

Abstract
CTXφ is a filamentous bacteriophage whose genome encodes cholera toxin, the principal virulence factor of Vibrio cholerae. We have found that the CTXφ-related element RS1 is a satellite phage whose transmission depends upon proteins produced from a CTX prophage (its helper phage). However, unlike other satellite phages and satellite animal viruses, RS1 can aid the CTX prophage as well as exploit it, due to the RS1-encoded protein RstC. RstC, whose function previously was unknown, is an antirepressor that counteracts the activity of the phage repressor RstR. RstC promotes transcription of genes required for phage production and thereby promotes transmission of both RS1 and CTXφ. Antirepression by RstC also induces expression of the cholera toxin genes, ctxAB, and thus may contribute to the virulence of V.cholerae. In vitro, RstC binds directly to RstR, producing unusual, insoluble aggregates containing both proteins. In vivo, RstC and RstR are both found at the cell pole, where they again appear to form stable complexes. The sequestration/inactivation process induced by RstC resembles those induced by mutant polyglutamine-containing proteins implicated in human neurodegenerative disorders.
Keywords: antirepressor/bacteriophage/satellite/Vibrio cholerae
Introduction
Vibrio cholerae is a Gram-negative bacterium that can colonize the human small intestine and cause the severe diarrheal disease cholera. The profuse watery diarrhea that is a hallmark of this disease is a response to the bacteria’s secretion of a potent exotoxin, cholera toxin (Kaper et al., 1995). Not all V.cholerae strains produce this toxin, as the toxin-encoding genes, ctxAB, are only present in the genomes of a subset of strains (Mekalanos, 1983; Waldor and Mekalanos, 1994). Toxigenic V.cholerae acquired the toxin genes as a consequence of infection by CTXφ, a filamentous phage whose genome includes ctxAB (Waldor and Mekalanos, 1996). Thus, infection by CTXφ was a key event in the evolution of pathogenic V.cholerae, and dissemination of CTXφ may contribute to the emergence of new pathogenic strains.
Dissemination of CTXφ to new hosts is a multistep process that requires proteins encoded within the phage genome and within the ancestral V.cholerae chromosome. Virion production is thought to require: (i) production of extrachromosomal phage DNA, mediated by the phage-encoded replication protein RstA and by chromosome-encoded replication factors (Waldor et al., 1997; Davis and Waldor, 2000; Moyer et al., 2001); (ii) packaging of the single-stranded phage genome within phage-encoded coat proteins (Waldor and Mekalanos, 1996; Russel et al., 1997); and (iii) secretion of virions, aided by the phage protein Zot and the chromosome-encoded protein EpsD (Waldor and Mekalanos, 1996; Davis et al., 2000a). Phage infection of a new host depends upon that host’s production of a toxin-coregulated pilus (TCP), which is CTXφ’s primary receptor (Waldor and Mekalanos, 1996), and on production of the periplasmic proteins TolQ, R and A (Heilpern and Waldor, 2000). Subsequently, chromosomal recombinases and the phage-encoded protein RstB usually enable site-specific integration of phage DNA into the new host’s chromosome (Waldor et al., 1997; Huber and Waldor, 2002). However, in strains lacking a CTXφ integration site, phage DNA is propagated in its replicative (plasmid) form, which is known as pCTX.
In all characterized isolates of V.cholerae from the ongoing seventh pandemic of cholera, CTXφ DNA is maintained as part of a chromosomal array found on the larger of the two V.cholerae chromosomes (Mekalanos, 1983; Waldor and Mekalanos, 1994) (Figure 1A). These arrays contain one or more prophages plus one or more copies of a related genetic element known as RS1. Both the CTX prophage and RS1 contain the genes rstR, rstA and rstB (Figure 1B). As mentioned above, RstA and RstB are required for replication of the phage genome (RstA) and integration of CTXφ DNA into the chromosome (RstB). RstR is a repressor that down-regulates the rstA promoter (Kimsey and Waldor, 1998); it appears to be the predominant phage gene product made by CTXφ lysogens (H.H.Kimsey, unpublished data) and thus to be responsible for CTXφ’s low titers compared with repressorless filamentous phages such as f1. The remaining genes of the CTXφ genome are referred to collectively as the ‘core’ region. The products of this region include cholera toxin (encoded by the genes ctxAB) and proteins required for secretion and packaging of CTXφ, such as the phage’s major coat protein, Cep. RS1 does not contain the genes of the core region; in their place it contains a single gene, rstC, which bears no significant homology to any other gene in the GenBank database and until now has been uncharacterized.

Fig. 1. The CTX prophage, the related element RS1 and their arrangement within chromosomal arrays. (A) Structures of RS1–CTX prophage arrays in natural (AS207, E7946 and P27459) (Mekalanos, 1983; Sharma et al., 1997) and engineered (ENK12 and PNK6) (Pearson, 1989) strains of V.cholerae. Dotted elements represent CTX prophages; solid elements represent RS1s. Each element is bounded by short direct repeats (black triangles). ENK12 and PNK6 are derivatives of E7946 and P27459, respectively. (B) Genetic structures of the CTX prophage and RS1. Bent arrows represent the previously characterized rstA promoter (left) (Kimsey and Waldor, 1998) and the ToxT-regulated ctxAB promoter (right) (Champion et al., 1997).
Tandem arrangements of CTX prophage and RS1 DNA, such as those shown in Figure 1A, are essential for production of extrachromosomal phage DNA and ultimately new CTXφ (Davis and Waldor, 2000). This is because extrachromosomal DNA is not produced by excision of a prophage from the chromosome; instead, it is generated by a replicative process in which the prophage array serves as a template for synthesis of new phage DNA. Replication initiates ∼500 bp from the 5′ end of one element in an array and terminates at an equivalent site in the adjacent downstream element (Moyer et al., 2001). This process yields phage genomes that are hybrids, composed primarily of sequences from the upstream phage but containing ∼500 bases from the downstream element. A complete extrachromosomal phage genome cannot be generated from a solitary prophage that lacks an adjacent RS1 or second prophage; consequently, strains with a solitary prophage do not produce CTXφ (Davis and Waldor, 2000). Thus, one role of RS1 is to enable replication of an adjacent CTX prophage.
We have now uncovered additional interactions between RS1 and CTXφ. RS1 is a satellite phage that depends upon proteins produced from a CTX prophage (its helper phage) for packaging and secretion of its genome. However, RS1 is not simply a parasitic element, as it can aid the CTX prophage while exploiting it. The previously uncharacterized RS1-encoded protein RstC is an antirepressor that promotes transcription of phage genes, including ctxAB. RstC acts by binding directly to RstR, resulting in the formation of large insoluble aggregates containing both proteins. RstC’s sequestration of RstR away from its operators promotes transmission both of RS1 and of CTXφ, and thus may contribute to emergence of new pathogenic vibrios.
Results
RS1 can be transmitted from RS1+ CTXφ+ donors
The replicative process used to generate extrachromosomal CTXφ DNA from a chromosomal template can also yield extrachromosomal RS1 in strains containing an appropriate array. This was shown by Southern blot analysis of extrachromosomal DNA isolated from several strains of V.cholerae, using the RS1-specific probe rstC. DNA containing rstC was present in plasmid DNA isolated from several clinical isolates, each of which has at least one RS1 followed by a CTX prophage in its genome (Figure 2A, donors a, c and e). Hybridizing DNA was also detected in a strain engineered to contain two tandem RS1s on its chromosome (BD153; data not shown). Two species of DNA were usually detected; restriction mapping and hybridization analyses using additional probes suggested that these correspond to double- and single-stranded forms of RS1 (data not shown). As expected, strains whose genomes contain a solitary RS1, not flanked by a related element, did not generate extrachromosomal (replicative) RS1 (Figure 2A, donors b and d). These data indicate that RS1, like the CTX prophage, must lie within a tandem array of related elements in order for synthesis of the replicative form of the element to occur. The factors that enable some strains (e.g. E7946) to produce relatively high levels of extrachromosomal RS1 DNA have not been identified; however, it seems likely that the structure of the CTX prophage/RS1 array can influence RS1 replication.

