

Abstract
The assembly of the Gal repressosome, a higher order nucleoprotein complex that represses transcription of the gal operon in Escherichia coli, involves the formation of a DNA loop encompassing the promoter segment. GalR dimers bound to two spatially separated operators, OE and OI, specifically interact with the histone-like protein HU and close the loop in supercoiled DNA. We isolated and characterized a GalR mutant containing an amino acid substitution (R282L) that can repress transcription in the absence of HU and supercoiled DNA both in vivo and in vitro. Repression involves the same DNA looping; deletion of either OE or OI makes the mutant GalR ineffective in repression. This and other results suggest that the R282L substitution increases the normal affinity between two DNA-bound GalR dimers, allowing looping. We conclude that GalR dimers interact directly and do not use HU as an adaptor in loop closure; HU and DNA supercoiling act in concert to stabilize the GalR tetramer. The stronger GalR–GalR interaction also made the gal transcription non-inducible, suggesting that the inducer binding acts by modulating tetramerization.
Keywords: gal regulation/protein–protein interface/transcription repression
Introduction
DNA–multiprotein complexes of different types participate in carrying out various DNA transaction reactions, transcription, replication and repair recombination (Echols, 1990). Many of these complexes have a common theme: they contain topologically independent DNA domains composed of loops formed by bringing two non-contiguous points of a DNA molecule together by sequence-specific DNA-binding proteins. As shown in Figure 1, the sequence-specific DNA-binding proteins, which close the DNA loop, may do so by using one of the following strategies: (I) two similar or dissimilar proteins bind to cognate DNA sites and interact with each other directly (Irani et al., 1983; Dunn et al., 1984; Su et al., 1990; Roy et al., 1991; de Beer et al., 2002); (II) a bidentate protein molecule simultaneously binds to two DNA sites (Kramer et al., 1987; Nash, 1996); (III) in example (I), the two DNA-bound proteins, instead of interacting directly, close the loop by both interacting with an adaptor (Kallipolitis et al., 1997); (IV) in the above examples, DNA looping may be aided energetically by another DNA-binding protein bound to an architecturally critical position (Santero et al., 1992; Aki et al., 1996; Nash, 1996; Wassem et al., 2000; Thomas and Travers, 2001; de Beer et al., 2002); or (V) the adaptor helps loop closure by acting as an architectural protein as well (Cosma et al., 2001; Kar and Adhya, 2001). In the Gal repressosome, which represses transcription in the gal operon in Escherichia coli, two GalR dimer proteins bind to two operator elements, OE and OI, and form a DNA loop encompassing a 113 bp DNA segment comprising the gal promoters (Irani et al., 1983; Majumdar and Adhya, 1984; Adhya et al., 1998; Figure 3A). Formation of the loop by the two operator-bound GalR dimers requires the architectural protein, HU, and supercoiled DNA (Aki et al., 1996; Lewis et al., 1999). The 113 bp DNA loop in the Gal repressome is refractory to transcription initiation from the two gal promoters, P1 and P2 (Choy et al., 1995). Repression is abolished when d-galactose binds to GalR (Nakanishi et al., 1973; Majumdar and Adhya, 1984). During repressosome formation, HU specifically interacts with a segment of the looped DNA, centered at position +6.5, and to GalR, thus suggesting the possibility of both an architectural and an adaptor role for HU (Aki and Adhya, 1997; Kar and Adhya, 2001). An adaptor role for HU raised the question of whether DNA-bound GalR directly interacts at all in loop closure. The results reported here show that operator-bound GalR forms the DNA loop by direct interaction, and that the inducer may act by targeting the tetramerization step.
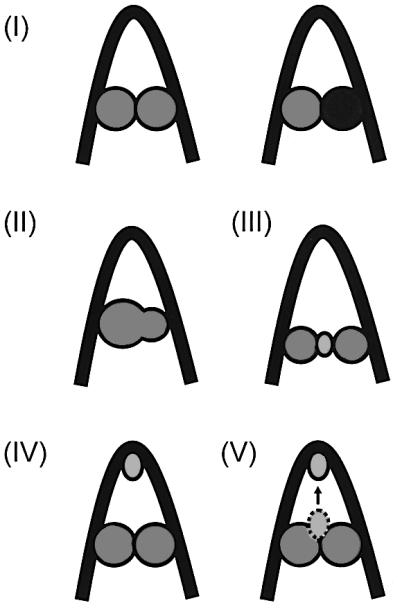
Fig. 1. Modes of DNA looping discussed in the text.

