

Abstract
Using a transactivation-defective p53 derivative as bait, STK15, a centrosome-associated oncogenic serine/threonine kinase, was isolated as a p53 partner. The p53–STK15 interaction was confirmed further by co-immunoprecipitation and GST pull-down studies. In co-transfection experiments, p53 suppressed STK15-induced centrosome amplification and cellular transformation in a transactivation-independent manner. The suppression of STK15 oncogenic activity by p53 might be explained in part by the finding that p53 inhibited STK15 kinase activity via direct interaction with the latter’s Aurora box. Taken together, these findings revealed a novel mechanism for the tumor suppressor function of p53.
Keywords: Aurora box/centrosome amplification/oncogenic kinase/p53/STK15
Introduction
The p53 tumor suppressor protein frequently is inactivated in human cancers. p53 plays a pivotal role in maintaining genome stability by responding to a number of environmental stresses consisting of DNA damage, ribonucleotide depletion, microtubule disruption and genetic stresses created by oncogene activation (Ko and Prives, 1996; Giaccia and Kastan, 1998; Mowat, 1998). These diverse stimuli lead to the stabilization of p53, allowing p53 to execute its functions, including nucleic acid modification as well as transcriptional activation of target genes participating in cell cycle control and apoptosis (Ko and Prives, 1996; Giaccia and Kastan, 1998; Mowat, 1998).
Furthermore, p53 is also distributed to the centrosome (Zantema et al., 1985; Blair and Blair, 1988; Brown et al., 1994; Morris et al., 2000; Ciciarello et al., 2001; Tarapore et al., 2001), which serves as the organizing center for the assembly of the mitotic spindle and therefore ensures the symmetric segregation of newly synthesized DNA into the two daughter cells. These observations strongly suggest that p53 participates in the centrosome biology. Indeed, downregulation of p53 activity correlates with centrosome amplification of wound cells during tissue repair (Antoniades et al., 1994; Oberringer et al., 1999). Furthermore, deletion of the p53 gene results in centrosome amplification (Fukasawa et al., 1996), which is reminiscent of one of the phenotypes induced by elevated expression of STK15 (Zhou et al., 1998). Nonetheless, how p53 regulates centrosome amplification remains to be addressed, although both transactivation-dependent and -independent mechanisms have been postulated (Jimenez et al., 2000; Tarapore et al., 2001). While modulation of CDK2 activity by p53 has been implicated in the regulation of the centrosome cycle via the transactivation-dependent process (Hinchcliffe et al., 1999; Lacey et al., 1999; Jimenez et al., 2000; Okuda et al., 2000), how p53 controls centrosome duplication via the transactivation-independent pathway is not fully understood.
STK15 (also named BTAK, Aurora 2, ARK1, Aik or HsAIRK1) is a centrosome-associated serine/threonine oncogenic kinase (Giet and Prigent, 1999), consisting of 403 amino acids with a mol. wt of 46 kDa. STK15 exhibits significant homology to Drosophila Aurora and yeast Ipl1 whose mutations are related to defects in centrosome cycle progression and chromosome segregation, respectively (Giet and Prigent, 1999). Although the precise function of STK15 is not known, amplification and overexpression of STK15 have been detected in primary breast tumors and colorectal cancers as well as in multiple human tumor cell types (Bischoff et al., 1998; Zhou et al., 1998). Recently, a pathological role for STK15 has been demonstrated by experiments showing that overexpression of STK15 evidently results in abnormal centrosome amplification and cellular transformation in mammalian cells (Bischoff et al., 1998; Zhou et al., 1998). Interestingly, the kinase function of STK15 is dispensable for centrosome amplification (Meraldi et al., 2002), although it is absolutely required for cellular transformation (Bischoff et al., 1998).
Using a transactivation-defective p53 derivative as bait, we isolated STK15 as a p53-interacting protein by a yeast two-hybrid screen. In conjunction with the observation that either loss of p53 function or overexpression of STK15 protein causes abnormal centrosome amplification and tumorigenesis, we set out to investigate whether p53 plays a role in regulating the STK15 oncogenic activity. Here we show that the p53–STK15 interaction resulted in the suppression of STK15 oncogenic activity in a p53 transactivation-independent manner.
Results
Identification and characterization of STK15 as a p53-interacting protein
Although p53 is organized in structure into three functional domains, an N-terminal region involved in transcriptional activation, a central region mediating specific DNA binding and a C-terminal region responsible for oligomerization, transcriptional repression and non- specific DNA binding (Ko and Prives, 1996), these domains are not totally independent. For example, both the N- and C-terminal domains are critical for the modulation of DNA-binding activity of the central domain (Hsu et al., 1995; Jayaraman and Prives, 1995; Ko and Prives, 1996; Selivanova et al., 1997; Hansen et al., 1998), and the conformation of the central domain has great influence on the transactivation activity of the N-terminal domain (Chuang et al., 1995; Tung et al., 1999). Thus, it should be desirable to use full-length p53 as bait for the isolation of p53 partners by yeast two-hybrid screening. However, to prevent the transactivation activity of p53 from interfering with the screening, a p53 mutant defective in transactivation activity, p53(L22Q/W23S) (Lin et al., 1994) (referred to as p53M hereafter), was used instead. Approximately 5.5 × 105 colonies were screened. Among several clones identified and sequenced, two of them contained cDNAs that encode derivatives of STK15 protein.
