

Abstract
To identify regulators of AU-rich element (ARE)-dependent mRNA turnover we have followed a genetic approach using a mutagenized cell line (slowC) that fails to degrade cytokine mRNA. Accordingly, a GFP reporter construct whose mRNA is under control of the ARE from interleukin-3 gives an increased fluorescence signal in slowC. Here we describe rescue of slowC by a retroviral cDNA library. Flow cytometry allowed us to isolate revertants with reconstituted rapid mRNA decay. The cDNA was identified as butyrate response factor-1 (BRF1), encoding a zinc finger protein homologous to tristetraprolin. Mutant slowC carries frame-shift mutations in both BRF1 alleles, whereas slowB with intermediate decay kinetics is heterozygous. By use of small interfering (si)RNA, independent evidence for an active role of BRF1 in mRNA degradation was obtained. In transiently transfected NIH 3T3 cells, BRF1 accelerated mRNA decay and antagonized the stabilizing effect of PI3-kinase, while mutation of the zinc fingers abolished both function and ARE-binding activity. This approach, which identified BRF1 as an essential regulator of ARE-dependent mRNA decay, should also be applicable to other cis-elements of mRNA turnover.
Keywords: AU-rich element/ERF-1/mRNA stability/siRNA/zinc finger protein
Introduction
While knockout technologies provide an excellent tool for determining the function of a given gene, the reverse task of finding the genes responsible for a given function remains a major challenge in mammalian cell biology. Functions that have only evolved in cells of higher organisms pose a particular problem as genetic approaches suitable for Saccharomyces cerevisiae are usually not applicable to mammalian cells. Problems include the efficiency of generating loss-of-function mutations with a diploid organism and the difficulty in back-selecting for the wild-type (wt) phenotype following gene transfer. Still, successful application of mammalian somatic genetics is possible as exemplified by the discovery of protein kinases essential for interferon-mediated signalling (Darnell et al., 1994).
In the work reported here, we have used somatic cell genetics to clone a gene required for AU-rich element (ARE)-dependent mRNA turnover. The ARE is a major cis-acting element located in the 3′ untranslated region (3′UTR) of cytokines, growth factors and immediate early genes, which targets the mRNA for rapid degradation. This ensures that the expression of these potent effectors remains very low in the resting cell. Stabilization of cytokine mRNAs upon extracellular stimulation contributes to the induction of cytokine expression and involves signalling by c-jun N-terminal protein kinase and p38 MAP-kinase, as well as phosphatidylinositol-3-kinase (PI3-kinase) pathways (Chen et al., 1998; Ming et al., 1998, 2001; Winzen et al., 1999). Following the activation phase, rapid decay takes over again and ensures return of the mRNA to basal levels. The importance of these mechanisms is emphasized by the fact that deregulated ARE-mRNA stability can contribute to oncogenic transformation or to inflammation (Schuler and Cole, 1988; Nair et al., 1994; Carballo et al., 1998).
Several proteins with affinity for the ARE have been identified, yet only a few of these AU-binding proteins (AUBPs) exert an effect on mRNA stability (for review, see Wilusz et al., 2001). Our strategy to identify regulators of ARE-dependent mRNA turnover was a functional approach based on the idea of translating changes in mRNA stability into a fluorescence signal. This would allow for both forward-selection for mutants defective in mRNA degradation and subsequent back-selection upon cDNA transfer. In a first step (Figure 1A), the diploid reporter cell line HT-GFPIL3-wt, herein referred to as ′wt′, was established by stably expressing a GFP/IL-3 hybrid reporter gene in HT1080 cells (Stoecklin et al., 2000). The ARE in the IL-3-derived 3′UTR ensures rapid reporter mRNA decay with low GFP expression (Figure 2A). In a second step (Figure 1B), the wt cells were subjected to multiple rounds of mutagenesis using frame-shift inducing compound ICR191. Clones overexpressing the GFP reporter were isolated by use of fluorescence activated cell sorting (FACS), subsequent cloning and inspection by fluorescence microscopy. Three mutant cell lines (slowA, slowB and slowC) could be identified which had lost the capacity to rapidly degrade the GFPIL3 transcript (Figure 2B). Analysis of these mutants revealed that their defect occurs in trans and causes inactivation of a major mRNA degradation pathway which targets the AREs of GM-CSF, TNFα, IL-2, IL-3 and IL-6 (Stoecklin et al., 2001). By somatic cell fusion experiments, the mutants were shown to be recessive and could be divided into two complementation groups (slowA and slowB/C), indicating loss of two distinct genetic functions required for cytokine ARE-mediated mRNA turnover (Stoecklin et al., 2000).

Fig. 1. Schematic representation of the strategy to clone a regulator of ARE-dependent mRNA degradation. (A) A GFP reporter gene linked to the 3′UTR of IL-3 was stably transfected into HT1080 cells, and a diploid subclone, referred to as wt, was chosen. GFPIL3 reporter mRNA decays rapidly in wt cells due to the ARE in the 3′UTR, and correspondingly, GFP expression is low. (B) Upon mutagenesis using frame-shift inducing compound ICR191, high GFP-expressing cells were isolated by FACS, subsequent cloning and direct fluorescence microscopy. Three mutant cell lines, in which GFP overexpression is due to a defect in degradation of GFPIL3 mRNA, were identified and termed slowA, slowB and slowC. (C) For rescue of mutant slowC, a cDNA library generated from wt cells was packaged into retrovirus and used for infection. Low GFP-expressing cells were isolated and screened for revertants with reconstituted decay of the GFPIL3 mRNA. (D) Provirally integrated cDNAs were recovered from the revertants by genomic PCR which allowed to clone the regulator.

Fig. 2. Analysis of GFPIL3 reporter gene expression in (A) unmutagenized wt cells; (B) slowC-R46, a clone derived from mutant slowC expressing the murine ecotropic retroviral receptor; and in the following clones selected for low GFP expression after cDNA library transfer: (C) CM81; (D) CM128; (E) CM1; (F) CM14. (G) slowC-BRF1-6 and (H) slowC-BRF1-13 are subclones of mutant slowC transfected with puCombi(SV40/BRF-1), a BRF1 cDNA expression vector. GFP levels were measured by flow cytometry (left panels), and stability of GFPIL3 mRNA was determined by actinomycin D chase experiments using northern blotting analysis (middle panels). Twenty micrograms of total RNA per lane was resolved by 1.1% agarose/formaldehyde gel electrophoresis. GFPIL3 mRNA was detected using a radiolabelled SP6-probe specific for the IL-3 3′UTR, and the membranes were rehybridized with a β-actin probe for loading control. GFPIL3 mRNA signal intensities were quantified by phosphoimager, normalized to the actin signal, and plotted as a percentage of the initial value against time (right panels).
We now report successful completion of the back-selection procedure. A retroviral cDNA library was transferred into mutant slowC (Figure 1C), and revertant cell lines were isolated by enrichment of low GFP-expressing clones followed by screening for reconstitution of rapid mRNA decay. The provirally integrated cDNAs were recovered by genomic PCR (Figure 1D) and identified as butyrate response factor-1 (BRF1). Both alleles of BRF1 in slowC were found to contain a frame-shift mutation, demonstrating the high specificity of our cloning strategy.