Fig. 3. Effect of the GalR mutant R282L on the transcription of the P1 and P2 promoters in vitro. (A) Schematic drawing of the gal regulatory region in plasmid pSA850 used as a template DNA in the in vitro transcription assay. The wild-type regulatory region, containing the external (OE) and the internal (OI) operator sites and the P1 and P2 promoters, is followed by the rpoC terminator. Transcription termination at the rpoC terminator results in a 125 nucleotide RNA from P1 and a 130 nucleotide RNA from P2. The numbering system used the +1 start site of P1 as reference. The HU-binding site (hbs) is centered at position 6.5. (B) In vitro transcription assay performed on supercoiled pSA850 DNA in the presence of wild-type (left panel) and R282L mutant (right panel) GalR protein. The RNA product was resolved on 8% polyacrylamide gels. The concentration of GalR and HU was 80 nM as shown on the top. The 80 nucleotide RNA1 transcript, which is independent of HU and GalR concentrations, served as an internal control between lanes. (C) Effect of operator deletions on the transcription of the gal promoters. Transcription on supercoiled pSA886 (OEOIN) DNA (left panel) and pSA887 (OENOI) DNA (right panel) in the presence of the wild-type and R282L mutant GalR protein. The concentration of the repressor proteins was 80 nM, as indicated on the top. Transcripts, marked P1 and P2, are RNAs made from the gal promoters. The transcript, marked RNA1, serves as an internal control. (D) Transcription of the gal promoters on linear pSA850 DNA. pSA850 was treated with the restriction enzyme HindIII. The HindIII restriction site is located 491 bp downstream of the OI operator site. The total length of the plasmid is 3.5 kb. Transcription was performed as described in Materials and methods. Proteins were used at the concentrations indicated on the top. Transcripts, marked P1 and P2, are RNAs made from the gal promoters. The transcript, marked RNA1, served as an internal control.
Results
GalR super-repressor mutants
In an in vivo system, the DNA looping and DNA binding activities of GalR were monitored by following the repression of the P2 and P1 promoters, respectively. This was based on the fact that the repression of P2 is entirely dependent upon the formation of a DNA loop, whereas binding of GalR to the OE operator is sufficient for repression of P1 (Aki et al., 1996; Choy et al., 1997). Repression of P2 requires the formation of the repressosome through cooperative binding of GalR to both OE and OI, and of HU to the interoperator segment. The tester strain, DM0026, with two corresponding reporter genes for the two promoters, contains a chromo somal OE+OI+P1–P2+–gusA transcriptional fusion, an OE+OI–P1+P2––lacZ translational fusion and a deletion of the galR gene (Lewis et al., 1999). Wild-type or mutant GalR protein was expressed from a plasmid (pSEM1029 or pSEM1050) in the cell. Assay of β-glucuronidase and β-galactosidase either on indicator plates or in cell extracts reflected the effect of the GalR protein on DNA looping and operator binding, respectively. By screening of site-directed substitutions in a defined surface area of GalR previously surmised to participate in a GalR–GalR interaction, we isolated a GalR mutant, R282L, which showed stronger repression of the P2 promoter relative to that of wild-type GalR without changing P1 repression on indicator plates. As shown in Figure 3A, the expression of either wild-type GalR or R282L mutant protein genes from a plasmid resulted in strong reduction of the β-glucuronidase activity compared with that in the total absence of GalR (ΔgalR). However, the R282L mutant reproducibly repressed the P2 expression more than that of the wild-type GalR. Efficient repression of the P2 promoter is totally dependent upon the presence of the HU protein (Lewis et al., 1999). Deletion of hupA (encoding HUα) and hupB (encoding HUβ) in the cells carrying wild-type galR in the chromosome fully derepresses the P2 promoter. We observed similar derepression of P2, by measuring β-glucuronidase synthesis when the galR+ gene was present in the plasmid (pSEM1029) in the strain deleted for the two hup genes (Figure 3B). However, the same strain showed a significantly lower β-glucuronidase level when the plasmid-borne galR gene was the mutant R282L. Thus, R282L mutant specifically repressed P2 in an HU-independent manner. Another arginine to leucine substitution, R279L, located in the vicinity of R282L in the GalR structure behaved like wild-type repressor, i.e. showed derepression of P2 in the absence of HU (data not shown).
In vitro properties of GalR R282L
To study the behavior of the R282L mutant in vitro, we overexpressed the wild-type and the R282L mutant GalR proteins, and purified them to >95% homogeneity as described in Materials and methods. In vitro transcription was performed using supercoiled DNA template as described previously (Geanacopoulos and Adhya, 1997). The results are shown in Figure 3B. The wild-type GalR protein, as expected, enhanced P2 and repressed P1 transcription. The presence of GalR (wild-type or R282L) and HU resulted in complete repression of both promoters. However, as was found in vivo, the purified R282L mutant GalR repressed transcription in the absence of HU. Addition of 80 nM HU did not interfere with the repression of P2 by R282L mutant.
DNA templates with an OE or OI substitution/deletion were used to study the nature of P2 repression by the mutant GalR protein. As mentioned, DNA looping- mediated P2 repression requires the binding of GalR to both gal operators (Irani et al., 1983; Choy and Adhya, 1992; Aki and Adhya, 1997). In the template pSA886, in which the OI operator was replaced by a random sequence, the R282L mutant did not show repression but activated transcription from P2 and repressed that from P1 as did the wild-type protein (Figure 3C). Neither the wild-type nor the mutant GalR had a significant effect on gal transcription in the DNA template in which OE was absent. These results demonstrate that the repression of P2 by R282L in the absence of HU involves DNA looping by the bipartite operators bound by the mutant GalR.
