

Abstract
PlcR is a pleiotropic regulator that activates the expression of genes encoding various virulence factors, such as phospholipases C, proteases and hemolysins, in Bacillus thuringiensis and Bacillus cereus. Here we show that the activation mechanism is under the control of a small peptide: PapR. The papR gene belongs to the PlcR regulon and is located 70 bp downstream from plcR. It encodes a 48-amino-acid peptide. Disruption of the papR gene abolished expression of the PlcR regulon, resulting in a large decrease in hemolysis and virulence in insect larvae. We demonstrated that the PapR polypeptide was secreted, then reimported via the oligopeptide permease Opp. Once inside the cell, a processed form of PapR, presumably a pentapeptide, activated the PlcR regulon by allowing PlcR to bind to its DNA target. This activating mechanism was found to be strain specific, with this specificity determined by the first residue of the penta peptide.
Keywords: cell–cell signaling/peptide/regulation/specificity/virulence
Introduction
Bacillus thuringiensis is a Gram-positive spore-forming bacterium belonging to the Bacillus cereus group (B.cereus, B.thuringiensis, Bacillus anthracis and Bacillus mycoides). Bacillus cereus is an opportunistic pathogen causing food-borne gastroenteritis due to the production of an emetic toxin (Agata et al., 1995) and enterotoxins such as Hbl and Nhe (Drobniewski, 1993; Granum and Lund, 1997). Bacillus cereus has also been found to be associated with more severe infections, such as pneumonia and endophthalmitis (Beecher et al., 1995; Miller et al., 1997). Bacillus thuringiensis is an entomopathogenic bacterium that produces a large variety of crystal proteins (Cry toxins) specifically active against insect larvae (Schnepf et al., 1998). However, B.thuringiensis spores have been reported to contribute to the overall virulence of the bacterium (Li et al., 1987; Dubois and Dean, 1995; Johnson and McGaughey, 1996). It is generally thought that the Cry proteins display toxic effects, killing or weakening susceptible insects and creating favorable conditions for the germination of spores and the development of septicemia. Previous work suggested that phospholipases C are involved in the entomopathogenic properties of the bacteria (Zhang et al., 1993).
Transcription of the plcA gene, encoding the phosphatidylinositol-specific phospholipase C, is activated at the end of the growth phase by a regulator known as PlcR (Lereclus et al., 1996). This gene codes for a 34 kDa protein presenting weak similarities to the PreL and NprA proteins that activate protease gene transcription in Lactobacillus and Bacillus stearothermophilus, respectively. The similarity between PlcR and these transcriptional activators is essentially due to the N-terminal part of the protein, presumably corresponding to the DNA-binding domain. PlcR positively regulates its own expression (Lereclus et al., 1996) and activates the transcription of at least 15 genes potentially involved in bacterial virulence (Agaisse et al., 1999). Deletion of the plcR gene in B.cereus and B.thuringiensis reduced hemolytic activity and virulence of the bacterium in insect larvae (Salamitou et al., 2000).
Analysis of the promoter region of PlcR-regulated genes has revealed the presence of a highly conserved palindromic sequence (TATGNAN4TNCATA), which probably constitutes the PlcR recognition site (Agaisse et al., 1999; Økstad et al., 1999). This sequence, the PlcR box, is situated between 20 and >200 nucleotides upstream from the –10 box of the promoter region. The –10 box of these promoters is similar to the –10 region recognized by the major sigma factor of the vegetative phase, σA. However the –35 box diverges from the –35 box recognized by this sigma factor (Lereclus et al., 1996). The transcription of plcR and of PlcR-regulated genes is activated at the end of the vegetative phase in cells grown in rich medium [e.g. Luria Broth (LB) medium]. In contrast, transcription is not activated in cells cultured in a sporulation-specific medium (Lereclus et al., 2000). This medium-dependent expression of plcR is controlled by the sporulation key-regulator, Spo0A∼P. This protein binds to two Spo0A boxes flanking the PlcR box upstream from plcR, and represses plcR expression, probably by preventing the binding of the activator to its recognition site (Lereclus et al., 2000). A B.thuringiensis Δspo0A mutant strain has been shown to overexpress plcR in cells grown in a sporulation-specific medium.