To determine whether the full-length STK15 interacts with p53 and to delineate the interaction domains, a series of experiments were performed. As summarized in Figure 1, STK15 interacted quite well with p53M (row 1). Control experiments confirmed the specificity of the p53–STK15 interaction: the introduction of pGAL4DB-p53M and pGAL4AD into yeast produced no colony (row 2). Similarly, no colony was observed upon co-transformation of pGAL4DB with pGAL4AD-STK15 into yeast (row 3). The p53 region required for the p53–STK15 interaction was mapped to the region spanning approximately residues 1–318 of the protein (row 4), since further deletions either destroying the p53 central region (row 5) or eliminating the p53 N-terminal region (rows 6 and 7) completely abolished the interaction. Thus, interaction with STK15 appeared to require most of the p53 protein, suggesting the possibility that p53 conformation plays a role in the p53–STK15 interaction. Further evidence to support this notion is provided below (Figure 2). As for STK15, the kinase domain appeared to be dispensable for the interaction (row 8). Interestingly, the N-terminal region of STK15 containing the Aurora box speculated to be a motif for protein–protein interaction (Giet and Prigent, 1999) was sufficient for the interaction with p53 (rows 9 and 10).

Fig. 1. Characterization of the p53–STK15 interaction in yeast. Left column: GAL4 DNA-binding domain (amino acids 1–147) hybrids. Middle column: GAL4 activation domain (amino acids 768–881) hybrids. The STK15 fragment fused to GAL4 activation domain is indicated on the right of the diagrams. The relative positions of the Aurora box and kinase domains of STK15 are also depicted. Right column: yeast colony color after transformation; the relative color is in parentheses. No colony indicates that no yeast colony was observed after incubation for a standard period of time.

Fig. 2. In vivo interaction of p53 with STK15. (A) Interaction of endogenous p53 and STK15 in 293 cells. Panels I and II: co-immunoprecipitation of p53 and STK15 using lysates prepared from 293 cells. The antibody used is indicated on the top of each lane. The position of p53 or STK15 is shown. (B) Interaction of p53 and STK15 derivatives in H1299 cells. Panel I: co-immunoprecipitation experiments using lysates prepared from H1299 cells transfected with HA-tagged STK15 alone (lanes 5 and 9), and plus p53 (lanes 6 and 10), p53(R175H) (lanes 7 and 11) or p53(R249S) (lanes 8 and 12), and then precipitated with pre-immune serum (lanes 5–8) or with an anti-HA antibody (lanes 9–12) followed by immunoblotting with an anti-p53 antibody. Cell extracts: one hundredth of the cell lysates used in the co-immunoprecipitation was loaded directly onto the gel. Panel II: as in panel I, except that HA-tagged STK15 was replaced with HA-tagged STK15(121–403) for transfection. Panel III: as in panel I, except that p53(R175H) and p53(R249S) were replaced with p53(R273H) and p53(R248W) for transfection.
Interaction of STK15 with p53 in vivo and in vitro
Due to technical considerations, a transactivation-defective p53 mutant, p53M, was used to demonstrate the interaction between p53 and STK15 in yeast (Figure 1). To address whether the interaction between wild-type p53 and STK15 exists under physiological conditions, a series of co-immunoprecipitation experiments were carried out. The 293 cell was chosen for the experiment because the p53 pathway seems to be intact in 293 cells. For example, p53-mediated biological activities, such as transactivation, induction of S and G2 checkpoints and apoptosis as well as post-translational modifications of p53, have been demonstrated frequently in 293 cells (Moore et al., 1996; Matsuzawa et al., 1998; Froesch et al., 1999; Kirch et al., 1999; Kwek et al., 2001; Alves et al., 2002). As shown in Figure 2A, when p53 was immunoprecipitated from 293 cell lysates, STK15 was also found in the precipitates (panel I). Similarly, a reciprocal immunoprecipitation experiment performed with an anti-STK15 antibody first and then blotted for the presence of p53 (panel II) led to the same conclusion that p53 associated with STK15 in vivo.
To characterize further the physical association between p53 and STK15, transient transfection studies were performed in 293 and H1299 cells. In these experiments, derivatives of p53 and a hemagglutinin (HA) epitope-tagged STK15 were ectopically expressed and their interaction was determined by co-immunoprecipitation. Since similar conclusions were drawn from both cell lines (data not shown) and since the p53-null H1299 cell line provides an ideal environment to study ectopically expressed p53 derivatives, results obtained from experiments done in H1299 cells are presented here (Figure 2B). As expected, wild-type p53 was detected in the STK15 immunoprecipitates (panel I, lane 10). The interaction appeared to be specific for wild-type p53, because neither p53(R175H) nor p53(R249S), though expressed to levels higher than that of wild-type (panel I, lanes 2–4), was co-precipitated with STK15 (panel 1, lanes 11 and 12). Among the hot-spot mutants of p53, the two used here are known to exhibit a denatured structure (Friend, 1994), suggesting that the conformation of p53 played an important role in the p53–STK15 interaction. Indeed, another two hot-spot p53 mutants retaining native structure and partial transactivation activity, p53(R248W) and p53(R273H) (Friend, 1994), were able to interact with STK15 (panel III, lanes 11 and 12). Consistent with data collected from the yeast two-hybrid experiments, the Aurora box was absolutely required for the interaction, since a STK15 derivative lacking the Aurora box, although expressed to a level similar to that of STK15 (data not shown), did not form a complex with p53 (panel II, lanes 10–12). Taken together, the interaction between p53 and STK15 was detected with endogenous proteins (Figure 2A) as well as with ectopically expressed p53 and STK15 (Figure 2B). Additionally, the conformation of both p53 and the Aurora box of STK15 were important determinants for the interaction. Further, since different antibodies (anti-p53 and anti-STK15 antibodies for Figure 2A and an anti-HA antibody for Figure 2B) were employed to detect the p53–STK15 interaction, the slight possibility that the p53–STK15 interaction was an artifact due to non-specific binding by the antibody was eliminated.