Results
Isolation of revertants upon cDNA library transfer
Our aim was to rescue the recessive mutant cell line slowC by transfer of a cDNA library. Size-fractionated cDNA from parental HT1080 cells was ligated into an ecotropic retroviral vector for production of infectious virus. In parallel, slowC was stably transfected with the ecotropic receptor ecoR and subcloned. slowC-R46, chosen for its high infection rate, was confirmed to express the mutant phenotype, i.e. high GFP levels and stable GFPIL3 reporter mRNA (Figure 2B). After infection of slowC-R46 cells with the viral library at an estimated m.o.i. of 0.3., low GFP-expressing cells were enriched by two rounds of FACS and subsequently cloned in microtiter plates. Colonies with weak GFP signals were then identified by direct fluorescence microscopy. Upon FACS analysis, clones with a broad or biphasic distribution of fluorescence intensity were eliminated, as they expressed stable GFPIL3 mRNA (exemplified by CM81 and CM128, Figure 2C and D). Out of 42 clones showing a single peak of low intensity, eight were true revertants which had regained the capacity to rapidly degrade the reporter transcript. Two of the revertants, CM1 and CM14, are represented in Figure 2E and F.
BRF1 identified by cDNA recovery
In order to recover the provirally integrated cDNAs from the revertants, PCR was performed on genomic DNA using primers specific for the retroviral vector. Sequencing of the amplicons revealed in all eight revertants integration of a cDNA encoding for the immediate early gene BRF1; also termed ERF-1 or Berg36 in man (Barnard et al., 1993; Bustin et al., 1994; Ning et al., 1996), Tis11b in mouse (Varnum et al., 1991) and cMG1 in rat (Gomperts et al., 1990). As three out of eight clones were copy isolates, the total number of independent BRF1 cDNA integration events was five. This strongly suggested that BRF1 was the activity responsible for rescuing the mutant phenotype. To formally prove this hypothesis, the BRF1 cDNA was cloned into an expression vector and stably transfected into slowC. Subclones of the transfected culture were tested by northern blotting for expression of the transgene (data not shown), and two BRF1-positive lines were chosen for further analysis. Indeed, slowC-BRF1-6 and -13 exhibited the wt characteristics: low levels of GFP expression and rapid decay of the reporter mRNA (Figure 2G and H).
Genetic analysis of slowC
Having identified BRF1 by its ability to rescue slowC, we examined whether the BRF1 locus was altered in slowC. The coding region was amplified from genomic DNA by PCR, and simultaneous sequencing of both alleles revealed two single nucleotide insertions. The first, an additional guanosine residue at position 144 of the open reading frame (Table I), produced an ambiguous electropherogram that was resolved after the second insertion (data not shown), a cytosine residue at position 289. From this pattern we concluded that both BRF1 alleles in slowC must be affected by a frame-shift mutation, one by insertion of +G(144), the other by +C(289).
Table I. Genotype and phenotype of ARE-mRNA decay mutants.
| wt | slowA | slowB | slowC | |
|---|---|---|---|---|
| BRF1 | wt/wt | wt/wt | wt/+C(289) | +G(144)/+C(289) |
| BRF2 | n.d. | wt/wt | n.d. | n.d. |
| TTP | wt/wt | wt/wt | n.d. | wt/wt |
| mRNA t1/2 | 1.0 h | 4.3 h | 3.5 h | 8.6 h |
Compared with the normal, 338 amino acid-long BRF1 protein, the aberrant proteins encoded by the mutated alleles are predicted to terminate at out-of-frame stop codons which occur after 73 and 123 amino acids in the +G(144) and +C(289) allele, respectively. As a BRF1-specific antibody was not available, we were unable to verify our genetic results at the protein level by western blotting. Instead, BRF1 cDNA was amplified from wt and slowC cells for in vitro RNA synthesis (Figure 3, lower panel), followed by translation in reticulocyte lysate using radiolabelled methionine (upper panel). BRF1 protein of normal size (∼45 kDa) was synthesized from wt cells (lane 4), but not from slowC (lane 3). Thus, lack of the full-length protein clearly confirmed our analysis of the slowC genotype. A smear at <20 kDa most likely represents the larger of the two truncated proteins.
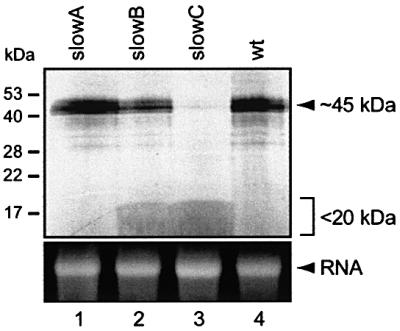
Fig. 3. In vitro transcription and translation of BRF1 cDNA amplified from slowA, slowB, slowC and wt cells. Use of a T7 promoter-linkered upstream primer allowed for in vitro RNA synthesis. Translation was carried out in reticulocyte lysate with 400 ng of RNA in the presence of [35S]methionine. Protein products were resolved through 15% SDS–PAGE and visualized by direct autoradiography (upper panel). For control, an equivalent amount of input RNA was analysed by 1% agarose/formaldehyde gel electrophoresis, and stained with ethidium bromide (lower panel).
Genetic analysis of slowA and slowB
In mutant slowB, which belongs to the same complementation group as slowC (Stoecklin et al., 2000), only the +C(289) mutation was detected in the genomic sequence, while the other allele was wt (Table I). Evidence for expression of the wt allele was obtained by sequencing of the BRF1 cDNA (data not shown), as well as by synthesis of the full-length protein from slowB cDNA in vitro (Figure 3, lane 2). Therefore, slowB appears to be heterozygous at the BRF1 locus.
Mutant slowA did not show any mutation in the coding region of its BRF1 alleles (Table I) and expressed the full-length protein in vitro (Figure 3, lane 1), which is in line with our cell fusion data indicating that slowA represents a second complementation group distinct from slowB/C (Stoecklin et al., 2000). BRF1, together with BRF2 and tristetraprolin (TTP), forms a family of RNA-binding proteins with high homology in the two zinc-finger domains of the unusual C3H composition (Varnum et al., 1991). We therefore investigated the other members of this gene family in slowA. Neither TTP (shown previously, Stoecklin et al., 2000) nor BRF2 (Table I) revealed any mutation in their coding region. Since promoter or enhancer mutations could also account for the loss of a genetic function, semi-quantitative PCR analysis was performed to assess the expression levels of these genes. Both BRF1 and TTP mRNA were found to be similarly abundant in wt cells and in the three mutants (Figure 4), while BRF2 mRNA could not be detected either by RT–PCR or by northern blotting analysis (data not shown). This indicated that differences in the expression levels of these genes do not account for the slowA phenotype. The genetic lesion of slowA remains to be elucidated.
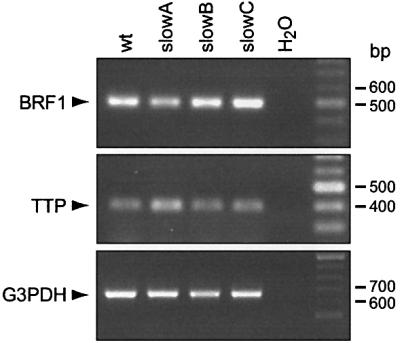
Fig. 4. Analysis of endogenous BRF1 and TTP mRNA expression. Semi-quantitative RT–PCR was carried out with RNA from wt, slowA, slowB and slowC cells using primers specific for BRF1 (upper panel), TTP (middle panel) and, as internal control, glyceraldehyde-3-phosphate dehydrogenase (G3PDH, lower panel). To allow for comparison of the expression levels, the number of PCR cycles was kept below saturation of the amplification reaction (25 cycles for BRF1, 25 for TTP and 20 for GAPDH).