Recent work demonstrated that the B.thuringiensis oligopeptide permease system (Opp) is required for plcR expression (Gominet et al., 2001). In Bacillus subtilis, Opp allows the uptake of a peptide (PhrA) that inactivates a phosphatase (RapA) involved in the Spo0A phosphorylation pathway, leading to an increase in the concentration of Spo0A∼P within the cell (Perego and Hoch, 1996; Perego, 1997). It was, therefore, thought likely that Opp exerts its effects on plcR by a mechanism involving the phosphorylation of Spo0A. However, plcR was found not to be expressed in a B.thuringiensis Δopp Δspo0A mutant, indicating that Opp is involved in plcR expression via a Spo0A-independent mechanism. As Opp is involved in the import of small peptides into the cell, these results suggest that peptide uptake is required for plcR expression.
Analysis of the nucleotide sequence surrounding the plcR gene revealed the presence of a short open reading frame, designated orf2, 70 bp downstream from plcR (Lereclus et al., 1996). The orf2 gene is positively regulated by PlcR and encodes a 48-amino-acid polypeptide with an N-terminal signal peptide sequence, suggesting that the mature Orf2 polypeptide is secreted (Agaisse et al., 1999). In addition, a study of PlcR activity in B.subtilis suggested that the orf2 gene was involved in the production or activity of PlcR (Lereclus et al., 1996). Thus, Opp may be involved in the uptake of the Orf2 peptide or in an interaction with this peptide that triggers a signaling pathway leading to activation of the PlcR regulon.
In this study, we focused on the effect of the polypeptide encoded by the orf2 gene on PlcR regulon expression. We first disrupted this gene in B.thuringiensis and analyzed the mutant phenotype under various conditions. We then investigated whether the polypeptide was functional within the cell and at which level it regulated expression of the PlcR regulon. Finally, we studied whether this activation was strain specific, using various B.thurin giensis strains and one strain each of B.anthracis, B.cereus and B.mycoides.
Results
Disruption of the orf2 gene prevents expression of the PlcR regulon
We investigated the possible involvement of the orf2 gene in expression of the PlcR regulon by disrupting this gene on the chromosome of the B.thuringiensis 407 Cry– [plcA′Z] strain, which carries a transcriptional fusion between the plcA promoter region and the lacZ gene (Gominet et al., 2001). As plcA expression is under the control of the transcriptional activator PlcR (Lereclus et al., 1996), it directly reflects plcR expression or PlcR activity in the cell. The orf2 gene was disrupted by homologous recombination (see Materials and methods). The B.thuringiensis 407 Cry– A′Z Δorf2 mutant strain gave white colonies (Lac–) on LB plates containing X-gal (5-bromo-4-chloro-3-indolyl-d-galactoside). Thus, unlike the parental B.thuringiensis 407 Cry– [plcA′Z] strain, the Δorf2 mutant strain did not express the lacZ gene. We investigated the kinetics of β-galactosidase activity in cells growing in liquid medium (Figure 1). The 407 Cry– [plcA′Z] cells displayed β-galactosidase activity at the beginning of the stationary phase, whereas the Δorf2 mutant cells did not. In addition, disruption of the orf2 gene in the B.thuringiensis 407 Cry– A′Z Δorf2 strain resulted in a drastic loss of hemolytic activity on sheep blood agar plates (not shown). The phenotype of this strain is similar to that of the ΔplcR mutant strain (Salamitou et al., 2000). These results suggested that the orf2 gene was positively involved in PlcR regulon expression.

Fig. 1. β-galactosidase activity of the B.thuringiensis 407 Cry– [plcA′Z] and 407 Cry– A′Z Δorf2 strains. The cells were grown at 30°C in LB medium. Time zero indicates the onset of the stationary phase, and tn is the number of hours before (–) or after time zero.
