

Abstract
The frequency of DNA transposition in transposition systems that employ a strand transfer step may be significantly affected by the occurrence of a disintegration reaction, a reaction that reverses the strand transfer event. We have asked whether disintegration occurs in the Tn10 transposition system. We show that disintegration substrates (substrates constituting one half of the strand transfer product) are assembled into a transpososome that mimics the strand transfer intermediate. This strand transfer transpososome (STT) does appear to support an intermolecular disintegration reaction, but only at a very low level. Strikingly, assembly of the STT is not dependent on IHF, a host protein that is required for de novo assembly of all previously characterized Tn10 transpososomes. We suggest that disintegration substrates are able to form both transposon end and target type contacts with transposase because of their enhanced conformational flexibility. This probably allows the conformation of DNA within the complex that prevents the destructive disintegration reaction, and is responsible for relaxing the DNA sequence requirements for STT formation relative to other Tn10 transpososomes.
Keywords: disintegration/DNA transposition/strand transfer/Tn10/transposase
Introduction
Many DNA transposition reactions involve a strand transfer step in which the 3′-OH termini of cleaved transposon DNA are joined to closely situated phosphate groups located on opposite strands of a target DNA molecule. This results in the insertion of the transposon into a new target site. However, in some of these systems, reversal of the strand transfer reaction, a process termed disintegration, has been shown to be a fairly robust reaction in vitro (Chow et al., 1992; Melek et al., 1998). In fact, given the thermodynamics that govern transposition reactions, if only the chemistry of the reaction is considered, there is no a priori reason for thinking that disintegration would not occur in transposition reactions that go through a strand transfer intermediate (Mizuuchi, 1997). It follows that there may have evolved specific mechanisms for preventing disintegration. Such mechanisms would be expected to significantly impact on the overall transposition frequency by protecting against the destruction of the strand transfer intermediate. Given the many levels of negative regulation that have been documented in transposition systems, it may be critical for transposon survival that once a transposition event is initiated there stands a high probability of the reaction going to completion. Tn10 is one of the most tightly negatively regulated bacterial transposition systems documented to date (Kleckner, 1989), and in our current work, we are interested in assessing whether the Tn10 strand transfer product is subject to disintegration.
Tn10 transposes by a cut-and-paste mechanism wherein the transposon is first cleanly excised from flanking donor DNA and then inserted by strand transfer into a new site (Bender and Kleckner, 1986; Haniford et al., 1991). Transposition involves four distinct chemical steps at each transposon end (Kennedy et al., 1998) and the available evidence indicates that each of these steps is catalyzed in precise succession by the single active site in the transposon-encoded transposase protein (Bolland and Kleckner, 1996; Kennedy et al., 2000). Prior to the initiation of the first chemical step, the two transposon end sequences synapse together to form a transpososome or ‘paired ends complex’ (PEC) (Sakai et al., 1995). This pairing reaction is mediated by molecules of transposase that bind to two transposon end sequences. If one of these ends is an outside end, then the Escherichia coli protein IHF can play an important role in transpososome assembly (Morisato and Kleckner, 1987; Sakai et al., 1995; Chalmers et al., 1998). IHF binds to a site located between ‘terminal’ and ‘subterminal’ transposase-binding sites and induces a bend in the DNA that permits transposase contacts to be made at both these sites (Figure 1A). This facilitates PEC formation presumably by stabilizing the transposase outside end interaction (Sakai et al., 2000; Crellin and Chalmers, 2001). Molecular modeling further suggests that while IHF can bind to both outside end sequences in a Tn10 PEC, transposase would make contacts with subterminal transposase-binding sites at one end at a time only (Crellin and Chalmers, 2001). Nevertheless, the IHF would permit the initial PEC to adopt a highly folded structure. This has in fact been confirmed by gel electrophoresis studies, where it was shown that the IHF-containing PEC has a significantly greater mobility than a PEC in which the IHF has been removed and thus unfolded (Sakai et al., 2000). Folded and unfolded forms of the PEC are referred to as bPECs and tPECs, respectively.

Fig. 1. Structures of IS10, the strand transfer intermediate, S-53 and possible disintegration products. (A) The structure of IS10-Right is shown, including the positions of the outside (OE) and inside (IE) ends. Black boxes represent terminal and subterminal transposase-binding sites with the former being closest to the outside end terminus and the open box represents the IHF-binding site. Half arrows indicate the 23 bp terminal inverted repeat. (B) The strand transfer reaction is shown along with one possible type of disintegration reaction. TS and NTS are transferred and non-transferred strands, respectively. (C) The sequence and expected conformation of the S-53 disintegration substrate is shown. The primary transposase-binding site and the HisG1 target core sequences are highlighted by boxes. S-92 is identical to S-53 except that it contains an additional 39 bp of the outside end. (D) Transesterification of the 3′OH of the target extension can be directed towards the transposon end-target junction of the same molecule generating a FB product or to the partner molecule generating SDs or DDs. Hydrolysis can also result in the separation of transposon end and target sequences. Short lines identify the position of paired and unpaired bases.
Outside end substrates that contain a nick at position 1 of the transferred strand, or no flanking DNA at all, still require IHF in order to be assembled into transpososomes (Bolland and Kleckner, 1996; Kennedy et al., 1998; Allingham et al., 2001). Thus, it is not possible to relieve the IHF requirement for transpososome assembly by simply bypassing any of the chemical steps. These results also indicate that transposase contacts with flanking donor DNA is not essential for transpososome assembly. While IHF is required for initial transpososome assembly, it must be removed from the PEC in order for the PEC to be able to participate in intermolecular target capture (Junop and Haniford, 1997; Sakai et al., 2000). Presumably, the subterminal contacts interfere with target DNA binding.
Strand transfer results in the formation of a transpososome in which each of the 3′ ends of the transposon is joined to a portion of the target DNA (Figure 1B). The strand transfer transpososome contains two 9 nucleotide (nt) gaps at each of the transposon–target DNA junctions (Benjamin and Kleckner, 1989). In the final stage of transposition, these gaps are repaired by host functions, generating a duplication of the original target core sequence. Thus, a portion of the target DNA becomes the flanking donor DNA in the next transposition event. Detailed characterization of the strand transfer transpososome (STT) has not been carried out in the Tn10 system previously, although transposase interactions with target DNA in the target capture complex have been defined by DNA footprinting and interference techniques. Trans posase appears to contact ∼25 bp of target DNA, including the target core and 8 bp on either side of the core. It is likely that contact over such a relatively large DNA segment is facilitated by the introduction of a pair of symmetrically localized kinks in the target DNA (Pribil and Haniford, 2000).