Inhibition of BRF1 by siRNA
We were concerned that the slowB and slowC mutants, having gone through extensive chemical mutagenesis, may have accumulated additional genetic alterations. As this might affect our conclusions, we sought independent evidence that the stable mRNA phenotype is solely due to mutation of BRF1. We therefore examined the effect of reducing BRF1 expression by small interfering (si)RNA, a technique that has recently been shown to work also in mammalian cells (Elbashir et al., 2001). A double-stranded (ds) 21-mer RNA oligonucleotide located in the BRF1 coding region was transfected into wt, slowB and slowC cells. This reduced BRF1 mRNA levels to 20–50% in all three cell lines, compared with cells transfected with medium alone (Figure 5A) or an unrelated control-oligo (not shown). Indeed, transfection of the BRF1-oligo, but not medium alone or a control-oligo, led to stabilization of the GFPIL3 mRNA in wt cells (Figure 5B). Concomitantly, the change in mRNA stability translated into enhanced GFP expression (Figure 5C). In heterozygous slowB with intermediate decay kinetics, the shift to the right of the GFP expression signal was less (Figure 5D), while reducing BRF1 levels in slowC, in agreement with its defective genotype, had no effect at all (Figure 5E). Although the shift in wt cells was relatively small, the response was very consistent as it occurred in 15/17 repeat experiments, as well as in 14/16 transfections using a second BRF1-oligo (data not shown). Thus, the results obtained with siRNA confirmed the requirement of BRF1 for ARE-mRNA decay in wt HT1080 cells.

Fig. 5. Inhibition of BRF1 expression by siRNA. (A) wt, slowB and slowC cells were transfected either with medium alone or with a 21-mer ds RNA BRF1-oligo. Cytoplasmic RNA was isolated 72 h later, and northern blotting analysis was performed to determine BRF1 mRNA expression levels. For quantification (bottom panel), signal intensities were measured by phosphoimager, normalized to actin and represented as a percentage of the control values from transfections with medium alone. (B) Upon transfection of wt cells with either medium alone, the BRF1-oligo or a control-oligo, stability of GFPIL3 reporter mRNA was monitored by actinomycin D treatment (5 µg/ml) for 0, 1 and 2 h. Northern blot analysis and quantification (right panel) was carried out as described for Figure 2. (C) GFP expression was measured by flow cytometry 72 h after transfection of medium alone or the BRF1-oligo into in wt cells, (D) slowB and (E) slowC.
BRF1 promotes degradation of ARE-containing mRNA
We proceeded to study the function of BRF1 by transient transfection in NIH 3T3 cells. In order to avoid actinomycin D we made use of a β-globin reporter gene that is under control of a tetracycline-sensitive promoter (Xu et al., 1998), and linked it to the IL-3 3′UTR in plasmid pSRL. Upon transient transfection into NIH 3T3 B2A2-23 cells which express the tTA (Tet-Off) transactivator, the reporter mRNA decayed rapidly after addition of tetracycline (Figure 6A, lanes 1–3, quantification in D). Cotransfection of BRF1wt slightly enhanced decay of the mRNA (Figure 6A, lanes 4–6). As previously reported (Ming et al., 2001), expression of rCD2-p110, a constitutively activated form of the PI3-kinase catalytic subunit, led to stabilization of the ARE-containing transcript (lanes 7–9). However, cotransfection of BRF1wt antagonized the PI3-kinase effect by promoting decay of the mRNA (lanes 10–12). A control mRNA lacking the ARE (ΔAU) was intrinsically stable, and cotransfection of BRF1 could not induce its decay (Figure 6B). BRF1 thus appears to act specifically on ARE-containing mRNAs. We then introduced mutations into BRF1 by replacing the first cysteine residue of either zinc finger domain with an arginine (C120R and C158R). Neither of the mutant BRF1 constructs was able to accelerate degradation of the ARE-mRNA (Figure 6C), demonstrating that integrity of both zinc finger domains is required for BRF1 activity.

Fig. 6. Influence of BRF1 on mRNA decay using a Tet-Off system. (A) Plasmid pSRL contains a β-globin reporter gene fused to the 3′UTR of IL-3 under transcriptional control of the tetracycline responsive element. NIH 3T3-B2A2-23 cells were transiently transfected with a constant amount of pSRL (12.5 µg) in combination with empty vector (7.5 µg, lanes 1–3), bsd-HisBRF1wt (1.5 µg, lanes 4–6), rCD2-p110 (6.5 µg, lanes 7–9) or rCD2-p110 and bsd-HisBRF1wt together (lanes 10–12). After 48 h, reporter gene transcription was blocked with tetracycline (2 µg/ml), and RNA was isolated at the time points indicated. Northern blotting analysis was performed as described in the legend of Figure 2 using a β-globin-specific probe for detection of the reporter mRNA. (B) The same co-transfections were carried out with 12.5 µg of reporter plasmid pSRL-ΔAU lacking the ARE. (C) Zinc-finger mutants of BRF1 were analyzed by cotransfection of pSRL together with bsd-HisBRF1C120R (lanes 1–3) or bsd-HisBRF1C158R (lanes 4–6). (D) For quantification of globin-IL3 mRNA decay, signal intensities from (A) and (C), as well as from independent repeat transfections were measured by phosphoimager, normalized to actin, and plotted as mean values ± SE against time. Number of repeat experiments: n = 12 for control, 8 for p110, 6 for p110+BRF1wt, and 4 for p110+BRF1C120R and p110+BRF1C158R.
BRF1 binds to the ARE
In order to determine whether BRF1 activity would correlate with binding to the ARE, gel mobility shift assays were performed. BRF1 was expressed as a His-tagged, recombinant protein in Escherichia coli. Increasing amounts (5–50 ng) were incubated with an in vitro-synthesized, radioactively labelled ARE+ RNA spanning a 259 nt-long portion of the IL-3 3′UTR. Efficient complex formation could be observed with 10 ng or more of BRF1wt protein (Figure 7, lanes 1–5), while 50 ng of mutant BRF1C120R showed no complex formation at all (lane 6). As a control for specificity, we also tested an ARE– RNA where the core sequence, the 33 nt-long AUUUA motif cluster, had been deleted. Indeed, BRF1wt showed hardly any complex formation with the ARE– RNA (lane 8), indicating that the AUUUA motif cluster is the target sequence of BRF1.
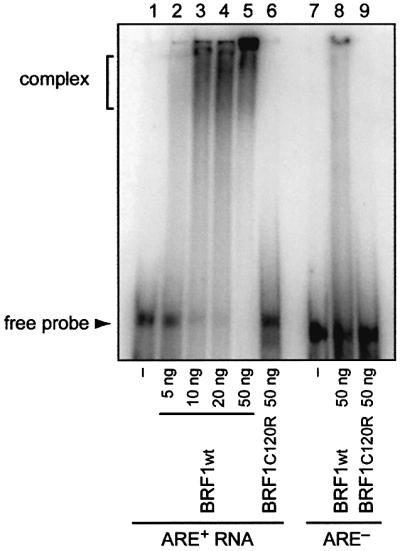
Fig. 7. In vitro binding of BRF1 to ARE-containing RNA. Gel mobility shift assays were performed with 5–50 ng of recombinant BRF1wt protein (lanes 2–5 and 8) or 50 ng of BRF1C120R protein (lanes 6 and 9). Radiolabelled ARE+ RNA (lanes 1–6) was synthesized from a 259 nt-long region of the IL-3 3′UTR containing the ARE. For ARE– RNA (lanes 7–9), the 33 nt-long ARE has been deleted from the same region. Samples were resolved by non-denaturing 4% PAGE.