Fig. 2. Transmission of unmarked and marked forms of RS1. (A) Southern blots of undigested extrachromosomal DNA from potential RS1 donors and recipients probed with the RS1-specific probe rstC. Left panel: donor strains a, c and e (AS207, E7946 and P27459, respectively), whose chromosomes all have at least one copy of RS1 followed by a CTX prophage (see Figure 1), produced extrachromosomal RS1 DNA. Donor strains b and d (ENK12 and PNK6), which each have a solitary RS1, did not produce a plasmid form of this element. Right panel: plasmid DNA was isolated from potential recipient cells cultured in filtered supernatants from donor strains. The TCP-expressing recipient strain O395 does not contain rstC-hybridizing DNA (–). O395 acquired RS1 DNA from supernatants of AS207, E7946 and P27459 (a, c and e), but not from supernatants of ENK12 or PNK6 (b and d). RS1 was not transmitted from the supernatant of E7946 into the TCP-deficient strain TCP2 (lane 7). Non-specific hybridization between the probe and abundant CTXφ DNA in some recipient cultures accounts for the extra band seen at the top of the gel in several lanes. DS RS1, double-stranded RS1; SS RS1, single-stranded RS1. (B) TCP-producing O395 were mixed with filtered supernatant from each donor strain (O395 containing one of the RS1 derivatives), then plated on kanamycin plates to select for infected recipients. pBD482, which contains an intact copy of rstC, was transmitted ∼100 times more frequently than were pBD485 and pBD153, which have mutations in rstC. The X in pBD485 represents the insertion in rstC of two stop codons followed by an 8 bp deletion. Frequencies are average results from experiments performed at least twice.
Although cis-acting sequences needed for packaging and secretion of CTXφ have not been mapped precisely, they appear to lie within the region at the 5′ end of the prophage genome that is almost identical to part of RS1 (Davis et al., 2000b). Consequently, it seemed likely that infectious RS1 particles could be formed in a manner similar to production of CTXφ. To test this possibility, filtered supernatants from potential donor strains were mixed with a recipient (O395) that expresses the CTXφ receptor, TCP, and plasmid DNA subsequently was extracted from the recipient cultures. No rstC-hybridizing species were detected in DNA from uninfected O395 cells or from O395 mixed with the supernatants of strains that do not produce extrachromosomal RS1 (Figure 2A, recipients). However, Southern blots clearly revealed that O395 can acquire RS1 from culture supernatants of CTX phage lysogens that produce extrachromosomal RS1 (Figure 2A). Levels of replicative RS1 were higher in the newly infected cells than in the donors, probably because O395 lacks a functional integration site, and consequently all the transmitted RS1 was maintained in O395 as an extrachromosomal element. RS1 was not transmitted from the CTXφ– strain BD153, which produces extrachromosomal RS1 but cannot produce CTXφ coat proteins (data not shown). RS1 also could not be transmitted to strain TCP2, an O395 derivative that does not produce the CTXφ receptor (Figure 2A). These data, which extend previous analyses of RS1 (Davis and Waldor, 1999; Faruque et al., 2002), suggest that RS1 is packaged using CTXφ coat proteins and that its uptake by a new host depends on the CTXφ receptor, TCP. Since RS1 can only give rise to infectious particles in the presence of CTXφ, its helper element, it is comparable with the ‘satellite’ phage P4, which transmits its genome using capsid and tail proteins encoded by its helper phage P2 (Lindqvist et al., 1993). Thus, RS1 can be considered a satellite phage of CTXφ.
RstC promotes transmission of RS1 and CTXφ
In order to study RS1 transmission quantitatively, marked derivatives of the replicative form of RS1 were generated by insertion of a KanR cassette at various sites (Figure 2B). These new plasmids were introduced into the strains Bah-2 and O395, which both lack functional phage integration sites, so the marked RS1s were maintained as extrachromosomal elements (Waldor and Mekalanos, 1996; Waldor et al., 1997). As expected, none of these plasmids could be transmitted from Bah-2, as Bah-2 does not contain CTX prophages and hence cannot supply phage coat proteins. In contrast, all three could be transmitted from O395, which contains several CTX prophages that previously were shown to encode functional phage proteins (Davis et al., 2000b). O395 does not transmit the CTXφ genome, even when transformed with plasmid RS1, due to the atypical structures of its prophage arrays (Davis et al., 2000b). These data, as well as subsequent results, confirmed that infectious RS1 contains only RS1 genes and that transmission of RS1 is not coupled to transmission of CTXφ. Unexpectedly, these experiments also revealed that the various RS1 derivatives yield infectious particles at significantly different frequencies. Supernatants from overnight cultures of O395(pBD482) yielded 3 × 104 KnR transductants/OD/ml, while supernatants from O395(pBD485) and O395(pBD153), which both lack functional copies of rstC, yielded ∼100-fold fewer transductants (Figure 2B). Thus, it appeared that production of RstC in the donor cells promoted transmission of RS1. As RstC was produced in these cells using its endogenous promoter on a naturally occurring plasmid, it seems unlikely that RstC’s effect is attributable to overexpression.
To test whether rstC also influences transmission of CTXφ, a plasmid encoding isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible RstC (pBD495) was introduced into several CTXφ lysogens. Synthesis of RstC increased CTXφ titers up to several thousand fold in a variety of strain backgrounds (Table I), although it did not induce phage production by CTXφ lysogens that ordinarily do not yield CTXφ (i.e. strains with a solitary prophage; data not shown). The increase in CTXφ production was accompanied by increased production of pCTX, the replicative form of the CTXφ genome (data not shown). Strains in which CTXφ is maintained as a plasmid [e.g. O395(pCTX-Kn) or Bah-2(pCTX-Kn)] also generated increased pCTX and increased CTXφ in response to rstC expression (Table I; data not shown). The phage DNA replication induced by RstC probably facilitates production of CTXφ, as it should provide more templates for phage gene transcription and more phage DNA for packaging. Not surprisingly, exogenous RstC also induced replication of RS1. Interestingly, RstC disproportionately increased the level of single-stranded phage and RS1 DNA, which is the form of DNA found within virions (data not shown); however, the mechanistic significance of the increases in phage and RS1 DNA has not been investigated directly.
Table I. RstC promotes transmission of CTXφ.
| Lysogen | Array structure | Reference | Transductants, no RstC (per OD/ml) | Transductants, + RstC (per OD/ml) | Fold increase due to RstC |
|---|---|---|---|---|---|
| BD81 | (CTX-Kn)(CTX-Kn) | Davis and Waldor (2000) | 1.2 × 104 | 2.1 × 107 | 1750 |
| BD206 (ΔrecA) | (CTX-Kn)(CTX-Kn) | Davis and Waldor (2000) | 1.4 × 103 | 6.8 × 106 | 4857 |
| E4 | (RS1)(CTX-Kn)(RS1) | Pearson (1989) | 1.2 × 105 | 4.5 × 106 | 38 |
| BD25 | (CTXcalc-Kn)(CTXcalc-Kn) | Davis and Waldor (2000) | 5.2 × 103 | 8.1 × 105 | 153 |
| Bah-2a | pCTX-Kn | Pearson et al. (1993) | 1.2 × 105 | 1.7 × 107 | 147 |
Each phage lysogen assayed contained either pBD495, which encodes an IPTG-inducible RstC (+ RstC) or a control plasmid, pGZ119 (no RstC). All cultures contained 0.1 mM IPTG as well as appropriate antibiotics. Transduction frequencies presented are averages from experiments performed at least twice.
aBah-2 does not contain a CTX prophage within its chromosome. Phage DNA in Bah-2(pCTK-Kn) is maintained as a double-stranded plasmid.