Effect of supercoiling
DNA looping-mediated repression of the P2 promoter requires a supercoiled DNA template (Aki et al., 1996). If GalR mutant R282L resulted in increased affinity of the bound dimers to form a tetrameric structure, one may expect to form the DNA loop and thus bring about P2 repression with reduced DNA supercoiling or improper helical phasing of the operator sites. Negative supercoiling can be decreased by the addition of coumermycin, an antibiotic that inactivates DNA gyrase (Gellert et al., 1976). Coumermycin inhibits P2 repression by GalR without affecting the intrinsic strength of the promoter (Lewis et al., 1999). Repression of P2 in cells expressing the wild-type and R282L mutant GalR was compared in the presence of coumermycin (Figure 2C and D). In contrast to wild-type, the mutant GalR showed repression of P2 in the presence of coumermycin. This suggests that DNA supercoiling may not be mandatory for DNA looping in the latter case. We tested this hypothesis by studying P2 repression in vitro on linear gal DNA templates with the R282L protein (Figure 3D). As expected, the wild-type GalR enhanced P2 transcription with a concurrent partial repression of P1. The presence of wild-type GalR and HU had the same effect, since the repressosome structure cannot be formed on linear template (Aki et al., 1996). In the presence of mutant GalR protein, however, transcription from both promoters decreased dramatically, in both the absence and presence of HU.

Fig. 2. (A) Effect of the GalR R282L mutation on the looping repression of the galP2 promoter. Differential rates of β-glucuronidase synthesis from P2–gusA in the presence of the plasmids pEM7/Zeo (ΔgalR, triangles), pSEM1029 (galR+, squares) and pSEM1050 (galR R282L, circles). Cells were grown in minimal medium supplemented with 0.4% (w/v) d-fructose, 0.1% (w/v) casamino acids and 0.004 (w/v) vitamin B1. (B) Effect of hup deletions on the looping repression of the P2 promoter in the presence of the wild-type GalR (squares) and R282L GalR (circles). DM0100 cells bearing the P2–gusA chromosomal fusion and lacking both the hupA and hupB genes were transformed with the pSEM1029 and pSEM1050 plasmids, carrying the wild-type and the R282L galR gene, respectively. Strains were assayed for the rates of β-glucuronidase synthesis in the P2–gusA fusions. Conditions for cell growth were as in (A). (C) Differential rates of β-glucuronidase synthesis from P2–gusA in DM0026 (pSEM1029) cells, expressing the wild-type galR gene, in the absence (squares) and presence (diamonds) of coumermycin A1. (D) Differential rates of β-glucuronidase synthesis from P2–gusA in DM0026 (pSEM1050) cells, expressing the R282L mutant galR gene, in the absence (circles) and presence (diamonds) of coumermycin A1. Cells were grown in minimal medium supplemented with 0.4% (w/v) d-fructose, 0.1% (w/v) casamino acids and 0.004% (w/v) vitamin B1. The arrowhead indicates the point of addition of 50 µg/ml coumermycin A1.
Effect of changing the DNA helical phasing
DNA looping-mediated repression of P2 depends on the correct helical phasing of the two operator sites (Lewis and Adhya, 2002). Derepression occurred if the relative angular orientation of OE and OI was changed by the addition or deletion of non-integral DNA helical turns between the two operators. We inserted 5 bp between OE and OI (at position +9) and tested the template for P2 repression by R282L GalR (Figure 4A). The improper helical phasing of the operators inhibited the looping-mediated repression of P2 with wild-type GalR in the presence of HU (Figure 4B). However, the mutant GalR protein, R282L, efficiently repressed transcription from the altered DNA template regardless of the presence or absence of HU. These results demonstrate the capability of the mutant protein to close the DNA loop in spite of improper helical phasing of the operators presumably because of its increased strength in tetramerization.

Fig. 4. The results of the in vitro transcription assay performed on DNA template with a 5 bp insertion. (A) Alteration of the topology of the gal regulatory region upon a 5 bp insertion. The sequence of 5 bp was inserted at position +9. The operator-bound GalR proteins occupy the opposite face of the DNA molecule. (B) Transcription of galP1 and galP2 on supercoiled pSA907 DNA. Proteins were used at the concentrations indicated on the top. Transcripts, marked P1 and P2, are RNAs made from the gal promoters. The transcript, marked RNA1, served as an internal control.
Interaction of the GalR dimers
GalR was shown to form a stable dimer in solution (Majumdar et al., 1987). To investigate the potential tetramerization of the GalR dimers, chemical cross-linking experiments were performed. The purified protein preparations were treated with dimethyl suberimidate (DMS), a homobifunctional imidoester that can react with primary amine groups to form stable covalent bonds. The products of the cross-linking reactions were separated by SDS– PAGE (Figure 5). In the presence of DMS, GalR resulted in protein bands with molecular weights of monomers, dimers, trimers and tetramers. The pattern and intensity of the bands representing the GalR oligomers were similar in the case of the wild-type and the R282L mutant protein preparations, clearly demonstrating the ability of both proteins to tetramerize under the conditions of cross-linking.
