

Abstract
SelB is an elongation factor needed for the co-translational incorporation of selenocysteine. Selenocysteine is coded by a UGA stop codon in combination with a specific downstream mRNA hairpin. In bacteria, the C-terminal part of SelB recognizes this hairpin, while the N-terminal part binds GTP and tRNA in analogy with elongation factor Tu (EF-Tu). We present the crystal structure of a C-terminal fragment of SelB (SelB-C) from Moorella thermoacetica at 2.12 Å resolution, solved by a combination of selenium and yttrium multiwavelength anomalous dispersion. This 264 amino acid fragment contains the entire C-terminal extension beginning after the EF-Tu-homologous domains. SelB-C consists of four similar winged-helix domains arranged into the shape of an L. This is the first example of winged-helix domains involved in RNA binding. The location of conserved basic amino acids, together with data from the literature, define the position of the mRNA-binding site. Steric requirements indicate that a conformational change may occur upon ribosome interaction. Struc tural observations and data in the literature suggest that this change happens upon mRNA binding.
Keywords: crystallography/elongation factor/RNA binding/SelB/selenocysteine
Introduction
Selenocysteine (Sec), the 21st amino acid, exists in organisms from all three kingdoms. It is the main biological form of selenium, and is used in the active site of redox enzymes such as formate dehydrogenase and glutathione peroxidase. Selenocysteine is the only genetically coded non-standard amino acid. The selenocysteine codon consists of a UGA stop codon in combination with a specific hairpin structure on the mRNA. This selenocysteine insertion sequence (SECIS) allows the stop codon to be read through instead of promoting termination.
SelB is a specialized elongation factor for delivery of selenocysteylated tRNASec to the ribosomal A-site when the two-component selenocysteine codon is present. It was first identified in Escherichia coli (Forchhammer et al., 1989). It forms a ternary complex with GTP and selenocysteylated tRNASec, in analogy with elongation factor Tu (EF-Tu), but it also binds to the SECIS, which in bacteria is located nearby, downstream of the selenocysteine UGA codon. Thus, in addition to the requirement of correct codon–anticodon interaction, SelB needs to recognize a SECIS structure to deliver Sec-tRNASec to the ribosomal A-site upon GTP hydrolysis.
The different tasks of SelB have been linked to different parts of the protein sequence (Kromayer et al., 1996). The N-terminal 342 amino acids display sequence homology to the three domains of EF-Tu, and contain the binding sites for tRNA and GTP. The C-terminal part is responsible for mRNA binding, and can be minimized to amino acids 472–634 (E.coli numbering) without losing binding affinity for the SECIS.
In E.coli, the determinants for RNA binding have been carefully dissected. SelB binds to the mRNA hairpin with a dissociation constant of ∼1 nM (Thanbichler et al., 2000). The recognition element of RNA can be minimized to a 17 nucleotide hairpin (Figure 1A; Kromayer et al., 1996). Extensive mutagenesis studies have shown that all the loop nucleotides as well as the closing base pairs and the bulged U are required for selenocysteine insertion (Heider et al., 1992). Another important feature is the distance between the UGA codon and the minimal hairpin, which in the E.coli selenoprotein mRNAs is 11 nucleotides. The hairpin sequence is not very well conserved between different bacterial species (Figure 1).
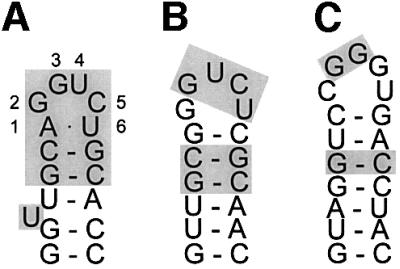
Fig. 1. (A) Escherichia coli fdhF mRNA hairpin (Hüttenhofer et al., 1996). Essential nucleotides are indicated by shading. (B) Predicted mRNA hairpin from the M.thermoacetica fdhA gene (GenBank accession No. U73807). Nucleotides identical to the essential nucleotides in E.coli are indicated by shading. (C) Predicted hairpin from the Aquifex aeolicus SelD gene (GenBank accession No. NC_000918). Shading as in (B). Hairpin structures were predicted using the mfold program (Zuker et al., 1999).
Here we describe the crystal structure of the C-terminal fragment 370–634 of Moorella thermoacetica (previously called Clostridium thermoaceticum) SelB (SelB-C) determined to a resolution of 2.12 Å. This fragment contains the complete C-terminal extension as compared with EF-Tu. It is the first structure of a component of the selenocysteine insertion machinery. In the light of functional data from the literature and structural constraints imposed by the ribosome, the SelB-C structure allows us to propose a functional model for SelB in selenocysteine incorporation.