The p53–STK15 interaction was also investigated by GST pull-down assays. In vitro translated 35S-labeled STK15 protein was incubated with agarose beads containing various GST–p53 derivatives. As shown in Figure 3A, STK15 bound to the GST–p53 beads, but not those with GST alone (compare lane 3 with lane 2). In agreement with the data obtained from the yeast study, the p53 domain required for interaction with STK15 was located to a region encompassing residues 1–318 (compare lanes 4–7). It is noteworthy that the central region encompassing residues 101–318 of p53 did not interact with the full-length STK15, but it bound avidly to a peptide with an apparent molecular weight smaller than that of STK15 (lane 7). This peptide probably represented a truncated form of STK15 because its appearance depended on the addition of STK15 template to the in vitro translation system (data not shown). In a reciprocal study shown in Figure 3B, a series of STK15 derivatives were translated in vitro and subjected to pull-down assays, using GST–p53(1–318) as a ligand. STK15 derivatives containing Aurora box 1 and 2 (Bischoff et al., 1998) of STK15 (lanes 3, 6 and 9) were retained by the beads, while one derivative comprising residues 121–403 that encompasses the kinase domain (Bischoff et al., 1998) failed to do so (lane 12). Thus, data collected from co-immunoprecipitation (Figure 2) and GST pull-down experiments (Figure 3), in conjunction with those obtained from the yeast two-hybrid study (Figure 1), demonstrated that p53 physically interacted with STK15.

Fig. 3. GST pull-down assay. (A) In vitro translated STK15 was incubated with GST (lane 2) and derivatives of GST–p53 (lanes 3–7). Input: one-tenth of the input protein directly loaded onto the gel. (B) As in (A), except that a series of STK15 derivatives were incubated with GST–p53 (1–318). Schematic diagrams of the human p53 and STK15 domain structure are shown below each of the autoradiograms.
Suppression of STK15-mediated centrosome amplification by p53
Next, the biological significance of p53–STK15 interaction was explored. Disappointingly, attempts to measure the influence of STK15 on the p53 transactivation activity failed to demonstrate any significant effect (data not shown). Nonetheless, the following observations prompted us to investigate an alternative possibility that p53 regulates STK15 activity regarding centrosome amplification and cellular transformation. (i) The malfunction of either protein results in centrosome amplification (Fukasawa et al., 1996; Zhou et al., 1998); (ii) both p53 and STK15 can localize to the centrosome (Zantema et al., 1985; Blair and Blair, 1988; Brown et al., 1994; Giet and Prigent, 1999; Morris et al., 2000; Ciciarello et al., 2001; Tarapore et al., 2001); and (iii) p53 specifically interacted with the Aurora box of STK15 (Figures 1, 2 and 3), a domain implicated in the modulation of STK15 activity (Giet and Prigent, 1999).
However, p53 is known to regulate centrosome amplification via both transactivation-dependent and -independent pathways (Jimenez et al., 2000; Tarapore et al., 2001). It has been shown that centrosome amplification observed in p53-null mouse embryonic fibroblasts (MEFs) is attributed largely to the loss of cellular activities dependent on the p53 transactivation function (Jimenez et al., 2000; Tarapore et al., 2001). Thus, the p53-null MEFs seem inappropriate for the planned experiments on the STK15-mediated centrosome amplification because the upregulated CDK2 activity due to the loss of p53 in p53-null MEFs (Hinchcliffe et al., 1999; Lacey et al., 1999; Jimenez et al., 2000; Okuda et al., 2000) may interfere with the study. Instead, the standard NIH-3T3 cell system (Zhou et al., 1998; Giet and Prigent, 1999) established for the study of centrosome amplification caused by overexpressed STK15 was adopted. A transactivation- and oligomerization-defective p53 derivative, named p53M(1–318), was used for the purpose. The rationales to eliminate the p53 oligomerization domain further were as follows. First, we aimed to prevent the formation of heterotetramers between transfected and endogenous p53 from complicating the interpretation of the results. Secondly, the p53 oligomerization domain was not required for the interaction with STK15 (Figures 1 and 3).
Evidently, p53M(1–318) appeared to be ideal for investigating the p53 transactivation-independent inhibition of STK15-mediated centrosome amplification in NIH-3T3 cells: it neither activated transcription driven by several p53-responsive promoters (Figure 4A, panels I and II, compare lanes 1, 2 and 4) nor appreciably interfered with the function of endogenous p53 (Figure 4A, panels I and II, compare lanes 1, 3 and 4), even though it was expressed to a level equivalent to those of p53 and p53M (Figure 4A, panel III). It was noteworthy that the failure of p53M(1–318) to activate transcription was in agreement with previous reports (Lin et al., 1994; Jimenez et al., 2000).

Fig. 4. Blocking of STK15-mediated centrosome amplification by a transactivation-defective p53 mutant. (A) Transactivation assay. Panel I: a luciferase reporter driven by three p53-binding sites was co-transfected with vector alone (lane 1), p53 (lane 2), p53M (lane 3) or p53M(1–318) (lane 4). Panel II: as in panel I, except that the reporter was driven by the p21 promoter. Panel III: protein expression levels of p53 derivatives. Lysates from NIH-3T3 cells transfected with vector alone (lane 1), p53 (lane 2), p53M (lane 3) or p53M(1–318) (lane 4) were blotted with an anti-p53 antibody. (B) Inhibition of STK15-mediated centrosome amplification by p53M(1–318). Panel I: centrosome numbers in 900 NIH-3T3 cells transfected with the specified plasmids were counted by immunostaining with an anti-γ-tubulin monoclonal antibody. The combination of plasmids transfected is indicated. Experiments were repeated three times. Panel II: protein expression levels of STK15. Lysates from NIH-3T3 cells transfected with vector alone (lane 1), HA-STK15 or HA-STK15 plus p53M(1–318) were blotted with an anti-HA antibody.