Localization of BRF1 and TTP
Finally, we determined the intracellular localization of BRF1 in comparison with its close homologue TTP. As prolonged overexpression of BRF1 and TTP is not well tolerated by many cells (Johnson et al., 2000; G.Stoecklin, B.Gross, X.F.Ming and C.Moroni, unpublished data), the myc-tagged cDNAs were placed under the control of a tetracycline-inducible promoter. Upon transient transfection together with the Tet-On rtTA transactivator into NIH 3T3 cells, expression was induced with doxycycline. Immunofluorescence revealed that BRF1 was localized in both the nucleus and the cytoplasm (Figure 8A). The relative level of nuclear and cytoplasmic staining showed considerable variation between individual cells, reflecting that the protein shuttles as reported recently (Phillips et al., 2002). Similarly, TTP was also found to be expressed in both compartments (Figure 8B). Specificity of the antibody was demonstrated with myc-tagged HuR (Figure 8C), which showed typical nuclear localization (Fan and Steitz, 1998; Peng et al., 1998). The occurrence of BRF1 and TTP in the cytoplasm is in agreement with the notion that ARE-mediated mRNA decay represents a cytoplasmic event.
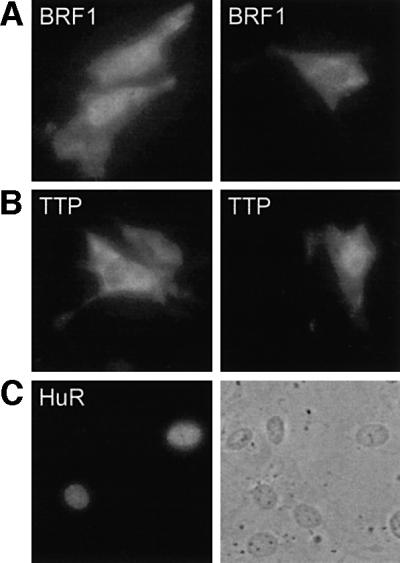
Fig. 8. Intracellular localization of BRF1 and TTP. (A) NIH 3T3 cells were transiently cotransfected with pTRE-BRF1 together with pTET-On. Expression of BRF1 was induced with 2 µg/ml doxycycline, and the myc-tagged protein was detected by immunofluorescence using 9E10 antibody at 60× magnification. (B) Localization of TTP was analyzed as in (A) by transient cotransfection of pTRE-TTP and pTET-On. (C) As a control for specificity of the 9E10 antibody, pcDNA-HuR-myc/His.B was transfected encoding myc-tagged HuR, a protein predominantly localized in the nucleus. The same area is shown by phase contrast in the right panel.
Discussion
To date, ARE-binding proteins with an established function in mRNA turnover have been identified by purification of an in vitro decay activity (AUF1; Zhang et al., 1993); by homology cloning (HuR; Ma et al., 1996); by examining the pathobiology of knockout mice (TTP; Carballo et al., 1998); or by purification of an RNA-binding activity (KSRP; Chen et al., 2001). We have developed a novel fluorescence-based strategy that should be applicable to any cis element mediating mRNA decay (Figure 1). The strategy includes a diploid reporter cell line where the element directs decay of a hybrid GFPIL3 reporter transcript, isolation of a mutant with a recessive defect correctable by cell fusion, and lastly phenotypic reversion by gene transfer. Here, by isolating BRF1, we validate this overall strategy and, using siRNA, provide independent evidence that BRF1 is an essential regulator of ARE-dependent mRNA turnover.
Initially, BRF1 was cloned from an epithelial rat cell line as an EGF-inducible cDNA of unknown function termed cMG1 (Gomperts et al., 1990). Later, the murine and human homologues were cloned as TIS11b (Varnum et al., 1991) and EGF-response factor 1 (ERF-1; Barnard et al., 1993), respectively. The human gene, renamed BRF1 by the Human Genome Database Nomenclature Committee, was located on chromosome 14q22 (Maclean et al., 1995). It contains a single intron (∼2.5 kb) in the coding region and a long 3′UTR of 1861 bp (Bustin et al., 1994). BRF1, together with its close homologue BRF2 (Varnum et al., 1991; Nie et al., 1995) and the more distantly related TTP (Varnum et al., 1989; Lai et al., 1990), belongs to a family of proteins defined by two characteristic zinc finger domains. The zinc fingers have the unusual C3H composition, an invariant RYKTEL sequence that precedes the first C, and a strict spacing between the zinc-binding moieties (CX8CX5CX3H). For TTP, which is the best characterized member of this family, the Blackshear laboratory has shown that it destabilizes TNFα mRNA by binding to the ARE via its zinc finger domains (Carballo et al., 1998; Lai et al., 1999). By overexpression studies, the same group has provided evidence that other members of this family including cMG1, the rat homologue of BRF1, are also ARE-binding proteins, and that they can suppress the level of an ARE-containing reporter transcript (Lai et al., 2000). This presumably occurred through destabilization of the mRNA, although formal proof has not been provided. Our report is the first documentation that another member of the TTP-family is a bona fide mediator of ARE-mRNA degradation.
Besides its role in mRNA turnover, antisense RNA experiments indicated that BRF1 is required for induction of apoptosis in response to treatment with calcium ionophore in a human B-cell line (Ning and Murphy, 1993; Ning et al., 1996). A recent study using transient transfection in NIH 3T3 cells supports the idea that BRF1 plays a role in the mitochondrial apoptosis pathway (Johnson et al., 2000). Somewhat contradictory is the finding that BRF1 levels are elevated in translocation t(8;21)-positive acute myeloid leukemia cells (Shimada et al., 2000). The corresponding AML1-ETO fusion transcription factor was found to induce expression of BRF1, and overexpression of BRF1 in turn was able to support myeloid cell proliferation in response to granulocyte colony-stimulating factor. The apparent link between ARE-dependent mRNA turnover control, apoptosis and leukemogenesis is a challenging topic for further studies.
The genetic approach described here identified BRF1 cDNA in each of eight phenotypic revertants. The occurrence of five independent integration events strongly argued that this cDNA is responsible for rescue of slowC, which was formally proven by reintroducing BRF1 into slowC (Figure 2). Sequencing of the endogenous BRF1 locus revealed independent nucleotide insertions in both BRF1 alleles of slowC (Table I). The mutations are located at positions 144 and 289 of the coding sequence, and both induce a frame shift upstream of the zinc finger domains which are crucial for RNA-binding activity. Also, both mutated mRNAs contain premature termination codons at nucleotides 219 and 369 and are thus expected to encode truncated proteins. Coupled in vitro transcription/translation reactions confirmed that the full-length protein is not generated from slowC cDNA (Figure 3). Thus, slowC has a homozygous loss of function at the BRF1 locus, which is in gratifying agreement with the observation that the mutant phenotype is recessive (Stoecklin et al., 2000). Cells prevent the expression of truncated proteins by subjecting an mRNA to nonsense-mediated decay if it contains a premature stop codon 50 nt or more upstream of the last exon–exon junction (Nagy and Maquat, 1998). This, however, is not the case with the BRF1 transcripts in slowC, as both premature stop codons occur downstream of the single exon–exon junction at position 57. Accordingly, BRF1 mRNA levels were not reduced in slowC (Figure 4). Of the predicted truncated proteins, at least the larger one could be detected by in vitro translation, although poorly resolved (Figure 3).
