001). Of particular interest are the recent results showing that NF-κB is important in the development of secondary lymphoid tissues (Fu and Chaplin, 1999). Thus, studies with knock-out (KO) mice demonstrate that p52 is required for the development of a normal splenic microarchitecture as well as B-cell responses (Caamano et al., 1998). In particular, p52–/– mice show impaired formation of follicular dendritic cells (FDCs), germinal centers and the marginal zone (Caamano et al., 1998). In addition, p50–/– mice show that NF-κB is required for the development of marginal zone B lymphocytes (Cariappa et al., 2000). In keeping with this, the genetic inactivation of receptors and cytokines that stimulate the NF-κB pathways leads to alterations in secondary lymphoid organs. Thus, for example, KO mice for RANK and RANKL demonstrate that these molecules seem important for development of lymph nodes (LNs) and Peyer’s patches (PPs) (Dougall et al., 1999; Kim et al., 2000). Signaling through lymphotoxin β receptor (LT-βR) is also required for development of LNs and PPs, as well as for a normal splenic microarchitecture (Koni et al., 1997; Futterer et al., 1998; Rennert et al., 1998), whereas tumor necrosis factor-α receptor-1 (TNFR1)–/– mice show intact spleen and LNs but are unable to form PPs properly (Neumann et al., 1996; Pasparakis et al., 1997; Futterer et al., 1998; Rennert et al., 1998). Interestingly, double TNFR1/RelA KO mice display more profound defects that include the lack of LNs, PPs and organized spleen microarchitecture (Alcamo et al., 2002).
We have recently characterized KO mice for the ζ isoform of protein kinase C and have found that the loss of this gene produces significant alterations in the development of secondary lymphoid organs (Leitges et al., 2001). These alterations were detected in very young (2-week-old) mice and were much less apparent in older animals. Thus, in the ζPKC–/– mice, although the overall structure of the spleen was preserved, there was a defect in the marginal zone together with smaller B-cell follicles in the white pulp as compared with age-matched wild-type controls (Leitges et al., 2001). In addition, defects were observed in peripheral and mesenteric LNs as well as in the PPs in which there was an impaired segregation between B- and T-cell zones and a decrease in FDCs (Leitges et al., 2001). These observations would be consistent, at least in part, with the proposed role of ζPKC in NF-κB signaling (Moscat and Diaz-Meco, 2000). In fact, embryonic fibroblasts (EFs) from ζPKC–/– mice display a severe impairment in the activation of NF-κB transcriptional activity (Leitges et al., 2001). In older (4- to 8-week-old) KO mice, the defects in the LNs nearly disappear and those in the PP, although still detectable, were much less dramatic, indicating that the loss of ζPKC causes a delay but not a complete blockade in the delivery of signals required for the proper development of these lymphoid organs (Leitges et al., 2001).
The development of lymphoid organs is controlled by a dynamic interplay between hematopoietic and non- hematopoietic cells. In this regard, young ζPKC–/– mice show a reduced relative percentage of B cells in peripheral and mesenteric LNs which was significantly enriched in an immature B220lowIgMhigh population. In PPs, although the relative percentage of T and B cells was normal in the ζPKC KO mice, there was a reproducible increase in immature B cells (Leitges et al., 2001). Of potential interest, an essential role in B-cell maturation and survival has been reported for RelA and c-Rel (Gerondakis et al., 1999; Grossmann et al., 2000; Gugasyan et al., 2000). Therefore, it is possible that the defects detected in B lymphocytes in the ζPKC–/– mice could be due to alterations in NF-κB signaling and cell survival of this hematopoietic cell type independently of the potential changes in the stroma of the lymphoid organs.
In this study, we have addressed in detail whether there is a defect in the signaling cascades that regulate the activation in vitro of splenic B cells from adult 4- to 6-week-old ζPKC–/– mice. We show here that the mitogenic activation and survival of isolated purified cultures of splenic B cells are severely impaired by the lack of ζPKC. These defects are not likely to be due to indirect stromal alterations or the maturation stage of the B cells and could potentially explain the deficiencies detected in the development of secondary lymphoid organs reported previously, and the evidence shown here that the ζPKC–/– mice are unable to mount an optimal immune response in vivo.
Results
Previous results from RelA and c-Rel KO mice demonstrate that B cells from these animals showed accelerated apoptosis in culture (Grossmann et al., 2000). To determine whether a similar defect could be observed in B cells from ζPKC–/– mice, we measured spontaneous apoptosis, by flow cytometric analysis, in cultures of splenic B lymphocytes from ζPKC-deficient and wild-type mice. Interestingly, the lack of ζPKC severely accelerates apoptosis in B cells (Figure 1A). Thus, whereas the percentage of apoptotic cells at 20 h is <10% in wild type, in KO B cells it is >20%. At 40 h of culture, the differences are even more dramatic (Figure 1A). Therefore, ζPKC seems to be required for B cells to survive efficiently in vitro. This defect is most probably intrinsic to the population of B cells and not due to potential indirect stromal alterations, since it is observed in in vitro cultures of purified B cells, and immunohistochemical analysis of spleens from ζPKC–/– mice reveals that the organ microarchitecture even in younger (2-week-old) ζPKC-deficient mice is not affected. Thus, staining for MAdCAM-1 on sinus-lining cells (Figure 2A, a and b) and MOMA-1 on metallophilic macrophages (Figure 2A, c and d) demonstrate intact marginal zone populations in the ζPKC–/– mice. Also, Figure 2A (e and f) shows normal CD11b and CD11c staining in the spleen of the ζPKC-deficient mice, indicating intact dendritic cell populations in the B-cell zone. Therefore, it seems that the deficiency detected in the in vitro experiments (Figure 1A) in the ability of isolated splenic B cells to survive seems to be an intrinsic alteration of the B-cell signaling properties and it is unlikely that it could be accounted for by indirect stromal defects.

Fig. 1. Impaired survival and proliferation of B cells from ζPKC–/– mice. B cells from either wild-type (empty bars) or ζPKC-deficient (black bars) mice were incubated for different times in culture medium, after which the percentage of apoptotic cells was determined by flow cytometry analysis (A). In other experiments, cells were stimulated for 72 h with either an anti-CD40 antibody or different concentrations of IgM (B), and the amount of [3H]thymidine incorporated was determined as described in Materials and methods. In a parallel experiment (C), cells were stimulated with 2 µg/ml IgM in the absence or presence of different concentrations of BAFF. This is a representative experiment of at least another two with incubations in duplicate.