Results and discussion
Crystal structure determination
The preparation and crystallization of M.thermoacetica SelB-C will be published elsewhere (M.Selmer, R.Wilting, D.Holmlund and X.-D.Su, submitted). The crystals belong to space group P212121 and contain one molecule in the asymmetric unit. The structure was determined by multiwavelength anomalous dispersion (MAD) to 2.12 Å resolution using a single crystal of selenomethionine-substituted protein grown in the presence of yttrium chloride (Table I). Since both Se and Y were expected to be present in the crystal, wavelengths were chosen to optimize the anomalous signal from both elements. In total, six wavelengths were collected on the same crystal (Table I). Two yttrium sites and one selenium site could be found using the respective peak wavelength data, and these sites subsequently were used for phasing. As seen in Table II, selenium made a slightly greater contribution to the phasing than did yttrium. Seventy-five percent of the residues were built into the experimental map. After phase combination, the rest of the chain could be traced. The structure was refined to an Rwork and Rfree of 21.5 and 25.8%, respectively, with good geometry (Table III). Two side chains are in the disallowed region of the Ramachandran diagram, Ala499 in the only poorly defined loop and Arg530 in a well-defined region of the map. The final model includes residues 380–634, one sulfate ion, two yttrium ions and 109 water molecules. The final 2Fo – Fc map around the sulfate ion is shown in Figure 2. Both yttrium ions are coordinated to residues from two neighbouring molecules, contributing to the crystal packing and explaining why the yttrium chloride additive was crucial for obtaining high quality crystals.
Table I. Data collection statistics.
| Wavelength (Å) | Resolution (Å) (last shell) | No. of observed/unique reflections | Completeness (%) overall/last shell | I/σ(I) overall/last shell | Rsyma (%) overall/last shell |
|---|---|---|---|---|---|
| 0.7250 (λ1) | 20–2.3 (2.38–2.3) | 112 124/12 635 | 100/99.9 | 24.4/3.3 | 7.7/34.2 |
| 0.7255 (λ2) | 20–2.5 (2.59–2.5) | 84 790/9887 | 100/100 | 21.5/3.7 | 8.1/33.2 |
| 0.9773 (λ3) | 20–2.12 (2.2–2.12) | 138 893/15 941 | 100/99.8 | 28.2/3.1 | 6.8/37.3 |
| 0.9783 (λ4) | 20–2.3 (2.38–2.3) | 104 450/12 575 | 100/99.9 | 23.5/3.3 | 7.7/39.1 |
| 0.9840 (λ5) | 20–2.3 (2.38–2.3) | 106 354/12 369 | 100/99.9 | 24.2/4.2 | 6.2/33.1 |
| 0.9050 (λ6) | 20–2.3 (2.38–2.3) | 107 038/12 969 | 100/99.9 | 23.8/2.8 | 7.4/37.9 |
aRsym = (Σ|I – <I>|/ΣI), where I is the observed intensity and <I> is the average intensity of symmetry-related reflections.
Table II. Phasing statistics.
| Yttrium phasing |
Selenium phasing |
|||||||
|---|---|---|---|---|---|---|---|---|
| peak (λ1) | ip (λ2) | rm1 (λ5) | rm2 (λ6) | peak (λ3) | ip (λ4) | rm1 (λ5) | rm2 (λ6) | |
| RCullisa | 0.68/0.69 | 0.53/0.64 | 0.95/0.93 | 10/0.83 | 0.64/0.61 | 0.50/0.58 | 0.70/0.70 | 10/0.71 |
| Phasing powerb | 1.71/1.60 | 2.01/1.88 | 0.66/0.64 | 0.85/0.76 | 2.22/2.09 | 2.58/2.30 | 1.76/1.62 | 1.51/1.38 |
All values are for centric/acentric phasing.
aRCullis = (Σ||FPH – FP| – FH(calc)|/Σ|FPH – FP|), where FP and FH are the protein and heavy-atom structure factors, respectively, and FH(calc) is the calculated heavy-atom structure factor.
bPhasing power = [FH(calc)/E], where E is the estimated lack-of-closure error.
Table III. Refinement statistics.
| Resolution (Å) | 19.2–2.12 |
| Rwork (%)* | 21.5 (24.7) |
| Rfree (%)* | 25.8 (28.4) |
| No. of reflections | |
| Working set | 25 755 |
| Test set | 2755 |
| No. of atoms | |
| Protein | 2073 |
| Solvent | 109 |
| Hetero | 7 |
| Deviations from ideal geometry (r.m.s.d.) | |
| Bond lengths | 0.006 Å |
| Bond angles | 1.2° |
| Dihedral angles | 20.7° |
| B-factors (Å2) | |
| Minimum/maximum | 12.5/53.2 Å2 |
| Average | 29.9 Å2 |
| Ramachandran plot | |
| Most favoured region | 92.3% |
| Additionally allowed | 6.4% |
| Generously allowed | 0.4% |
| Disallowed | 0.9% |
aRwork = Σh||Fobs| – |Fcalc||/Σh|Fobs|, where Fcalc and Fobs are calculated and observed structure factors; Rfree = Rwork for a 10% subset of the reflections that were not included in the refinement.
Numbers in parentheses represent the value for the highest resolution shell.

Fig. 2. Section of the final 2Fo – Fc map showing the coordination of the sulfate ion by Arg599, Ser605 and Arg606, prepared using O (Jones et al., 1991).
Overall structure
The SelB-C structure is L-shaped and has the overall dimensions 70 × 50 × 20 Å (Figure 3). Each arm of the L consists of two globular winged-helix domains. The N-terminal part of SelB presumably consists of three domains (I–III) in analogy to EF-Tu. The SelB-C domains are therefore numbered IV–VII. Domain IV consists of amino acids Gly377–Ser436, domain V of Thr437– Phe511, domain VI of Ser512–His573 and domain VII of Arg574–Asn634.