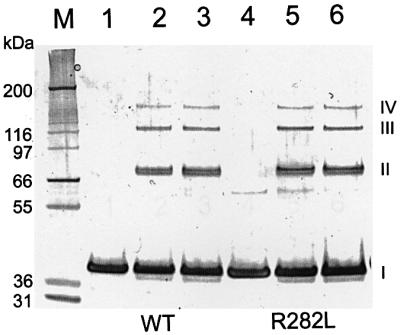
Fig. 5. Cross-linking of the GalR protein. Wild-type (lanes 1–3) and R282L mutant (lanes 4–6) proteins were cross-linked using DMS in the presence of 250 mM (lanes 2 and 5) or 500 mM (lanes 3 and 6) NaCl. In lanes 1 and 4, no DMS was added. Reaction products were separated by SDS–PAGE on a 4–12% gel. The positions of monomers (I), dimers (II), trimers (III) and tetramers (IV) of GalR are marked. Mark 12™ Standard (M; Invitrogen) was used to estimate the molecular weight of the products.
Non-inducibility of GalR R282L
The addition of inducer d-galactose to cells expressing the wild-type GalR derepressed P2 transcription, as expected (Lewis et al., 1999; Figure 6A). However, the R282L mutant GalR showed a non-inducible phenotype; the mutant protein maintained repression of P2 in the presence of d-galactose (Figure 6B). Because of this non-inducible nature, we tested the effect of the inducer on the OE–GalR complex in vitro by electrophoretic mobility shift assay (EMSA; Majumdar and Adhya, 1984). GalR was bound to the operator DNA, followed by the addition of different amounts of d-galactose. Compared with the wild type, the R282L mutant GalR protein bound to OE with ∼10% higher efficiency in the absence of the inducer (data not shown). The percentage of DNA remaining bound to GalR after the addition of different concentrations of d-galactose is shown in Figure 7A and B. The amount of d-galactose needed to reduce the amount of OE–GalR complexes by 50% was ∼0.12 mM for wild-type GalR and 8 mM for R282L.

Fig. 6. (A) Differential rates of β-glucuronidase synthesis in the DM0026 (pSEM1029) cells expressing the wild-type galR gene in the absence (squares) and presence (diamonds) of the inducer d-galactose. (B) Differential rates of β-glucuronidase synthesis in the DM0026 (pSEM1050) cells expressing the R282L mutant galR gene in the absence (circles) and presence (triangles) of the inducer d-galactose. The arrow indicates the time of addition of the inducer at 5 mM final concentration.

Fig. 7. Effect of d-galactose on the stability of the GalR–OE complex. (A) GalR binding to the 32P-labeled OE operator sequence in the presence of varying amounts of d-galactose in a gel retardation assay. (B) Percentage of the OE DNA bound by the wild-type (squares) and the R282L mutant (circles) GalR proteins in the presence of the indicated amounts of d-galactose.
Discussion
In our attempt to define the structure of the Gal re pressosome, we previously described the isolation and characterization of GalR mutants that bind to DNA operators normally but are defective in potential GalR–GalR or GalR–HU contacts during DNA loop formation (Geanacopoulos et al., 1999, 2001). These looping-defective amino acid substitutions in the GalR dimer were located in the DNA-distal domain, which preferably defined a GalR–GalR rather than a GalR–HU contact area. Further genetic investigations of the potential GalR–GalR contacts by isolation and characterization of intragenic suppressors and by alanine scanning of looping-defective GalR mutations suggested the existence of a GalR tetramerization interface (Geanacopoulos and Adhya, 2002). In this study, we report the physiological and biochemical characterization of a GalR mutant protein, which can repress gal transcription and thus form the Gal repressosome in the absence of HU and supercoiled DNA. Since the mutant GalR behaved in this way not only in vivo, but also in assays with purified proteins, the repressosome formation in cells by HU and supercoiled DNA independent of GalR was not because of substitutions of HU by an unknown protein capable of working in concert with the mutant protein. We also showed the ability of both the wild-type and the mutant GalR to tetramerize by chemical cross-linking experiments. These results demonstrate that tetramerization is an intrinsic property of GalR; it closes the DNA loop by direct GalR–GalR contact. Since the substituted amino acid residue (R282L) is located within the genetic ally defined area of the tetramerization interface (Geanacopoulos et al., 2001), and the GalR containing both R282L and another looping-defective substitution failed to form the repressosome (data not shown), we conclude that the R282L change did not create a new GalR–GalR tetramer interface, but enhanced the strength of the pre-existing interface. This conclusion is consistent with the results that GalR with the single R282L substitution: (i) showed stronger repression than the wild-type protein; (ii) could withstand a distortion of the relative angular alignment of the two operator-bound GalR dimers on DNA; and (iii) was not dependent upon the architectural protein HU and supercoiled DNA to stabilize energetically a presumably weaker GalR–GalR interaction. The R282A substitution also showed the same phenotype as the R282L change. It seems likely that a non-hydrophobic residue at position 282 in wild-type GalR contributes to the reversibility of the system by making the dimer–dimer interaction weaker. Strong hydrophobic interactions are not favored in reversible protein–protein interactions (Jones and Thornton, 1996; Gerk et al., 2000). The R282L/A changes in the mutant GalR did just that. The predicted quantitative difference in the dimer–dimer interaction between the wild type and mutant in the transcription system was not investigated.