As shown in Figure 4B, an overwhelming majority of NIH-3T3 cells transfected with vector revealed one or two centrosomes (panel I, lanes 1, 9 and 17), while ∼40% of STK15-transfected cells revealed more than two centrosomes (panel I, lanes 2, 10 and 18). Although the expression level of STK15 remained constant (panel II, compare lanes 2 and 3), co-expression of p53M(1–318) with STK15 substantially reduced the number of cells with amplified centrosomes to ∼15% (panel I, lanes 3, 11 and 19). In contrast, p53(101–318), a p53 derivative unable to interact with STK15 (Figures 1 and 3), failed to block the STK15-mediated centrosome amplification (panel I, lanes 4, 12 and 20). Interestingly, an STK15 derivative lacking the Aurora box, STK15(121–403), exhibited only some residual activity to augment centrosome amplification (panel I, lanes 5, 13 and 21), and the presence of p53M(1–318) had little effect on the residual activity (panel I, lanes 6, 14 and 22). Moreover, cells transfected with an STK15 derivative containing the Aurora box alone, STK15(1–63), in the absence or presence of p53M(1–318) behaved just like those transfected with the vector alone (panel I, compare lanes 7 and 8 to lane 1). Taken together, four conclusions could be drawn. (i) Blockage of STK15-mediated centrosome amplification by p53 was transactivation independent. (ii) The ability of p53 derivatives to bind STK15 was essential for this blockage. (iii) The Aurora box was required, but not sufficient, for the STK15-mediated centrosome amplification. (iv) The kinase activity alone was not sufficient for STK15-mediated centrosome amplification.
Suppression of STK15 oncogenic activity by p53
In addition to causing centrosome amplification, the elevated expression of STK15 also leads to transformation of NIH-3T3 cells (Bischoff et al., 1998; Zhou et al., 1998). To test whether p53 also suppresses STK15-mediated cellular transformation, the transactivation-defective p53 derivative, p53M(1–318), was again employed, because wild-type p53 is known to suppress transformation by activating the expression of target genes with growth arrest or apoptosis activity. As described previously (Zhou et al., 1998), the number of soft agar colonies obtained from STK15-transfected NIH-3T3 cells increased ∼200-fold, compared with that obtained from cells transfected with the vector alone (Figure 5, lanes 1 and 2). Co-transfection with p53M(1–318) reduced the number of colonies growing in soft agar by ∼20-fold (Figure 5, lane 3). Like the blocking of centrosome amplification, suppression of cellular transformation also depended on the p53 derivative’s ability to bind STK15 (Figure 5, lane 4). As controls, transformation of NIH-3T3 cells either by the kinase domain of STK15 or by another totally unrelated onco gene, H-ras(Q61L), was not affected by p53M(1–318) or p53(101–318) (Figure 5, lanes 5–10). On the basis of these studies, we concluded that the specific suppression of STK15-mediated cellular transformation required a p53 transactivation-independent function and that the ability of p53 derivatives to bind STK15 was essential for the suppression.
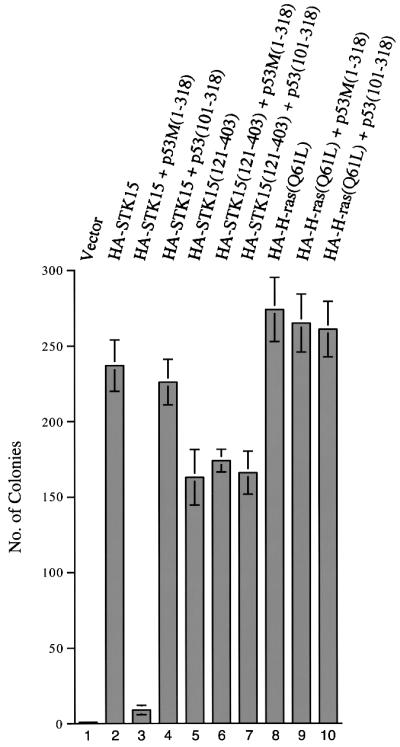
Fig. 5. Inhibition of STK15-mediated cellular transformation by a transactivation-defective p53 mutant. NIH-3T3 cells were transfected with vector alone (lane 1), HA-tagged STK15 (lane 2), HA-tagged STK15 plus p53M(1–318) (lane 3), HA-tagged STK15 plus p53(101–318) (lane 4), HA-tagged STK15(121–403) (lane 5), HA-tagged STK15(121–403) plus p53M(1–318) (lane 6), HA-tagged STK15(121–403) plus p53(101–318) (lane 7), HA-tagged H-ras(Q61L) (lane 8), HA-tagged H-ras(Q61L) plus p53M(1–318) (lane 9) or HA-tagged H-ras(Q61L) and p53(101–318) (lane 10). Colonies were counted from three independent experiments.
Inhibition of STK15 kinase activity by p53
The above experiments demonstrated that p53 blocked STK15-mediated centrosome amplification and cellular transformation in a transactivation-independent manner. Partly because the Aurora box of STK15, implicated to play a role in the modulation of the protein’s activity (Giet and Prigent, 1999), was involved in the interaction with p53 (Figures 1 and 3) and partly because the kinase activity of STK15 is thought to be indispensable for its oncogenic function (Bischoff et al., 1998; Tatsuka et al., 1998; Zhou et al., 1998; Giet and Prigent, 1999), it was speculated that attenuation of STK15 kinase activity by p53 might provide an explanation for the suppression of STK15 oncogenic activity. Thus, experiments shown in Figure 6 were carried out to test this possibility. Using myelin basic protein (MBP) as a substrate (Zhou et al., 1998), the kinase activity of STK15 was greatly inhibited by GST–p53 (panel I, compare lane 3 with lane 6). The inhibition could not be explained by the slight possibility that STK15 was degraded by a contaminating protease from the GST–p53 preparation, since the STK15 protein remained intact after the kinase reaction (data not shown) and an STK15 derivative containing only the kinase domain was not inhibited (panel II, lane 5). Among the p53 derivatives tested, only GST–p53(1–318), which was capable of binding STK15 (Figures 1 and 3) and blocking the STK15 oncogenic activity (Figure 5), could inhibit the STK15 kinase activity (Figure 6, panel I, lanes 7–10). Several lines of evidence further supported the notion that inhibition of STK15 by p53 was specific. First, neither GST alone nor GST fused to a tumor-derived p53 mutant, p53(R175H), inhibited the STK15 kinase activity (panel I, lanes 4 and 5). Secondly, neither the kinase activity of an STK15 derivative, STK15(121–403), devoid of p53-binding activity (panel II, lane 5) nor that of the protein kinase A catalytic subunit (data not shown) was affected by p53, ruling out the possibility that p53 has a general negative effect on the activity of protein kinases.