Fig. 3. Overall structure of SelB-C. Colour coding: domain IV, blue; domain V, green; domain VI, yellow; domain VII, red. The view in (B) is rotated 90° from the view in (A). The image was prepared using Molmol (Koradi et al., 1996).
The winged-helix domain is an α/β structure consisting of three α-helices and a twisted three-stranded antiparallel β-sheet. The connectivity is α–β–α–α–β–β, and the secondary structure elements are indicated in Figure 4. The domains are arranged in a consecutive manner so that domains IV and V as well as domains VI and VII have approximately the same orientation. In the surfaces between domains IV and V and between domains VI and VII, the β-hairpin from one domain packs against the third α-helix of the following domain. In the interface between domains V and VI, the turn between H4 and S4 in domain V contacts H9 in domain VI as well as the hinge between domains V and VI (between S6 and H7).

Fig. 4. Sequence alignment of the bacterial SelB sequences present in the Swissprot, Trembl and GenBank databases. White, light grey and dark grey boxes indicate increasing degrees of conservation. The positions of compensatory mutations in response to loop mutations are shown by filled arrows (Kromayer et al., 1999). The positions of compensatory mutatons in response to bulge mutations are shown by open arrows (Li et al., 2000). Abbreviations of species names (accession Nos): MOOTH, Moorella thermoacetica (Swissprot Q46455); ECOLI, Escherichia coli (Swissprot P14081); SALEN, Salmonella enterica serovar typhi (GenBank NP_458244); YERPE: Yersina pestis (GenBank 16120353); HAEIN, Haemophilus influenza (Swissprot P43927); PASMU, Pasteurella multocida (Trembl Q9CK68); PSEAE, Pseudomonas aeruginosa (Trembl Q9HV02); RHIME, Rhizobium meliloti (Trembl Q931D5); DESBA, Desulfomicrobium baculatum (Swissprot Q46497); EUBAC, Eubacterium acidaminophilum (Trembl Q9S3K2); AQUAE, Aquifex aeolicus (Trembl O67141); CAMJE, Campylobacter jejuni (Trembl Q9PMS1).
Comparison of domains
Structural alignment of domains IV–V with domains VI–VII using TOP (Lu, 2000) gives a root mean square deviation (r.m.s.d.) of 1.5 Å for 98 residues. These 98 residues (392–426, 433–454, 463–471, 473–476 and 478–505) include all secondary structure elements except the N-terminal helix. The sequence identity over this stretch is 11% and the sequence similarity 32%. Multiple structural alignment of all four domains using the MAPS program (http://bioinfo1.mbfys.lu.se/TOP/maps.html) re sults in the superpositioning shown in Figure 5A. The domains are remarkably similar, the major differences being the length of the helices corresponding to H1 and of the β-hairpin corresponding to S2–S3. Domain IV is structurally most similar to domain VI, while domain V is most similar to domain VII. There are three equivalent structural segments present in all four domains (Figure 5B). These segments are (with numbering from domain IV): amino acids 397–401 (central part of H2), 405–426 (H3–S2) and 433–436 (S3). Within these 31 residues from each of the four domains that superimpose with an r.m.s.d. of 1.6 Å, only two leucine residues in H3 are conserved throughout the four domains. In the hydrophobic core of the domain, Leu413 packs against H1, and Leu414 packs against H2 and S3. The low sequence homology between the four domains explains why their common fold was not detected earlier. It also shows that this is a fold where a large sequence diversity can be tolerated.

Fig. 5. Superpositioning of domains IV–VII using MAPS (http://bioinfo1.mbfys.lu.se/TOP/maps.html). (A) Ribbon model. Colour coding as in Figure 3. The image was prepared using Molmol (Koradi et al., 1996). (B) Aligned sequences for the equivalent structural segments found in all four domains.
Sequence conservation of SelB
A sequence alignment of the 12 bacterial SelB sequences present in the databases is shown in Figure 4. The rather low sequence similarity between SelB from different bacterial species may reflect the same kind of divergence as the low sequence similarity between the four domains. The conserved amino acids are mapped to the structure in Figure 6A and B. The conserved exposed residues are localized in two regions of the protein. Most of the conserved residues are located in the C-terminal part of domain VII, which is rich in basic amino acids, and 11 out of 24 residues are conserved. In domain V, there is a small charged patch next to a small hydrophobic area (H5–H6).

Fig. 6. (A) Location of conserved residues in SelB-C (see Figure 5). SelB-C is seen from below domains VI–VII, in parallel with helix H12, which is surrounded by conserved residues. (B) A 90° rotated view. (C) Positions of single compensatory mutations isolated in response to loop changes in the mRNA hairpin (Kromayer et al., 1999). (D) Positions of single compensatory mutations isolated in response to bulge changes in the mRNA hairpin (Li et al., 2000). The image was prepared using Rasmol (Bernstein, 2000).