Notably, mutant slowB, which was found heterozygous at the BRF1 locus, shows an intermediate phenotype with a GFPIL3 mRNA half-life of 3.5 h, compared with 8.6 h in slowC (Table I). Analysis of slowA suggested that other genes may also be responsible for the slow decay phenotype. In this cell we did not detect mutations or abnormal expression levels of either BRF1, BRF2 or TTP (Figure 4), which is consistent with our previous cell fusion data showing that slowA belongs to a complementation group different from slowB/C. We hope to identify the genetic function lacking in slowA using the same approach that has proven successful in slowC.
By means of siRNA we were able to verify our genetic data in the defined background of HT1080 wt cells by generating a transient and partial ‘knockout’ of BRF1. Importantly, reducing endogenous BRF1 expression to 20–50% caused stabilization of GFPIL3 mRNA and a corresponding shift of the fluorescence signal (Figure 5). This indicated that BRF1 plays an essential role as a limiting factor for constitutive ARE-mediated mRNA decay in unstimulated cells. The fact that the BRF1-oligo did not change fluorescence in slowC, and gave an intermediate shift with heterozygous slowB, elegantly confirmed our genotype analysis. In future, the siRNA technique might become a powerful tool to probe the function of other regulators of mRNA turnover as well.
BRF1 was also examined by overexpression in NIH 3T3 cells where regulatory pathways influencing the stability of ARE-mRNA can be conveniently analysed (Ming et al., 2001). A β-globin/IL-3 reporter gene under control of the TET-Off system (Xu et al., 1998) allowed us to study mRNA decay without using actinomycin D which, as a general inhibitor of transcription, can perturb mRNA decay kinetics (Shyu et al., 1989), and the response to activation of signalling pathways. Transient transfection of BRF1 accelerated reporter mRNA decay and antagonized the stabilizing effect of a cotransfected activated form of PI3-kinase, but had no effect on the stability of reporter transcript lacking the ARE (Figure 6). This confirmed the function of BRF1 as an activator of ARE-dependent mRNA decay. Mutation of either zinc finger domain resulted in both loss of function and loss of ARE-binding activity (Figure 7). Corresponding mutations in TTP were shown to have the same effect (Lai et al., 1999), indicating that the highly conserved zinc finger domains of BRF1 and TTP are functionally equivalent. The N- and C-terminal parts of the proteins, however, show only limited homology (Varnum et al., 1991), suggesting that BRF1 and TTP may differ with respect to RNA specificity or response to regulation.
In resting cells, transcription of ARE-containing cytokine mRNAs is low and the transcripts are degraded constitutively. Upon cell activation, transcription is induced and stabilizing mechanisms set in to ensure that the nascent transcripts do not succumb to ARE-mediated degradation. Stabilization is a consequence of signalling through c-jun N-terminal protein kinase and p38 MAP-kinase, as well as PI3-kinase pathways (Chen et al., 1998; Ming et al., 1998, 2001; Winzen et al., 1999). A possible downstream target of p38 MAP-kinase is TTP (Mahtani et al., 2001) which, upon phosphorylation, shows reduced ARE-binding capacity (Carballo et al., 2001). Changes in mRNA stability may also involve direct competition between stabilizing and destabilizing AUBPs such as HuR and BRF/TTP, respectively. Such a model is supported by a UV-crosslinking study using primary T-cell extracts, where increased binding of HuR to ARE-RNA was observed as early as 1 h after cell stimulation, whereas TTP binding occurred 6 h after stimulation (Raghavan et al., 2001). Interestingly, the post-activation type of decay appears to be different from the constitutive decay, as binding of TTP could not be detected in resting T-cells prior to stimulation.
In vitro studies demonstrated that the ARE, in a AUBP-dependent fashion, acts as a potent stimulator of deadenylation and decapping in cytoplasmic HeLa S100 extracts (Ford et al., 1999; Gao et al., 2001). By analogy to yeast, this would render the mRNA accessible to 5′–3′ exonucleolytic decay. Recent data from the Karin laboratory suggest an alternative mechanism. Using S100 extracts from Jurkat cells, Chen et al. (2001) showed that ARE-RNA is specifically degraded by the exosome in a 3′–5′ direction. Targeting to the exosome seems to be carried out by AUBPs such as KSRP or TTP which co-precipitate with exosomal components. Further experiments will have to show whether BRF1 promotes ARE-mRNA degradation also by recruiting the exosome. The challenge ahead is to find the functional targets of the stabilizing kinases in order to understand how phosphorylation events regulate the switch from decay to stabilization and back.
Materials and methods
Cell culture and transfection
All cell lines were grown in Iscove’s Modified Dulbecco Medium (IMDM) supplemented with 10% fetal calf serum (FCS), 50 µM 2-mercaptoethanol, 2 mM glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin. Transfection of the ecotropic receptor (ecoR; Albritton et al., 1989) was performed in 6-well plates with 1 µg of plasmid DNA and 3 µl of Lipofectamine 2000 (Life Technologies), following the manufacturer’s protocol. G418 (1 mg/ml) was added 48 h later for selection of stably transfected cells. Subsequently, cells were subcloned by limiting dilution in 72-well Terazaki plates. B2A2-23 cells constitutively expressing the tTA (Tet-Off) transactivator were derived from NIH 3T3 B2A2 cells (Xu et al., 1998) by subcloning and selection of a clone with high tetracycline response.
cDNA library
For cDNA synthesis, 800 µg of total RNA (RNeasy, Qiagen) were isolated from wt HT1080 cells and enriched for poly-A+ RNA using oligo-dT25-linked dynabeads (Dynal Biotech). As measured by incorporation of [32P]dCTP during second strand synthesis, 4.4 µg of EcoRI-linkered ds cDNA were obtained using Choice system reagents (Life Technologies) and Qiaquick columns (Qiagen) for purification. The cDNA was then resolved by 1.5% agarose gel electrophoresis and fractionated into three sublibraries (0.5–1.5 kb, 1.5–3 kb and >3 kb). Upon purification from the agarose (Qiaex II, Qiagen), the cDNAs were ligated into the EcoRI site of retroviral vector pMX (Onishi et al., 1996), E.coli DH10B cells (Sure, Stratagene) were transformed by electroporation (1700 V, 25 µF, Easyjec T plus pulser, Equibio) and directly expanded in liquid culture for large-scale plasmid isolation (Endofree maxi preparation, Qiagen). As an indicator of complexity, the primary titre (determined by plating an aliquot of the bacteria directly after transformation) reached 1.6 × 106 for the three sublibraries together. PCR was performed on 40 colonies using vector-specific primers M2143 and M2144 to determine size and frequency of inserts, which showed that the proportion of religated vector was below 15%.
Retrovirus production and infection
For production of retroviral supernatants, 107 Plat-E packaging cells (Morita et al., 2000) were transiently transfected with 10 µg of each sublibrary using Lipofectamine 2000 (Life Technologies). Fresh medium was given 6 and 16 h post-transfection, and supernatants (10 ml) were harvested after another 24 h of incubation. slowC-R46 cells were infected with 1/20-diluted supernatants supplemented with 10 µg/ml polybrene (hexadimethrine bromide, Sigma) corresponding to an estimated m.o.i. of 0.3. After 6 h, the medium was changed and cells were expanded for 6 days prior to freezing or selection.
Flow cytometry and selection of low GFP-expressing cells
FACS analysis was performed as described previously (Stoecklin et al., 2000). For selection, low GFP-expressing cells were enriched with help of a FACSorter (Becton Dickinson) from 1 × 107 library-infected slowC-R46 cells. 3 × 104 cells were recovered, expanded, and subjected to a second round of sorting 11 days later. 6 × 103 cells were then directly subcloned in 72-well Terazaki plates (two cells/well) using medium supplemented with 20% FCS. 741 colonies were obtained, and inspection by fluorescence microscopy indicated that 334 had weak GFP signals. One hundred and fifty-three of these clones could be further expanded and analyzed by FACS. Forty-two clones showed a single peak of low intensity, and eight clones were true revertants with rapid GFPIL3 mRNA turnover.