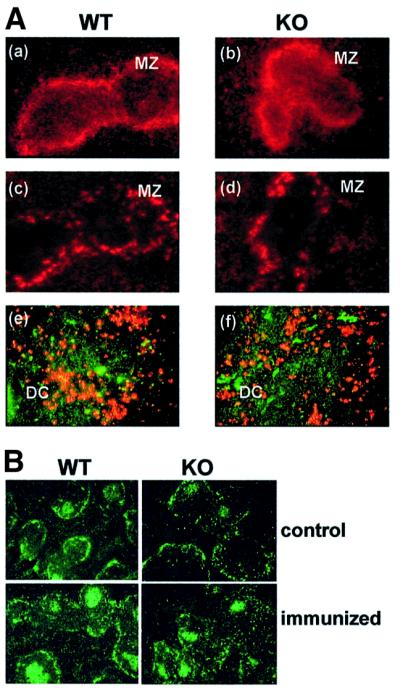
Fig. 2. Immunofluorescent analysis of the splenic architecture in ζPKC–/– mice. (A) Sections of spleen from wild-type mice (a, c and e) and ζPKC–/– mice (b, d and f) were analyzed by immunofluorescent staining for MAdCAM-1 on marginal sinus-lining cells (a and b), MOMA-1 on marginal zone metallophilic macrophages (c and d) and CD11b/CD11c on T-cell zone dendritic cells (e and f). MZ, marginal zone; DC, dendritic cells. (B) Mice (either wild type or KO) were either not immunized or immunized intraperitoneally with 100 µg of DNP–OVA. Eleven days after the immunization, frozen splenic sections were stained with PNA.
It should be noted that the splenic B-cell populations used in this study were from 4- to 6-week-old ζPKC–/– mice which have a composition similar to that of age-matched wild-type animals. Thus, three-color flow cytometric analysis of splenocytes (Figure 3A; Table I) demonstrates that the percentages of mature (CD21int IgMlowCD23+), marginal zone (CD21hiIgMhiCD23–), T1 (CD21lowIgMhiCD23–) and T2 (CD21hiIgMhiCD23+) B-cell subpopulations were not significantly different between the wild-type and the ζPKC-deficient mice. The same results were obtained when this analysis was performed in purified B cells (Figure 3B; Table I). It must be stressed that the B-cell purification procedure involves indirect magnetic labeling using a cocktail of antibodies against CD43, CD4 and Ter-119 which remove most of the T lymphocytes as well as natural killer (NK) and dendritic cells, macrophages, granulocytes and erythroid cells. This is consistent with our previous observations demonstrating that there were only minor alterations in the splenic B-cell populations in the 2-week-old ζPKC–/– mice and that these alterations were undetectable in older animals (Leitges et al., 2001). Therefore, the higher apoptosis observed in splenic B cells from ζPKC KO mice (Figure 1A) cannot be explained by differences in the percentage of B-cell subpopulations in KO in vitro cultures as compared with the wild type.
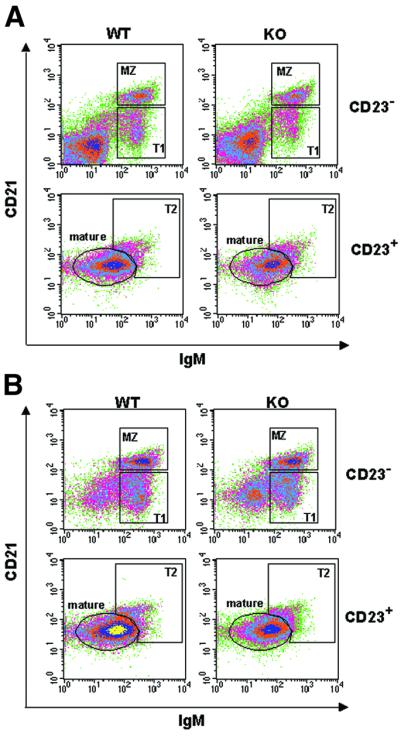
Fig. 3. Analysis of B-cell subpopulations. Three-color flow cytometric analyses of splenocytes (A) or purified splenic B cells (B) were carried by staining cells with antibodies to IgM, CD21 and CD23. Cells were separated into CD23-positive and -negative. MZ (CD21hiIgMhiCD23–), marginal zone B cells; mature B cells characterized by (CD21intIgMlow CD23+); T1 (CD21l°wIgMhiCD23–), transitional 1 immature B cells; T2 (CD21hiIgMhiCD23+), transitional 2 immature B cells. This is a representative experiment of another two with similar results.
Table I. Analysis of B-cell subpopulations.
| Splenocytes | Purified B cells | |||
|---|---|---|---|---|
| |
Wild type |
KO |
Wild type |
KO |
| Mature | 25 | 22 | 44 | 43 |
| MZ | 6 | 6 | 12 | 12 |
| T1 | 6 | 5 | 12 | 11 |
| T2 | 3 | 3 | 6 | 6 |
MZ (CD21hiIgMhiCD23–), marginal zone B cells; mature B cells characterized by (CD21intIgMlowCD23+); T1 (CD21lowIgMhiCD23–), transitional 1 immature B cells; T2 (CD21hiIgMhiCD23+), transitional 2 immature B cells. Numbers indicate the relative percentage of total cells. This is a representative experiment of another two with similar results.
In the next series of experiments, we determined whether the lack of ζPKC would impair the mitogenic activation of B cells in response to different stimuli. Thus, we measured the amount of thymidine incorporation in B cells from ζPKC–/– and wild-type mice in response to different concentrations of IgM to activate the B-cell receptor (BCR). The results in Figure 1B show that addition of different concentrations of IgM provokes a robust increase in thymidine incorporation in the wild-type B-cell cultures. This response is significantly inhibited in the ζPKC KO B cells (Figure 1B). When these cell cultures were challenged with lipopolysaccharide (LPS; data not shown) or an agonistic anti-CD40 antibody (Figure 1B), no differences were observed between wild-type and KO cells. Therefore, it seems that the loss of ζPKC significantly impairs the mitogenic activation of B cells through the BCR. Recently, BAFF (B-cell activating factor belonging to the TNF family), also known as Blys, has been shown to play an essential role in B-cell proliferation in vivo and in vitro as a co-stimulant of BCR signaling (Thompson et al., 2001). The data in Figure 1C demonstrate that the addition of BAFF cannot overcome the defect in BCR-induced proliferation observed in the ζPKC–/– cells. Flow cytometric analysis of B cells from wild-type and ζPKC–/– mice stimulated with different concentrations of IgM demonstrates that the KO cells display an impaired response not only to cell cycle entry (Figure 4A) but also to the inhibition of apoptosis (Figure 4B). Recent evidence has shown a critical role for the novel PKC, θPKC, in the proliferation of T lymphocytes (Sun et al., 2000). Interestingly, T cells from ζPKC KO mice did not have any defect in proliferation when challenged with different concentrations of anti-CD3 antibody in either the absence or presence of CD28 stimulation (Figure 5).