Structural comparison with other proteins
The winged-helix motif is a subfamily within the helix– turn–helix family (HTH), which is used extensively in DNA-binding proteins (Gajiwala and Burley, 2000). In searches for structurally similar proteins with DALI (Holm and Sander, 1993) and TOP (Lu, 2000) using all four domains or two domains as search model, no hit is found that displays similarity for more than a single domain. Thus, the double winged-helix with the conserved packing between domains IV and V and domains VI and VII is not found elsewhere in the database. There is only one previous example in the literature of a protein containing two consecutive winged-helix domains, the replication initiator protein Repe54 (Komori et al., 1999). This protein has domain–domain packing with a pseudo 2-fold axis between the domains instead of the translational repeat seen in SelB-C.
Searching with domain VII, the most similar proteins found by DALI are the transcriptional repressor Smtb (Cook et al., 1998) with an r.m.s.d. of 1.6 Å for 60 Cα-atoms, the Z-DNA-binding domain of double-stranded RNA-specific adenosine deaminase (Schwartz et al., 1999) with an r.m.s.d. of 2.0 Å for 58 residues, and human replication protein A (Mer et al., 2000) with an r.m.s.d. of 1.7 Å for 60 residues. There is a large number of winged-helix proteins where >50 residues match, including many DNA-binding proteins but no RNA-binding protein. However, in the large family of HTH proteins, there are examples of RNA-binding proteins.
Location of the RNA-binding site
By deletion mutagenesis, it was shown that the C-terminal fragment of 163 amino acids of E.coli SelB binds to the mRNA hairpin with the same affinity as intact SelB (Kromayer et al., 1996). This was the smallest construct that was assayed for RNA binding. In M.thermoacetica SelB, this fragment corresponds to domains VI–VII with 17 extra N-terminal amino acids. The short N-terminal tail will most probably be unstructured, and domains VI–VII thus constitute the mRNA-binding part of SelB.
The most conserved part of the SelB-C sequence is domain VII (Figures 4 and 6). There are five conserved basic residues (Arg599, Arg606, Lys607, Arg624 and Arg629) and one acidic residue (Asp617). These residues are all located on one side of the domain, defining a surface between helices H11 and H12 and the β-hairpin S11–S12 (Figure 6A and B). The size of this surface, which probably interacts with RNA, is ∼20 × 10 Å along H12. Since the predicted mRNA hairpins are quite diverse in different bacterial species (Figure 1), we predict these conserved amino acids to make mostly backbone contacts with the RNA. Similarly to most of the contacts between ribosomal proteins and rRNA (Brodersen et al., 2002), SelB may rely on surface shape and charge complementarity for RNA recognition.
Sulfate ion suggests a mode of RNA binding
In our structure, Arg599, Ser605 and Arg606 coordinate a sulfate ion (Figure 2). Both arginine residues are conserved. We believe that the sulfate mimics an RNA backbone phosphate and that this is a conserved phosphate-binding site. In SelB from other species, other residues can probably substitute for Ser605. Sulfate ions present in the crystallization buffer often go into phosphate-binding sites (Su et al., 1994) since the two ions are similar. In this case, no sulfate was present intentionally in the crystallization, and the ion must have stayed bound to SelB-C when the sulfate-containing buffer was exchanged for storage buffer (M.Selmer, R.Wilting, D.Holmlund and X.-D.Su, submitted). This phosphate is probably located in the vicinity of the loop or the bulge of the hairpin structure, both of which have been shown by chemical probing to be in contact with SelB (Hüttenhofer et al., 1996). Since the bulge is not conserved throughout SECIS sequences (Figure 1), our best guess is that a phosphate available for interaction would be located in the loop region, and that the loop will be directed towards the sulfate side of domain VII.
The yttrium ions are also bound in this region of the protein, and could mimic cations involved in RNA binding. The yttrium ions are coordinated with five oxygen atoms each, in magnesium-like geometry. Some of the amino acids involved in coordination of the yttrium ions (Asp600 and Asp627) are in close proximity to the conserved residues in domain VII.
Mapping of mutational data on the structure
RNA recognition by SelB also involves sequence-specific interactions. Mutations of the loop nucleotides are detrimental to binding (Heider et al., 1992), while these nucleotides are not conserved in SECIS from many other bacteria (Figure 1). Also, protection from chemical probing has shown that the bulged U as well as the G3–U4 of the loop is involved directly in binding to E.coli SelB (Hüttenhofer et al., 1996). Two different genetic studies looking for compensatory mutations in E.coli SelB when essential elements of the SECIS are mutated have been performed. When the bulged U (Li et al., 2000) or the loop nucleotides (Kromayer et al., 1999) are mutated, most compensatory single mutations are located in domain VII (Figures 4, 6C and D). One exception is E437K, which will be discussed below. None of the mutations involve conserved residues, and only two involve surface-exposed residues. The mutation A569V in E.coli (corresponding to Leu595 in M.thermoacetica) compensates a C5A change in the loop rather specifically, and the mutation M556I (M.thermoacetica Val583) compensates the same change, as well as a U to C change of the bulged nucleotide. Thus, out of these mutations, the only amino acid that could potentially involve a side chain contact with the mutated base is Ala569 in E.coli (M.thermoacetica Leu595). The conclusion of mapping these results on our structure is that there are probably few sequence-specific contacts between protein and RNA.