In our current work, we examined the ability of Tn10 to undergo a disintegration reaction using substrates analogous to those that have previously been used to detect disintegration in retroviral integration and recombination activating gene (RAG)-mediated transposition systems (Chow et al., 1992; Chow and Brown, 1994; Melek and Gellert, 2000). We provide evidence that while Tn10 disintegration substrates are assembled into a transpososome that mimics the strand transfer intermediate, this transpososome has very limited reactivity. Importantly, transpososome assembly in this system is not IHF dependent, providing the first example of a Tn10 transpososome that does not require IHF for de novo assembly. A detailed structural and functional characterization of this novel Tn10 transpososome is presented.
Results
Disintegration substrates
To test for disintegration in the Tn10 system, we designed substrates (Figure 1C) that, when assembled into a higher order protein–DNA complex, would mimic the Tn10 strand transfer intermediate. Substrates S-92 and S-53 contain 65 and 26 bp of the outside end of IS10-Right, respectively, plus a 27 nt extension on the ‘transferred’ strand. S-92 but not S-53 contains the full outside end sequence required for transpososome assembly in the linear fragment assay. This includes the IHF-binding site and the sub-terminal transposase-binding site. The 27 nt extension contains part of the HisG1 hotspot for Tn10 insertion (Halling and Kleckner, 1982). This is expected to generate an 8 bp duplex immediately adjacent to a 9 nt single-strand segment, a conformation whose formation is assisted by the inclusion of a T-T dinucleotide capable of facilitating hairpin formation (Blommers et al., 1989). The substrate S(T-D)-92 contains 65 bp of the outside end and ∼29 bp of flanking donor DNA. This is a standard substrate for bPEC formation and provides a point of comparison with respect to the dependencies of transpososome formation and chemical reactivity for the various substrates.
A number of chemical reactions could theoretically be supported by the STT. This includes three different disintegration products (Figure 1D). Two of these, the single disintegrant (SD) and the double disintegrant (DD), arise from intermolecular transesterification events. The third, the fold back (FB) product results from an intramolecular transesterification event. In addition, hydrolysis may occur instead of disintegration. All three disintegration products described in Figure 1D have been detected previously at reasonably high levels in the HIV system, where substrates similar to those described here were employed (Chow and Brown, 1994; Mazumder et al., 1994).
Disintegration analysis
All of the chemical steps in Tn10 transposition take place in the context of a transpososome and it was therefore important to establish that our disintegration substrates are assembled into a higher order complex. In Figure 2A, we show that both S-92 and S-53 are able to form a higher order complex, and that in each case the formation of this complex is not dependent on the presence of IHF (compare lanes 7 and 8, and lanes 11 and 12). This is in contrast to the situation for S(T-D)-92, where transpososome formation is almost completely dependent on IHF (compare lanes 3 and 4). Notably, the mobility of the S-92 complex is comparable to that of the S(T-D)-92 transpososome and the mobility of the S-53 complex is only slightly faster. These results are consistent with S-53 and S-92 being assembled into transpososomes in an IHF-independent manner. Also, assembly efficiencies are comparable to that of pre-cleavage transpososomes where typically 10–30% of starting substrate is converted to transpososome (Sakai et al., 1995; Allingham et al., 2001). Interestingly, the mobility of the S-92 complex is the same with or without IHF, indicating that an IHF-independent assembly pathway for higher order complex formation is the predominant assembly pathway. Further evidence that transpososome assembly is occurring with disintegration substrates is presented below.

Fig. 2. Complex formation with disintegration substrates and disintegration activity. (A) The indicated substrates were mixed with either IHF, transposase or IHF + transposase as described in Materials and methods, and analyzed on a native 5% polyacrylamide gel. Positions of S-92 and S-53 higher order complexes are indicated along with the bPEC, IHF complex and unreacted substrate (URS). The set of bands within the bracket (lane 4) has not been characterized and could be indicative of an extremely low level of transpososome formed in the absence of IHF. (B) PEG precipitated S-53 complex or S(T-D)-92 PEC was incubated in reaction buffer either without a divalent metal ion or with MgCl2 (5 mM) or MnCl2 (0.18 mM) as indicated. Reactions were terminated by phenol extraction and the purified DNA analyzed on an 8% denaturing gel. S-53 DNA was labeled at the 5′ terminus of the transferred strand with 32P and thus it was only possible to detect SD and cleaved end products. S(T-D)-92 DNA was 5′ end labeled with 32P on both transferred and non-transferred strands and transposase-directed cleavage generates a flanking donor fragment of 31 nt and a transposon fragment of 59 nt. FD is flanking donor DNA.
Initially, we looked for evidence of disintegration using substrates labeled with 32P at the 5′ terminus of the transferred strand. Labeling in this way permits detection of SD and/or cleaved ends (CEs), but not DD or FB (Figure 1D). Notably, in the HIV system, SD and DD are formed at roughly equivalent levels after 90 min (Chow and Brown, 1994). To optimize the sensitivity of the reaction, we selectively precipitated the S-53 complex with polyethylene glycol. The solubilized complex was then incubated for 2 or 16 h in reaction buffer containing either MgCl2 or MnCl2. Reactions were analyzed on a sequencing-type denaturing gel (Figure 2B). The SD and CE were expected to be 80 and 26 nt in length, respectively. A species of the size expected for SD was observed with both Mg2+ and Mn2+. At 2 h, ∼0.2% of input complex was converted to this species in the Mg2+ reaction, while there was an ∼5% conversion in the Mn2+ reaction. At 16 h, putative SD increased ∼5-fold to 1% of total input complex in the Mg2+ reaction. At both time points, there was ∼4-fold more CE than putative SD, indicating that hydrolysis is the more efficient reaction. Notably, this level of hydrolysis is very low compared with what is typically seen with a standard substrate such as S(T-D)-92, where up to 85% of PEG-enriched complex underwent double-strand cleavage. In contrast, the amount of putative SD and CE is roughly the same after 2 h in the Mn2+ reaction, suggesting that disintegration is the only significant reaction taking place here. At 16 h, the putative SD level dropped and the CE level increased. This could result from conversion of SD to DD as only the former would be detected in this assay, and this would coincide with an increase in the CE level.
In comparison, SD formation in the HIV system in the presence of Mn2+, and in the V(D)J system in the presence of Mg2+, can constitute up to 20% of total input substrate (Chow and Brown, 1994; Melek and Gellert, 2000). No significant increase in reactivity was observed in our analysis when the assembly reaction contained both S-53 and S-53* (data not shown). The latter is identical to S-53 except that the target core of S-53* is the complementary sequence of the target core of S-53; consequently, it is possible for the two substrates to form base pairs in their core regions. Similarly, substrate complementarity failed to increase the efficiency of disintegration in the HIV system (Chow and Brown, 1994).