The R282L/A substitutions also make the GalR protein, as shown above, insensitive to the effect of d-galactose, which acts by interacting at a distant domain of the protein. In the closely related LacI protein, the mechanism of inducer binding was explained by the allosteric transition model (Lewis et al., 1996; Bell and Lewis, 2000). In this model, inducer and DNA binding exert their effects by shifting the protein between two basic states. These states differ in the relative rotation of the two highly similar globular subunits of the monomer core. Sugar binding in the cleft between these two globular domains causes a rotation of the two subunits with respect to one another that alters the position of the DNA-binding domain so that in the dimeric protein the orientation of the two headpieces or DNA-binding domains is less favorable for operator binding. In the GalR mutant R282L, non-inducibility resulting from strong tetramerization was coupled with a slight increase in the stability of the GalR–OE complex, strongly suggesting that the R282L mutation acts by stabilizing the DNA-binding conformation and increasing the energy required for the structural rearrangement in the sugar-binding state. The multiple effects of the R282L substitution indicate that tetramerization as well as DNA and inducer binding are coupled properties of the GalR protein. In LacI, inducer binding does not affect protein tetramerization because the two protein dimers are linked by the relatively flexible four helix bundle (Alberti et al., 1993). The V-shaped stacked structure in the modeled GalR tetramer does not allow such flexibility of the dimers (Geanacopoulos et al., 2001). Based on our results, we propose that the allosteric transition resulted from inducer binding affecting not only the DNA binding but also tetramerization of the GalR protein. Because of the very weak dimer–dimer interaction, induction by d-galactose is likely to involve breaking of the loop by altering the tetramerization interface before GalR is removed from the operators. Because, in the absence of DNA looping, GalR activates transcription from the P2 promoter without the inducer being present (Choy and Adhya, 1992; Aki and Adhya, 1997), induction of the gal operon by this scenario would be considerably more rapid than in the case where the stability of the GalR–GalR interface is not modulated by inducer binding.
In summary, we report that the DNA loop in the Gal repressosome closes by tetramerization of two DNA-bound GalR dimers [model (I); see Introduction] without using HU as an adaptor in between [model (III)] although the latter is recruited by GalR [model (V)] (Kar and Adhya, 2001). Wild-type GalR, because of weak dimer– dimer interaction, needs the aid of supercoiled DNA and binding of HU as an architectural protein during DNA loop formation. A weak dimer–dimer interaction makes the DNA looping-mediated transcription repression, as argued above, quickly responsive to cellular need for gal operon expression.
Materials and methods
DNA manipulation methods
Plasmid manipulations followed protocols described in Sambrook and Russel (2001). Transformations were performed with Max Efficiency DH5α competent cells (Invitrogen). Restriction endonucleases were purchased from Invitrogen, DNA oligonucleotide primers from BioServe Biotechnologies, PCR (GeneAmp XL) and sequencing (ABI Prism) kits from Applied Biosystems, and DNA purification kits from Qiagen. DNA sequencing reactions were analyzed in a Perkin-Elmer/Applied Biosystems (model 373A) automated sequencer.
Plasmid constructions
To create plasmid pSEM1026, the galR gene was PCR amplified to allow for the addition of a sequence encoding the His6 tag (GSSHHHHHHSS) at the C-terminal end of the galR open reading frame. The PCR product contained an NcoI site coincident with the ATG start codon and an XbaI site downstream of the stop codon of the galR gene. The amplified DNA fragment was inserted into the pBAD24 vector using the NcoI and XbaI sites (Guzman et al., 1995). The vector contained a Shine–Dalgarno sequence upstream of the translation start codon of the galR gene. The R282L mutation in the galR gene in plasmid pSEM1051 was created by PCR. The DNA sequence of the gene in the plasmid was verified. To create pSEM1029, the NheI–PstI fragment of pSEM1026 containing the ribosome-binding site and the galR gene was inserted between the XbaI and PstI sites of plasmid pEM7/Zeo (Invitrogen). Similarly, the NheI–PstI fragment containing the R282L substitution from pSEM1051 was cloned into pEM7/Zeo to generate pSEM1050. Plasmid pSA886 (OEOIN) was generated by replacing the BstEII–HindIII fragment of pSA850 (Lewis and Adhya, 2002) by a PCR fragment, which contained a random 16 bp sequence (5′-CACTATGGCGAACGTC-3′) at the OI operator site. This site will be referred to as ON. It was shown by gel retardation assay that GalR does not bind to this sequence (data not shown). Similarly, the OE operator of pSA850 was replaced by the same random 16 bp sequence to generate pSA887 (OENOI). Plasmid pSA907 contained a 5 bp (GATCT) insertion at +9 to generate a BglII site in pSA850. It was constructed by two overlapping PCR amplifications: (i) pSA509-1 (5′-GAGCTCGTCGACCCGGGTACCG-3′) and DLinsert 5D (5′-AATTCGCTCCATTAGGCAGATCTTATGGTATGAAATAACC ATAGCA-3′); and (ii) DLinsert 5C (5′-TTTCATACCATAAGAT CTGCCTAATGGAGCGAATTATGAG-3′) and BAMH-1 (5′-AAG ACTCTATGGGATCCAGATAAGCGATAAGTTTGCTCAACATCT TCTCGG-3′). Both PCR fragments were mixed and amplified with pSA509-1 and BAMH-1. The resulting product, which was digested with EcoRI and PstI, was cloned into pSA850, which was also digested beforehand with EcoRI and PstI.