The introduction into the Δorf2 mutant strain of pHT315Ωxyl′-orf2, a plasmid carrying a transcriptional fusion between the xylose-inducible promoter pxylA and the orf2 gene (see Materials and methods), restored lacZ gene expression in the presence of xylose (not shown). Therefore, the loss of plcA expression in the B.thuringiensis 407 Cry– A′Z Δorf2 strain was due to orf2 disruption. As the Orf2 polypeptide is involved in expression of the PlcR regulon, we propose to call this protein PapR, for peptide activating PlcR. We therefore also refer to the recombinant strain, B.thuringiensis 407 Cry– A′Z Δorf2, as B.thuringiensis 407 Cry– A′Z ΔpapR.
Disruption of the papR gene decreases the virulence of B.thuringiensis against insects
We evaluated the effect of papR disruption on the virulence of B.thuringiensis against insects by assessing the synergistic effect of spores on the insecticidal activity of the crystal proteins. Some insects, such as Galleria mellonella, are not susceptible to the ingestion of either spores or crystal proteins alone, but are susceptible to ingestion of a spore–crystal mixture (Li et al., 1987; Salamitou et al., 2000). Spores from the wild type or mutant strain were fed to the larvae together with the insecticidal toxin Cry1C.
The mean mortality rate of the G.mellonella larvae 2 days after ingestion of the B.thuringiensis spore–crystal mixture was ∼67% (47–83%, depending on the assay) if wild-type B.thuringiensis 407 Cry– spores were used. However, we recorded a mean mortality rate of only 18% (7–27%, depending on the assay) if we used ΔpapR mutant spores and of only 9% (0–17%, depending on the assay) if we used ΔplcR mutant spores. The induced mortality rate varies significantly among the tested strains, with no replication effect. The spores of the ΔpapR mutant are significantly less virulent against insect larvae than those of the wild-type strain (d.f. = 1; χ2 = 13.59, P > 0.0002). Similarly, the mean mortality rate obtained with the ΔplcR mutant spores is significantly lower than that obtained with the wild-type spores (d.f. = 1; χ2 = 17.51, P > 0.0000). The virulence of the two mutant strains is not significantly different (d.f. = 1; χ2 = 1.1, P > 0.2864).
A secreted factor restores PlcR regulon expression in the ΔpapR mutant
We investigated whether the papR gene product was secreted from the cell by performing a complementation experiment on solid medium. The B.thuringiensis 407 Cry– strain (the wild type) was streaked close to the B.thuringiensis 407 Cry– A′Z ΔpapR mutant strain on LB agar plates containing X-gal (Figure 2A). ΔpapR mutant cells grown close to the wild-type strain displayed a blue coloration, indicating that lacZ expression was restored in these cells. In contrast, ΔpapR mutant cells grown close to ΔpapR mutant cells did not express the lacZ gene. Thus, the B.thuringiensis 407 Cry– A′Z ΔpapR strain was complemented by a diffusible factor secreted from the wild-type strain and dependent on the papR gene.
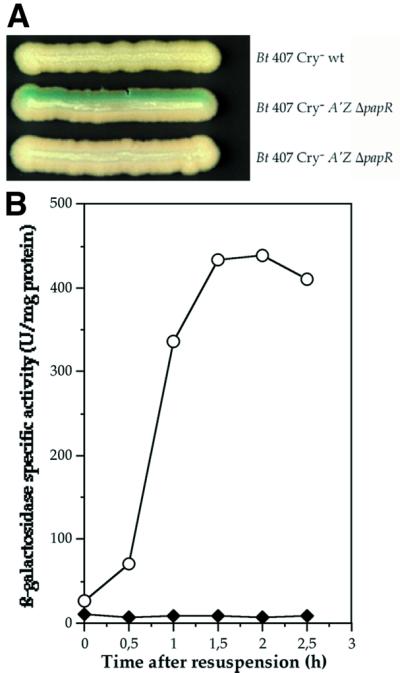
Fig. 2. Intercellular activation of PlcR regulon expression in the B.thuringiensis 407 Cry– A′Z ΔpapR mutant strain. (A) The cells were grown on LB plates containing X-gal for 24 h at 37°C. The name of the strain is indicated beside each streak. (B) β-galactosidase activity of the B.thuringiensis 407 Cry– A′Z ΔpapR cells resuspended in filtered B.thuringiensis 407 Cry– (wild type) culture supernatant (circles) or in their own filtered culture supernatant (diamonds). The cells were grown at 37°C in LB medium.