Fig. 6. p53 inhibits the STK15 kinase activity in vitro. Myelin basic protein (MBP) was used as a substrate for STK15 (panel I) or STK15(121–403) (panel II). The absence (–) or presence of GST derivatives is indicated.
Discussion
In this report, we show that p53 interacts with a centrosomal oncogenic kinase STK15. The interaction is detected in vivo with endogenous or ectopically expressed proteins in different cell lines by different antibodies (Figure 2). Similar conclusions are also drawn from experiments conducted in yeast as well as in vitro by GST pull-down assays (Figures 1 and 3). Moreover, a potential mechanism underlying the p53 tumor suppressor activity is uncovered. Suppression of STK15-mediated centrosome amplification and cellular transformation does not require the transactivation and oligomerization activities of p53 (Figures 4 and 5). Rather, inhibition of STK15 kinase activity by p53 via interaction with the STK15 N-terminal Aurora box, a domain implicated in modulating STK15 function (Giet and Prigent, 1999), may provide, in part, an explanation for the suppression (Figure 6).
Our model thus predicts that the activity of endogenous STK15 differs significantly between matched sets of cells differing only in their p53 status. However, technical problems, such as the low abundance of endogenous STK15, the lack of a specific in vitro STK15 substrate and other contaminating kinases that are co-immunoprecipitated by the anti-STK15 antibody (our unpublished results), prevent the prediction from being tested. In addition, there is one discrepancy between our hypothesis and the observation of STK15-mediated centrosome amplification and cellular transformation in NIH-3T3 cells known to express wild-type p53 protein. We propose that the endogenous p53 protein concentration in NIH-3T3 cells might be too low to keep the activity of ectopically overexpressed STK15 protein in check, thus resulting in centrosome amplification and cellular transformation. Moreover, although we favor the idea that inactivation of STK15 kinase activity is responsible for the suppression of STK15 oncogenic activity, the influence of p53M(1–318) on other p53 activities or p53-interacting proteins remains an open possibility for the suppression.
The ability of an STK15 derivative lacking the Aurora box, STK15(121–403), to transform NIH-3T3 cells (Figure 5) is in contrast to the failure of the same STK15 derivative to promote centrosome amplification efficiently (Figure 4), strongly suggesting that STK15-mediated centrosome amplification and cellular transformation may proceed through distinct mechanisms. In this regard, it is interesting to note that the kinase activity of STK15, although required for transformation (Bischoff et al., 1998), seems dispensable for centrosome amplification (Meraldi et al., 2002). Nonetheless, the current study indicates that this putative kinase-independent activity of STK15 should still be subjected to the suppression by p53 in a transactivation-independent manner (Figure 4).
The p53 transactivation-independent function has been shown to play a role in apoptosis, growth arrest, suppression of homologous recombination and regulation of centrosome amplification (Caelles et al., 1994; Del Sal et al., 1995; Haupt et al., 1997; Ruaro et al., 1997; Jimenez et al., 2000; Willers et al., 2000; Tarapore et al., 2001). Here we provide evidence to support the notion that this p53 function specifically suppresses STK15-mediated centrosome amplification and cellular transformation. In light of this, it is interesting to note that the mouse equivalent of the transactivation-defective human p53M reduces tumor incidence as well as affects tumor tissue distribution in transgenic mice (Jimenez et al., 2000).
We also note that the N-terminal region of p53 appears to be a substrate of STK15 and its derivative STK15(121–403) in vitro (e.g. see Figure 6, panel I, lanes 6–8, and panel II, lane 5). Whether p53 is phosphorylated by STK15 in vivo and whether the phosphorylation has any biological significance currently are unknown. Furthermore, identification of STK15 target(s) involved in centrosome amplification and cellular transformation is important for understanding the role of STK15 in tumorigenesis. In this regard, it must be pointed out that the localization of overexpressed STK15 is not restricted to the centrosome (data not shown; see Meraldi et al., 2002). Consequently, the question of whether any non-centrosomal function of STK15 may contribute to its oncogenic activity remains open.
STK15 is not the only kinase whose activity is regulated by interaction with p53. Previous experiments demonstrate that p53 may stimulate the trkA signal transduction pathway through its association with the trkA kinase (Brown et al., 2000). Also, it has been shown that STK15 localizes to centrosome in p53-null cells (Meraldi et al., 2002) and that p53R175H and p53R249S, which failed to interact with STK15 (Figure 2B), retain the ability to associate with the centrosome (Tarapore et al., 2001). Taken together, the physical interaction between p53 and STK15 is dispensable for their centrosomal localization. Finally, many post-translational modifications, such as phosphorylation, acetylation and ribosylation, activate p53 as well as dictate its subcellular localization (Jimenez et al., 1999). It would be interesting to dissect the signal transduction pathway(s) responsible for transporting p53 to the centrosome.
Materials and methods
Plasmid construction
For the yeast two-hybrid screening, p53M and its derivatives were cloned between the EcoRI and BamHI sites of pAS2-1 (Clontech), while STK15 and its derivatives were cloned between the BamHI and XhoI sites of pACT2 (Clontech). For mammalian expression constructs, STK15 derivatives were cloned between the BamHI and XhoI sites of pcDNA3-HA (Invitrogen). H-ras(Q61L) (Pan et al., 1994) was cloned between the BamHI and EcoRI sites of pcDNA3-HA. Constructs for the expression of p53 derivatives in mammalian cells were described previously (Mietz et al., 1992; Hsu et al., 1995). GST–p53 derivatives were cloned by inserting DNA fragments encoding corresponding p53 regions between the BamHI and EcoRI sites of pGEX-1 (Smith and Johnson, 1988). His-tagged STK15 and STK15(121–403) were constructed by inserting the corresponding DNA fragments between the BamHI and PstI sites of pRSET B (Invitrogen). p3PBS-Luc was constructed by inserting three copies of the p53 consensus binding site (el-Deiry et al., 1992) between the XhoI and HindIII sites of pGL3-basic vector (Promega).