We have also carried out a two dimensional gel electrophoresis experiment to evaluate the reactivity of the S-53 complex. In this approach, the S-53 complex is separated from unreacted substrate in the first (native) dimension and then the gel strip is soaked in Mg2+ buffer and electrophoresis is carried out in a second (denaturing) dimension. Thus, any products arising from the S-53 complex will have the same mobility as the S-53 complex in the first dimension. In this case, the substrate contained an internal 32P label in the target extension, as well as a 5′ end label on the transferred strand. This labeling scheme allowed detection of FB and DD products as well as SD and CE. We also carried out this experiment without enrichment of the S-53 complex by PEG precipitation a step that, in principle, could selectively affect the reactivity of this complex.
The results of such an experiment are presented in Figure 3. In one control experiment, Mg2+ was left out of the soak buffer (–Mg2+) and in another, the S(T-D)-92 substrate containing a 5′ end label on both DNA strands was used in the assembly reaction. The latter provided an indication of the efficiency of ‘in-gel’ cleavage. There is a light spot above the S-53 complex that is only seen in the presence of Mg2+, consistent with the low level of putative SD formation in Figure 2B. In addition, there is a spot of similar intensity below the S-53 complex at the expected position for the CE fragment. There was no evidence of a 27 nt circle, which would be indicative of FB formation; this species is expected to run slower in the second dimension than a 26 nt linear fragment (Chow and Brown, 1994; Mazumder et al., 1994). It is less clear whether the DD product is formed because it is possible that the corresponding 54 nt circle would co-migrate with the 53 nt linear fragment. The efficiency of in-gel cleavage is estimated at 25% as judged by the amount of CE generated from the bPEC (note that the CE contains half the number of end labels as the bPEC).

Fig. 3. Two-dimensional gel assay for disintegration products. The indicated assembly reactions were subjected to native gel electrophoresis in the absence of MgCl2 treatment. Gel strips were excised, soaked in reaction buffer with or without MgCl2 as indicated, and then polymerized to a second gel. Electrophoresis, in a second dimension, was then carried out under denaturing conditions. Labels on the top of each gel indicate the position of various species in the first dimensions of electrophoresis. Light spots above and below the S-53 complex have mobilities expected for the SD and CE, respectively. Asterisks indicate cleavage that took place during assembly of substrate.
Taken together, the results in this section demonstrate that complexes formed with Tn10 disintegration substrates are much less reactive than the corresponding complexes formed with the analogous retroviral and V(D)J disintegration substrates (Chow and Brown, 1994; Melek and Gellert, 2000). Also, of the limited reactivity observed, intermolecular transesterification appears to be favored over intramolecular transesterification, and can be as efficient as hydrolysis.
S-53 and S-92 complexes are transpososomes
We were interested in further characterizing the S-53 and S-92 complexes for a number of reasons. First, the conclusion that Tn10 does not support an efficient disintegration reaction depends upon knowing whether, in the above reactions, the strand transfer intermediate is assembled. Secondly, prior to the work presented here, it has not been possible to form significant amounts of a Tn10 transpososome de novo in the absence of IHF.
We examined the ability of S-53 and S-92 to participate in transpososome formation by performing assembly reactions with an equimolar mixture of 5′ end-labeled S-53 and S-92 substrates. If these substrates are capable of forming a transpososome, then substrate mixing should result in the formation of a higher order complex with a novel mobility in a native gel relative to reactions containing only S-53 or S-92. The results presented in Figure 4 show that ‘substrate mixing’ results in the formation of a novel higher order complex, which has a mobility intermediate to the higher order complexes formed in reactions with S-53 or S-92 alone. To prove directly that the complex with intermediate mobility actually contains both S-53 and S-92, bands in lane 5 were cut out of the gel and the purified DNA was run on a denaturing gel. The results presented in Figure 4B (lane 3) clearly show that both S-53 and S-92 are components of the intermediate mobility species. It is also evident from the relative intensities of S-53 and S-92 that the two substrates are present in the complex at close to a 1:1 ratio; the labeling efficiency of the two substrates was not significantly different (data not shown). We therefore conclude that disintegration substrates assemble into a transpososome when mixed with transposase and hereafter refer to this transpososome as the STT.
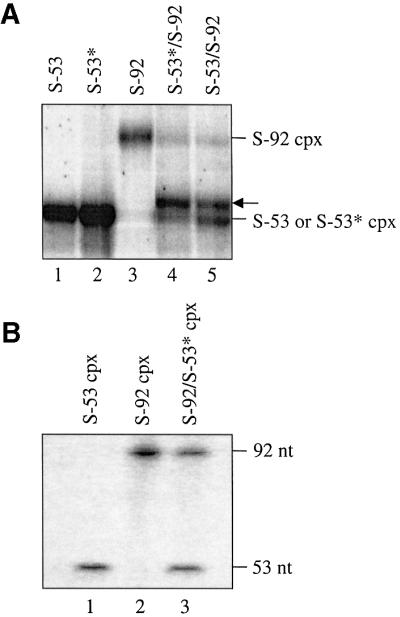
Fig. 4. Complex formation with mixtures of disintegration substrates. (A) Assembly reactions containing the indicated substrate or an equimolar mixture of substrates were analyzed on a 5% native polyacrylamide gel in the absence of a divalent metal ion. S-53* and S-92 contain complementary target cores while S-53 and S-92 contain identical target cores. The position of the hybrid complex is indicated with an arrow. Only the portion of the gel containing higher order complexes is shown. (B) DNA was eluted from the indicated bands of the native gel in (A), purified and subjected to electrophoresis on an 8% denaturing polyacrylamide gel.
S-53 and S-92 contain identical core sequences, and we were interested in determining whether STT formation is enhanced by substrates containing complementary core sequences. Presumably a short double helix formed between single-strand regions could help stabilize the transpososome. In Figure 4A (lane 4), we show that when S-92 was mixed with S-53*, the hybrid STT was formed almost exclusively. Thus, while STT formation is not dependent on complementary base pairing between target cores, the ability to form such base pairs may facilitate STT formation. However, as noted previously the potential for base pairing of target cores does not influence the reactivity of the STT.
DNA sequence dependencies for STT formation
To further characterize the STT, we investigated the DNA sequence dependencies of its formation. We generated a number of variants of S-53 and tested these substrates for STT formation and, in some cases, chemical reactivity. Effects of these changes on STT formation are shown in Figure 5, where levels of STT are presented as a percentage of total input substrate. Several important features of STT formation are revealed by this analysis and are summarized below.