We determined the kinetics of PlcR regulon activation by this secreted factor in liquid medium. Bacillus thuringiensis 407 Cry– A′Z ΔpapR cells grown in LB medium were harvested at t1 and resuspended in conditioned medium (filtered supernatant from an early stationary phase culture of B.thuringiensis 407 Cry– cells). lacZ expression assays (Figure 2B) showed that β-galactosidase was produced in the ΔpapR cells immediately after their resuspension in the conditioned medium. This confirms that PlcR regulon expression in the ΔpapR mutant strain is activated by a factor present in the supernatant of wild-type cells.
The papR gene product activates expression of the PlcR regulon
To determine whether the diffusible factor responsible for the rescue of plcA-directed transcription was the product of the papR gene, we used synthetic peptides corresponding to the C-terminus of the PapR polypeptide. The following peptides were added to a B.thuringiensis 407 Cry– A′Z ΔpapR culture at t1: OS3, OS4, OS5, OS7 and OS9, consisting of three, four, five, seven and nine amino acids, respectively (Figure 3A). We then assayed β-galactosidase activity (Figure 3B). The lacZ gene was not expressed in ΔpapR mutant cells grown in the presence of OS3 or OS4. However, lacZ expression was restored if the cells were cultured in the presence of OS5, OS7 or OS9. Thus, the secreted factor allowing the activation of PlcR regulon expression corresponds to the C-terminal part of PapR and is at least five amino acids long.

Fig. 3. Complementation of the B.thuringiensis 407 Cry– A′Z ΔpapR mutant strain with synthetic peptides. (A) Amino acid sequence of the PapR protein and of the synthetic peptides used for the complementation experiment. The characters in bold correspond to the N-terminal signal peptide sequence. The potential cleavage site was predicted with the SignalP VI.I program (Nielsen et al., 1997). (B) β-galactosidase activity of the B.thuringiensis 407 Cry– A′Z ΔpapR mutant strain. The cells were grown at 37°C in LB medium and each peptide (OS3, OS4, OS5, OS7 and OS9) was added to a final concentration of 5 µM at t1 (1 h after the onset of the stationary phase).
The pentapeptide encoded by papR is functional within the cell
Given that the oligopeptide permease Opp is required for plcR expression and that the Opp system can transport small peptides (Detmers et al., 2001) or act as a receptor in a signal transduction pathway (Wanner, 1993), we investigated whether the OS5 pentapeptide was functional within the cell. We introduced pHT304Ωos5i (see Materials and methods) into the B.thuringiensis 407 Cry– ΔoppB and B.thuringiensis 407 Cry– A′Z ΔpapR strains. The os5i gene is under the control of the xylose-inducible promoter pxylA in this plasmid, and encodes MLPFEF, which corresponds to the OS5 peptide preceded by an initiator methionine.
We assessed the expression of the os5i gene within ΔoppB mutant cells on sheep blood agar plates containing xylose (Figure 4A). The hemolytic activity of the ΔoppB strain carrying pHT304Ωos5i was restored when os5i was expressed. In the absence of xylose, no increase in hemolytic activity was observed (not shown). The transformant strain 407 Cry– A′Z ΔpapR (pHT304Ωos5i) was grown in LB medium, and β-galactosidase activity was assayed at various stages of growth between t–1 and t3 (Figure 4B). lacZ expression was restored in ΔpapR (pHT304Ωos5i) cells in the presence of xylose when os5i was expressed. Thus, the OS5 pentapeptide is able to function within the cell, activating PlcR regulon expression.