Fig. 4. Impaired cell cycle entry and survival of ζPKC–/– B cells. B cells from either wild-type (empty bars) or ζPKC-deficient (black bars) mice were stimulated with different concentrations of IgM for 72 h, after which the percentage of cells undergoing proliferation (S + G2/M; A) or apoptosis (sub-G0; B) was determined by flow cytometry. This is a representative experiment of at least another two with incubations in duplicate.

Fig. 5. Normal proliferative responses of ζPKC–/– T lymphocytes. T lymphocytes from either wild-type or ζPKC-deficient mice were incubated for 48 h with increasing concentrations of anti-CD3 antibody (0, 1 and 10 µg/ml) with or without 1 µg/ml anti-CD28, after which [3H]thymidine incorporation was determined as above. This is a representative experiment of at least another two with incubations in duplicate.
BCR activation triggers a myriad of signaling pathways, some of which are essential for growth and survival (Kurosaki, 1999). Of particular interest is the activation of extracellular signal-regulated kinase (ERK) in which ζPKC has been implicated using co-transfection and overexpression experiments in several cell systems (Berra et al., 1995; Moscat and Diaz-Meco, 2000). However, in EFs and lungs from ζPKC–/– mice challenged with different stimuli, ERK activation was not affected (Leitges et al., 2001). Therefore, it was of great interest to test whether ERK activation is impaired in B cells from ζPKC-deficient mice. The results in Figure 6A clearly demonstrate that triggering the BCR with 20 µg/ml IgM potently activates ERK at 10 but not at 20 min, and that this is dramatically impaired by the lack of ζPKC. When the stimulation of the stress-activated mitogen-activated protein kinases (MAPKs), p38 (Figure 6B) and JNK/SAPK (data not shown), was investigated in this system, no defects were detected in the B cells from the ζPKC–/– mice as compared with controls. Therefore, it seems that ζPKC is important in B cells for the activation of ERK but not for other MAPKs. This is in keeping with early observations from co-transfection experiments, but is in marked contrast to what has been observed in ζPKC–/– EFs. Since the mechanism whereby ζPKC has been proposed to participate in ERK activation is through MEK (Berra et al., 1995), in the next experiments B cells from either wild-type or ζPKC-deficient mice were stimulated for 10 min (the time at which ERK is activated) with two concentrations of IgM, and the stimulation of MEK was determined using an anti-phospho-MEK antibody. The results in Figure 6C show that the activation of MEK by 20 µg/ml IgM is completely impaired in the ζPKC–/– B cells, whereas it is not affected when they are stimulated with 50 µg/ml IgM. Interestingly, the activation of ERK by 50 µg/ml IgM is not affected by the lack of ζPKC (data not shown). Collectively, these results indicate that although at high concentrations of IgM, the MEK/ERK pathway is activated through ζPKC-independent mechanisms, at lower concentrations of the stimulant, ζPKC is essential for the activation of that signaling cascade. It should be noted that the maximal activation of B-cell proliferation is achieved at 10 µg/ml IgM (Figure 1B) and, therefore, 50 µg/ml is clearly a supraphysiological concentration, indicating that the defect detected in the ERK pathway at 20 µg/ml IgM is most likely of physiological relevance. Importantly, when B cells were activated with 50 µg/ml IgM, the difference between the wild-type and ζPKC–/– B cells in terms of mitogenic activation was diminished dramatically (data not shown). This suggests a very good correlation between the impairment in the activation of MEK/ERK and the defect in B-cell proliferation in the KO B cells.
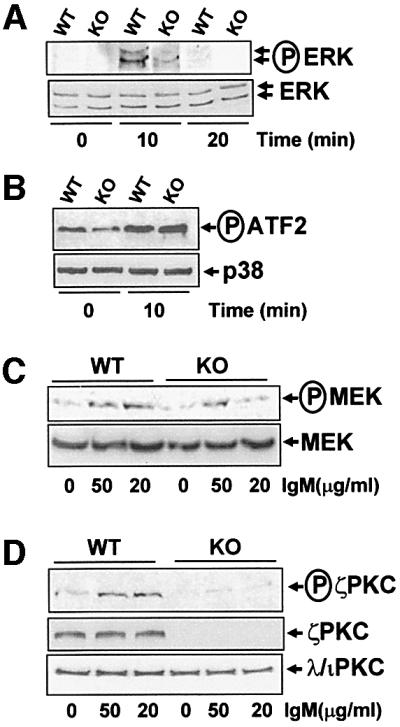
Fig. 6. Impaired ERK signaling in ζPKC–/– B cells. B cells from wild-type or ζPKC–/– mice were stimulated with 20 µg/ml IgM (A and B) for different times or with different concentrations of IgM for 10 min (C and D), after which cell extracts were analyzed by immunoblotting with anti-phospho-ERK and anti-ERK (A), anti-phospho-MEK and anti-MEK (C), or anti-phospho-aPKC and anti-ζPKC and anti-λ/ιPKC (D) antibodies. Cell extracts were also immunoprecipitated with an anti-phospho-p38 antibody, and the enzymatic activity of the immunoprecipitated enzyme was determined as phosphorylation of its substrate ATF-2 with the corresponding anti-phospho-ATF-2 antibody, and the loading control was performed with an anti-p38 antibody (B). These are representative experiments of at least another three with similar results.
If ζPKC is important for the activation of B-cell proliferation and signaling, it should be activated upon BCR stimulation. Therefore, we next determined whether this was actually the case. It has been documented extensively that the activation of ζPKC, and other PKC isoforms, correlates with the phosphorylation of Ser410 in the kinase activation loop through a PDK1-dependent mechanism (Le Good et al., 1998; Standaert et al., 1999). Interestingly, the results in Figure 6D demonstrate that ζPKC is activated in wild-type but not in ζPKC–/– B cells in response to IgM stimulation. As the B cells from the ζPKC-deficient mice have intact levels of the other atypical PKC (aPKC), λ/ιPKC (Figure 6D, lower panel), and the anti-phospho410-aPKC antibody does not discriminate between both aPKC isoforms, the results in Figure 6D strongly suggest that ζPKC but not λ/ιPKC is activated selectively during BCR stimulation. Further more, when activated B-cell extracts from wild-type or KO mice were immunoprecipitated with the anti-λ/ιPKC antibody and immunoblotted with the anti-phospho410-aPKC antibody, no signal was detected, indicating that λ/ιPKC was not activated (data not shown). When B cells were stimulated with the anti-CD40 antibody, there was no detectable phosphorylation with the anti-phospho410-ζPKC antibody (data not shown), indicating that neither ζPKC nor λ/ιPKC are activated by that pathway. Collectively, all these results are consistent with the concept that ζPKC is an important intermediary in the BCR signaling pathway for the activation of ERK. Such an inhibition of ERK activation in the ζPKC KO B cells could give a significant reduction in AP-1 (activating protein-1) stimulation. However, the data in Figure 7A and B show that, although significant and reproducible, the impact that the loss of ζPKC has in AP-1 activation in BCR-stimulated cells is relatively minor. The classical components of the AP-1 transcription factor are c-Fos and c-Jun (Karin et al., 1997). When the nuclear extracts of IgM-activated B cells from wild-type and ζPKC–/– mice were pre-incubated with an anti-c-Fos antibody, the amount of the supershifted band in the KO nuclear extracts was significantly smaller than that in the wild type (Figure 7C). This indicates that c-Fos induction is severely reduced in ζPKC–/– B cells, consistent with the fact that ERK is also inhibited in that system, but that other components of the AP-1 complex may compensate for the c-Fos defect.