Some of the mutations, e.g. V578A (M.thermoacetica Ser604) and F572Y (M.thermoacetica Ala598) make subtle changes to the hydrophobic core that may slightly change the orientation of the exposed amino acids contributing to the RNA-binding site, or change the stability of the hydrophobic core. They may adjust the shape complementarity to a mutated RNA fragment. Notably, several mutations are found to be the same in both these studies, despite the fact that different parts of the RNA hairpin are changed (Figure 4), suggesting that these changes make the binding site more tolerant or less specific, unless the different mutations in the RNA change the shape of the RNA similarly.
Winged-helix interaction with RNA
To our knowledge, there are no previous examples in the literature of winged-helix domains that bind RNA. However, there are protein–DNA complex structures available for a number of winged-helix proteins. In the canonical mode of DNA binding, the recognition helix, the third helix in the winged-helix domain, interacts with the major groove of a DNA double-helix (as in the structure of Sap1, Figure 7). These domains can also interact with DNA in different ways, but the recognition helix is always involved (Gajiwala and Burley, 2000). Helix H12 is the corresponding helix in domain VII of SelB (similar view in Figure 6A). This helix is surrounded by conserved basic residues, suggesting that SelB uses the same part of the winged-helix structure for RNA binding as is normally used for DNA binding. However, a SelB–SECIS complex structure is needed to clarify how similar these interactions are.

Fig. 7. Canonical binding of double-stranded DNA to a winged-helix domain in the structure of Sap1 bound to the E74 promoter (Protein Data Bank code 1bc8; Mo et al., 1998). The image was prepared using Rasmol (Bernstein, 2000).
Steric requirements imposed by the ribosome
The distance between the UGA codon and the minimal SelB-binding site in the E.coli selenoprotein mRNAs is 11 nucleotides, and in the M.thermoacetica fdhA mRNA, the corresponding distance is 12 nucleotides. In the recent structure of a 70S ribosome bound to mRNA, the length of the mRNA tunnel between the entrance and A-site codon is ∼7–9 nucleotides (Yusupova et al., 2001). Thus, we envisage that SelB can stay bound to the recognition site on the mRNA until Sec-tRNASec is delivered under GTP hydrolysis to the A-site.
When SelB delivers Sec-tRNASec to the ribosomal A-site, the orientation of its N-terminal part or G-domain before GTP hydrolysis should be very similar to the corresponding state of EF-Tu. The kirromycin-stalled EF-Tu complex, which represents the location of the factor after codon recognition and GTP hydrolysis, but supposedly is similar to the GTP state, has been localized on the 70S subunit by single particle cryo-electron microscopy (Stark et al., 1997). If the ternary complex structure (Nissen et al., 1995) is docked to the A-site of the 70S ribosome (Yusupov et al., 2001) in this position, the distance from the C-terminus of EF-Tu to the entrance of the mRNA tunnel described above is ∼90–100 Å. This corresponds to the distance from the C-terminal end of the EF-Tu homologous part of SelB to the start of the mRNA hairpin when a selenocysteine codon is to be decoded. Thus, SelB-C and the hairpin together have to span a distance of 90–100 Å from domain III of SelB to the mRNA tunnel entrance.
The length of a 17 nucleotide RNA hairpin would be ∼25–30 Å, as judged from other hairpin structures in the Protein Data Bank. Despite the substantial length of the RNA hairpin, SelB-C has to bridge most of the distance, since a direct contact between SelB and the bulged U (Figure 1A) has been shown by chemical footprinting (Hüttenhofer et al., 1996). Thus, the maximum contribution of RNA will be two single-stranded nucleotides and two base pairs of RNA spacer, or ∼10 Å. On the protein side, the EF-Tu-homologous sequence is followed by a stretch of 2–5 basic residues, and a predicted helix before the start of domain IV. The diagonal of the SelB structure is ∼75 Å. Thus, the L-shaped molecule may need to open up to bridge this 90–100 Å distance in complex with the RNA hairpin.
Indications of communication between the tRNA- and mRNA-binding sites of SelB
There are several previous indications of interdomain communication or conformational changes in SelB. In analogy with EF-Tu, SelB has a low intrinsic GTPase activity that is stimulated upon addition of ribosomes even when tRNA is absent (Parmeggiani and Sander, 1981; Hüttenhofer and Böck, 1998). In the presence of ribosomes, but still without tRNA, the addition of a 17 nucleotide SECIS stimulates the GTP hydrolysis of SelB further (Hüttenhofer and Böck, 1998), suggesting that mRNA binding to SelB-C leads to a more active SelB GTPase or a more favourable interaction with the ribosome.
Interdomain communication in the opposite direction, between the tRNA- and mRNA-binding sites, was shown in another study. Isolated domains VI–VII bind more tightly to SECIS than does intact SelB (dissociation constants of 0.14 and 1.26 nM, respectively). When Sec-tRNASec is bound to SelB, the affinity for the SECIS increases to the same level as for the isolated mRNA-binding domains (Thanbichler et al., 2000).