Genomic- and RT–PCR
In order to recover the proviral cDNAs from the revertants, cellular DNA was extracted (DNeasy, Qiagen) for genomic PCR using Hotstartaq polymerase (Qiagen) and the vector-specific primers M2142 and M2144. All amplicons were sequenced with primers M2142 and M2144. For amplification of genomic BRF1 (exon 1, intron 1 and exon 2), PCR was performed with primers M2248 and M2245. Amplification of genomic BRF2 required primary PCR with primers M2295 and M2296, and secondary PCR with M2275 and M2280.
For semi-quantitative RT–PCR, cDNA was generated from 5 µg of total RNA with oligo-dT and MMLV reverse transcriptase (New England Biolabs). Amplification was performed using Taq polymerase (Qiagen) and the following primers: M2243 and M2245 for human BRF1; M1935 and M2311 for human TTP; and M2353 and M2352 for G3PDH.
In vitro transcription/translation
BRF1 cDNA was amplified from 2 µg of total RNA of wt and mutant cells using primer TV76 for reverse transcription and primers TV96 and TV77 for PCR. A T7-promoter was introduced by single-strand (ss) DNA synthesis using primer TV79, containing T7 sequences at the 5′ and TV96 sequences at the 3′ terminus. Upon further amplification with primers TV80 and TV77, the DNA served as template for in vitro transcription of sense RNA using Megascript reagents (Ambion). Protein translation was carried out in reticulocyte lysate (TNT, Promega) using 400 ng of RNA and [35S]methionine, followed by 15% SDS–PAGE and autoradiography.
Northern blotting analysis
Actinomycin D chase experiments, RNA extraction and northern blotting analysis have been described previously (Stoecklin et al., 2000). GFPIL3 mRNA was detected with a [32P]GTP-labelled SP6 RNA-probe from the XbaI–EcoRI fragment of murine IL-3 cDNA. For detection of BRF1 mRNA, a 564 nt-long portion of the cDNA was amplified by PCR with primers M2329 and M2245, and used as template for a [32P]dCTP labelled, M2245-primed DNA probe. The probes for β-globin and actin mRNA have been described previously (Stoecklin et al., 2001).
siRNA
Expression of BRF1 was inhibited by transfection of ds RNA oligos as recently described (Elbashir et al., 2001). Briefly, after passaging cells the previous day, 20 pmol of ds RNA oligos, or medium alone as control, were transfected into each well of a 24-well plate using Oligofectamine (Invitrogen). Cells were subjected to FACS analysis 72 h after transfection. The following BRF1 RNA duplex was employed: sense 5′-CAAGAUGCUCAACUAUAGUdTdT-3′ (nt 63–81; Bustin et al., 1994); antisense 5′-ACUAUAGUUGAGCAUCUUGdTdT-3′. An RNA duplex from the coding region of murine IL-3 cDNA served as non-specific control-oligo: sense 5′-CCAGAACGUUGAAUUGCAGdTdT-3′; antisense 5′-CUGCAAUUCAACGUUCUGGdTdT-3′.
RNA decay using Tet-Off system
To measure RNA decay by using the Tet-Off system, a previously published protocol (Xu et al., 1998) was slightly modified. NIH 3T3-B2A2-23 cells expressing the tTA (Tet-Off) transactivator were seeded into a 15-cm dish 1 day prior to transient transfection with reporter plasmid pSRL or pSRL-ΔAU by calcium phosphate precipitation. In addition, rCD2-p110 (Reif et al., 1997), bsd-HisBRF1wt, bsd-HisBRF1C120R or bsd-HisBRF1C158R were cotransfected either alone or in combination. After 1 day, cells were split into three 10-cm dishes and incubated in IMDM supplemented with 0.5% Tet-system approved FCS (Clontech). Twenty-four hours later, cells were treated with 2 µg/ml tetracycline, and total cytoplasmic RNA was isolated after 0, 1 and 2 h.
Gel mobility shift assay
Recombinant His-tagged BRF1 was produced in E.coli strain M15 upon transformation with pQE-30-BRF1wt and pQE-30-BRF1C120R. The denatured protein was purified and renatured on Ni-NTA columns (Qiagen) according to the manufacturer’s protocol. 32P-labelled RNA was synthesized in vitro using T7 RNA polymerase (Promega) and plasmids T7-ARE+ or T7-ARE– as templates. Analysis of RNA–protein interaction was performed as described previously (You et al., 1992). Five to fifty nanograms of purified BRF1 protein were incubated with 105 c.p.m. of RNA at room temperature for 20 min in 10 µl of buffer containing 10 mM HEPES pH 7.6, 3 mM MgCl2, 40 mM KCl, 5% glycerol, 0.5% NP-40, 2 mM dithiothreitol, 0.5 mg/ml heparin and 50 ng/µl yeast tRNA. The reaction mixture was separated by non-denaturing 4% PAGE.
Immunofluorescence
NIH 3T3 cells were seeded in LabTek 16-well chamber slides (Life Technologies) 1 day prior to transfection with 0.1 µg of plasmid using 0.15 µl of Lipofectamine 2000 per well. Twenty-four hours later, 2 µg/ml doxycycline were added, and cells were fixed after 24 h for 10 min in 4% paraformaldehyde followed by 2 min in 0.2% Triton X-100. The myc-tagged proteins were detected with mouse monoclonal 9E10 antibody and secondary Cy3-coupled goat anti-mouse antibody. Pictures were taken by digital camera (Hamamtsu C4742–95) on a fluorescence microscope (Nicon Eclipse TE200).
Plasmids
For expression vector bsd-HisBRF1wt, BRF1 cDNA was amplified with primers M2240 and M2245, and ligated as a BamHI blunt fragment into the BamHI–EcoRV sites of pcDNA6/HisA (Invitrogen). Plasmids bsd-HisBRF1C120R and bsd-HisBRF1C158R were constructed by site-directed mutagenesis using the primers M2265 and M2267, respectively. The BRF1 expression vector puCombi(SV40/BRF-1) was derived from a modified plasmid co-expressing the tetracylin-dependent tTA transactivator under the SV40 promotor (Schultze et al., 1996; U.Certa, unpublished data), and BRF-1 under control of the tet operator. pQE-30-BRF1wt and pQE-30-BRF1C120R were generated by insertion of a BamHI(blunt)–XhoI fragment from the corresponding bsd-HisBRF1wt and bsd-HisBRF1C120R plasmids into the SmaI and SalI sites of pQE-30 (Qiagen). Plasmid pTRE-BRF1 contains a XbaI–KpnI(blunt) fragment from bsd-HisBRF1wt in the XbaI–SalI(blunt) sites of pTRE-Myc-GFP (M.Schmidlin and C.Moroni, unpublished data). Similarly, pTRE-TTP was constructed by inserting a XbaI(blunt)–HindIII(blunt) fragment of mTTP.tag (Stoecklin et al., 2000) into the HindIII(blunt) site of pTRE-Myc-GFP. pcDNA-HuR-myc/His.B has been described previously (Ming et al., 2001).