RstC increases transcripts of phage genes
One mechanism by which RstC might cause increased phage DNA replication and phage production is by promoting expression of rstA and other phage genes. Northern blots revealed that strains producing RstC had dramatically higher levels of transcripts for rstA and cep than strains lacking RstC (Figure 3A and B). RstC induced phage gene transcription both in CTXφ lysogens that produce CTXφ and in lysogens that do not (e.g. strains with solitary prophages; data not shown); thus, RstC can increase transcript levels without increasing levels of pCTX. It is not yet known which phage gene products usually limit phage production; consequently, we cannot determine how many step(s) of phage production (e.g. replication, assembly and/or secretion) are augmented by RstC.

Fig. 3. Levels of phage gene transcripts are elevated in response to RstC. RNA was isolated from V.cholerae containing a plasmid- encoded, IPTG-inducible rstC that were grown in the presence (+) or absence (–) of IPTG. Northern blots were probed with rstA (A), cep (B) and ctxAB (C and D). Strains used for analysis of ctxAB expression contained either wild-type copies of toxT (wt), toxT with an inactivating deletion mutation (Δ; Champion et al., 1997), or a wild-type chromosomal gene and a plasmid-encoded gene induced by IPTG (pMT5; DiRita et al., 1996) (p). The strain containing the toxT plasmid did not contain the rstC plasmid. Some strains in (D) contain a transcription terminator (term.) in rstA of the CTX prophage. The strain in (A) and (B) is a derivative of BD81 (see Table I); strains in (C) and (D) are derivatives of E7946 and P27459, respectively (see Figure 1). Ethidium bromide staining of gels was used to confirm that the lanes of each gel contained equal amounts of total RNA.
RstC production also resulted in increased transcript levels for ctxAB. ctxAB are not generally transcribed in cultures grown at 37°C in LB, as ToxT, the transcriptional activator that regulates the ctxAB promoter (see Figure 1B), typically is not made under these conditions (DiRita et al., 1996). However, northern blots revealed that RstC dramatically elevated ctxAB transcript levels in cultures grown in LB at 37°C (Figure 3C). ToxT does not mediate RstC’s effect on ctxAB transcript levels, as RstC’s effect was apparent in a strain lacking a functional copy of toxT. These data suggested that the RstC-induced ctxAB transcripts might not initiate at the ToxT-regulated ctxAB promoter.
RstC is an antirepressor that counteracts RstR
RstC’s effect upon phage promoters, in particular the rstA promoter, subsequently was assayed using transcription reporters. Three rstA::lacZ fusions were utilized, as several variants of CTXφ have been identified, each with a distinct rstR and corresponding rstA promoter (Kimsey and Waldor, 1998; Davis et al., 1999). We have worked with three phages known as CTXETφ, CTXcalcφ and CTXclφ; their respective repressors and rstA::lacZ reporters are also denoted by the ET, calc and cl superscripts. Each of these three reporters can only be regulated by the corresponding repressor (Kimsey and Waldor, 1998; Davis et al., 1999). RstC was expressed in V.cholerae together with each reporter in the presence and absence of the appropriate phage-encoded repressor. In all cases, production of RstC caused increased synthesis of β-galactosidase from previously repressed rstA::lacZ fusions (Table II). An RstC-induced increase in β-galactosidase activity was also detected when plasmid-encoded RstRET and RstC were produced in Escherichia coli containing an rstAET::lacZ reporter (data not shown); thus, RstC’s effect is not dependent upon any Vibrio-specific factors. However, RstC did not increase β-galactosidase activity from the rstA::lacZ reporters when a corresponding rstR was not present (Table II). Thus, RstC did not appear to be a transcriptional activator. Instead, these data suggested that RstC’s role is to counteract RstR’s repression of the rstA promoter, i.e. that RstC is an antirepressor. The fact that RstC counteracted repression by three distinct variants of RstR is consistent with previous observations that RstC stimulates phage production and/or phage gene expression from lysogens regulated by each of the three repressors (Table I; data not shown). It should be noted, however, that to date RS1 has only been found to encode one of the RstR variants: RstRET.
Table II. RstC counteracts RstR-mediated repression of rstA::lacZ transcription reporters.
| Lysogen | Repressor | Reporter | β-gal., no RstC (Miller units) | β-gal. + RstC (Miller units) | Fold increase due to RstC |
|---|---|---|---|---|---|
| BD81 | RstRET | RstAET::lacZ | 2.4 | 40 | 17 |
| 2740-80 | – | RstAET::lacZ | 91 | 83 | 0.9 |
| BD25 | RstRcalc | RstAcalc::lacZ | 20 | 2313 | 116 |
| 2740-80 | – | RstAcalc::lacZ | 5625 | 5091 | 0.9 |
| O395 | RstRcl | RstAcl::lacZ | 1.2 | 122 | 101 |
| BD81 | RstRET | RstAcl::lacZ | 457 | 710 | 1.5 |
β-galactosidase activity generated from the reporters was assayed both in V.cholerae (above) and in E.coli (not shown). In V.cholerae, the various forms of RstR were produced from CTX prophages, while both RstC and β-galactosidase were plasmid encoded. A control plasmid (pGZ119) was present in strains that did not contain the RstC-encoding plasmid (pBD495). Strain 2740-80 does not contain any CTX prophages and hence does not produce any form of RstR (Pearson et al., 1993). BD81 and BD25 are lysogens of 2740-80. β-galactosidase levels presented are averages from experiments performed at least twice.
β-galactosidase assays using chromosomal and plasmid-encoded ctxA::lacZ fusions suggested that the ctxAB transcripts induced by RstC might also initiate at the rstA promoter (data not shown). This possibility was explored using a strain with a transcriptional terminator inserted into rstA of its only prophage. Such a terminator should block transcription of any downstream genes that are usually co-transcribed with rstA. Using northern blots, we found that this terminator prevented the increase in cep and ctxAB transcripts typically induced by RstC (Figure 3D; data not shown). The mutant strain has two intact copies of rstA in the RS1s that flank the prophage; consequently, the terminator’s effect on cep and ctxAB transcripts should not reflect a deficiency in RstA. Furthermore, the terminator did not impair the activity of the ToxT-regulated toxin promoter immediately upstream of ctxAB, as this promoter still functioned in response to ToxT (Figure 3D). These data suggest that all the transcripts synthesized in response to RstC initiate at the rstA promoter, presumably because RstC prevents RstR-mediated repression at this site.