Yeast two-hybrid screen
GAL4DB-p53M was used to screen a human K562 activation domain cDNA library (MATCHMAKER Two-Hybrid System; Clontech). Approximately 5.5 × 105 transformants of the yeast Y190 strain were screened according to the manufacturer’s protocol. Several cDNA clones from the activation domain library encoding proteins that interacted with p53M were isolated and sequenced. Two of them were found to contain partial clones of the STK15 gene.
Cell culture and transfection
293, H1299 and NIH-3T3 cells were maintained in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum. Approximately 1 × 106 cells were seeded 12 h before transfection. Calcium phosphate-mediated DNA transfection was performed as described previously (Tsai et al., 1996).
Immunoprecipitation and western blotting
Co-immunoprecipitation and western blotting were performed as described previously (Huang et al., 2000). An anti-p53 antibody that recognizes human but not murine p53 was described previously (Hsu et al., 1995). Anti-HA monoclonal antibody (12CA5) was purchased from Roche. Polyclonal anti-STK15 antibodies were prepared from rabbits according to standard protocols (Hsu et al., 1995) using His-tagged STK15 expressed in and purified from bacteria as the antigen.
GST pull-down
GST pull-down was performed as described previously (Tsai et al., 1996), except that 20 µl of an in vitro translated STK15 derivative was incubated with 40 µl of beads containing individual GST derivative in the presence of 2 mg of proteins from Escherichia coli lysates. In vitro transcription and translation was performed with the TNT T7 Quick Coupled Transcription/Translation System (Promega) according to the manufacturer’s protocol. [35S]Methionine (Amersham) was included in the reaction so that the synthesized proteins were labeled. The ligand concentration was 3.2, 1.4, 1.2, 0.7, 0.6 and 0.8 mg/ml for GST, GST–p53, GST–p53(1–318), GST–p53(1–100), GST–p53(51–100) and GST–p53(101–318), respectively. Purification of GST derivatives was performed as described previously (Tsai et al., 1996).
Centrosome staining and soft agar assay
NIH-3T3 cells were transfected with a combination of vectors expressing HA-tagged STK15 (10 µg), p53M(1–318) (10 µg), p53(101–318) (10 µg) and enhanced green fluorescent protein (EGFP; 2 µg) as indicated. At 24 h post-transfection, cells were sorted for EGFP-positive cells. Subse quently, sorted cells were grown in medium for another 24 h before the number of their centrosomes was counted by staining with an anti-γ-tubulin monoclonal antibody, GTU-88 purchased from Sigma, as described previously (Zhou et al., 1998). Soft agar assays were performed as described previously (Zhou et al., 1998) except that 700 µg/ml of G418 and 400 µg/ml of hygromycin B were included as selection agents. At 24 h post-transfection, cells were trypsinized and mixed with 0.3% soft agar and poured onto a bed of 0.5% agarose. Four weeks later, colonies were visualized by staining with 1% crystal violet and counted from three independent experiments.
Transactivation assay
A luciferase reporter driven by three p53-binding sites (Hsu et al., 1995) or by the p21 promoter (Gaiddon et al., 1999) and a plasmid expressing individual p53 derivatives were transfected into NIH-3T3 cells. The β-galactosidase expression vector, pCH110 (Amersham Pharmacia Biotech), was included as an internal control. Luciferase activity was measured and quantified 24 h after transfection according to the manufacturer’s suggestions (Promega).
In vitro kinase assay
Kinase assays were performed as described previously (Zhou et al., 1998), except that 600 ng of His-tagged STK15 were used. His-tagged STK15 proteins were expressed in and purified from E.coli strain BL21(DE3). The amount of GST derivatives added into the kinase reaction was 2.2, 6.0, 6.0, 5.4, 3.2, 2.5 and 3.4 µg for GST, GST–p53(R175H), GST–p53, GST–p53(1–318), GST–p53(1–100), GST–p53(51–100) and GST–p53(101–318), respectively.
Acknowledgments
Acknowledgements
We would like to thank Drs Nan-Shih Liao, Wen Chang, Woan-Yuh Tarn and Carmay Lim for critical reading of the manuscript. We also acknowledge Dr Sheau-Yann Shieh for the p21-luciferase reporter. This work was supported by grants from the National Science Council of Taiwan and the Academia Sinica to Y.S.L.