Fig. 7. Effect of the lack of ζPKC on AP-1 and NF-κB activation in B cells. Nuclear extracts from either wild type (empty bars) or ζPKC-deficient (black bars) B cells that have been stimulated or not with 20 µg/ml IgM for 90 min were analyzed by EMSA using an AP-1 probe (A). Gels of three experiments like the one shown in (A) were quantified in an InstantImager, and the mean ± SD of the CPMs of the AP-1 bands are shown in (B). The experiment shown in (C) corresponds to nuclear extracts from stimulated B cells as above but which have been pre-incubated or not with an anti-c-Fos antibody. The supershifted band corresponding to the c-Fos-containing AP-1 complex is shown as SS. These are representative experiments of another two with similar results. In a parallel experiment, nuclear extracts from either wild-type or ζPKC-deficient B cells that have been stimulated or not with 20 µg/ml IgM with or without 10 ng/ml BAFF for 90 min were analyzed by EMSA using a κB probe (D). This is a representative experiment of at least another three with similar results.
Since ζPKC has been shown to be important during NF-κB activation in EFs (Leitges et al., 2001), we next determined whether this was also true in B cells. Therefore, B-cell cultures, either wild type or ζPKC-deficient, were stimulated or not with IgM with or without BAFF, and the nuclear levels of NF-κB were determined by electrophoretic mobility shift assays (EMSAs). Consistent with the data from ζPKC–/– EFs, no differences in NF-κB activation by EMSA were detected between the wild-type and mutant B cells (Figure 7D). It is of note that BAFF was unable to activate NF-κB or to potentiate IgM actions, either in the wild-type or in the KO B cells. In the ζPKC–/– EFs, it seems clear that, although NF-κB nuclear translocation is only a little or not affected in response to TNF-α and interleukin (IL)-1, the activation of IκB-dependent transcription is severely impaired (Leitges et al., 2001). Therefore, we determined whether the transcriptional activation of IκB, a classical and prototypic NF-κB-dependent gene, in B cells was affected by the loss of ζPKC. The data in Figure 8A demonstrate by RT–PCR, using 18S RNA as an internal control, that the activation of IκB transcription is severely inhibited in ζPKC–/– B cells activated through the BCR. The transcription of two other NF-κB-dependent genes was also impaired in the ζPKC-deficient B cells. The results in Figure 8B and C demonstrate that the transcription of IL-6 and Bcl-XL, respectively, was diminished in B cells from the ζPKC KO mice. The data on Bcl-XL are particularly relevant in light of the role of this gene in cell survival.

Fig. 8. Effect of the lack of ζPKC on the transcription of NF-κB-dependent genes. Wild-type (empty bars) and ζPKC-deficient (black bars) B cells were stimulated with 20 µg/ml IgM for 1.5 and 4 h, after which RNA was extracted and the levels of IκB (A), IL-6 (B), Bcl-XL (C) and 18S (A, B and C) RNAs were determined by RT–PCR with the appropriate primers. The inset panels show representative experiments of another two with similar results, and the lower panels show the quantification of that experiment.
Based on the above results, and the published evidence that different Rel family members are essential for T-dependent immune responses (Gerondakis et al., 1999), one can predict that the ζPKC–/– mice will have a defect in mounting an optimal adaptative immune response. To determine whether this was actually the case, we initially measured the basal Ig production in resting unimmunized, 7- and 16-week-old age-matched wild-type and ζPKC-deficient mice by enzyme-linked immunosorbent assay (ELISA) (Figure 9A). Although both groups of animals made all Ig isotypes, there was a slight but significant decrease in the amount of IgG1, IgG2a, IgG2b, IgA and IgE produced in the ζPKC–/– mice as compared with the controls at 7 weeks of age. The differences were even more significant in older (16-week-old) animals for the same isotypes, except for IgG1. To assay whether humoral immune responses were impaired in the ζPKC KO mice, 7-week-old age-matched wild-type and ζPKC-deficient mice were immunized with dinitrophenyl (DNP)–LPS and DNP–ovalbumin (OVA) as T-independent type I (TI-I) and T-dependent (TD) antigen, respectively, and the specific anti-DNP antibodies in sera were measured at several time points after immunization. Of note ζPKC-deficient mice were able to mount humoral immune responses to TI-I and to TD antigens (Figure 9B and C), but the level of the TD response was significantly lower in ζPKC-deficient mice for IgG1, IgG2a and IgG2b isoptypes, while the levels of IgA and IgG3 (data not shown) antibodies were within the normal range in TD responses, as was the level of IgM in TI-I responses. Interestingly, despite the deficiency in mounting a TD response, peanut agglutinin (PNA)+ clusters, representing germinal center B cells, were readily detected in the spleens from immunized ζPKC–/– mice (Figure 2B).

Fig. 9. Impaired adaptative immune response in ζPKC–/– mice. (A) Serum Ig isotype levels in unimmunized mice were determined by ELISA. The data represent the mean ± SD of serum Ig isotype levels in unimmunized, 7- and 16-week-old age-matched wild-type (open squares) and ζPKC-deficient (filled circles) mice (n = 4). P-values (asterisks) were calculated according to the Student’s t-test. Serum DNP-specific isotype levels in wild-type (open squares, n = 4) and ζPKC-deficient (filled circles, n = 3) mice immunized with DNP– LPS (B) and DNP–OVA (C) were determined by ELISA, and are represented as mean ± SD. The curves represent the best-fit Gaussian distributions. Asterisks denote P-values calculated according to the F-test applied to compare the entire immunization response curves using the GraphPad Prism software. The data are representative of two independent analyses.