Fig. 5. DNA sequence dependencies for STT formation. S-53 and variants of this substrate were mixed with transposase in the absence of a divalent metal ion, and the ability of these substrates to form STT was determined as in Figure 2A. STT levels are presented as the percentage of total input substrate converted to STT. Asterisks indicate the approximate position of base pair substitutions. S-53 OE-S, OE-D and OE-T contain single (9TA to CG), double (8AT to CG, 9TA to CG) and triple (8AT to CG, 9TA to CG, 13CG to TA) mutations, respectively, in the outside end. S-53 TC-T contains a triple mutation (–2AT to TA, –3CG to GC, –8CG to AT) in the target core. Substrates are illustrated as in Figure 1D.
As expected, STT formation is sensitive to the DNA sequence in the primary transposase-binding site, the region between base pairs 6–13, as single (S-53 OE-S), double (S-53 OE-D) and triple (S-53 OE-T) mutations in this region all significantly decreased the level of STT formed. However, it is interesting that mutations in this region did not have as severe an effect on STT formation as on bPEC formation. For example, the single mutation at base pair 9 in S-53 OE-S reduced STT formation by about one-third of the wild-type level, whereas this same mutation completely inhibited bPEC formation (Sakai et al., 1995). Formation of the STT is also much less dependent on the sequence of the target core than is the formation of the target capture transpososome. S-53 TC-T contains three base pair changes in the conserved region of the target core and this was found to have no effect on STT formation. In contrast, any one of the three mutations present in S-53 TC-T is sufficient to significantly block formation of the target capture transpososome (P.Pribil and D.Haniford, unpublished results). Together, these results suggest that STT formation is less dependent on sequence-specific contacts than the formation of other Tn10 transpososomes is.
While the sequence of the target core does not appear to be important in STT formation, the length of the target core, as well as the target arm, is important. Complete removal of the arm flanking the target core, as in S-35, reduced STT formation by 32-fold, whereas retaining 9 nt of the arm, as in S-43, reduced STT formation by 3.5-fold. A contributing factor to the reduced levels of STT supported by S-35 and S-43 may be the absence of the bottom strand of the target.
We also asked whether it is important to maintain the 9 nt of single-stranded DNA by extending the bottom strand. S-56, S-59 and S-62 all exhibited significant reductions in SST levels (ranging from 3.2- to 8-fold), indicating that maintaining the 9 nt gap is relatively important. Taken together, the above results indicate that STT formation has unique dependencies with regard to transposase–DNA interactions, in accord with this transpososome being distinct from other transpososomes in the Tn10 pathway.
The observation that substrates with longer 3′ termini than S-53 do form STT, albeit at reduced levels, gave us the opportunity to test whether the low reactivity of the S-53 transpososome might be due to suboptimal positioning of the 3′ terminus. Thus, we assembled 5′ end-labeled S-56, S-59 and S-62 into STTs and tested these STTs (without enrichment) for reactivity in the presence of MgCl2 on a denaturing gel. We failed to detect any reaction products with these substrates after a 2 h incubation (data not shown), consistent with the idea that low reactivity of STTs is not simply due to inappropriate positioning of the 3′-OH terminus of the target extension.
Mapping of protein–DNA contacts within the STT
We have obtained more detailed information regarding transposase–DNA contacts within the STT by performing 1,10-phenathroline-copper (or OP-Cu) chemical nuclease and P1 nuclease footprinting on the STT. The OP-Cu reagent intercalates into the minor groove of DNA and initiates oxidative attack on the nearest deoxyribose ring, leading to cleavages, both 5′ and 3′ of the ring (Sigman et al., 1991). Footprinting with this reagent has provided information relating to transposase–DNA contacts with the outside end portion of the STT and has defined regions in the STT that have a distorted structure. P1 nuclease acts as a random endonuclease that cleaves both single- and double-stranded DNA, although it is much more active on single-stranded DNA, including regions of duplex DNA that are unpaired. P1 nuclease footprinting has provided information relating to the zone of contact within the target portion of the STT.
The results of OP-Cu footprinting on STT generated with S-92 are presented in Figure 6. Only interactions between transposase and the transferred strand were defined. The footprint reveals one major zone of contact spanning roughly nucleotides 9–18 of the outside end. The same segment is also protected against OP-Cu cleavage in the bPEC (Allingham et al., 2001). Three zones of OP-Cu hypersensitvity are also apparent. These zones are located at the transposon–target junction (base pairs 1 and –1), base pairs 25–26 of the outside end and base pairs –11 and –12 of the target extension. The first two zones of OP-Cu hypersensitivity match those found in the tPEC (Allingham and Haniford, 2002). The remaining zone is displaced 3 nt in the 5′ direction from the site of OP-Cu hypersensitivity in the target capture transpososome formed with the HisG1 target fragment (Pribil and Haniford, 2000).

Fig. 6. OP-Cu footprinting of the STT. (A) In situ OP-Cu footprinting was carried out as described in Materials and methods. The thick line indicates a region of protection against OP-Cu cleavage, and arrows indicate sites of OP-Cu hypersensitivity, with the size of the arrow indicating the relative level of hypersensitivity. The terminal base of the transferred strand is defined as the +1 position and all negative numbers are base positions in the target extension. (B) A densitometric trace of the gel in (A) is shown. (C) Summary of the results of OP-Cu footprinting.
For P1 nuclease footprinting, an S-53 assembly reaction was treated with P1 nuclease prior to subjecting the reaction to electrophoresis on a native gel. DNA was extracted from bands corresponding to STT and free substrate and then run on a denaturing gel. The results in Figure 7 show that none of the free DNA was full length and the sizes of the fragments are indicative of P1 cleaving the ‘free’ DNA up to the junction of single- and double-stranded DNA. In contrast, none of these cleavage sites is detected in the STT present in the same reaction. The absence of any strong cleavage sites suggests that the entire target extension (17 bp) is largely protected against P1 cleavage. An assumption implicit in this interpretation is that P1 cleavage anywhere in the target extension will not cause the STT to fall apart. However, when this experiment was repeated with PEG-enriched S-53 STT, essentially the same pattern of protection was observed (data not shown), alleviating the above concern. Note that complementary base pairing is not a possibility in the target core of the S-53 STT and thus cannot be responsible for the absence of P1 cleavage in this region. The minor cleavage sites that are apparent (indicated by the small arrows) do coincide with P1 cleavage at the hairpin turn of the target extension, indicating somewhat weaker protection in this region compared with the target core. This fairly broad footprint over the target segment of the S-53 STT is quite similar to that reported previously for the target segment of the target capture complex (Pribil and Haniford, 2000).