References
- Alves D.C., Paitel,E., Mattson,M.P., Amson,R., Telerman,A., Ancolio,K. and Checler,F. (2002) Wild-type and mutated presenilins 2 trigger p53-dependent apoptosis and down-regulate presenilin 1 expression in HEK293 human cells and in murine neurons. Proc. Natl Acad. Sci. USA, 99, 4043–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniades H.N., Galanopoulos,T., Neville-Golden,J., Kiritsy,C.P. and Lynch,S.E. (1994) p53 expression during normal tissue regeneration in response to acute cutaneous injury in swine. J. Clin. Invest., 93, 2206–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff J.R. et al. (1998) A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J., 17, 3052–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair Z.M. and Blair,G.E. (1988) The intracellular distribution of the transformation-associated protein p53 in adenovirus-transformed rodent cells. Oncogene, 2, 579–584. [PubMed] [Google Scholar]
- Brown A., Browes,C., Mitchell,M. and Montano,X. (2000) c-abl is involved in the association of p53 and trk A. Oncogene, 19, 3032–3040. [DOI] [PubMed] [Google Scholar]
- Brown C.R., Doxsey,S.J., White,E. and Welch,W.J. (1994) Both viral (adenovirus E1B) and cellular (hsp 70, p53) components interact with centrosomes. J. Cell Physiol., 160, 47–60. [DOI] [PubMed] [Google Scholar]
- Caelles C., Helmberg,A. and Karin,M. (1994) p53-dependent apoptosis in the absence of transcriptional activation of p53-target genes. Nature, 370, 220–223. [DOI] [PubMed] [Google Scholar]
- Chuang J.Y., Lin,C.T., Wu,C.W. and Lin,Y.S. (1995) A movable and regulable inactivation function within the central region of a temperature-sensitive p53 mutant. J. Biol. Chem., 270, 23899–23902. [DOI] [PubMed] [Google Scholar]
- Ciciarello M., Mangiacasale,R., Casenghi,M., Zaira,L.M., D’Angelo,M., Soddu,S., Lavia,P. and Cundari,E. (2001) p53 displacement from centrosomes and p53-mediated G1 arrest following transient inhibition of the mitotic spindle. J. Biol. Chem., 276, 19205–19213. [DOI] [PubMed] [Google Scholar]
- Del Sal G., Ruaro,E.M., Utrera,R., Cole,C.N., Levine,A.J. and Schneider,C. (1995) Gas1-induced growth suppression requires a transactivation-independent p53 function. Mol. Cell. Biol., 15, 7152–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Deiry W.S., Kern,S.E., Pietenpol,J.A., Kinzler,K.W. and Vogelstein,B. (1992) Definition of a consensus binding site for p53. Nat. Genet., 1, 45–49. [DOI] [PubMed] [Google Scholar]
- Friend S. (1994) p53: a glimpse at the puppet behind the shadow play. Science, 265, 334–336. [DOI] [PubMed] [Google Scholar]
- Froesch B.A., Aime-Sempe,C., Leber,B., Andrews,D. and Reed,J.C. (1999) Inhibition of p53 transcriptional activity by Bcl-2 requires its membrane-anchoring domain. J. Biol. Chem., 274, 6469–6475. [DOI] [PubMed] [Google Scholar]
- Fukasawa K., Choi,T., Kuriyama,R., Rulong,S. and Vande,W.G. (1996) Abnormal centrosome amplification in the absence of p53. Science, 271, 1744–1747. [DOI] [PubMed] [Google Scholar]
- Gaiddon C., Moorthy,N.C. and Prives,C. (1999) Ref-1 regulates the transactivation and pro-apoptotic functions of p53 in vivo. EMBO J., 18, 5609–5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaccia A.J. and Kastan,M.B. (1998) The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev., 12, 2973–2983. [DOI] [PubMed] [Google Scholar]
- Giet R. and Prigent,C. (1999) Aurora/Ipl1p-related kinases, a new oncogenic family of mitotic serine-threonine kinases. J. Cell Sci., 112, 3591–3601. [DOI] [PubMed] [Google Scholar]
- Hansen S., Lane,D.P. and Midgley,C.A. (1998) The N terminus of the murine p53 tumour suppressor is an independent regulatory domain affecting activation and thermostability. J. Mol. Biol., 275, 575–588. [DOI] [PubMed] [Google Scholar]
- Haupt Y., Rowan,S., Shaulian,E., Kazaz,A., Vousden,K. and Oren,M. (1997) p53 mediated apoptosis in HeLa cells: transcription dependent and independent mechanisms. Leukemia, 11 (Suppl. 3), 337–339. [PubMed] [Google Scholar]
- Hinchcliffe E.H., Li,C., Thompson,E.A., Maller,J.L. and Sluder,G. (1999) Requirement of Cdk2–cyclin E activity for repeated centrosome reproduction in Xenopus egg extracts. Science, 283, 851–854. [DOI] [PubMed] [Google Scholar]
- Hsu Y.S., Tang,F.M., Liu,W.L., Chuang,J.Y., Lai,M.Y. and Lin,Y.S. (1995) Transcriptional regulation by p53: functional interactions among multiple regulatory domains. J. Biol. Chem., 270, 6966–6974. [DOI] [PubMed] [Google Scholar]
- Huang C.F., Wang,Y.C., Tsao,D.A., Tung,S.F., Lin,Y.S. and Wu,C.W. (2000) Antagonism between members of the CNC-bZIP family and the immediate-early protein IE2 of human cytomegalovirus. J. Biol. Chem., 275, 12313–12320. [DOI] [PubMed] [Google Scholar]
- Jayaraman J. and Prives,C. (1995) Activation of p53 sequence-specific DNA binding by short single strands of DNA requires the p53 C-terminus. Cell, 81, 1021–1029. [DOI] [PubMed] [Google Scholar]
- Jimenez G.S., Khan,S.H., Stommel,J.M. and Wahl,G.M. (1999) p53 regulation by post-translational modification and nuclear retention in response to diverse stresses. Oncogene, 18, 7656–7665. [DOI] [PubMed] [Google Scholar]
- Jimenez G.S., Nister,M., Stommel,J.M., Beeche,M., Barcarse,E.A., Zhang,X.Q., O‘Gorman,S. and Wahl,G.M. (2000) A transactivation-deficient mouse model provides insights into Trp53 regulation and function. Nat. Genet., 26, 37–43. [DOI] [PubMed] [Google Scholar]