Discussion
The recent characterization of the ζPKC KO mouse has provided valuable information on the role that this kinase plays in the control of the immune and inflammatory responses (Leitges et al., 2001). The results from these mice clearly establish that ζPKC is critically involved in the control of NF-κB activation, most probably at two different levels. In tissues where it is poorly expressed, it plays a non-redundant role in the control of NF-κB transcriptional activity with no effects in more upstream steps of the pathway. However, in lung, where ζPKC is greatly expressed, it seems to be required also for the activation of the IKK complex and the nuclear translocation of NF-κB. The lack of ζPKC produces mainly a significant delay in the development of LNs and the PPs which display a reduced content of the more mature subpopulations of B cells (Leitges et al., 2001). This defect is corrected in adult KO mice in which the content of the different major B-cell splenic subpopulations is normal (Leitges et al., 2001; this study). In this study, we show that ζPKC is important for B-cell proliferation and survival and for NF-κB-dependent transcriptional activation, as determined by measuring the transcription of at least three NF-κB-dependent genes. However, the loss of ζPKC does not impair NF-κB nuclear translocation in this cell system. In this regard, from a mechanistic point of view, ζPKC requirements for B-cell function and NF-κB activation are more similar to those of EFs than those of lung. The fact that there are not any changes in the relative percentages and absolute content of the different B-cell subpopulations in the KO splenic B cells used in this study as compared with age-matched wild-type controls suggests that these defects are most probably due to intrinsic impairment of the B-cell signaling capabilities. These results are of great physiological relevance because they genetically prove that ζPKC is important for B-cell function, and may explain the alterations detected in the development of secondary lymphoid organs in ζPKC KO mice (Leitges et al., 2001), as well as why ζPKC is required for an optimal TD immune response. Interestingly, alterations in the ability to mount a normal humoral response in vivo have been observed in mice mutant for different Rel proteins (Gerondakis et al., 1999). Thus, for example, NF-κB1–/– mice display a cell-autonomous defect in heavy chain isotype switching (Snapper et al., 1996) with a relatively minor defect in germinal center PNA+ staining (Pohl et al., 2002). Experiments in which fetal liver cells from RelA KO mice were transplanted into SCID mice revealed that, although B cells have the ability to develop normally, the secretion of IgG1 and IgA was dramatically impaired in RelA-deficient B cells (Doi et al., 1997). In addition, studies in c-Rel–/– B cells suggest a role for this protein in germline CH transcription (Gerondakis et al., 1999). The generation of mutant mice with combined deletions of different Rel genes gives rise to even more profound phenotypes (Gerondakis et al., 1999). Therefore, the intrinsic deficiency detected here in the ability of ζPKC–/– B cells to proliferate and survive in vitro may account for the in vivo defect in mounting an optimal TD immune response. It is possible that through some of the Rel proteins, ζPKC controls critical steps in the isotype switching process (Snapper et al., 1997). In this regard, defects in the ability of B cells to divide may be important for an appropriate isotype switching (Hodgkin et al., 1996). However, other possibilities cannot be ruled out yet. For example, a deficiency in T cell cytokine secretion in vivo could also explain the defect in the TD response in the ζPKC-deficient B cells. However, the lack of alterations in T-cell activation in the ζPKC–/– mice suggests that this would not be linked to the ability of T cells to proliferate but could be due to more subtle alterations in T-cell activation to produce certain cytokines. In this regard, it is noteworthy that ζPKC and NF-κB signaling are required for IL-6 transcription (Leitges et al., 2001; this study) and that IL-6 is an important cytokine in, for example, the regulation of IgA production (Ramsay et al., 1994), whose basal serum levels are dramatically diminished in ζPKC–/– mice. On the other hand, although a stromal cell defect cannot yet be ruled out completely as a potential explanation for these in vivo alterations, the evidence that B cells from ζPKC–/– mice display a clear impairment in their response to BCR activation in vitro, together with the lack of alterations in the spleen microarchitecture in the ζPKC KO mice, suggest that the contribution of the stroma to the changes reported here is unlikely. The fact that germinal center formation was unaltered in immunized ζPKC-deficient mice despite their inability to mount an optimal TD immune response may indicate that the loss of ζPKC results in either the failure to switch (which requires cell division and survival) or the death (failure to survive) of plasmacytes and antibody-forming cells coming out of the germinal center. Future studies will address these different possibilities.
There is an important difference between B cells and EFs from the point of view of the involvement of ζPKC in cell signaling. Thus, whereas ζPKC is not necessary in EFs for the activation of the MEK/ERK pathway in response to different stimuli (Leitges et al., 2001), in B cells this PKC seems to be essential for ERK and MEK activation. These results would be consistent with previous observations from this and other laboratories that using overexpression of active and dominant-negative mutants has implicated ζPKC as a potential activator of ERK through MEK (Berra et al., 1995; Moscat and Diaz-Meco, 2000). The degree of inhibition that we see in ERK activation in the ζPKC–/– B cells does not correlate with the reduction that should be expected in AP-1 activation. A possible explanation for this apparent paradox is that the ζPKC-deficient cells have devised mechanisms to compensate for the reduction of ERK activity. In this regard, it should be noted that the activation of other MAPKs, particularly p38 and SAPK/JNK, is not affected by the lack of ζPKC. However, when the content of c-Fos, a direct target of ERK activation, is determined in the AP-1 complex in supershift experiments, it is apparent that there is a better correlation between the diminution of ERK activation and that of the c-Fos-containing complex. This is particularly relevant in the light of the role that c-Fos plays in cell survival and growth (Shaulian and Karin, 2001).
The possible involvement of different PKC isoforms in the immune system has attracted great interest lately. For example, there is genetic evidence based in the information from θPKC KO mice that this kinase is essential for the proliferation of mature T lymphocytes and for the activation of both AP-1 and NF-κB transcription factors (Sun et al., 2000). We show here that T lymphocyte proliferation, in marked contrast to B cells, is not affected by the lack of ζPKC. This would be in keeping with the notion that there are cell type-specific functions for different PKC isoforms. Recent data in B cells using overexpression experiments and pharmacological inhibitors suggest that either δPKC or θPKC could be important in the activation of NF-κB and SAPK/JNK in the BCR pathway, through an as yet not fully characterized mechanism (Krappmann et al., 2001). If the genetic evidence using KO models for these two novel PKC isoforms confirms these suggestions, one can envisage a model according to which different PKC isotypes may control distinct steps in the NF-κB cascade. Thus, whereas ζPKC will control NF-κB transcriptional activation, other isoforms could regulate the IKK complex and the subsequent nuclear translocation of NF-κB. Based on our previous data, the other atypical isoform, λ/ιPKC, also could be critically involved in IKK activation (Lallena et al., 1999). However, according to the results presented here, it seems that in B cells ζPKC is the major aPKC activated by IgM, suggesting that λ/ιPKC many not be important in this pathway. It should be noted that λ/ιPKC is expressed ubiquitously and that it may compensate for the lack of ζPKC in the KO mice due to the extremely high degree of conservation between both aPKCs. This may explain the relatively mild phenotype of the ζPKC–/– mice. A more definitive response to these questions must await the characterization of cells from conditional λ/ιPKC KO mice when available.