RstC binds directly to RstR and renders it insoluble
We next investigated the mechanism by which RstC blocks repression of the rstA promoter. Several phage antirepressors operate by binding to their cognate repressors, thereby preventing interaction between the repressor and its operator (Poteete, 1988; Heinzel et al., 1992; Liu et al., 1998; Eriksson et al., 2000). Conse quently, we examined whether a physical interaction between RstR and epitope-tagged RstC (H6RstC) could be detected. H6RstC (which, like untagged RstC, can induce production of CTXφ by CTX lysogens) and endogenous RstR could not be co-immunoprecipitated from lysates of V.cholerae; however, this appears to have been a consequence of H6RstC’s insolubility, as centrifugation removed most of this protein from bacterial lysates, along with cellular debris, prior to addition of antibody. Under non-denaturing conditions, H6RstC only remained soluble in the presence of detergents, such as 0.1–1.0% N-dodecyl-N,N-dimethylamine-n-oxide (LDAO). (How ever, algorithms such as PSORT and the GES hydrophobicity scale predict that RstC is not a membrane protein.) H6RstC was therefore affinity purified in the presence of at least 0.1% LDAO, then diluted into a detergent-free mixture of purified proteins that included epitope-tagged RstR (RstRH6). Reduction of the LDAO concentration caused H6RstC to become insoluble, allowing for separation of H6RstC from the solution by centrifugation. Analysis of the precipitated protein on an acrylamide gel revealed that RstRH6 had precipitated along with H6RstC and that all the control proteins (e.g. myoglobin, ovalbumin, γ-globulin and thyroglobulin) had remained soluble (Figure 4A, lane 2). An E.coli lambdoid phage repressor (434 CI) also did not become insoluble in the presence of H6RstC (data not shown). RstRH6 could not be removed from the protein mixture by centrifugation when H6RstC was not added (Figure 4A, lanes 3 and 4). RstRH6 and H6RstC also did not form insoluble complexes if they were mixed in the presence of 0.1% LDAO (data not shown). These results suggest that, given appropriate reaction conditions, H6RstC and RstRH6 can interact specifically and directly and that RstRH6 in such protein complexes has different properties (e.g. different solubility) than RstRH6 in solutions lacking H6RstC. As dimers and tetramers of purified RstRH6 are soluble (H.H.Kimsey, unpublished data), it is likely that complexes of RstRH6 and H6RstC are significantly larger structures.

Fig. 4. RstC converts RstR into an isoluble form. (A) A mixture of RstR6H and several control proteins (1) was combined with (2) H6RstC, (3) dilution buffer for H6RstC or (4) nothing. Samples in lanes 1, 5 and 6 were not centrifuged; they show sizes and total quantities of each protein used. After incubation for 2 h at 4°C, samples in lanes 2, 3 and 4 were centrifuged for 30 min at 16 100 g; the pellet fractions were solubilized in loading buffer and analyzed on an acrylamide gel, while soluble proteins were discarded. (B) Lysates of E.coli expressing GFP or GFP fusion proteins were mixed with H6RstC (C), dilution buffer for H6RstC (B) or nothing (–); insoluble proteins were isolated by centrifugation and loaded on the gel, while soluble proteins were discarded. Uncentrifuged lysates (L) are shown for comparison. Gels in (A) and (B) were stained with Coomassie Blue. (C) Western blots of gels identical to those shown in (B) were probed with an anti-GFP antibody to confirm that precipitation of RstR–GFP fusions was enhanced by mixing with RstC and that the solubility of GFP and ToxT–GFP was unaffected by RstC.
We used fusion proteins consisting of RstR and green fluorescent protein (GFP) to assess whether RstC could also induce aggregation of RstR within a cell. When fusions of RstRET and RstRcalc to GFP were produced in V.cholerae and/or E.coli that were not producing RstC, the fusion proteins were distributed generally throughout the cytoplasm, as was GFP alone (Figure 5A, C and D). An RstRcl–GFP fusion and a ToxT–GFP control fusion protein were also distributed diffusely, although in some cells they formed foci as well (Figure 5B and E). However, in the presence of RstC produced from pBD1293 (which lacks an epitope tag), all three RstR–GFP fusion proteins formed large polar foci in the majority of cells (Figure 5F–H and K–N). Each cell generally contained a single large focus, and many cells lacked detectable diffusely distributed fluorescent protein. In contrast, the distribution of GFP and ToxT–GFP was not affected by co-expression of RstC (Figure 5I and J). Thus, RstC appears specifically to induce aggregation of RstR in vivo as well as in vitro. H6RstC also forms large polar foci (Figure 5O), although their formation is not dependent on the presence of RstR. The mechanism by which RstC and the proteins that associate with it are targeted to a single cell pole has not yet been determined.

Fig. 5. Intracellullar localization of RstR–GFP fusion proteins, RstC and control proteins. Proteins were visualized using epifluorescence microscopy (A–J, P and Q) or epifluorescence and phase microscopy combined (K–O). Escherichia coli transformed with plasmids encoding proteins to be assayed were grown either without arabinose (A–E and O) or with 1 mM arabinose (F–L), which induced production of RstC from pBD1293. Fluorescent proteins assayed were RstRET–GFP (A, F and K), RstRcl–GFP (B, G and L), RstRcalc–GFP (C and H), GFP (D and I) and ToxT–GFP (E and J). Focal localization of RstR–GFP fusion proteins in response to RstC was observed both in E.coli (F–H, K and L) and in V.cholerae (M and N; both show strain E7946 expressing plasmid-encoded RstRET–GFP and RstC). H6RstC was expressed in E.coli from pBD686 and visualized with indirect immunofluorescence microscopy using an anti-His5 antibody (O). Aggregation of RstRET–GFP was induced by addition of purified H6RstC to cell lysates containing the fusion protein. Small aggregates formed in <20 min (P); larger aggregates were detectable within 40 min (Q). The minimum time needed for visible aggregate formation was not assessed. The magnification is the same for all images presented.
The RstR–GFP aggregates that formed in response to RstC were very stable. They remained intact despite cell lysis, freezing and thawing, sonication, exposure to reducing agents and sedimentation through high osmolarity solutions (data not shown). Their formation did not require active cellular processes, as they could also be generated simply by addition of purified H6RstC to bacterial lysates containing unaggregated RstR–GFP (Figure 5P and Q). RstR–GFP was the only protein in these lysates whose solubility was altered detectably by the presence of RstC (Figure 4B and C); thus, RstC appears to interact specifically with RstR even in the complex milieu of a cell lysate.
RstC prevents RstR from binding to its operator
Formation of protein complexes containing RstR and RstC altered the DNA binding capacity of RstR as well as RstR’s solubility. In the absence of RstC, almost all of a probe containing an RstRET operator was bound by RstRET (Figure 6, lane 1). However, when the same quantity of RstRET was pre-incubated with affinity-purified H6RstC prior to mixing with the probe, far fewer probe–protein complexes were detected; this inhibition of DNA binding appeared to be dose dependent (Figure 6, lanes 2 and 3). Equal quantities of RstC did not influence binding of the chromosome-encoded repressor SetR to its operator (data not shown). In addition, no binding of RstC to either probe was observed (Figure 6, lane 5; data not shown). These data suggest (i) that RstC does not bind to the rstA promoter or compete with RstR for binding sites there; and (ii) that interactions between RstC and RstR directly and specifically inhibit binding of RstR to its operators.
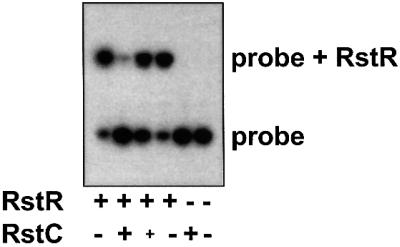
Fig. 6. RstC prevents binding of RstR to DNA. Almost all of the labeled probe containing an RstR operator was bound by RstR following addition of ∼30 ng of purified protein (lane 1). Pre-incubation of RstR with purified RstC (300 and 150 ng) prior to mixing with the probe prevented binding of RstR to DNA (lanes 2 and 3). Pre-incubation of RstR with RstC dilution buffer had no effect on RstR binding (lane 4). Probe incubated with 300 ng of RstC (lane 5) migrated the same as did probe with no added proteins (lane 6).