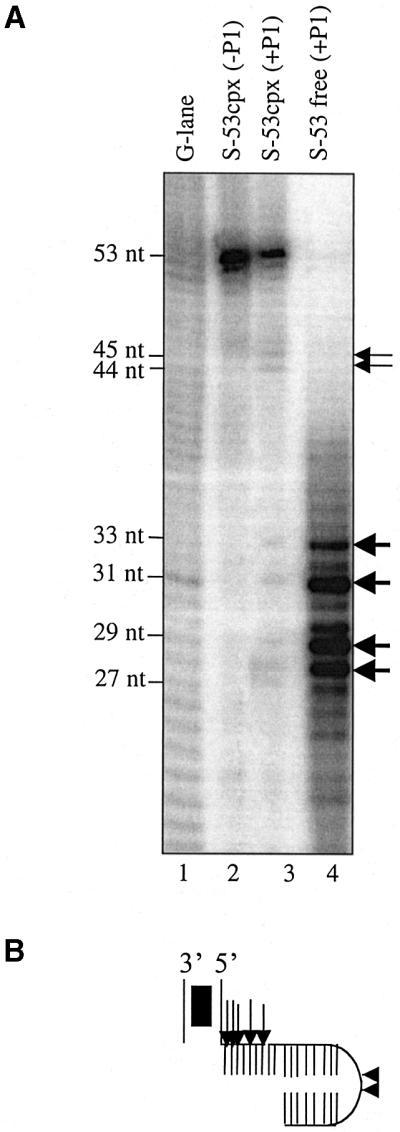
Fig. 7. P1 nuclease footprinting of the STT. (A) A P1 nuclease-treated S-53 assembly reaction was fractionated on a 5% native gel. DNA corresponding to S-53 STT and unreacted substrate was purified from gel slices and then applied to an 8% polyacrylamide gel. (B) The major sites of P1 cleavage in the unreacted substrate (large arrows) and in the S-53 STT (small arrows) are indicated.
Discussion
Disintegration, the reverse reaction of strand transfer (Gerton et al., 1999), may have a significant negative impact on transposition frequencies. The evolution of a mechanism to limit disintegration may therefore be important in ensuring the survival of transposons, particularly those that are subject to multiple levels of negative regulation. We have asked whether disintegration occurs in the Tn10 transposition system by assembling disintegration substrates into a transpososome and testing the chemical reactivity of this transpososome. We provide evidence that an authentic STT is formed under our assembly conditions and that, relative to other systems that do support disintegration reactions, the reactivity of the Tn10 STT is extremely low (Chow et al., 1992; Chow and Brown, 1994; Melek and Gellert, 2000). We have also shown that assembly of the STT occurs in an IHF-independent manner, making this the first transpososome in the Tn10 system that does not require IHF for de novo assembly. Detailed characterization of the STT illustrates that while it has some features in common with both pre-cleavage and the target capture transpososomes, it also has some novel properties.
Factors favoring de novo assembly of a STT
The transpososome formed with the disintegration substrates might have exhibited properties of a pre-cleavage transpososome. The only major difference between S-92 and S(T-D)-92 is the 9 nt gap in the DNA flanking the outside end in S-92. Strikingly, this difference is sufficient to direct transposase to form a transpososome that does not support efficient donor cleavage. This conclusion is based on our observation that the levels of the CE observed were very low for the S-53 compared with the S(T-D)-92 substrate (Figure 2B). It is known that in the pre-cleavage transpososome, contacts are made at base positions –4 to –7 on the continuation of the non-transferred strand (Sakai et al., 2000; Crellin and Chalmers, 2001). This region is not present in the disintegration substrates, and this could certainly contribute to the absence of bPEC formation in the presence of IHF. However, this is probably not the only factor in favoring STT formation because when the size of the gap was reduced, as in substrates S-59 and S-62, there was no rescue of donor cleavage. In addition, the above contacts are not critical for transpososome assembly as transpososome assembly still occurs efficiently with outside end substrate lacking flanking donor DNA. We think it is more likely that it is the combination of missing contacts and an increase in conformational flexibility afforded by the discontinuity of the non-transferred strand that favors SST formation.
The formation of a stable transpososome de novo in the Tn10 system appears to require that transposase contacts two different DNA segments. In the case of formation of the bPEC and post-cleavage transpososomes such as the double-end break complex, the two different DNA segments contacted are the terminal and the subterminal transposase-binding sites of the outside end (Sakai et al., 2000; Crellin and Chalmers, 2001). Formation of contacts at both sites is facilitated by IHF binding to the outside end, where it is expected to generate a U-shaped bend (Rice et al., 1996). The conformational flexibility afforded by the 9 nt gap might permit transposase to make alternative interactions with the target DNA that are not accessible when the junction region is double stranded. These interactions might then replace the requirement for the formation of the subterminal contacts. This would nicely account for the absence of an IHF requirement for STT assembly. By taking up a different orientation in the active site relative to the donor flank in a pre-cleavage transpososome, the target DNA may be protected against hydrolysis as well (see below).
De novo assembly of the STT also suggests that the target DNA-binding domain of transposase is accessible either prior to transposase interacting with DNA or immediately after transposase interacts with the transposon end. The occurrence of the chemical steps that normally precede target capture are therefore not required to induce a conformational change in transposase that subsequently exposes an otherwise buried target binding domain. However, with a substrate lacking the conformational flexibility of the disintegration substrates, this pathway for transpososome assembly is presumably not available, as entry of DNA into the target binding pocket is expected to be blocked by the presence of other DNA strands in the active site. In both Mu and Tn7 systems, target DNA is able to enter the transpososome at an early stage (Bainton et al., 1991; Naigamwalla and Chaconas, 1997), indicating that the target DNA-binding domain within each of the respective transposases is also accessible at an early stage of transpososome development. However, it is not known whether the target DNA is actually engaged in the active site in these systems.
While V(D)J and Tn10 systems differ significantly in their abilities to support disintegration reactions, the two systems are similar in that they do not require DNA-bending proteins for de novo assembly of the strand transfer intermediate. As in Tn10 transposition, formation of a pre-cleavage complex in the V(D)J system is greatly stimulated by the presence of a DNA-bending protein, in this case HMG1 (van Gent et al., 1997). However, there is no stimulation of disintegration by HMG1 in this system (Melek and Gellert, 2000). Presumably, substrate flexibility is also a key factor in permitting de novo assembly of the V(D)J STT, and as discussed above, this transpososome must also have a target binding domain that is accessible at an early stage of assembly.
The position of the target DNA in the STT may prevent efficient reaction chemistry
What may account for the inability of the Tn10 STT to support an efficient disintegration or cleavage reaction? In previous work, we have provided evidence that the target DNA enters the active site in a different configuration than the non-transferred strand does during hairpin formation (Figure 8) (Kennedy et al., 2000). This is significant because hairpin resolution, but not disintegration, is an efficient reaction. Hairpin resolution utilizes a configuration of the active site that is also used for first strand nicking, and this could reflect a requirement for a very specific active site configuration for hydrolysis reactions. Thus, the unique positioning of the target DNA in the active site could prevent hydrolysis of the strand transfer product, perhaps by preventing proper positioning of the OH nucleophile.