Finally, both aPKCs have been implicated as downstream targets of phosphatidylinositol (PI) 3-kinase. It is noteworthy that mice with genetic inactivation of the PI 3-kinase regulatory subunit, p85α, display B-cell proliferative defects in response to several stimuli (Fruman et al., 1999). Therefore, it can be proposed that whereas ζPKC is important for signaling through the BCR, other PI 3-kinase targets may be responsible for the activation of ζPKC-independent pathways, such as those triggered by CD40 or LPS.
Materials and methods
Isolation of B and T cells
The generation of ζPKC–/– mice was described previously (Leitges et al., 2001). Spleens were removed from 4-week-old mice, and single-cell suspensions were prepared by crushing organs between glass slides. Red blood cells were lysed with a hypotonic solution (NH4Cl buffer). Afterwards, splenocytes were incubated with anti-mouse CD43 (Ly-48) microbeads (Miltenyi Biotec, Auburn, CA), and resting B cells were separated from the CD43-negative fraction by using an autoMACS magnetic cell sorter (Miltenyi Biotec). The purity of B cells was >95%, as indicated by the percentage of B220+ cells by flow cytometry analysis. T-cell isolation was performed from the CD43-positive fraction by a second positive selection after B-cell separation, and the purity of T cells was >90% as indicated by the percentage of Thy-1+ cells.
Flow cytometric analysis
Spleens were removed from 4- and 6-week-old mice. Single-cell suspensions were made and washed in phosphate-buffered saline (PBS) supplemented with 5% fetal calf serum (FCS) and 5 mM EDTA. Subsequently, cells were analyzed after staining with the following monoclonal antibodies (mAbs): fluorescein isothiocyanate (FITC)-conjugated anti-CD21 (clone 7G6; PharMingen, San Diego CA), phycoerythrin (PE)-conjugated anti-CD23 (clone B3B4; PharMingen) and biotin-conjugated anti-IgM (clone R6-60.2; PharMingen) followed by streptavidin–Tricolor (Caltag, Burlingame, CA). Analysis was performed on a FACScalibur flow cytometer (Becton Dickinson, Mountain View, CA) with CELLQuest software. Dead cells were excluded from the analysis on the basis of low forward light scatter. B cells purified with the MACS B cell isolation kit (Miltenyi Biotec) were analyzed as above.
Proliferation of B and T cells
Purified B cells were cultured in 96-well plates at 2 × 105 cells per well in 100 µl of RPMI 1640 medium supplemented with 10% FCS, 2 mM l-glutamine, 50 U/ml penicillin/streptomycin and 50 µM 2-mercaptoethanol, and were stimulated for 72 h with the indicated concentrations of F(ab′)2 fragments of goat anti-mouse IgM antibody (Jackson Immuno research Laboratories) and/or different concentrations of BAFF, 0.5 µg/ml anti-CD40 (clone HM40-3; PharMingen) or 20 µg/ml LPS (Escherichia coli; Sigma). Purified T cells were cultured at 1 × 105 cells per well in 160 µl of medium and were stimulated for 48 h in anti-CD3ε antibody (clone 145-2C11; PharMingen) on plates pre-coated for 1 h (either 1 or 10 µg/ml). For co-stimulation of T cells, soluble anti-CD28 mAb (clone 37.51; PharMingen) was also added at 1 µg/ml. Proliferation was assessed by the incorporation of [3H]thymidine added (1 µCi/well) during the last 6 h of culture in triplicate wells. Cells were collected using a cell harvester, and [3H]thymidine incorporation was quantified by scintillation counting (Wallac Oy 1450 Microbeta).
Cell cycle analysis
Cell cycle analysis was performed by flow cytometry by incubation of cells in a buffer consisting of 0.1% sodium citrate, 0.6% NP-40, 20 µg/ml RNase and 50 µg/ml propidium iodide, for 30 min at 4°C in the dark. Analysis was performed on a FACScalibur flow cytometer (Becton Dickinson) with CELL-Quest software on the basis of FL-2 area/FL-2 weight parameters. The percentage of apoptotic cells was determined by calculating the fraction of cells with sub-G1 DNA content.
Western blotting
Purified B cells were stimulated with IgM and lysed in buffer PD [40 mM Tris–HCl pH 8.0, 500 mM NaCl, 0.1% NP-40, 6 mM EDTA, 6 mM EGTA, 10 mM β-glycerophosphate, 10 mM NaF, 10 mM PNPP, 300 µM Na3VO4, 1 mM benzamidine, 2 μM phenylmethylsulfonyl fluoride (PMSF), 10 µg/ml aprotinin, 1 µg/ml leupeptin, 1 µg/ml pepstatin, 1 mM dithiothreitol (DTT)]. Proteins corresponding to 20 µg were electrophoresed in 8% SDS–polyacrylamide gels and transferred onto ECL nitrocellulose membranes (Amersham). The following antibodies were used in this study: anti-phospho-ERK (sc-7383; Santa Cruz), anti-phospho-MEK (cat. 9110; Cell Signaling Technology), anti-phospho-PKCζ (cat. 9378; Cell Signaling Technology), anti-ERK (sc-94; Santa Cruz), anti-MEK (cat. 9122; Cell Signaling Technology) and anti-λ/ιPKC (cat. P22520; Transduction Laboratories). The anti-ζPKC was a rabbit affinity-purified polyclonal antibody raised against a peptide corresponding to residues 5–19 of rat ζPKC.
P38 kinase assay
The p38 kinase assay from Cell Signaling Technology (cat. 9820) was used according to the manufacturer’s procedures. Briefly, IgM-stimulated purified B cells were lysed and immunoprecipitated with an immobilized phospho-p38 mAb. Afterwards, an in vitro kinase assay was performed using activating transcription factor-2 (ATF-2) as substrate, and ATF-2 phosphorylation was detected by western blot using a phospho-ATF-2 antibody. Loading control was performed by immunoblot analysis with an anti-p38 antibody (cat. 9212; Cell Signaling Technology).