Expression and purification of the His6-tagged GalR protein
Escherichia coli strain DH5α bearing pSEM1026 or pSEM1051 was grown in superbroth containing 50 µg/ml ampicillin. After the optical density at 600 nm reached 0.4, the culture was induced with 0.2% arabinose for 5 h. The cells were harvested by centrifugation and stored at –80°C. The frozen cells were resuspended in 1/40 volume of lysis I buffer (50 mM sodium phosphate pH 8.0, 0.5 mg/ml lysozyme) and stored on ice for 30 min. An equal volume of lysis II buffer (50 mM sodium phosphate pH 8.0, 2 M NaCl, 8 mM imidazole, 20% glycerol, 1% Triton X-100) was added and incubated for 30 min on ice. The cell debris was removed by centrifugation at 10 000 g for 1 h. Addition of 3% Ni-NTA slurry (Qiagen) to the solution was followed by 1 h incubation at 4°C. A Poly-Prep Chromatography Column (Bio-Rad) was used to collect the protein bound to Ni-NTA–agarose from the mixture. Twenty column volumes of washing buffer (50 mM sodium phosphate pH 8.0, 600 mM NaCl, 60 mM imidazole, 10% glycerol) were allowed to flow through the column. GalR was eluted by four column volumes of elution buffer (50 mM sodium phosphate pH 8, 600 mM NaCl, 10% glycerol) containing 250 and 500 mM imidazole for wild-type and mutant GalR, respectively, and stored at –80°C in 100 µl aliquots. Approximately 2.5 mg of protein was eluted from 1 ml of Ni-NTA–agarose in each case.
Chemical cross-linking
Repressor proteins were cross-linked using DMS (Pierce). Reactions were performed in HEPES pH 8.0 buffer containing 250 or 500 mM NaCl. The final concentration of the protein in the cross-linking reaction was 0.1 mg/ml. DMS was added at 2.5 mM concentration and the reactions were incubated at room temperature for 60 min. Reactions were terminated by addition of NuPAGE LDS sample buffer and Sample reducing agent (Invitrogen). Reaction products were separated on a 4–12% SDS– polyacrylamide gel and stained using the SilverQuest Silver Staining Kit (Invitrogen).
In vitro transcription
Transcription reactions were performed as described previously (Geanacopoulos and Adhya, 1997). The reaction mixture (50 µl) contained 20 mM Tris–acetate pH 7.8, 10 mM magnesium acetate, 100 mM potassium glutamate, 2 nM DNA template and 20 nM RNA polymerase. After incubation of the reactions at 37°C for 5 min, transcription was started by the addition of 1.0 mM ATP, 0.1 mM GTP, 0.1 mM CTP, 0.01 mM UTP and 5 µCi (for supercoiled template) or 20 µCi (for linear template) of [α-32P]UTP (3000 Ci/mmol). The reaction was terminated after 10 min by addition of an equal volume of transcription loading buffer (0.025% bromophenol blue, 0.025% xylene cyanol, 0.01 M EDTA and 90% deionized formamide). After heating at 90°C for 3 min, the samples were loaded onto 8% polyacrylamide–urea DNA sequencing gels. To generate linear templates for transcription, pSA850 DNA was digested with HindIII, which cuts the plasmid 491 bp downstream of OI. Successful linearization was verified by agarose gel electrophoresis.
DNA binding of GalR in the absence and presence of inducer
The 88 bp EcoRI–BglII DNA fragment containing the OE operator from pSA907 was labeled with [α-32P]dATP by end filling, separated by PAGE and purified using a Qiaex II gel extraction kit (Qiagen). Proteins (2.5 µg/ml) and labeled DNA fragments (2 pM) were mixed in the same buffer used for the in vitro transcription assay together with 10% glycerol and 2.5 ng/µl sonicated salmon sperm DNA. Aliquots of 19 µl of the binding reaction mixture were added to Eppendorf tubes containing 1 µl of d-galactose solutions to give the final concentrations of the inducer as indicated in Figure 6, and incubated at room temperature for 6 min. The reaction mixtures were loaded on a 4% polyacrylamide gel and electrophoresed with 1.5 mA/cm of gel in 1× TBE buffer for 2 h at room temperature. DNA bands were quantified using the ImageQuant™ PhosphorImager (Molecular Dynamics).