Thus, the binding of Sec-tRNASec to the N-terminal part and SECIS to the C-terminal part of SelB affect each other. One possibility is that the two parts interfere with each other’s function in the absence of RNA. The other possibility is that RNA binding induces conformational changes in one part that contribute favourably to the activity of the other part in terms of GTPase activity or mRNA binding, respectively.
In a SELEX (systematic evolution of ligands by exponential enrichment) study selecting RNA fragments that would bind to SelB (Klug et al., 1997), several sequences that bound with similar affinity to SelB, and as judged by chemical and enzymatic probing data in a similar binding mode, did not promote selenocysteine read through. There was no clear connection between affinity and function. Rather, a specific SelB-mRNA interaction seemed to be needed to trigger a conformational change necessary to achieve UGA read through, or to orientate SelB in a proper way for functional interaction with the ribosome. In agreement with this, it was shown that overproduction of SelB and the other components of the selenocysteine insertion machinery fails to induce any detectable selenocysteine incorporation in the absence of the proper hairpin structure on the mRNA (Suppmann et al., 1999). Thus, the function of the mRNA hairpin is more than increasing the local concentration of SelB in proximity to the stop codon. Some kind of conformational change seems to be induced by the proper SelB–SECIS interaction.
Structural indication of switch
What kind of conformational change can happen in SelB upon mRNA binding? The most likely site for a conformational change is the elbow of the L-shaped structure. It is possibly flexible, as indicated by the small contact area between domains V and VI. The surface area that is buried in this interaction is 383 Å2 per domain as compared with the contact areas between domains IV and V, and VI and VII, where 655 Å2 and 706Å2 of the domain area is buried, respectively (calculated using areaimol; CCP4, 1994).
In our structure, there is a salt bridge between Nε of Arg461 and a carboxyl oxygen atom of Glu552 across the cleft between domains V and VI. These two residues are not conserved throughout the bacterial sequences, but they display a pattern of co-variation so that the salt bridge is conserved. It can be seen in Figure 4 that when residue 460 or 461 is negatively charged, 551 or 552 is positively charged, and vice versa. In genetic studies (Kromayer et al., 1999), it has been shown that a mutation altering the charge of residue 461 (E437K in E.coli, corresponding to M.thermoacetica Arg461) makes SelB less stringent, allowing near-perfect SECIS to work in vivo. This may suggest that mRNA binding leads to a hinge movement, which in turn may signal to the N-terminal part of SelB that mRNA is bound. A hinge movement would cost less energy when the salt bridge is absent (mutational data), and therefore a near-perfect mRNA–protein interaction could be tolerated to promote read through.
Functional model
Summarizing our interpretation of the SelB-C structure in the light of the available literature data, we can conclude with a functional model (Figure 8). As depicted in this figure, SelB binds to GTP and Sec-tRNASec in solution. It binds further to the SECIS element with subnanomolar affinity (Thanbichler et al., 2000), using mainly domain VII. This induces a conformational change of SelB (Klug et al., 1997; Hüttenhofer and Böck, 1998) that may involve an opening of the hinge between domains V and VI. This conformational change is indicated by the large distance that SelB-C has to bridge from the tRNA-binding domains to the mRNA hairpin, and by the effect of breaking a salt bridge across the interdomain cleft. When the selenocysteine UGA codon reaches the A-site of a translating ribosome, the quaternary complex of SelB with GTP and Sec-tRNASec is in the proper conformation and position to achieve codon–anticodon interaction. When a correct codon–anticodon match occurs, GTP is hydrolysed and tRNA is released. In the absence of tRNA, SelB has lower, but still nanomolar, affinity for the mRNA hairpin (Thanbichler et al., 2000), and the combined translocation and unwinding of the mRNA may be needed to release it from SelB.

Fig. 8. Proposed model for SelB function in stop codon read through. (A) SelB binds GTP. (B) SelB·GTP binds Sec-tRNASec. (C) SelB·GTP·Sec-tRNASec binds to an mRNA hairpin in a selenoprotein gene. Upon binding, SelB-C undergoes a conformational change so that the L is opened. SelB is ready to deliver Sec-tRNASec to the A-site of the approaching translating ribosome. (D) When the UGA codon is available in the A-site, SelB-N swings in the tRNA. If a correct codon–anticodon match occurs, GTP is hydrolysed. (E) SelB changes conformation to the GDP form and releases the tRNA that is accommodated in the peptidyl-transfer site. (F) SelB leaves the mRNA hairpin, perhaps simultaneously with the combined unwinding and translocation of mRNA.
In conclusion, the structure of SelB-C shows that winged-helix domains can be used for RNA binding and that the mRNA-binding site is located in the part most distant from the tRNA-binding site. Furthermore, selenocysteine incorporation most probably involves a conformational change between domains V and VI.
Materials and methods
Protein preparation and crystallization
Selenomethionine-substituted SelB was prepared using the methionine pathway inhibition method (Van Duyne et al., 1993). Purification and crystallization were performed as described (M.Selmer, R.Wilting, D.Holmlund and X.-D.Su, submitted). The crystals belong to space group P212121 with the cell dimensions a = 37.84 Å, b = 67.01 Å, c = 105.36 Å, and contain one molecule per asymmetric unit.