Electrophoretic mobility shift assays
EMSA experiments for NF-κB were performed as described previously (Leitges et al., 2001). For AP-1, the following oligonucleotide was used: 5′-CGCTTGATGACTCAGCCGGAA-3′; with the AP-1 5× binding buffer: 50 mM Tris–HCl pH 7.5, 500 mM KCl, 2.5 mM MgCl2, 0.5 mM EDTA, 50% glycerol, 250 µg/ml poly(dI:dC), 5 mM DTT and 1 mg/ml bovine serum albumin (BSA).
RT–PCR analysis of NF-κB-dependent genes
Purified splenic B cells were incubated with or without 20 µg/ml IgM for 1.5 or 4 h. Extraction of total RNAs, reverse transcription and PCR analysis were performed as recommended by the Gene Specific Relative RT–PCR kit from Ambion. The primers used were: for IκB, 5′-GCCTTCCTCAACTTCCAGAACAAC-3′ and 5′-CAGACGCTGGCCTCCAAACACACAG-3′; for IL-6, 5′-TTGCCTTCTTGGGACTGATG-3′ and 5′-CTGAAGGACTCTGGCTTTGT-3′; and for Bcl-XL, 5′-GAGTTTGAACTGCGGTACCGG-3′ and 5′-GTGTCTGGTCATTTCCGACTG-3′. A 2:8 ratio of 18S primers to competimers was used in the PCR for quantification analysis.
Ig serum detection and immunization
Ig serum detection was performed by ELISAs as described previously (Martinez et al., 2001). Total serum Igs were detected by capture ELISA using plates coated with a goat anti-mouse IgS (10 µg/ml; Southern Biotechnology Associates) as capture antibody. For IgE determination, the plates were coated with rat anti-mouse IgE (10 µg/ml; clone EM-95). Plates used for the anti-DNP ELISAs were coated with DNP–BSA (10 µg/ml). The assays were revealed with biotinylated goat anti-mouse IgM, IgG1, IgG2a, IgG2b and IgG3 (Southern Biotechnology Associates, Birmingham, AL), and biotinylated rat anti-mouse κ chain (clone 187.1) for IgE determination. Plates were then incubated with streptavidin-conjugated peroxidase (Southern Biotechnology Associates) and developed with 0.5 M o-phenylenediamine (Sigma). The absorption values were read at 405 nm. A normalized Ig isotype titer was calculated for each serum using the GraphPad Prism 3.0 software (GraphPad Software Inc., San Diego, CA), with the dilution value (D50) representing 50% of the absorbance obtained by each serum at the top plateau. Mice were immunized intraperitoneally with 100 µg of DNP–LPS and with 100 µg of DNP–OVA in complete Freund’s adjuvant (CFA). Specific Ab titers were tested in serum samples obtained at 3, 7 and 11 days from DNP–LPS-immunized mice, and at day 3 and weekly from DNP–OVA-immunized mice, respectively.
Immunofluorescent analyses
Immunofluorescent staining was performed using 8 µm frozen sections that were air dried and fixed in ice-cold acetone. Antibodies used were MOMA-1 (Serotec, Oxford, UK), anti-CD11c and anti-CD11b (all from PharMingen). Sections were also stained with biotin-conjugated PNA to detect germinal center formation in immunized mice.
Acknowledgments
Acknowledgements
We thank Susan Kalled of Biogen, Inc. for reagents. This work was supported by grants SAF1999-0053, 2FD97-1429 and SAF2000-0175 from MCYT, and 08.1/0060/2000 from CAM, and has benefited from an institutional grant from Fundación Ramón Areces to the CBM. J.M. is the recipient of the ‘II Ayuda de la Fundación Juan March a la Investigación Básica’.
References
- Alcamo E., Hacohen,N., Schulte,L.C., Rennert,P.D., Hynes,R.O. and Baltimore,D. (2002) Requirement for the NF-κB family member RelA in the development of secondary lymphoid organs. J. Exp. Med., 195, 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin A.S. Jr, (1996) The NF-κB and IκB proteins: new discoveries and insights. Annu. Rev. Immunol., 14, 649–683. [DOI] [PubMed] [Google Scholar]
- Berra E., Diaz-Meco,M.T., Lozano,J., Frutos,S., Municio,M.M., Sanchez,P., Sanz,L. and Moscat,J. (1995) Evidence for a role of MEK and MAPK during signal transduction by protein kinase Cζ. EMBO J., 14, 6157–6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caamano J.H., Rizzo,C.A., Durham,S.K., Barton,D.S., Raventos-Suarez,C., Snapper,C.M. and Bravo,R. (1998) Nuclear factor (NF)-κB2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J. Exp. Med., 187, 185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cariappa A., Liou,H.C., Horwitz,B.H. and Pillai,S. (2000) Nuclear factor κB is required for the development of marginal zone B lymphocytes. J. Exp. Med., 192, 1175–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi T.S., Takahashi,T., Taguchi,O., Azuma,T. and Obata,Y. (1997) NF-κB RelA-deficient lymphocytes: normal development of T cells and B cells, impaired production of IgA and IgG1 and reduced proliferative responses. J. Exp. Med., 185, 953–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougall W.C. et al. (1999) RANK is essential for osteoclast and lymph node development. Genes Dev., 13, 2412–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman D.A., Snapper,S.B., Yballe,C.M., Davidson,L., Yu,J.Y., Alt,F.W. and Cantley,L.C. (1999) Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85α. Science, 283, 393–397. [DOI] [PubMed] [Google Scholar]
- Fu Y.X. and Chaplin,D.D. (1999) Development and maturation of secondary lymphoid tissues. Annu. Rev. Immunol., 17, 399–433. [DOI] [PubMed] [Google Scholar]
- Futterer A., Mink,K., Luz,A., Kosco-Vilbois,M.H. and Pfeffer,K. (1998) The lymphotoxin β receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity, 9, 59–70. [DOI] [PubMed] [Google Scholar]
- Gerondakis S., Grossmann,M., Nakamura,Y., Pohl,T. and Grumont,R. (1999) Genetic approaches in mice to understand Rel/NF-κB and IκB function: transgenics and knockouts. Oncogene, 18, 6888–6895. [DOI] [PubMed] [Google Scholar]
- Grossmann M., O‘Reilly,L.A., Gugasyan,R., Strasser,A., Adams,J.M. and Gerondakis,S. (2000) The anti-apoptotic activities of Rel and RelA required during B-cell maturation involve the regulation of Bcl-2 expression. EMBO J., 19, 6351–6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gugasyan R., Grumont,R., Grossmann,M., Nakamura,Y., Pohl,T., Nesic,D. and Gerondakis,S. (2000) Rel/NF-κB transcription factors: key mediators of B-cell activation. Immunol. Rev., 176, 134–140. [DOI] [PubMed] [Google Scholar]