Assay of β-glucuronidase and β-galactosidase activities
Cells were grown overnight in LB medium and diluted 50-fold for further growth in M63 supplemented with 0.4% (w/v) d-fructose, 0.1% (w/v) casamino acids and 0.004% (w/v) vitamin B1. At various times, aliquots of cells were removed, pelleted and resuspended in M63 medium containing 100 µg/ml chloramphenicol, and stored on ice (Wilson et al., 1992). The optical densities of the cells were measured at 600 nm. The activity of β-glucuronidase from the P2–gusA fusion was determined by the Softmax microplate spectrophotometer system. Aliquots of 50 µl of permeabilization buffer [100 mM Tris pH 7.8, 32 mM sodium phosphate, 8 mM dithiothreitol (DTT), 8 mM CDTA and 4% Triton X-100] containing 200 µg/ml polymyxin B (Schupp et al., 1995) were placed in the wells of a microtiter plate, followed by the addition of 100 µl of cells. Cells were allowed to permeabilize at room temperature for 15 min before 50 µl aliquots of GUS assay buffer (0.5 mM DTT, 1 mM EDTA, 50 mM sodium phosphate pH 7) containing 1.25 mM α-p-nitrophenyl β-d-glucuronide were added (Wilson et al., 1992). The rate of β-glucuronide hydrolysis was determined at 405 nm and 37°C. The activity of β-galactosidase from the P1–lacZ fusion was measured as described above for β-glucuronidase activity except that 50 µl of o-nitrophenyl β-d-galactoside solution (4 mg/ml in 2 mM sodium citrate) was used as a substrate. The rate of β-galactoside hydrolysis was determined at 420 nm and 28°C.
References
- Adhya S., Geanacopoulos,M., Lewis,D.E., Roy,S. and Aki,T. (1998) Transcription regulation by repressosome and by RNA polymerase contact. Cold Spring Harbor Symp. Quant. Biol., 63, 1–9. [DOI] [PubMed] [Google Scholar]
- Alberti S., Oehler,S., von Wilcken-Bergmann,B. and Muller-Hill,B. (1993) Genetic analysis of the leucine heptad repeats of Lac repressor: evidence for a 4-helical bundle. EMBO J., 12, 3227–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aki T. and Adhya,S. (1997) Repressor induced site-specific binding of HU for transcriptional regulation. EMBO J., 12, 3666–3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aki T., Choy,H.E. and Adhya,S. (1996) Histone-like protein HU as a specific transcriptional regulator: co-factor role in repression of gal transcription by Gal repressor. Genes Cells, 1, 179–188. [DOI] [PubMed] [Google Scholar]
- Bell C.E. and Lewis,M. (2000) A closer view of the conformation of the Lac repressor bound to operator. Nat. Struct. Biol., 7, 209–214. [DOI] [PubMed] [Google Scholar]
- Choy H.E. and Adhya,S. (1992) Control gal transcription through DNA looping; inhibition of the initial transcribing complex. Proc. Natl Acad. Sci. USA, 89, 11264–11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy H.E., Park,S.W., Parrack,P. and Adhya,S. (1995) Transcription regulation by inflexibility of promoter DNA in a looped complex. Proc. Natl Acad. Sci. USA, 92, 7327–7331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy H.E., Hanger,R.R., Aki,T., Mahoney,M., Murakami,K., Ishihama,A. and Adhya,S. (1997) Repression and activation of promoter-bound RNA polymerase activity by Gal repressor. J. Mol. Biol., 272, 293–300. [DOI] [PubMed] [Google Scholar]
- Cosma M.P., Panizza,S. and Nasmyth,K. (2001) Cdk1 triggers association of RNA polymerase to cell cycle promoters only after recruitment of the mediator by SBF. Mol. Cell, 7, 1213–1220. [DOI] [PubMed] [Google Scholar]
- de Beer T. et al. (2002) Insights into specific DNA recognition during the assembly of a viral genome packaging machine. Mol. Cell, 9, 981–991. [DOI] [PubMed] [Google Scholar]
- Dunn T.M., Hahn,S., Ogden,S. and Schleif,R.F. (1984) An operator at –280 base pairs that is required for repression of araBAD operon promoter: addition of DNA helical turns between the operator and promoter cyclically hinders repression. Proc. Natl Acad. Sci. USA, 81, 5017–5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echols H. (1990) Nucleoprotein structures initiating DNA replication transcription and site-specific recombination. J. Biol. Chem., 265, 14697–14700. [PubMed] [Google Scholar]
- Geanacopoulos M. and Adhya,S. (1997) Functional characterization of roles of GalR and GalS as regulators of the gal regulon. J. Bacteriol., 179, 228–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geanacopoulos M. and Adhya,S. (2002) Genetic analysis of GalR tetramerization in DNA looping during repressosome assembly. J. Biol. Chem., in press. [DOI] [PubMed] [Google Scholar]
- Geanacopoulos M., Vasmatzis,G., Lewis,D.E.A., Roy,S., Lee,B.K. and Adhya,S. (1999) GalR mutants defective in repressosome formation. Genes Dev., 13, 1251–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geanacopoulos M., Vasmatzis,F., Zhurkin,V.B. and Adhya,S. (2001) Gal repressosome contains an antiparallel DNA loop. Nat. Struct. Biol., 8, 432–436. [DOI] [PubMed] [Google Scholar]