The Tet-Off β-globin reporter construct pSRL was generated from pTet-BBB-AREGMCSF (Xu et al., 1998) by exchanging the BamHI– BstXI(blunt) fragment for the BamHI–SphI(blunt) fragment of plasmid pMXh-β-IL3(UTR)wt (Ming et al., 1998, 2001), thereby replacing the ARE of GM-CSF with the 3′UTR of IL-3. In pSRL-ΔAU, the ARE was removed from the 3′UTR by excision of a 220 nt NcoI–StyI fragment.
For plasmid T7-ARE+, a 259 nt-long region of the IL-3 3′UTR was amplified using primers M527 and M992, and ligated into vector pGEM-T (Promega). To generate plasmid T7-ARE–, the 33 nt-long ARE was deleted from T7-ARE+ and replaced with an EcoRI site using primers M2196 and M2197.
DNA oligonucleotides
M527: 5′-CATGGTCAAAAGGATTTTACA-3′;
M992: 5′-TCCACAAGGGACAAATGAAC-3′;
M1935: 5′-TGTCCTCCAGCTCCTTC-3′;
M2142: 5′-GGTGGACCATCCTCTAGACT-3′;
M2143: 5′-CATCGCAGCTTGGATACACG-3′;
M2144: 5′-CCAAAATGGCGTTACTTAAGC-3′;
M2196: 5′-ATGAATTCTTGCCTTCTG-3′;
M2197: 5′-ATGAATTCAGCCTTAATGG-3′;
M2240: 5′-AACGCACAGGATCCCCACC-3′;
M2243: 5′-CATCCACAACGCTGAAGAGC-3′;
M2245: 5′-GGCTTAGTCATCTGAGATGG-3′;
M2248: 5′-CAGCCCGGAGTCAGAAAGG-3′;
M2265: 5′-GCTGCGCCGCCCCTTTGAGGAAAAC-3′;
M2267: 5′-GCTGCGCCGCACCTTCCACACCATC-3′;
M2275: 5′-TCCTCCAACATCTCTGAACC-3′;
M2280: 5′-TTATTCGTCGTGGCTCAAGC-3′;
M2295: 5′-TCTGGCATTTTGCAATTCCC-3′;
M2296: 5′-CAGAGTTCGAGTCCAAGTGC-3′;
M2311: 5′-TGAGATCCAGCTGATCTGACC-3′;
M2329: 5′-ATTTAGGTGACACTATAGAATACAAGACGGAG-3′;
M2352: 5′-TGATGGTACATGACAAGGTGC-3′;
M2353: 5′-ACAGTCCATGCCATCACTGC-3′;
TV76: 5′-GACTTGTCCTTATGTTAGGTGG-3′;
TV77: 5′-TTGTTAATGTAGGGCCTGTGG-3′;
TV79: 5′-CGATTAGGTGACACTATAGAATACGTAATACGACTCACTATAGGGCGAATTCGCGACACACCAGATCC-3′;
TV80: 5′-CGATTTAGGTGACACTATAGAATACG-3′;
TV96: 5′-TGCCGACACACCAGATCCTC-3′.
Acknowledgments
Acknowledgements
We are especially grateful to Verena Jaeggin who performed flow cytometry-based cell sorting. We also thank Adrian Wyss and Stefan Mahlmann for help with the retroviral library, Ulrich Certa for providing vectors, and Helena Côrte-Real for advice on the manuscript. G.S. was the recipient of a fellowship from the Roche Research Foundation. C.M. was supported by grant No. 31-57065.99 from the Swiss National Science Foundation.
References
- Albritton L.M., Tseng,L., Scadden,D. and Cunningham,J.M. (1989) A putative murine ecotropic retrovirus receptor gene encodes a multiple membrane-spanning protein and confers susceptibility to virus infection. Cell, 57, 659–666. [DOI] [PubMed] [Google Scholar]
- Barnard R.C., Pascall,J.C., Brown,K.D., McKay,I.A., Williams,N.S. and Bustin,S.A. (1993) Coding sequence of ERF-1, the human homologue of Tis11b/cMG1, members of the Tis11 family of early response genes. Nucleic Acids Res., 21, 3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin S.A., Nie,X.F., Barnard,R.C., Kumar,V., Pascall,J.C., Brown,K.D., Leigh,I.M., Williams,N.S. and McKay,I.A. (1994) Cloning and characterization of ERF-1, a human member of the Tis11 family of early-response genes. DNA Cell Biol., 13, 449–459. [DOI] [PubMed] [Google Scholar]
- Carballo E., Lai,W.S. and Blackshear,P.J. (1998) Feedback inhibition of macrophage tumor necrosis factor-α production by tristetraprolin. Science, 281, 1001–1005. [DOI] [PubMed] [Google Scholar]
- Carballo E., Cao,H., Lai,W.S., Kennington,E.A., Campbell,D. and Blackshear,P.J. (2001) Decreased sensitivity of tristetraprolin-deficient cells to p38 inhibitors suggests the involvement of tristetraprolin in the p38 signaling pathway. J. Biol. Chem., 276, 42580–42587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.Y., Del Gatto-Konczak,F., Wu,Z. and Karin,M. (1998) Stabilization of interleukin-2 mRNA by the c-Jun NH2-terminal kinase pathway. Science, 280, 1945–1949. [DOI] [PubMed] [Google Scholar]
- Chen C.Y. et al. (2001) AU binding proteins recruit the exosome to degrade ARE-containing mRNAs. Cell, 107, 451–464. [DOI] [PubMed] [Google Scholar]
- Darnell J.E. Jr, Kerr,I.M. and Stark,G.R. (1994) Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science, 264, 1415–1421. [DOI] [PubMed] [Google Scholar]
- Elbashir S.M., Harborth,J., Lendeckel,W., Yalcin,A., Weber,K. and Tuschl,T. (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature, 411, 494–498. [DOI] [PubMed] [Google Scholar]
- Fan X.C. and Steitz,J.A. (1998) Overexpression of HuR, a nuclear–cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J., 17, 3448–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford L.P., Watson,J., Keene,J.D. and Wilusz,J. (1999) ELAV proteins stabilize deadenylated intermediates in a novel in vitro mRNA deadenylation/degradation system. Genes Dev., 13, 188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M., Wilusz,C.J., Peltz,S.W. and Wilusz,J. (2001) A novel mRNA-decapping activity in HeLa cytoplasmic extracts is regulated by AU-rich elements. EMBO J., 20, 1134–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomperts M., Pascall,J.C. and Brown,K.D. (1990) The nucleotide sequence of a cDNA encoding an EGF-inducible gene indicates the existence of a new family of mitogen-induced genes. Oncogene, 5, 1081–1083. [PubMed] [Google Scholar]
- Johnson B.A., Geha,M. and Blackwell,T.K. (2000) Similar but distinct effects of the tristetraprolin/TIS11 immediate-early proteins on cell survival. Oncogene, 19, 1657–1664. [DOI] [PubMed] [Google Scholar]
- Lai W.S., Stumpo,D.J. and Blackshear,P.J. (1990) Rapid insulin-stimulated accumulation of an mRNA encoding a proline-rich protein. J. Biol. Chem., 265, 16556–16563. [PubMed] [Google Scholar]
- Lai W.S., Carballo,E., Strum,J.R., Kennington,E.A., Phillips,R.S. and Blackshear,P.J. (1999) Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor α mRNA. Mol. Cell. Biol., 19, 4311–4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai W.S., Carballo,E., Thorn,J.M., Kennington,E.A. and Blackshear,P.J. (2000) Interactions of CCCH zinc finger proteins with mRNA. Binding of tristetraprolin-related zinc finger proteins to Au-rich elements and destabilization of mRNA. J. Biol. Chem., 275, 17827–17837. [DOI] [PubMed] [Google Scholar]
- Ma W.-J., Cheng,S., Campbell,C., Wright,A. and Furneaux,H. (1996) Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. J. Biol. Chem., 271, 8144–8151. [DOI] [PubMed] [Google Scholar]
- Maclean K.N., See,C.G., McKay,I.A. and Bustin,S.A. (1995) The human immediate early gene BRF1 maps to chromosome 14q22-q24. Genomics, 30, 89–90. [DOI] [PubMed] [Google Scholar]