- Hodgkin P.D., Lee,J.H. and Lyons,A.B. (1996) B cell differentiation and isotype switching is related to division cycle number. J. Exp. Med., 184, 277–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel A. (2000) The IKK complex: an integrator of all signals that activate NF-κB? Trends Cell Biol., 10, 129–133. [DOI] [PubMed] [Google Scholar]
- Karin M. and Ben-Neriah,Y. (2000) Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu. Rev. Immunol., 18, 621–663. [DOI] [PubMed] [Google Scholar]
- Karin M., Liu,Z. and Zandi,E. (1997) AP-1 function and regulation. Curr. Opin. Cell Biol., 9, 240–246. [DOI] [PubMed] [Google Scholar]
- Kim D. et al. (2000) Regulation of peripheral lymph node genesis by the tumor necrosis factor family member TRANCE. J. Exp. Med., 192, 1467–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koni P.A., Sacca,R., Lawton,P., Browning,J.L., Ruddle,N.H. and Flavell,R.A. (1997) Distinct roles in lymphoid organogenesis for lymphotoxins α and β revealed in lymphotoxin β-deficient mice. Immunity, 6, 491–500. [DOI] [PubMed] [Google Scholar]
- Krappmann D., Patke,A., Heissmeyer,V. and Scheidereit,C. (2001) B-cell receptor- and phorbol ester-induced NF-κB and c-Jun N-terminal kinase activation in B cells requires novel protein kinase Cs. Mol. Cell. Biol., 21, 6640–6650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaki T. (1999) Genetic analysis of B cell antigen receptor signaling. Annu. Rev. Immunol., 17, 555–592. [DOI] [PubMed] [Google Scholar]
- Lallena M.J., Diaz-Meco,M.T., Bren,G., Pay,C.V. and Moscat,J. (1999) Activation of IκB kinase β by protein kinase C isoforms. Mol. Cell. Biol., 19, 2180–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Good J.A., Ziegler,W.H., Parekh,D.B., Alessi,D.R., Cohen,P. and Parker,P.J. (1998) Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science, 281, 2042–2045. [DOI] [PubMed] [Google Scholar]
- Leitges M. et al. (2001) Targeted disruption of the ζPKC gene results in the impairment of the NF-κB pathway. Mol. Cell, 8, 771–780. [DOI] [PubMed] [Google Scholar]
- Martinez M.J., Minguet,S., Gonzalo,P., Soro,P.G., de Andres,B., Izcue,A., Marcos,M.A. and Gaspar,M.L. (2001) Long-lived polyclonal B-cell lines derived from midgestation mouse embryo lymphohematopoietic progenitors reconstitute adult immunodeficient mice. Blood, 98, 1862–1871. [DOI] [PubMed] [Google Scholar]
- Moscat J. and Diaz-Meco,M.T. (2000) The atypical protein kinase Cs. Functional specificity mediated by specific protein adapters. EMBO rep., 1, 399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann B., Luz,A., Pfeffer,K. and Holzmann,B. (1996) Defective Peyer’s patch organogenesis in mice lacking the 55-kD receptor for tumor necrosis factor. J. Exp. Med., 184, 259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasparakis M., Alexopoulou,L., Grell,M., Pfizenmaier,K., Bluethmann,H. and Kollias,G. (1997) Peyer’s patch organogenesis is intact yet formation of B lymphocyte follicles is defective in peripheral lymphoid organs of mice deficient for tumor necrosis factor and its 55-kDa receptor. Proc. Natl Acad. Sci. USA, 94, 6319–6323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl T., Gugasyan,R., Grumont,R.J., Strasser,A., Metcalf,D., Tarlinton,D., Sha,W., Baltimore,D. and Gerondakis,S. (2002) The combined absence of NF-κB1 and c-Rel reveals that overlapping roles for these transcription factors in the B cell lineage are restricted to the activation and function of mature cells. Proc. Natl Acad. Sci. USA, 99, 4514–4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay A.J., Husband,A.J., Ramshaw,I.A., Bao,S., Matthaei,K.I., Koehler,G. and Kopf,M. (1994) The role of interleukin-6 in mucosal IgA antibody responses in vivo. Science, 264, 561–563. [DOI] [PubMed] [Google Scholar]
- Rennert P.D., James,D., Mackay,F., Browning,J.L. and Hochman,P.S. (1998) Lymph node genesis is induced by signaling through the lymphotoxin β receptor. Immunity, 9, 71–79. [DOI] [PubMed] [Google Scholar]
- Shaulian E. and Karin,M. (2001) AP-1 in cell proliferation and survival. Oncogene, 20, 2390–2400. [DOI] [PubMed] [Google Scholar]
- Silverman N. and Maniatis,T. (2001) NF-κB signaling pathways in mammalian and insect innate immunity. Genes Dev., 15, 2321–2342. [DOI] [PubMed] [Google Scholar]
- Snapper C.M., Zelazowski,P., Rosas,F.R., Kehry,M.R., Tian,M., Baltimore,D. and Sha,W.C. (1996) B cells from p50/NF-κB knock out mice have selective defects in proliferation, differentiation, germ-line CH transcription and Ig class switching. J. Immunol., 156, 183–191. [PubMed] [Google Scholar]
- Snapper C.M., Marcu,K.B. and Zelazowski,P. (1997) The immuno globulin class switch: beyond ‘accessibility’. Immunity, 6, 217–223. [DOI] [PubMed] [Google Scholar]
- Standaert M.L. et al. (1999) Insulin activates protein kinases C-ζ and C-λ by an autophosphorylation-dependent mechanism and stimulates their translocation to GLUT4 vesicles and other membrane fractions in rat adipocytes. J. Biol. Chem., 274, 25308–25316. [DOI] [PubMed] [Google Scholar]
- Sun Z. et al. (2000) PKC-θ is required for TCR-induced NFκB activation in mature but not immature T lymphocytes. Nature, 404, 402–407. [DOI] [PubMed] [Google Scholar]
- Thanos D. and Maniatis,T. (1995) NFκB: a lesson in family values. Cell, 80, 529–532. [DOI] [PubMed] [Google Scholar]
- Thompson J.S. et al. (2001) BAFF-R, a newly identified TNF receptor that specifically interacts with BAFF. Science, 293, 2108–2111. [DOI] [PubMed] [Google Scholar]
- Verma I.M., Stevenson,J.K., Schwarz,E.M., Van Antwerp,D. and Miyamoto,S. (1995) Rel/NFκB/IκB family: intimate tales of association and dissociation. Genes Dev., 9, 2723–2735. [DOI] [PubMed] [Google Scholar]