- Gellert M., O’Dean,M.H., Itoh,T. and Tomizawa,J.-I. (1976) Novobiocin and coumermycin inhibit DNA supercoiling catalyzed by DNA gyrase. Proc. Natl Acad. Sci. USA, 73, 4474–4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerk L.P., Leven,O. and Muller-Hill,B. (2000) Strengthening the dimerisation interface of Lac repressor increases its thermostability by 40°C. J. Mol. Biol., 299, 805–812. [DOI] [PubMed] [Google Scholar]
- Guzman L.-M., Belin,D., Carson,M.J. and Beckwith,J. (1995) Tight regulation, modulation and high level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol., 177, 4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irani M., Orosz,L. and Adhya,S. (1983) A control element within a structural gene: the gal operon of Escherichia coli. Cell, 32, 783–788. [DOI] [PubMed] [Google Scholar]
- Jones S. and Thornton,J.M. (1996) Principles of protein–protein interactions. Proc. Natl Acad. Sci. USA, 93, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallipolitis B.H., Norregaard-Madsen,M. and Valentin-Hansen,P. (1997) Protein–protein communication: structural model of the repression complex formed by CytR and the global regulator CRP. Cell, 89, 1101–1109. [DOI] [PubMed] [Google Scholar]
- Kar S. and Adhya,S. (2001) Recruitment of HU by piggyback: a special role of GalR in repressosome assembly. Genes Dev., 15, 2273–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer H., Niemoller,M., Amouyal,M., Revet,B., von Wilcken-Bergmann,B. and Muller-Hill,B. (1987) Lac repressor forms loops with linear DNA carrying two suitably placed lac operators. EMBO J., 6, 1481–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis D.E.A. and Adhya,S. (2002) In vitro repression of the gal promoters by GalR and HU depends on the proper helical phasing of the two operators. J. Biol. Chem., 277, 2498–2504. [DOI] [PubMed] [Google Scholar]
- Lewis D.E.A., Geanacopoulos,M. and Adhya,S. (1999) Roles of HU and DNA supercoiling in transcription repression: specialized nucleo protein repression complex at gal promoters in Escherichia coli. Mol. Microbiol., 31, 451–462. [DOI] [PubMed] [Google Scholar]
- Lewis M., Chang,G., Horton,N.C., Kercher,M.A., Pace,H.C., Schumacher,M.A., Brennan,R.G. and Lu,P. (1996) Crystal structure of the lactose operon repressor and its complexes with DNA and inducer. Science, 271, 1247–1254. [DOI] [PubMed] [Google Scholar]
- Majumdar A. and Adhya,S. (1984) Demonstration of two operator elements in gal: in vitro repressor binding studies. Proc. Natl Acad. Sci. USA, 81, 6100–6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar A., Rudikoff,S. and Adhya,S. (1987) Purification and properties of Gal repressor:pL-galR fusion in pKC31 plasmid vector. J. Biol. Chem., 262, 2326–2331. [PubMed] [Google Scholar]
- Nakanishi S., Adhya,S., Gottesman,M.E. and Pastan,I. (1973) In vitro repression of the transcription of gal operon by purified gal repressor. Proc. Natl Acad. Sci. USA, 70, 334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash H.A. (1996) The E.coli HU and IHF proteins; accessory factors for complex protein–DNA assemblies. In Lin,E.C.C. and Lynch,A.S. (eds), Regulation of Gene Expression in E.coli. R.G. Landes Company, Georgetown, TX, pp. 149–179.
- Roy A.L., Meisterernst,M., Pognonec,P. and Roeder,R.G. (1991) Cooperative interaction of an initiator-binding transcription initiation factor and the helix–loop–helix activator USF. Nature, 394, 245–248. [DOI] [PubMed] [Google Scholar]
- Sambrook J. and Russel,D.D. (2001) Molecular Cloning. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Santero E., Hoover,T.R., North,A.K., Berger,D.K., Porter,S.C. and Kustu,S. (1992) Role of integration host factor in stimulating transcription from the σ54-dependent nifH promoter. J. Mol. Biol., 227, 602–620. [DOI] [PubMed] [Google Scholar]
- Schupp J.M., Travis,S.E., Price,L.B., Shand,R.F. and Keim,P. (1995) Rapid bacterial permeabilization reagent useful for enzyme assays. Biotechniques, 19, 18–20. [PubMed] [Google Scholar]
- Su W., Porter,S., Kustu,S. and Echols,H. (1990) DNA-looping and enhancer activity, association between DNA-bound NtrC activator and RNA polymerase at the bacterial glnA promoter. Proc. Natl Acad. Sci. USA, 87, 5504–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas J.O. and Travers,A.A. (2001) HMG1 and 2 and related ‘architectural’ DNA-binding proteins. Trends Biochem. Sci., 26, 167–174. [DOI] [PubMed] [Google Scholar]
- Wassem R., deSose,E.M., Yates,M.G., Pedrosa,F.D. and Buck,M. (2000) Two roles for integration host factor at an enhancer-dependent nifA promoter. Mol. Microbiol., 35, 756–764. [DOI] [PubMed] [Google Scholar]
- Wilson K.J., Hughes,S.G. and Jefferson,R.A. (1992) The Escherichia coli gus operon: induction and expression of the gus operon in Escherichia coli and the occurrence and use of the gus in other bacteria. In Gallagher,S.R. (ed.), Gus Protocols: Using the gus Gene as a Reporter of Gene Expression. Academic Press, San Diego, CA, pp. 7–22