Fig. 8. Stereochemical model of hairpin formation/resolution and target strand transfer steps in Tn10 transposition. The indicated chemical steps are shown focusing on the scissile phosphates and the entry and exit of different DNA strands into the active site. Positions of bottom (Tn-BS) and top (Tn-TS) transposon strands, the flanking donor (Flank TS) portion of the top strand and a single strand of target DNA are shown. The attacking nucleophile, either OH or C3′O, is indicated with an arrow. Also shown are the non-bridging oxygen atoms (proR and proS) and the possible positions of two Mg2+ ions (dashed lines indicate coordination of Mg2+ ions to oxygen atoms). This figure was adapted and reprinted from Kennedy et al. (Cell, 101, 295–305, 2000) with permission from Elsevier Science.
The true reversal of strand transfer involves transesterification as compared with hydrolysis and consideration of the illustrations in Figure 8 reveals that transesterification reactions occurring in hairpin formation and disintegration require nucleophilic attack from different sides of the active site. This, together with the different DNA strand configurations in the hairpin versus strand transfer intermediates, may contribute to the very low level of true disintegration in the Tn10 system. Perhaps the ability of both retroviral integrases and the V(D)J recombinase to support high levels of disintegration reflects substantially different active site organization relative to that of the Tn10 transposase.
Properties of the STT
The structure of the strand transfer intermediate has not been well studied in the Tn10 transposition system. In previous work, it was possible to infer that the 9 bp single-strand gaps at the transposon–target junctions are bound by transposase and that the synaptic complex remains intact (Benjamin and Kleckner, 1989; Haniford et al., 1991). However, more precise details of the transposase– DNA interactions in this intermediate were not defined. De novo assembly of a transpososome with disintegration substrates has given us an opportunity to learn more about this reaction intermediate. This assumes that the transpososome we have characterized is an accurate representation of the authentic strand transfer intermediate.
OP-Cu footprinting studies revealed nearly identical patterns of protection within the terminal transposase-binding site in the outside end in the STT and the pre-cleavage transpososomes (Sakai et al., 2000; Allingham et al., 2001; Crellin and Chalmers, 2001). This observation implies that, with respect to the terminal transposase-binding site, progression through the reaction does not result in a major change in the way transposase contacts this region. Comparison of DNase I footprints obtained from cleaved donor and STT in the Mu transposition system have similarly led to the conclusion that transposase–transposon end interactions are largely retained in the Mu strand transfer transpososome, despite the presence of target DNA and the MuB protein (Mizuuchi et al., 1991). It follows that the preservation of the primary transposase–transposon end interactions that are formed within the initial synaptic complex may be a feature of many different DNA transposition systems.
While the zone of contact within the primary transposase-binding site did not change in the STT relative to pre-cleavage transpososomes, it is notable that STT formation is less sensitive to mutations in the primary transposase-binding site than is the bPEC. This could indicate that utilization of the target DNA as a binding partner permits a more stable transposase end interaction than is achieved when the binding partner is the subterminal transposase-binding site. This would be consistent with the observation that the STT is more stable than earlier transpososomes formed in the pathway (Haniford et al., 1991). Interestingly, disintegration in the V(D)J system shows a similar relaxation in the DNA sequence requirements within the primary recombinase binding site compared with the initial chemical steps (Melek and Gellert, 2000).
The combined data from P1 and OP-Cu footprinting revealed a contact map in the target DNA of the STT that is very similar to that observed for the target DNA in the target capture complex (Pribil and Haniford, 2000). This is consistent with the idea that it is the engagement of the target binding domain of transposase that is responsible for making the second set of contacts with transposase in the STT. We anticipate that the demand for sequence-specific target contacts becomes less stringent in STT formation because of the covalent linkage between the Tn10 terminus and the target segment. In other words, there is a lower energy cost for binding the DNA segment that is already linked to the transposon terminus compared with the cost of binding a separate DNA segment from solution.
In a bPEC, where HisG1 makes up part of the flanking donor DNA, transposase makes contacts out to ∼8 bp of the flank (Crellin and Chalmers, 2001) compared with ∼17 bp of the HisG1 sequence in the STT. This difference is significant because it shows that transposase interacts with the disintegration substrates in a way that is distinct from its interaction with standard linear substrates. This is consistent with disintegration substrates not being good substrates for hydrolysis. The difference in HisG1 contacts noted above could also be an indication that the target DNA and the donor flank do not occupy the same position in the active site. This was suggested previously based on the observation that hairpin formation and target strand transfer exhibit opposite phosphorothioate stereoselectivities (Kennedy et al., 2000).
We have shown that disintegration substrates are assembled into a STT, even when IHF and an appropriate outside end sequence are missing. This is strong evidence that target binding can compete with the subterminal contacts required for the normal pathway of transpososome assembly. Moreover, in the resulting transpososome with the properly positioned target DNA segment, which is normally produced through target strand transfer, the active sites are rendered inefficient to catalyze further reaction chemistry. One message from these observations is that by requiring both terminal and subterminal contacts for stabilizing transposase binding to the outside end, a particular ordered pathway for transpososome assembly is set and this in turn helps to ensure that the chemical steps in Tn10 transposition occur in their appropriate order. In addition, demand for the subterminal contact at the start of the reaction and the maintenance of this contact, perhaps until release from the original donor site, would help to avoid commitment to nearby target sites and suicidal intra-transposon transposition.
Materials and methods
Assembly of disintegration substrates and transpososomes
DNA oligos used in this work are listed in Table I and were PAGE purified before use. Where indicated, oligos were labeled at their 5′ termini by treatment with polynucleotide kinase and [γ-32P]ATP. To generate a disintegration substrate with an internal label, oligo T27 was 5′ end-labeled and then mixed with oligo TS26 in the presence of the bridge oligo B17 in 1× DNA buffer. After heating to 85°C and then cooling to 25°C over a period of ∼1 h, 10× DNA ligase buffer was added followed by T4 DNA ligase, and ligation was carried out overnight at 16°C. The ligated strand was then gel purified from an 8% denaturing gel. Transferred strand and non-transferred strand oligos were annealed as described above using roughly equimolar amounts of DNA. Disintegra tion substrate concentrations were evaluated using an ethidium bromide-based fluorescence assay (Bio-Rad) and adjusted to 0.2 pmol/µl. The S(T-D)-92 fragment was generated by BbsI digestion of pWY1005 (details available upon request). To assemble transpososomes, disintegration or standard substrates (0.2 pmol) were mixed with 0.3 pmol of purified transposase (1 µl) and/or, where indicated, 0.35 pmol of purified IHF (1 µl) in transposase reaction buffer (Sakai et al., 1995), giving a final volume of 20 µl. This is defined as a 1× volume reaction. Incubation was at 25°C for 2 h. Where indicated, transpososome enrichment was achieved by adding polyethylene glycol-8000 (Sigma) to 31% (w/v) to the assembly reaction. The transpososome was pelleted by centrifugation at 20°C in a microfuge at 14 K for 20 min, and after removal of the supernatent, the pellet was resuspended in 25 µl of 1× reaction buffer.