X-ray data collection and structure solution
X-ray data were collected under cryo conditions (M.Selmer, R.Wilting, D.Holmlund and X.-D.Su, submitted) on a single crystal at BW7A, EMBL outstation, DESY, Hamburg using a 165 mm marCCD detector. Fluorescence scans were used to find the absorption edges for yttrium and selenium. Data were collected at peak and inflection point wavelengths of both elements plus at a low energy remote wavelength and a remote wavelength between the two peaks. Data were indexed and integrated using Denzo and scaled using Scalepack (Otwinowski and Minor, 1997). Data collection statistics are shown in Table I. Two yttrium sites and one selenium site were found using Solve (Terwilliger and Berendzen, 1999). Phase calculation to 3.5 Å, where good anomalous signal existed, and cross checking of the sites in anomalous Fourier maps were performed in CNS (Brünger et al., 1998). Using the same program, yttrium and selenium phases were combined, and solvent flipping and phase extension to 2.12 Å were performed to improve the quality of the experimental map. Phasing statistics are summarized in Table II.
Model building and refinement
Model building was performed with the program O (Jones et al., 1991). About 75% of the residues could be built in the experimental map. After cycles of simulated annealing and combination of model and experimental phases, the full chain except the first nine amino acids could be built. The coordinates and individual restrained B-factors were refined against the mlhl target (maximum-likelihood with Hendrickson–Lattman coefficients) with CNS (Brünger et al., 1998) and, finally, water molecules were added. Refinement statistics are summarized in Table III. Difference densities possibly indicate double conformations of some side chains but, due to the limited resolution, no double conformations were modelled.
The atomic coordinates and structure factors have been deposited in the RCSB Protein Data Bank with accession number 1lva.
Sequence alignment
Sequence alignment was performed with Clustal X (Thompson et al., 1997) and presented with Alscript (Barton, 1993). The alignment was edited manually to avoid gaps inside the conserved structural elements (Figure 5B).
Acknowledgments
Acknowledgements
We thank Christopher Enroth and Emkhe Pohl, EMBL, Hamburg for help during data collection, and Professor Anders Liljas for stimulating discussions and valuable comments on the manuscript. M.S. and X.-D.S. are recipients of financial support from the Swedish foundation for Strategic research through SBNet. This work was supported by grants from the Swedish Research Council (NFR) to Anders Liljas.
References
- Barton G.J. (1993) ALSCRIPT: a tool to format multiple sequence alignments. Protein Eng., 6, 37–40. [DOI] [PubMed] [Google Scholar]
- Bernstein H.J. (2000) Recent changes to RasMol, recombining the variants. Trends Biochem. Sci., 25, 453–455. [DOI] [PubMed] [Google Scholar]
- Brodersen D.E., Clemons,W.M., Carter,A.P., Wimberly,B.T. and Ramakrishnan,V. (2002) Crystal structure of the 30S ribosomal subunit from Thermus thermophilus: structure of the proteins and their interactions with 16S RNA. J. Mol. Biol., 316, 725–768. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Cook W.J., Kar,S.R., Taylor,K.B. and Hall,L.M. (1998) Crystal structure of the cyanobacterial metallothionein repressor SmtB: a model for metalloregulatory proteins. J. Mol. Biol., 275, 337–346. [DOI] [PubMed] [Google Scholar]
- Forchhammer K., Leinfelder,W. and Böck,A. (1989) Identification of a novel translation factor necessary for the incorporation of seleno cysteine into protein. Nature, 342, 453–456. [DOI] [PubMed] [Google Scholar]
- Gajiwala K.S. and Burley,S.K. (2000) Winged helix proteins. Curr. Opin. Struct. Biol., 10, 110–116. [DOI] [PubMed] [Google Scholar]
- Heider J., Baron,C. and Böck,A. (1992) Coding from a distance: dissection of the mRNA determinants required for the incorporation of selenocysteine into protein. EMBO J., 11, 3759–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L. and Sander,C. (1993) Protein structure comparison by alignment of distance matrices. J. Mol. Biol., 233, 123–138. [DOI] [PubMed] [Google Scholar]
- Hüttenhofer A. and Böck,A. (1998) Selenocysteine inserting RNA elements modulate GTP hydrolysis of elongation factor SelB. Biochemistry, 37, 885–890. [DOI] [PubMed] [Google Scholar]