Discussion
RS1 can replicate in the absence of CTXφ, but RS1 virions are only produced by V.cholerae that are also infected with CTXφ. Therefore, RS1 can be thought of as a satellite phage that depends on CTXφ, its helper phage, to enable its transfer. Only one additional class of satellite phages has been identified in prokaryotes: phage P4 and related satellites of phage P2. These satellite phages are best described simply as parasites of P2 (Bertani and Six, 1988); in contrast, there is a reciprocal relationship between RS1 and CTXφ. RS1 promotes CTXφ replication and transfer, due to the activity of RstC, the RS1-encoded antirepressor. RstC binds and sequesters the repressor of RS1 and CTXφ (RstR) and thereby enables transcription of genes needed for transmission of both elements. RstC also increases production of cholera toxin, as transcripts initiating at the derepressed rstA promoter can extend through ctxAB, which lie downstream. Thus, RstC may contribute both to the evolution of new pathogenic vibrios and to the virulence of existing strains.
Although exogenous production of RstC induces phage gene transcription, under most in vitro growth conditions RstC does not appear to be an initiator of phage and toxin production. Rather, it seems to augment their synthesis following a separate phage-inducing stimulus. Production of RstC does not appear to be the primary event that promotes phage production due to the fact that rstC expression, like that of most genes in RS1 and CTXφ, depends upon the rstA promoter, which typically is repressed by RstR (B.M.Davis, unpublished data). As with many other phages, repression of CTXφ gene expression can be reduced by DNA-damaging agents such as mitomycin C, which decrease the level of repressor and increase phage production (Waldor and Mekalanos, 1996; H.H.Kimsey and B.M.Davis, unpublished data). However, induction of CTXφ differs from that of non-filamentous phages in that CTXφ induction is not a terminal event for the host cell, since phage production is not coupled to cell lysis. Thus, phage induction can be a transient event; repression should be re-established if RstR returns to its pre-induction level. The role of RstC appears to be to prolong the process of phage production by sequestering RstR and impeding re-establishment of repression. More CTXφ (and more infectious RS1) should be produced per derepressed cycle when RstC is present than when it is absent. Consistent with this model, we have observed that RstC is a more potent phage-inducing stimulus than is mitomycin C. It is not yet known how or if repression is re-established following production of RstC.
Most CTXφ genes transcribed from the rstA promoter yield more transcripts when CTXφ is maintained in plasmid form (H.H.Kimsey, unpublished data). Simi larly, expression of rstC is correlated with production of extrachromosomal RS1 (B.M.Davis, unpublished data). Thus, it appears likely that the primary template for transcription of rstC is plasmid, rather than chromosomal, RS1. Although RS1 is a low-copy plasmid, it enables sufficient synthesis of RstC to promote phage gene expression (Figure 2). Furthermore, in strain O395, the increases in phage gene transcription caused by RstC produced under the control of its native promoter from an RS1-derived plasmid and by RstC produced from an inducible promoter appear to be of comparable magnitude (Figure 2; Table II). Thus, the dramatic consequences of RstC production that we have observed are unlikely to be artifacts resulting from unnatural levels of the antirepressor.
It has been difficult to define precisely the parameters governing the RstR–RstC interaction. In particular, it has not been possible to identify a minimum concentration of H6RstC below which H6RstC does not induce aggregation of RstR, as the formation of RstR–RstC complexes increases with increasing incubation periods. Thus, the reductions in aggregate formation that we have observed in response to decreasing concentrations of H6RstC may simply reflect a slower rate of aggregate formation. Furthermore, as not all visible aggregates can be isolated via centrifugation, RstR insolubility is probably an imperfect indicator of RstR–RstC interactions. Some RstR may be sequestered in relatively small complexes that do not precipitate yet still prevent RstR’s binding to DNA. Additional assays need to be developed to characterize more precisely the nature and kinetics of the RstC-induced conversion of RstR into an inactive form.
Aspects of antirepression by RstC resemble the effects of other phage antirepressors; however, several features of RstC’s activity are unusual. First, RstC is atypical among phage antirepressors in that it routinely promotes transmission of genetic elements other than the element in which it is encoded. Other antirepressors function primarily to foster their own transmission. Even the antirepressor E of phage P4, which activates gene expression from the helper phages P2 and WΦ, does not induce production of P2 or WΦ, as not all necessary functions of these phages become derepressed (Bertani and Six, 1988). (P2 production is also prevented by the P4 sid gene product, which induces formation of capsids too small to package P2 DNA; Bertani and Six, 1988.) Secondly, the activation of cholera toxin synthesis by RstC appears to be unprecedented; other phage antirepressors have not been shown to influence expression of virulence genes. Thirdly, although several phage antirepressors bind their cognate repressors to inactivate them, RstC is unique in that it can bind to and counteract repression by multiple unrelated repressors. The antirepressors of phages P4 and P22 are both active against multiple targets, but the partners for each antirepressor have significant sequence similarity (e.g. expected values from pairwise BLAST of 1e-22 to 3e-56; identities of 48–53%), and the antirepressors are thought to target conserved domains within them (Liu and Haggard-Ljungquist, 1999). In contrast, RstRET and RstRcl show limited sequence similarity (pairwise BLAST expected value, 4e-08; 28% identity) and no similarity is detected between RstRcalc and the other forms of RstR. It is likely that RstC recognizes a structural motif present in all three proteins, but such interaction domains within the RstRs have yet to be defined.
Finally, the complexes that RstC forms with the RstRs appear to differ from those formed by most antirepressors. To our knowledge, RstC is the only phage antirepressor reported to cause intracellular aggregation and relocalization of its cognate repressor. RstC–RstR complexes also appear to lack a defined stoichiometry, while other repressor–antirepressor complexes have been shown to contain a precise number of subunits. C1 and Coi of phage P1, for example, form complexes containing one molecule of each protein (Heinzel et al., 1992), and SinI and SinR (which are not phage encoded) form SinI2SinR2 tetramers (Lewis et al., 1998). In contrast, our preliminary analyses suggest that the relative levels of RstR and RstC are not fixed, and that the ratio of precipitated proteins can be as unbalanced as 18:1 (RstR:RstC; data not shown). The unexpectedly high level of RstR in these complexes raises the possibility that all molecules of RstR in the complexes may not interact directly with RstC. It is conceivable that RstC nucleates formation of RstR–RstC complexes by interacting with RstR and changing its conformation, and that additional molecules of RstR are recruited by, and bind directly to, the altered RstR. Such a mechanism would be similar to those underlying conversion of mammalian PrP and yeast Sup35p into their insoluble prion forms (Scheibel and Lindquist, 2001). However, it should be noted that prion proteins have only been shown to mediate conformational change in prion precursor proteins; they have not been shown to interact with heterologous proteins.
RstC–RstR aggregates may also share attributes with complexes formed by mutant proteins that contain extended polyglutamine [poly(Q)] tracts. Such proteins increasingly are recognized to be responsible for a variety of human neurodegenerative disorders (Ross, 1997). The mutant poly(Q)-containing proteins form aggregates that have been shown to sequester other proteins and restrict their activity, as we have observed for RstC and RstR (Kazantsev et al., 1999; Steffan et al., 2000). Furthermore, poly(Q)-containing aggregates are insoluble and highly resistant to disruption, and contain apparently non-stoichiometric amounts of proteins (Kazantsev et al., 1999). GFP-tagged proteins sequestered in such aggregates are, like RstR–GFP, still fluorescent, indicating that at least the GFP moiety has retained its native conformation and has not been denatured. However, it is not known whether the GFP fusion partners have retained a native conformation or, as discussed above for RstR, whether they have assumed novel structures. Such domain-restricted alterations appear to be present in aggregates containing the yeast prion protein Sup35 fused to GFP (Serio and Lindquist, 2000). Further investigation is needed to characterize more precisely the similarities and distinctive features of all these protein complexes.
In prokaryotes, protein aggregation is an atypical mechanism of protein inactivation. Given the specificity of the RstR–RstC interaction, RstC may be useful as a biological tool, as RstC may be able to block the activity of heterologous proteins tagged with RstR.