Table I. Oligonucleotides used in the assembly of STTs.
| Group | Oligo | Substrate | Sequence (5′-3′) |
|---|---|---|---|
| A | NT26 | CTGATGAATCCCCTAATGATTTTGGT | |
| TT31 | S-31 | ACCAAAATCATTAGGGGATTCATCAGCGCTG | |
| TT35 | S-35 | ACCAAAATCATTAGGGGATTCATCAGCGCTGAGTG | |
| TT39 | S-39 | ACCAAAATCATTAGGGGATTCATCAGCGCTGAGTGTGTA | |
| TT43 | S-43 | ACCAAAATCATTAGGGGATTCATCAGCGCTGAGTGTGTAAATT | |
| TT53 | S-53 | ACCAAAATCATTAGGGGATTCATCAGCGCTGAGTGTGTAAATTTTAATTTACA | |
| TTC53 | S-53* | ACCAAAATCATTAGGGGATTCATCAGCACTCAGCGTGTAAATTTTAATTTACA | |
| TMT53 | S-53 TC-T | ACCAAAATCATTAGGGGATTCATCAGCTGTGAGAGTGTAAATTTTAATTTACA | |
| TT56 | S-56 | ACCAAAATCATTAGGGGATTCATCAGCGCTGAGTGTGTAAATTTTAATTTACACAC | |
| TT59 | S-59 | ACCAAAATCATTAGGGGATTCATCAGCGCTGAGTGTGTAAATTTTAATTTACACACTCA | |
| |
TT62 |
S-62 |
ACCAAAATCATTAGGGGATTCATCAGCGCTGAGTGTGTAAATTTTAATTTACACACTCAGCG |
| B | NT26 | S-53 | CTGATGAATCCCCTAATGATTTTGGT |
| TS26 | ACCAAAATCATTAGGGGATTCATCAG | ||
| T27 | CGCTGAGTGTGTAAATTTTAATTTACA | ||
| |
B19 |
|
CACTCAGCGCTGATGAATC |
| C | NTS26 | S-53 OE-S | CTGATGAACCCCCTAATGATTTTGGT |
| |
TTS53 |
|
ACCAAAATCATTAGGGGGTTCATCAGCGCTGAGTGTGTAAATTTTAATTTACA |
| D | NTD26 | S-53 OE-D | CTGATGACTCCCTTAATGATTTTGGT |
| |
TTD53 |
|
ACCAAAATCATTAAGGGAGTCATCAGCGCTGAGTGTGTAAATTTTAATTTACA |
| E | NTT26 | S-53 OE-T | CTGATGACCCCCTTAATGATTTTGGT |
| |
TTT53 |
|
ACCAAAATCATTAAGGGGGTCATCAGCGCTGAGTGTGTAAATTTTAATTTACA |
| F | NT65 | S-92 | CTGATGAATCCCCTAATGATTTTGGTAAAAATCTTAAGTTAAGGTGGATACACATCTTGTCATAT |
| TT92 | ATATGACAAGATGTGTATCCACCTTAACTTAATGATTTTTACCAAAATCATTAGGGGATTCATCAGCGCTGAGTGTGTAAATTTTAATTTACA |
Group designations define oligos that were mixed together to generate the indicated substrates. For group A, NT26 was mixed with only one of the other indicated oligos.
Detection of transpososomes and disintegration products
Transpososome assembly was monitored by subjecting reactions to electrophoresis on a 5% native polyacrylamide gel (Sakai et al., 1995). Disintegration was assessed in two different ways. In one approach, transpososome-enriched assembly reactions were mixed with either MgCl2 or MnCl2 and after 2 or 16 h, reactions were terminated by phenol extraction. The purified DNA was then analyzed on an 8% denaturing gel. In the second approach, a native gel slice containing an assembly reaction was incubated in transposase reaction buffer (plus 5 mM MgCl2) for 2 h and then in denaturing gel buffer (8 M urea, 30% formamide, 1× TBE) for 2 h. The gel slice was then placed on top of a second (denaturing) gel and electrophoresis was carried out in a second dimension for 2 h at 200 V. Gels were subjected to PhosphorImaging analysis, and band intensities were determined using ImagQuant software (Molecular Dynamics).
OP-Cu footprinting
A 100× volume assembly reaction was carried out by mixing the appropriate amounts of S-92 (5′ end-labeled on the transferred strand) and transposase in the absence of a divalent metal ion. Following incubation for 2 h at 25°C, the reaction was loaded onto a 5% native gel where the transpososome was separated from unreacted substrate. The entire gel was treated with the OP-Cu footprinting reagent (Sigman et al., 1991), and DNA corresponding to unreacted substrate and STT was purified from the gel and loaded (∼100 000 c.p.m.) onto an 8% denaturing gel. A G-cleavage reaction prepared from the long continuous strand of S-92 was used as a size standard.
P1 nuclease footprinting
A 1× volume assembly reaction was carried out by mixing S-53 (5′ end-labeled on the transferred strand) and transposase in the absence of a divalent metal ion. Following incubation for 2 h at 25°C, zinc sulfate was added to a final concentration of 0.1 mM and then 2 units of P1 nuclease (Bethesda Research Labs) was added. The reaction was then subjected to electrophoresis on a 5% native gel where transpososome was separated from unreacted substrate. S-53 transpososome and unreacted substrate were excised from the gel, the corresponding DNA was purified and then subjected to electrophoresis on an 8% denaturing polyacrylamide gel.
Acknowledgments
Acknowledgements
We thank K.Mizuuchi, J.Allingham and P.Pribil for comments on the manuscript and helpful discussions during the course of this work. This work was funded by a grant to D.B.H. from the Canadian Institutes of Health Research. B.J.S. was supported in part by an Ontario Graduate Scholarship.