- Kirch H.C., Flaswinkel,S., Rumpf,H., Brockmann,D. and Esche,H. (1999) Expression of human p53 requires synergistic activation of transcription from the p53 promoter by AP-1, NF-κB and Myc/Max. Oncogene, 18, 2728–2738. [DOI] [PubMed] [Google Scholar]
- Ko L.J. and Prives,C. (1996) p53: puzzle and paradigm. Genes Dev., 10, 1054–1072. [DOI] [PubMed] [Google Scholar]
- Kwek S.S., Derry,J., Tyner,A.L., Shen,Z. and Gudkov,A.V. (2001) Functional analysis and intracellular localization of p53 modified by SUMO-1. Oncogene, 20, 2587–2599. [DOI] [PubMed] [Google Scholar]
- Lacey K.R., Jackson,P.K. and Stearns,T. (1999) Cyclin-dependent kinase control of centrosome duplication. Proc. Natl Acad. Sci. USA, 96, 2817–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J., Chen,J., Elenbaas,B. and Levine,A.J. (1994) Several hydrophobic amino acids in the p53 amino-terminal domain are required for transcriptional activation, binding to mdm-2 and the adenovirus 5 E1B 55-kD protein. Genes Dev., 8, 1235–1246. [DOI] [PubMed] [Google Scholar]
- Matsuzawa S., Takayama,S., Froesch,B.A., Zapata,J.M. and Reed,J.C. (1998) p53-inducible human homologue of Drosophila seven in absentia (Siah) inhibits cell growth: suppression by BAG-1. EMBO J., 17, 2736–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraldi P., Honda,R. and Nigg,E.A. (2002) Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53–/– cells. EMBO J., 21, 483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mietz J.A., Unger,T., Huibregtse,J.M. and Howley,P.M. (1992) The transcriptional transactivation function of wild-type p53 is inhibited by SV40 large T-antigen and by HPV-16 E6 oncoprotein. EMBO J., 11, 5013–5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M., Horikoshi,N. and Shenk,T. (1996) Oncogenic potential of the adenovirus E4orf6 protein. Proc. Natl Acad. Sci. USA, 93, 11295–11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris V.B., Brammall,J., Noble,J. and Reddel,R. (2000) p53 localizes to the centrosomes and spindles of mitotic cells in the embryonic chick epiblast, human cell lines and a human primary culture: an immunofluorescence study. Exp. Cell Res., 256, 122–130. [DOI] [PubMed] [Google Scholar]
- Mowat M.R. (1998) p53 in tumor progression: life, death and everything. Adv. Cancer Res., 74, 25–48. [DOI] [PubMed] [Google Scholar]
- Oberringer M., Lothschutz,D., Jennewein,M., Koschnick,M., Mutschler,W. and Hanselmann,R.G. (1999) Centrosome multiplication accompanies a transient clustering of polyploid cells during tissue repair. Mol. Cell. Biol. Res. Commun., 2, 190–196. [DOI] [PubMed] [Google Scholar]
- Okuda M. et al. (2000) Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell, 103, 127–140. [DOI] [PubMed] [Google Scholar]
- Pan B.T., Chen,C.T. and Lin,S.M. (1994) Oncogenic Ras blocks cell cycle progression and inhibits p34cdc2 kinase in activated Xenopus egg extracts. J. Biol. Chem., 269, 5968–5975. [PubMed] [Google Scholar]
- Ruaro E.M., Collavin,L., Del Sal,G., Haffner,R., Oren,M., Levine,A.J. and Schneider,C. (1997) A proline-rich motif in p53 is required for transactivation-independent growth arrest as induced by Gas1. Proc. Natl Acad. Sci. USA, 94, 4675–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selivanova G., Iotsova,V., Okan,I., Fritsche,M., Strom,M., Groner,B., Grafstrom,R.C. and Wiman,K.G. (1997) Restoration of the growth suppression function of mutant p53 by a synthetic peptide derived from the p53 C-terminal domain. Nat. Med., 3, 632–638. [DOI] [PubMed] [Google Scholar]
- Smith D.B. and Johnson,K.S. (1988) Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene, 67, 31–40. [DOI] [PubMed] [Google Scholar]
- Tarapore P., Tokuyama,Y., Horn,H.F. and Fukasawa,K. (2001) Difference in the centrosome duplication regulatory activity among p53 ‘hot spot’ mutants: potential role of Ser315 phosphorylation-dependent centrosome binding of p53. Oncogene, 20, 6851–6863. [DOI] [PubMed] [Google Scholar]
- Tatsuka M., Katayama,H., Ota,T., Tanaka,T., Odashima,S., Suzuki,F. and Terada,Y. (1998) Multinuclearity and increased ploidy caused by overexpression of the aurora- and Ipl1-like midbody-associated protein mitotic kinase in human cancer cells. Cancer Res., 58, 4811–4816. [PubMed] [Google Scholar]
- Tsai H.L., Kou,G.H., Chen,S.H., Wu,C.W. and Lin,Y.S. (1996) Human cytomegalovirus immediate-early protein IE2 tethers a transcriptional repression domain to p53. J. Biol. Chem., 271, 3534–3540. [PubMed] [Google Scholar]
- Tung S.F., Chuang,J.Y., Lin,C.T., Lai,M.Y., Wu,C.W. and Lin,Y.S. (1999) Inhibition of hTAFII32-binding implicated in the transcriptional repression by central regions of mutant p53 proteins. J. Biol. Chem., 274, 7748–7755. [DOI] [PubMed] [Google Scholar]
- Willers H., McCarthy,E.E., Wu,B., Wunsch,H., Tang,W., Taghian,D.G., Xia,F. and Powell,S.N. (2000) Dissociation of p53-mediated suppression of homologous recombination from G1/S cell cycle checkpoint control. Oncogene, 19, 632–639. [DOI] [PubMed] [Google Scholar]
- Zantema A., Schrier,P.I., Davis,O.A., van-Laar,T., Vaessen,R.T. and van-der-Eb,A.J. (1985) Adenovirus serotype determines association and localization of the large E1B tumor antigen with cellular tumor antigen p53 in transformed cells. Mol. Cell. Biol., 5, 3084–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H., Kuang,J., Zhong,L., Kuo,W.L., Gray,J.W., Sahin,A., Brinkley,B.R. and Sen,S. (1998) Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat. Genet., 20, 189–193. [DOI] [PubMed] [Google Scholar]