Materials and methods
Bacterial strains and culture conditions
All bacteria were cultured in LB at 37°C unless otherwise noted. Antibiotics were used as follows: ampicillin (Ap), 50 µg/ml (V.cholerae); Ap, 100 µg/ml (E.coli); kanamycin (Kn), 50 µg/ml; chloramphenicol (Cm), 5 µg/ml (V.cholerae); Cm, 20 µg/ml ml (E.coli); streptomycin (Sm), 200 µg/ml; and spectinomycin (Sp), 100 µg/ml. E7946 toxT contains a deletion in toxT’s helix–turn–helix domain; it was obtained from Vic DiRita (strain VJ739) (Champion et al., 1997). TCP2 contains a deletion within tcpA (Herrington et al., 1988). P27459 with a terminator in rstA was constructed by allele exchange with pBD873 (a sacB ApR counterselectable plasmid; see below). BD163 was constructed by integration of pBD153 (see Figure 2) downstream of the single RS1 in strain ENK12. Additional strain descriptions can be found in Table III.
Table III. Strains of V.cholerae used in this study.
| Strain |
Relevant characteristics; CTX prophage/RS1 arraya |
Source/reference |
| E7946 | O1 El Tor clinical isolate; (RS1)(CTX)(RS1)(CTX)(RS1) | Mekalanos (1983) |
| E4 | E7946 derivative; (RS1)(CTX-Kn)(RS1)b | Pearson (1989) |
| ENK12 | E7946 derivative; (RS1) | Pearson (1989) |
| P27459 | O1 El Tor clinical isolate; (RS1)(CTX)(RS1) | Mekalanos (1983) |
| PNK6 | P27459 derivative; RS1 | Pearson (1989) |
| AS207 | O139 clinical isolate; (RS1)(CTX)(CTXcalc)(CTXcalc) | Sharma et al. (1997) |
| Bah-2 | E7946 ΔattRS Δ(RS1) Δ(CTX); NA | Pearson et al. (1993) |
| O395 | O1 classical clinical isolate; (CTXcl’ ‘CTXcl)[chrI]; (CTXcl) [chrII]c | Mekalanos (1983) |
| TCP-2 | O395 ΔtcpA; (CTXcl’ ‘CTXcl)[chrI]; (CTXcl) [chrII]c | Herrington et al. (1988) |
| 2740-80 | US Gulf Coast isolate, attRS+, CTXφ–; NA | Pearson et al. (1993) |
| BD81 | 2740–80 derivative; (CTX-Kn)(CTX-Kn)b | Davis and Waldor (1999) |
| BD206 | BD81ΔrecA; (CTX-Kn)(CTX-Kn)b | Davis and Waldor (1999) |
| BD25 | 2740-80 derivative; (CTXcalc-Kn) (CTXcalc-Kn)b | Davis et al. (1999) |
aCTX prophages (CTX) are CTXETφ unless noted otherwise. NA, not applicable.
bCTX-Kn and CTXcalc-Kn contain a kanamycin resistance cassette in place of ctxAB.
cClassical biotype strains contain CTX prophages on both their large [chrI] and small [chrII] chromosomes. (CTX’ ‘CTX) represents two truncated, fused prophages that do not yield CTXφ.
Phage production and infection assays
Donor cultures were grown to late log phase. Cultures of recipient O395 and TCP2 were grown overnight at 30°C. Marked and unmarked RS1 and CTXφ infection assays were performed as described (Davis et al., 1999).
Molecular biology methods
Plasmid/extrachromosomal DNA was purified using the QIAprep Spin Miniprep Kit (Qiagen). Southern hybridization was carried out using horseradish peroxidase-labeled DNA probes, prepared and hybridized using the ECL direct nucleic acid labeling and detection system (Amersham Pharmacia). Northern blotting was carried out using the NorthernMax-Gly system (Ambion). Riboprobes were synthesized using the Strip-EZ RNA synthesis kit (Ambion). β-galactosidase assays were performed basically as described (Miller, 1992). Other techniques were performed using standard protocols (Ausubel et al., 1995).
H6RstC purification
6HRstC was expressed in V.cholerae from pBD686 according to standard manufacturer’s recommendations. The cell pellet was lysed in 50 mM phosphate pH 8.0, 0.3 M NaCl, 1% LDAO, 30 mM imidazole, 2 mM phenylmethylsulfonyl fluoride (PMSF), aprotinin 1:10 000. The lysate was cleared by centrifugation at 18 000 r.p.m. and loaded on Ni-NTA resin (Qiagen) equilibrated with 50 mM phosphate pH 8.0, 0.3 M NaCl, 0.1% LDAO, 30 mM imidazole. The column was washed with buffer adjusted to 50 mM imidazole, then 6HRstC was eluted with buffer adjusted to 200 mM imidazole. The eluate was passed over an Amicon YM30 membrane in a stirred cell, and the retentate was washed with copious 50 mM phosphate pH 8.0 with 0.1% LDAO (RstC dilution buffer). The filtrate was concentrated on a YM10 filter in a stirred cell, then on a Centricon YM10.
RstRH6 purification
RstRH6 was expressed in E.coli from plasmid pHK300. It was affinity purified on Ni-NTA resin (Qiagen), largely according to the manufacturer’s protocol.
Co-precipitation of RstC and RstR
A 1.5 µg aliquot of purified RstRH6 was mixed with similar quantities of control proteins (Gel Filtration Standard; Bio-Rad) in 20 µl of lysis buffer [20 mM Tris pH 8.0, 250 mM NaCl, 0.05% Tween-20, 10 mM dithiothreitol (DTT)] and insoluble proteins were pre-cleared by centrifugation at 16 100 g for 30 min at 4°C. A 0.3 µg aliquot of purified H6RstC was added to the cleared supernatant, and the mixture was incubated on ice for 2 h. RstC dilution buffer (50 mM phosphate pH 8.0 with 0.1% LDAO) was added to control reactions in place of H6RstC. Insoluble proteins were isolated by centrifugation as above, resuspended in gel loading buffer and run on NUPAGE polyacrylamide gels (Invitrogen). Analyses with cell lysates were performed in a similar fashion. Cell lysates were generated from E.coli (XL1Blue) expressing plasmid-encoded GFP or GFP fusion proteins; cell pellets from late log phase cultures were frozen, resuspended in lysis buffer, sonicated and pre-cleared by centrifugation prior to mixing with H6RstC.
Fluorescence microscopy
Escherichia coli (XL1Blue) or V.cholerae (E7946) were co-transformed with plasmids encoding GFP derivatives (ApR) and arabinose-inducible RstC (pBD1293; SpR). Overnight cultures were diluted into selective media with or without 1 mM arabinose and grown at 30°C for several hours. Vibrio cholerae strains contained a high copy number plasmid containing lacIq; consequently, production of GFP derivatives in V.cholerae was induced with 0.1 mM IPTG. Cells were fixed with 2% formaldehyde in phosphate-buffered saline (PBS) onto microscope slides; Vectashield mounting media with 4′,6-diamidino-2-phenylindole (Vector Laboratories) was used to preserve cells and fluorescence. Cells were visualized at 1000× magnification using a Nikon epifluorescence microscope and photographed with a Nikon 35 mM camera.
Immunofluorescence microscopy
XL1Blue(pBD686), which produces IPTG-inducible H6RstC, was induced for 2 h with 100 µM IPTG at 30°C, fixed onto microscope slides as above, and permeabilized with lysozyme (2 mg/ml in 50 mM glucose, 10 mM EDTA, 20 mM Tris pH 7.5). Cells were incubated in PBS/0.01% Tween-20/10% goat serum prior to addition of antibodies and during incubations with antibodies. Primary antibody (anti-His5; Qiagen) and secondary antibody [fluorescein isothiocyanate-conjugated goat anti-mouse IgG; Sigma] were each bound for 1 h at room temperature.