References
- Allingham J.S. and Haniford,D.B. (2002) Mechanisms of metal ion action in Tn10 transposition. J. Mol. Biol., 319, 53–65. [DOI] [PubMed] [Google Scholar]
- Allingham J.S., Wardle,S.J. and Haniford,D.B. (2001) Determinants for hairpin formation in Tn10 transposition. EMBO J., 20, 2931–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainton R., Gamas,P. and Craig,N.L. (1991) Tn7 transposition in vitro proceeds through an excised transposon intermediate generated by staggered breaks in DNA. Cell, 65, 805–816. [DOI] [PubMed] [Google Scholar]
- Bender J. and Kleckner,N. (1986) Genetic evidence that Tn10 transposes by a nonreplicative mechanism. Cell, 45, 801–815. [DOI] [PubMed] [Google Scholar]
- Benjamin H.W. and Kleckner,N. (1989) Intramolecular transposition by Tn10. Cell, 59, 373–383. [DOI] [PubMed] [Google Scholar]
- Blommers M.J.J., Walters,J.A., Haasnoot,C.A., Aelen,J.M., van der Marel,G.A., van Boom,J.H. and Hilbers,C.W. (1989) Effects of base sequence on the loop folding in DNA hairpins. Biochemistry, 28, 7491–7498. [DOI] [PubMed] [Google Scholar]
- Bolland S. and Kleckner,N. (1996) The three chemical steps of Tn10/IS10 transposition involve repeated utilization of a single active site. Cell, 84, 223–233. [DOI] [PubMed] [Google Scholar]
- Chalmers R., Guhathakurta,A., Benjamin,H. and Kleckner,N. (1998) IHF modulation of Tn10 transposition: sensory transduction of supercoiling status via a proposed protein/DNA molecular spring. Cell, 93, 897–908. [DOI] [PubMed] [Google Scholar]
- Chow S.A. and Brown,P.A. (1994) Juxtaposition of two viral DNA ends in a bimolecular disintegration reaction mediated by multimers of human immunodeficiency virus type I or murine leukemia virus integrase. J. Virol., 68, 7869–7878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow S.A., Vincent,K.A., Ellison,V. and Brown,P.O. (1992) Reversal of integration and DNA splicing mediated by integrase of human immunodeficiency virus. Science, 255, 723–726. [DOI] [PubMed] [Google Scholar]
- Crellin P. and Chalmers,R. (2001) Protein–DNA contacts and conformational changes in the Tn10 transpososome during assembly and activation for cleavage. EMBO J., 20, 3882–3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerton J.L., Herschlag,D. and Brown,P.O. (1999) Stereospecificity of reactions catalyzed by HIV-1 integrase. J. Biol. Chem., 274, 33480–33487. [DOI] [PubMed] [Google Scholar]
- Halling S.M. and Kleckner,N. (1982) A symmetrical six-base-pair target site sequence determines Tn10 insertion specificity. Cell, 28, 155–163. [DOI] [PubMed] [Google Scholar]
- Haniford D.B., Benjamin,H.W. and Kleckner,N. (1991) Kinetic and structural analysis of a cleaved donor intermediate and a strand transfer intermediate in Tn10 transposition. Cell, 64, 171–179. [DOI] [PubMed] [Google Scholar]
- Junop M.S. and Haniford,D.B. (1997) Factors responsible for target site selection in Tn10 transposition: a role for the DDE motif in target DNA capture. EMBO J., 16, 2646–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy A.K., Guhathakurta,A., Kleckner,N. and Haniford,D.B. (1998) Tn10 transposition via a DNA hairpin intermediate. Cell, 95, 125–134. [DOI] [PubMed] [Google Scholar]
- Kennedy A.K., Haniford,D.B. and Mizuuchi,K. (2000) Single active site catalysis of the successive phosphoryl transfer steps by DNA transposases: insights from phosphorothioate stereoselectivity. Cell, 101, 295–305. [DOI] [PubMed] [Google Scholar]
- Kleckner N. (1989) Transposon Tn10. In Berg,D.E. and Howe,M.M. (eds), Mobile DNA. Vol. 1, American Society for Microbiology, Washington, DC, pp. 227–268.
- Mazumder A., Engelman,A., Craigie,R., Fesen,M. and Pommier,Y. (1994) Intermolecular disintegration and intramolecular strand transfer activities of wild-type and mutant HIV-1 integrase. Nucleic Acids Res., 22, 1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melek M. and Gellert,M. (2000) RAG1/2-mediated resolution of transposition intermediates: two pathways and possible conse quences. Cell, 101, 625–633. [DOI] [PubMed] [Google Scholar]
- Melek M., Gellert,M. and van Gent,D.C. (1998) Rejoining of DNA by the RAG1 and RAG2 proteins. Science, 280, 301–303. [DOI] [PubMed] [Google Scholar]
- Mizuuchi K. (1997) Polynucleotidyl transfer reactions in site-specific DNA recombination. Genes Cells, 2, 1–12. [DOI] [PubMed] [Google Scholar]
- Mizuuchi M., Baker,T.A. and Mizuuchi,K. (1991) DNase protection analysis of the stable synaptic complexes involved in Mu transposition. Proc. Natl Acad. Sci. USA, 88, 9031–9035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisato D. and Kleckner,N. (1987) Tn10 transposition and circle formation in vitro. Cell, 51, 101–111. [DOI] [PubMed] [Google Scholar]
- Naigamwalla D.Z. and Chaconas,G. (1997) A new set of DNA transposition intermediates: alternate pathways of target capture preceding strand transfer. EMBO J., 16, 5227–5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribil P.A. and Haniford,D.B. (2000) Substrate recognition and induced DNA deformation by transposase at the target capture stage of Tn10 transposition. J. Mol. Biol., 303, 145–159. [DOI] [PubMed] [Google Scholar]
- Rice P.A., Yang,W., Mizuuchi,K. and Nash,H.A. (1996) Crystal structure of an IHF–DNA complex: a protein-induced DNA U-turn. Cell, 87, 1295–1306. [DOI] [PubMed] [Google Scholar]
- Sakai J., Chalmers,R.M. and Kleckner,N. (1995) Identification and characterization of a pre-cleavage synaptic complex that is an early intermediate in Tn10 transposition. EMBO J., 14, 4374–4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai J.S., Kleckner,N., Yang,X. and Guhathakurta,A. (2000) Tn10 transpososome assembly involves a folded intermediate that must be unfolded for target capture and strand transfer. EMBO J., 19, 776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigman D.S., Kuwabara,M.D., Chen,C.-H.B. and Bruice,T.W. (1991) Nuclease activity of 1,10-phenanthroline-copper in study of protein–DNA interaction. Methods Enzymol., 208, 414–433. [DOI] [PubMed] [Google Scholar]
- van Gent D.C., Hiom,K., Paull,T.T. and Gellert,M. (1997) Stimulation of V(D)J cleavage by high mobility group proteins. EMBO J., 16, 2665–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]