- Hüttenhofer A., Westhof,E. and Böck,A. (1996) Solution structure of mRNA hairpins promoting selenocysteine incorporation in Escherichia coli and their base-specific interaction with special elongation factor SELB. RNA, 2, 354–366. [PMC free article] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.-Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Klug S.J., Hüttenhofer,A., Kromayer,M. and Famulok,M. (1997) In vitro and in vivo characterization of novel mRNA motifs that bind special elongation factor SelB. Proc. Natl Acad. Sci. USA, 94, 6676–6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori H., Matsunaga,F., Higuchi,Y., Ishiai,M., Wada,C. and Miki,K. (1999) Crystal structure of a prokaryotic replication initiator protein bound to DNA at 2.6 Å resolution. EMBO J., 18, 4597–4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koradi R., Billeter,M. and Wuthrich,K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graphics, 14, 51–55. [DOI] [PubMed] [Google Scholar]
- Kromayer M., Wilting,R., Tormay,P. and Böck,A. (1996) Domain structure of the prokaryotic selenocysteine-specific elongation factor SelB. J. Mol. Biol., 262, 413–420. [DOI] [PubMed] [Google Scholar]
- Kromayer M., Neuhierl,B., Friebel,A. and Böck,A. (1999) Genetic probing of the interaction between the translation factor SelB and its mRNA binding element in Escherichia coli. Mol. Gen. Genet., 262, 800–806. [DOI] [PubMed] [Google Scholar]
- Li C., Reches,M. and Engelberg-Kulka,H. (2000) The bulged nucleotide in the Escherichia coli minimal selenocysteine insertion sequence participates in interaction with SelB: a genetic approach. J. Bacteriol., 182, 6302–6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G. (2000) TOP: a new method for protein structure comparisons and similarity searches. J. Appl. Crystallogr., 33, 176–183. [Google Scholar]
- Mer G., Bochkarev,A., Gupta,R., Bochkareva,E., Frappier,L., Ingles, C.J., Edwards,A.M. and Chazin,W.J. (2000) Structural basis for the recognition of DNA repair proteins UNG2, XPA and RAD52 by replication factor RPA. Cell, 103, 449–456. [DOI] [PubMed] [Google Scholar]
- Mo Y., Vaessen,B., Johnston,K. and Marmorstein,R. (1998) Structures of SAP-1 bound to DNA targets from the E74 and c-fos promoters: insights into DNA sequence discrimination by Ets proteins. Mol. Cell, 2, 201–212. [DOI] [PubMed] [Google Scholar]
- Nissen P., Kjeldgaard,M., Thirup,S., Polekhina,G., Reshetnikova,L., Clark,B.F.C. and Nyborg,J. (1995) Crystal structure of the ternary complex of Phe-tRNA-Phe, elongation factor Tu and a GTP analogue. Science, 270, 1464–1472. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. In Carter,C.W.,Jr and Sweet,R.M. (eds), Macromolecular Crystallography, Part A. Academic Press, New York, NY, pp. 307–326. [DOI] [PubMed]
- Parmeggiani A. and Sander,G. (1981) Properties and regulation of the GTPase activities of elongation factors Tu and G and of initiation factor 2. Mol. Cell. Biochem., 35, 129–158. [DOI] [PubMed] [Google Scholar]
- Schwartz T., Rould,M.A., Lowenhaupt,K., Herbert,A. and Rich,A. (1999) Crystal structure of the Zα domain of the human editing enzyme ADAR1 bound to left-handed Z-DNA. Science, 284, 1841–1845. [DOI] [PubMed] [Google Scholar]
- Stark H., Rodnina,M.V., Rinke-Appel,J., Brimacombe,R., Wintermeyer,W. and van Heel,M. (1997) Visualization of elongation factor Tu on the Escherichia coli ribosome. Nature, 389, 403–406. [DOI] [PubMed] [Google Scholar]
- Su X.D., Taddei,N., Stefani,M., Ramponi,G. and Nordlund,P. (1994) The crystal structure of a low-molecular-weight phosphotyrosine protein phosphatase. Nature, 370, 575–578. [DOI] [PubMed] [Google Scholar]
- Suppmann S., Persson,B.C. and Böck,A. (1999) Dynamics and efficiency in vivo of UGA-directed selenocysteine insertion at the ribosome. EMBO J., 18, 2284–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger T.C. and Berendzen,J. (1999) Automated MAD and MIR structure solution. Acta Crystallogr. D, 55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanbichler M., Böck,A. and Goody,R.S. (2000) Kinetics of the interaction of translation factor SelB from Escherichia coli with guanosine nucleotides and selenocysteine insertion sequence RNA. J. Biol. Chem., 275, 20458–20466. [DOI] [PubMed] [Google Scholar]
- Thompson J.D., Gibson,T.J., Plewniak,F., Jeanmougin,F. and Higgins,D.G. (1997) The ClustalX Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res., 25, 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Duyne G.D., Standaert,R.F., Karplus,P.A., Schreiber,S.L. and Clardy,J. (1993) Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol., 229, 105–124. [DOI] [PubMed] [Google Scholar]
- Yusupov M.M., Yusupova,G.Z., Baucom,A., Lieberman,K., Earnest,T.N., Cate,J.H. and Noller,H.F. (2001) Crystal structure of the ribosome at 5.5 Å resolution. Science, 292, 883–896. [DOI] [PubMed] [Google Scholar]
- Yusupova G.Z., Yusupov,M.M., Cate,J.H. and Noller,H.F. (2001) The path of messenger RNA through the ribosome. Cell, 106, 233–241. [DOI] [PubMed] [Google Scholar]
- Zuker M., Mathews,D.H. and Turner,D.H. (1999) Algorithms and thermodynamics for RNA secondary structure prediction: a practical guide. In Barciszewski,J. and Clark,B.F.C. (eds), RNA Biochemistry and Biotechnology, Kluwer Academic Publications, Dordrecht, The Netherlands, pp. 11–43. [Google Scholar]