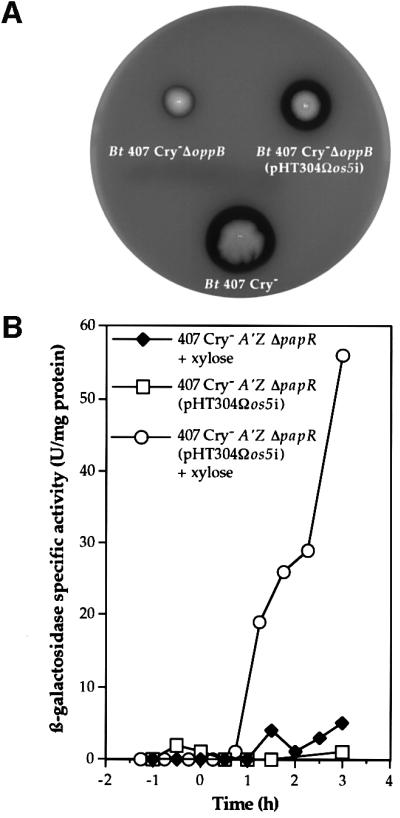
Fig. 4. Effect of the intracellular production of OS5 in the ΔoppB and ΔpapR mutant strains. (A) The expression of os5i in ΔoppB mutant cells restores hemolytic activity in the mutant. Colonies were grown on sheep blood agar plates in the presence of 20 mM xylose for 24 h at 37°C. Bt 407 Cry–, B.thuringiensis 407 Cry– (wild type); Bt 407 Cry– ΔoppB, B.thuringiensis 407 Cry–ΔoppB mutant strain; Bt 407 Cry– ΔoppB (pHT304Ωos5i), B.thuringiensis 407 Cry–ΔoppB (pHT304Ωos5i). (B) Expression of os5i in ΔpapR mutant cells restores plcA-directed transcription in the mutant. β-galactosidase activity of the B.thuringiensis 407 Cry– A′Z ΔpapR mutant strain carrying, or not carrying, pHT304Ωos5i, and in the presence or absence of 20 mM xylose. The cells were grown at 30°C in LB medium.
The papR gene product activates the PlcR protein
As PapR is required for expression of the PlcR regulon, PapR may activate transcription of the plcR gene or, alternatively, it may directly activate the PlcR protein. We constructed pHT304.18Ωxyl′-plcR, a plasmid carrying the plcR gene under the control of the xylose-inducible promoter pxylA (see Materials and methods), to split plcR transcription from the activity of its product. pHT304.18Ωxyl′-plcR was introduced into the B.thuringiensis 407 Cry– A′Z and 407 Cry– A′Z ΔpapR strains. Cells were grown in a sporulation-specific medium (HC medium) in the presence and absence of xylose. In this medium, plcR is not expressed in the wild-type strain (Lereclus et al., 2000). We then assayed β-galactosidase activity (Figure 5). β-galactosidase activity was detected only in the B.thuringiensis 407 Cry– A′Z (pHT304. 18Ωxyl′-plcR) strain grown in the presence of xylose. Since plcA transcription requires PlcR, this indicates that plcR is expressed and active in this strain. However, lacZ was not expressed in B.thuringiensis 407 Cry– A′Z ΔpapR (pHT304.18Ωxyl′-plcR). Thus, PlcR requires the presence of the papR gene product to be functional as a transcriptional activator.

Fig. 5. Expression of the plcA′-lacZ transcriptional fusion in the B.thuringiensis 407 Cry– A′Z and B.thuringiensis 407 Cry– A′Z ΔpapR strains carrying pHT304.18Ωxyl′-plcR. The cells were grown at 30°C in HC medium in the presence or absence of 1 mM xylose. As determined using a xyl′-lacZ transcriptional fusion, this concentration of xylose induces a level of transcriptional activity similar to that induced by the native plcR promoter (result not shown).
PlcR requires the OS5 pentapeptide for binding to the PlcR box
The plcR gene was overexpressed in Escherichia coli strain BL21λDE3 harboring pET28.16ΩplcR (see Materials and methods). After induction and protein extraction, PlcR was purified and analyzed by SDS–PAGE. It migrated with the expected apparent molecular mass, calculated from the amino acid sequence of His-tagged PlcR (34 744 Da) (not shown).