Gel shift assays of DNA binding
A DNA restriction fragment derived from the rstA promoter and containing an RstR operator (H.H.Kimsey, in preparation) was labeled with 32P using T4 DNA polymerase then used as a probe in gel shift assays. Reactions consisting of purified RstR and/or RstC and 80 mM NaCl, 25 mM Tris pH 8.0, 1.1 mM EDTA, 550 µg/ml bovine serum albumin (BSA), 12 mM DTT, 50 µg/ml poly(dI/dC) and 100 µg/ml salmon sperm DNA were incubated at 4°C for 1 h. Then 20 000 c.p.m. of probe were added and reactions incubated at room temperature for 30 min. Reactions were analyzed on 6% DNA retardation gels (Invitrogen).
Plasmid construction
To construct pBD482, unmarked plasmid RS1 DNA from strain E7946 was PCR amplified with primers Ig1R1 (5′-ACGACTGCCCCT CAACATAATG-3′) and 657F (5′-GAGGAGACGGAATTTCTACAG-3′) and the product was cloned into the TA cloning vector pCRII-TOPO (Invitrogen). An EcoRI fragment containing the PCR product was then ligated to a Kn resistance gene flanked by EcoRI sites to generate pBD482. pBD153 was constructed in similar fashion using primers RstC3 (5′-GCTTTATATCTGCGTTCAGGCG-3′) and RstC4 (5′-GCTGTT GCATCAGTTACGGGC-3′). pBD485 was generated by PCR amplification of pBD482 with primers RstC3 and RstC5 (5′-TCAGTC TTATAGGCTATCGTAAAC-3′) followed by phosphorylation and circularization of the PCR product. pBD495 (CmR), which encodes IPTG-inducible rstC, contains an ∼380 bp SacI–EcoRI fragment from pBD482 cloned into SacI–EcoRI-digested pGZ119 (Lessl et al., 1992). The rstAET::lacZ, rstAcl::lacZ and rstAcalc::lacZ reporters have been described previously (Kimsey and Waldor, 1998; Davis et al., 1999). pBD873 consists of the ApR sacB counterselectable suicide vector pCVD442 (Donnenberg and Kaper, 1991) containing an ‘rstA–rstB’ fragment of the CTX prophage with the transcriptional terminator rrnB T1 in the EcoRV site of rstA. The ‘rstA–rstB’ fragment was PCR amplified from pCTXKn with RstAF3 (5′-CGGAATTCGCTAGGG GGAAACCGTAATACCTG-3′) and RstBR3 (5′-GCTCTAGATTGC GTGATGGGTCTTCTGG-3′). The terminator was amplified from pQE60 (Qiagen) with the primers TermF1 (5′-TCCCCCGGGTTG GTGCCCTTAAACGCCTG-3′) and TermR1 (5′-TCCCCCGGGTTT TCACCGTCATCACCGAAAC-3′) and digested with SmaI prior to insertion into the rstA EcoRV site. Plasmids encoding most GFP fusion proteins were generated by ligation of a BamHI–PstI-digested PCR product to similarly digested pEGFP (Clontech). rstRcl was amplified using RstR23 (5′-AACTGCAGGTTTAGTTCAAAAATTAGG GATTTAAG-3′) and RstR24 (5′-CGGGATCCCTAAATTCTTTTT GTATTTCTCG-3′). rstRcalc was amplified using RstR25 (5′-AAC TGCAGGGCAACAAAGCACATTAAAGACAC-3′) and RstR26 (5′-CGGGATCCGCTTTTTTTTGCTTTATCTTATGG-3′). toxT was amplified using ToxTF1 (5′-ACCTGCAGGATTGGGAAAAAATCTTT TCAAAC-3′) and ToxTR1 (5′-CGGGATCCGCTTTTTCTGCAA CTCCTGTCAAC-3′). RstR–GFP was generated by amplifying rstRET with RstR10 (5′-GGAAGCTTGAAGATAAAAGAAAGGC-3′) and RstRR10 (5′-GGTACCCGAGCACCATGATTTAAGATG-3′), cloning the PCR product into the TA cloning vector pCRII-TOPO, then ligating a HindIII–KpnI fragment containing rstRET to similarly digested pEGFP. pBD686, an H6RstC expression vector, is pQE30 (Qiagen) ligated to a BamHI–HindIII fragment from pCRII-TOPO-RstC, which was generated using RstC6 (5′-TTGAAACCATACACTTTAATGGATG-3′) and RstC7 (5′-TTACAGTGATGGCTCAGTCAATG-3′). To generate arabinose-inducible rstC, rstC was amplified with RstC8 (5′-GTTAACCATG GGTTTGAAACCATACAC-3′) and RstC7 and cloned into the TA cloning vector pBAD-TOPO (Invitrogen). The resulting ApR plasmid was digested with NcoI to release the enterokinase domain, religated, then digested with NsiI–XmnI to release araC and pBAD::rstC. The NsiI–XmnI fragment was ligated to PstI–HincII-digested pGB2 (SpR) (Churchward et al., 1984), yielding pBD1293. pHK300, the RstRH6 expression vector, was generated with PCR primers RstR1 (5′-CCCATGGCGAAGATAAAAGAAA-3′) and RstR4 (5′-GCGGATCC AGCACCATGATTT-3′). The PCR product was cloned into pCRII-TOPO, released from the resulting plasmid with NcoI and BamHI, and cloned into the same sites in the expression vector pQE60 (Qiagen). All plasmid constructs were checked by DNA sequencing.
Acknowledgments
Acknowledgements
We thank Drs A.Camilli, D.Friedman, S.McLeod, D.Raychaudhuri, L.Sonenshein, P.Wagner, P.Watnick and A.Wright for helpful suggestions and critical reading of the manuscript. We thank J.Koudelka for 434 CI, J.Beaber for SetR, K.Skorupski for pMT5, V.DiRita and J.Mekalanos for strains, S.Lu for technical assistance, and the NEMC GRASP Center for preparation of plates and media. This work was supported by NIH AI-42347 to M.K.W., GM20483-01 to B.M.D., P30DK-34928 for the NEMC GRASP Digestive Center, and the Howard Hughes Medical Institute. M.K.W. is a Pew Scholar in the Biomedical Sciences.
References
- Ausubel F.M., Brent,R., Kingston,R.E., Moore,D., Seidman,J.G., Smith,J.A. and Struhl,K. (eds) (1995) Current Protocols in Molecular Biology. John Wiley and Sons, New York, NY.
- Bertani L.E. and Six,E.W. (1988) The P2-like phages and their parasite, P4. In Calendar,R. (ed.), The Bacteriophages. Vol. 2. Plenum Publishing, New York, NY, pp. 73–143.
- Champion G.A., Neely,M.N., Brennan,M.A. and DiRita,V.J. (1997) A branch in the ToxR regulatory cascade of Vibrio cholerae revealed by characterization of toxT mutant strains. Mol. Microbiol., 23, 323–331. [DOI] [PubMed] [Google Scholar]
- Churchward G., Belin,D. and Nagamine,Y. (1984) A pSC101-derived plasmid which shows no sequence homology to other commonly used cloning vectors. Gene, 31, 165–171. [DOI] [PubMed] [Google Scholar]
- Davis B.M. and Waldor,M.K. (1999) The Vibrio cholerae RS1 element is both a parasite and a helper. In 35th US–Japan Cholera and Other Bacterial Enteric Infections Joint Panel Meeting. US–Japan Cooperative Science Panels, Baltimore, MD, pp. 100–102.