- Mahtani K.R., Brook,M., Dean,J.L., Sully,G., Saklatvala,J. and Clark,A.R. (2001) Mitogen-activated protein kinase p38 controls the expression and posttranslational modification of tristetraprolin, a regulator of tumor necrosis factor α mRNA stability. Mol. Cell. Biol., 21, 6461–6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming X.F., Kaiser,M. and Moroni,C. (1998) c-jun N-terminal kinase is involved in AUUUA-mediated interleukin-3 mRNA turnover in mast cells. EMBO J., 17, 6039–6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming X.F., Stoecklin,G., Lu,M., Looser,R. and Moroni,C. (2001) Parallel and independent regulation of interleukin-3 mRNA turnover by phosphatidylinositol 3-kinase and p38 mitogen-activated protein kinase. Mol. Cell. Biol., 21, 5778–5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita S., Kojima,T. and Kitamura,T. (2000) Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther., 7, 1063–1066. [DOI] [PubMed] [Google Scholar]
- Nagy E. and Maquat,L.E. (1998) A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem. Sci., 23, 198–199. [DOI] [PubMed] [Google Scholar]
- Nair A.P., Hahn,S., Banholzer,R., Hirsch,H.H. and Moroni,C. (1994) Cyclosporin A inhibits growth of autocrine tumour cell lines by destabilizing interleukin-3 mRNA. Nature, 369, 239–242. [DOI] [PubMed] [Google Scholar]
- Nie X.F., Maclean,K.N., Kumar,V., McKay,I.A. and Bustin,S.A. (1995) ERF-2, the human homologue of the murine Tis11d early response gene. Gene, 152, 285–286. [DOI] [PubMed] [Google Scholar]
- Ning Z.Q. and Murphy,J.J. (1993) Calcium ionophore-induced apoptosis of human B cells is preceded by the induced expression of early response genes. Eur. J. Immunol., 23, 3369–3372. [DOI] [PubMed] [Google Scholar]
- Ning Z.Q., Norton,J.D., Li,J. and Murphy,J.J. (1996) Distinct mechanisms for rescue from apoptosis in Ramos human B cells by signaling through CD40 and interleukin-4 receptor: role for inhibition of an early response gene, Berg36. Eur. J. Immunol., 26, 2356–2363. [DOI] [PubMed] [Google Scholar]
- Onishi M. et al. (1996) Applications of retrovirus-mediated expression cloning. Exp. Hematol., 24, 324–329. [PubMed] [Google Scholar]
- Peng S.S., Chen,C.Y., Xu,N. and Shyu,A.B. (1998) RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. EMBO J., 17, 3461–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips R.S., Ramos,S.B. and Blackshear,P.J. (2002) Members of the tristetraprolin family of tandem CCCH zinc finger proteins exhibit CRM1-dependent nucleocytoplasmic shuttling. J. Biol. Chem., 277, 11606–11613. [DOI] [PubMed] [Google Scholar]
- Raghavan A., Robison,R.L., McNabb,J., Miller,C.R., Williams,D.A. and Bohjanen,P.R. (2001) HuA and tristetraprolin are induced following T cell activation and display distinct but overlapping RNA binding specificities. J. Biol. Chem., 276, 47958–47965. [DOI] [PubMed] [Google Scholar]
- Reif K., Lucas,S. and Cantrell,D. (1997) A negative role for phosphoinositide 3-kinase in T-cell antigen receptor function. Curr. Biol., 7, 285–293. [DOI] [PubMed] [Google Scholar]
- Schuler G.D. and Cole,M.D. (1988) GM-CSF and oncogene mRNA stabilities are independently regulated in trans in a mouse monocytic tumor. Cell, 55, 1115–1122. [DOI] [PubMed] [Google Scholar]
- Schultze N., Burki,Y., Lang,Y., Certa,U. and Bluethmann,H. (1996) Efficient control of gene expression by single step integration of the tetracycline system in transgenic mice. Nat. Biotechnol., 14, 499–503. [DOI] [PubMed] [Google Scholar]
- Shimada H., Ichikawa,H., Nakamura,S., Katsu,R., Iwasa,M., Kitabayashi,I. and Ohki,M. (2000) Analysis of genes under the downstream control of the t(8;21) fusion protein AML1-MTG8: overexpression of the TIS11b (ERF-1, cMG1) gene induces myeloid cell proliferation in response to G-CSF. Blood, 96, 655–663. [PubMed] [Google Scholar]
- Shyu A.-B., Greenberg,M.E. and Belasco,J.G. (1989) The c-fos transcript is targeted for rapid decay by two distinct mRNA degradation pathways. Genes Dev., 3, 60–72. [DOI] [PubMed] [Google Scholar]
- Stoecklin G., Ming,X.F., Looser,R. and Moroni,C. (2000) Somatic mRNA turnover mutants implicate tristetraprolin in the interleukin-3 mRNA degradation pathway. Mol. Cell. Biol., 20, 3753–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoecklin G., Stoeckle,P., Lu,M., Muehlemann,O. and Moroni,C. (2001) Cellular mutants define a common mRNA degradation pathway targeting cytokine AU-rich elements. RNA, 7, 1578–1588. [PMC free article] [PubMed] [Google Scholar]
- Varnum B.C., Lim,R.W., Sukhatme,V.P. and Herschman,H.R. (1989) Nucleotide sequence of a cDNA encoding TIS11, a message induced in Swiss 3T3 cells by the tumor promoter tetradecanoyl phorbol acetate. Oncogene, 4, 119–120. [PubMed] [Google Scholar]
- Varnum B.C., Ma,Q.F., Chi,T.H., Fletcher,B. and Herschman,H.R. (1991) The TIS11 primary response gene is a member of a gene family that encodes proteins with a highly conserved sequence containing an unusual Cys-His repeat. Mol. Cell. Biol., 11, 1754–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilusz C.J., Wormington,M. and Peltz,S.W. (2001) The cap-to-tail guide to mRNA turnover. Nat. Rev. Mol. Cell Biol., 2, 237–246. [DOI] [PubMed] [Google Scholar]
- Winzen R. et al. (1999) The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J., 18, 4969–4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N., Loflin,P., Chen,C.Y. and Shyu,A.B. (1998) A broader role for AU-rich element-mediated mRNA turnover revealed by a new transcriptional pulse strategy. Nucleic Acids Res., 26, 558–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Y., Chen,C.-Y.A. and Shyu,A.-B. (1992) U-rich sequence-binding proteins (URBPs) interacting with a 20-nucleotide U-rich sequence in the 3′ untranslated region of the c-fos mRNA may be involved in the first step of c-fos mRNA degradation. Mol. Cell. Biol., 12, 2931–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Wagner,B.J., Ehrenman,K., Schaefer,A.W., DeMaria,C.T., Crater,D., DeHaven,K., Long,L. and Brewer,G. (1993) Purification, characterization and cDNA cloning of an AU-rich element RNA-binding protein, AUF1. Mol. Cell. Biol., 13, 7652–7665. [DOI] [PMC free article] [PubMed] [Google Scholar]