- Davis B.M. and Waldor,M.K. (2000) CTXφ contains a hybrid genome derived from tandemly integrated elements. Proc. Natl Acad. Sci. USA, 97, 8572–8577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis B.M., Kimsey,H.H., Chang,W. and Waldor,M.K. (1999) The Vibrio cholerae O139 Calcutta bacteriophage CTXφ is infectious and encodes a novel repressor. J. Bacteriol., 181, 6779–6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis B.M., Lawson,E.H., Sandkvist,M., Ali,A., Sozhamannan,S. and Waldor,M.K. (2000a) Convergence of the secretory pathways for cholera toxin and the filamentous phage, CTXφ. Science, 288, 333–335. [DOI] [PubMed] [Google Scholar]
- Davis B.M., Moyer,K.E., Boyd,E.F. and Waldor,M.K. (2000b) CTX prophages in classical biotype Vibrio cholerae: functional phage genes but dysfunctional phage genomes. J. Bacteriol., 182, 6992–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiRita V.J., Neely,M., Taylor,R.K. and Bruss,P.M. (1996) Differential expression of the ToxR regulon in classical and E1 Tor biotypes of Vibrio cholerae is due to biotype-specific control over toxT expression. Proc. Natl Acad. Sci. USA, 93, 7991–7995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnenberg M.S. and Kaper,J.B. (1991) Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect. Immun., 59, 4310–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson S.K., Liu,T. and Haggard-Ljungquist,E. (2000) Interacting interfaces of the P4 antirepressor E and the P2 immunity repressor C. Mol. Microbiol., 36, 1148–1155. [DOI] [PubMed] [Google Scholar]
- Faruque S.M., Asadulghani, Kamruzzaman,M., Nandi,R.K., Ghosh,A.N., Nair,G.B., Mekalanos,J.J. and Sack,D.A. (2002) RS1 element of Vibrio cholerae can propagate horizontally as a filamentous phage exploiting the morphogenesis genes of CTXφ. Infect. Immun., 70, 163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilpern A.J. and Waldor,M.K. (2000) CTXφ infection of Vibrio cholerae requires the tolQRA gene products. J. Bacteriol., 182, 1739–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzel T., Velleman,M. and Schuster,H. (1992) C1 repressor of phage P1 is inactivated by noncovalent binding of P1 Coi protein. J. Biol. Chem., 267, 4183–4188. [PubMed] [Google Scholar]
- Herrington D.A., Hall,R.H., Losonsky,G., Mekalanos,J.J., Taylor,R.K. and Levine,M.M. (1988) Toxin, toxin-coregulated pili and the toxR regulon are essential for Vibrio cholerae pathogenesis in humans. J. Exp. Med., 168, 1487–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber K.E. and Waldor,M.K. (2002) Filamentous phage integration requires the host recombinases XerC and XerD. Nature, 417, 656–659. [DOI] [PubMed] [Google Scholar]
- Kaper J.B., Morris,J.G.,Jr and Levine,M.M. (1995) Cholera. Clin. Microbiol. Rev., 8, 48–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantsev A., Preisinger,E., Dranovsky,A., Goldgaber,D. and Housman,D. (1999) Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc. Natl Acad. Sci. USA, 96, 11404–11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimsey H.H. and Waldor,M.K. (1998) CTXφ immunity: application in the development of cholera vaccines. Proc. Natl Acad. Sci. USA, 95, 7035–7039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessl M., Balzer,D., Lurz,R., Waters,V.L., Guiney,D.G. and Lanka,E. (1992) Dissection of IncP conjugative plasmid transfer: definition of the transfer region Tra2 by mobilization of the Tra1 region in trans. J. Bacteriol., 174, 2493–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis R.J., Brannigan,J.A., Offen,W.A., Smith,I. and Wilkinson,A.J. (1998) An evolutionary link between sporulation and prophage induction in the structure of a repressor:anti-repressor complex. J. Mol. Biol., 283, 907–912. [DOI] [PubMed] [Google Scholar]
- Lindqvist B.H., Deho,G. and Calendar,R. (1993) Mechanisms of genome propagation and helper exploitation by satellite phage P4. Microbiol. Rev., 57, 683–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T. and Haggard-Ljungquist,E. (1999) The transcriptional switch of bacteriophage WΦ, a P2-related but heteroimmune coliphage. J. Virol., 73, 9816–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T., Renberg,S.K. and Haggard-Ljungquist,E. (1998) The E protein of satellite phage P4 acts as an anti-repressor by binding to the C protein of helper phage P2. Mol. Microbiol., 30, 1041–1050. [DOI] [PubMed] [Google Scholar]
- Mekalanos J.J. (1983) Duplication and amplification of toxin genes in Vibrio cholerae. Cell, 35, 253–263. [DOI] [PubMed] [Google Scholar]
- Miller J.H. (1992) A Short Course in Bacterial Genetics: A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Moyer K.E., Kimsey,H.H. and Waldor,M.K. (2001) Evidence for a rolling-circle mechanism of phage DNA synthesis from both replicative and integrated forms of CTXφ. Mol. Microbiol., 41, 311–323. [DOI] [PubMed] [Google Scholar]
- Pearson G.D.N. (1989) The cholera toxin genetic element: a site specific transposon. PhD thesis, Harvard University Medical School, Boston, MA.
- Pearson G.D., Woods,A., Chiang,S.L. and Mekalanos,J.J. (1993) CTX genetic element encodes a site-specific recombination system and an intestinal colonization factor. Proc. Natl Acad. Sci. USA, 90, 3750–3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poteete A.R. (1988) Bacteriophage P22. In Calendar,R. (ed.), The Bacteriophages. Vol. 2. Plenum Publishing, New York, NY, pp. 647–682.
- Ross C.A. (1997) Intranuclear neuronal inclusions: a common pathogenic mechanism for glutamine-repeat neurodegenerative diseases? Neuron, 19, 1147–1150. [DOI] [PubMed] [Google Scholar]
- Russel M., Linderoth,N.A. and Sali,A. (1997) Filamentous phage assembly: variation on a protein export theme. Gene, 192, 23–32. [DOI] [PubMed] [Google Scholar]
- Scheibel T. and Lindquist,S.L. (2001) The role of conformational flexibility in prion propagation and maintenance for Sup35p. Nat. Struct. Biol., 8, 958–962. [DOI] [PubMed] [Google Scholar]
- Serio T.R. and Lindquist,S.L. (2000) Protein-only inheritance in yeast: something to get [PSI+]-ched about. Trends Cell Biol., 10, 98–105. [DOI] [PubMed] [Google Scholar]
- Sharma C. et al. (1997) Unique organization of the CTX genetic element in Vibrio cholerae O139 strains which reemerged in Calcutta, India, in September 1996. J. Clin. Microbiol., 35, 3348–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan J.S. et al. (2000) The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl Acad. Sci. USA, 97, 6763–6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldor M.K. and Mekalanos,J.J. (1994) Emergence of a new cholera pandemic: molecular analysis of virulence determinants in Vibrio cholerae O139 and development of a live vaccine prototype. J. Infect. Dis., 170, 278–283. [DOI] [PubMed] [Google Scholar]
- Waldor M.K. and Mekalanos,J.J. (1996) Lysogenic conversion by a filamentous phage encoding cholera toxin. Science, 272, 1910–1914. [DOI] [PubMed] [Google Scholar]
- Waldor M.K., Rubin,E.J., Pearson,G.D., Kimsey,H. and Mekalanos,J.J. (1997) Regulation, replication and integration functions of the Vibrio cholerae CTXφ are encoded by region RS2. Mol. Microbiol., 24, 917–926. [DOI] [PubMed] [Google Scholar]