Purified PlcR was used in DNase I footprinting assays with DNA fragments corresponding to the promoter region of plcA (see Materials and methods). No labeled fragment incubated with PlcR was protected, whatever the PlcR concentration was, using this PlcR preparation (Figure 6A, lanes 3 and 4). If OS5 was added to the reaction mixture (in the presence of the highest concentration of PlcR), a protected region appeared, extending from position –50 to –73 (lanes 8 and 9). This region corresponds to the PlcR box of the plcA gene (Figure 6C). This result indicates that OS5 allows the binding of PlcR to its recognition site.

Fig. 6. DNase I footprinting analysis of PlcR binding to the plcA promoter region. Radiolabeled fragments (50 000 c.p.m.) were incubated with various concentrations of PlcR and OS5. (A) Lane 1, G+A Maxam and Gilbert reaction; lane 2, no protein; lane 3, 0.1 µg of PlcR; lane 4, 10 µg of PlcR; lane 5, 20 µg of OS5; lane 6, 0.1 µg of PlcR and 10 µg of OS5; lane 7, 0.1 µg of PlcR and 20 µg of OS5; lane 8, 10 µg of PlcR and 10 µg of OS5; lane 9, 10 µg of PlcR and 20 µg of OS5. The region protected by PlcR is indicated by a bracket. (B) Lane 1, G+A Maxam and Gilbert reaction; lanes 2–8, 50 pmol of PlcR (1.7 µg) and OS5: lane 2, 1000 pmol (0.65 µg); lane 3, 500 pmol; lane 4, 150 pmol; lane 5, 70 pmol; lane 6, 35 pmol; lane 7, 10 pmol; lane 8, 5 pmol; lane 9, 50 pmol of PlcR; lane 10, neither protein nor peptide. (C) plcA promoter region. The DNase I-protected area is indicated by a bracket and the PlcR box is in bold. Positions are relative to the transcription start point. The –35 and –10 promoter regions, and the transcription start (+ 1) of the plcA gene, are indicated as described previously (Lereclus et al., 1996).
The probe was then incubated with increasing amounts, ranging from 200 (6.5 µg) to 1 pmol, of a new PlcR preparation. A very weak protection appeared when the probe was incubated with quantities ≥100 pmol (3.25 µg) of the protein (data not shown). Based on this result, and to determine the precise quantity of OS5 needed to allow PlcR binding to its recognition site, the plcA probe was incubated with a fixed quantity of PlcR (50 pmol) and decreasing quantities of OS5 ranging from 1000 to 5 pmol (Figure 6B). The protected region appeared when OS5 was added in amounts from 1000 to 70 pmol (Figure 6B, lanes 2–5). In these experimental conditions, the binding of PlcR to the PlcR box occurs efficiently when the amount of OS5 in the mixture is ≥70 pmol, a quantity comparable to that of PlcR.
Specificity of the PlcR–PapR complex
Our results suggest that a peptide (presumably a pentapeptide) generated by the processing of PapR interacts with PlcR to form a functional transcriptional activator. Such a mechanism of action, involving a protein–protein interaction, led us to investigate whether the two components of the protein complex dis played strain-dependent specificity. We streaked 15 B.thuringiensis strains (serotypes 1–14 and 34) and B.cereus ATCC 14579, B.anthracis Sterne and a B.mycoides type strain near the B.thuringiensis 407 Cry– A′Z ΔpapR mutant strain on LB plates containing X-gal (not shown). The expression of lacZ (blue colonies) was restored in the ΔpapR mutant if it was streaked close to B.thuringiensis serovars thuringiensis (serotype 1 strain berliner 1715), finitimus (serotype 2), dendrolimus (serotype 4), morrissoni (serotype 8), tolworthi (serotype 9), darmstadiensis (serotype 10), pakistani (serotype 13) and israelensis (serotype 14) or B.cereus strain ATCC 14579. The ΔpapR mutant did not express the plcA′-lacZ fusion (white colonies) when streaked close to the B.thuringiensis serovars kurstaki KT0 (serotype 3), canadensis (serotype 5a), entomocidus (serotype 6), aizawai (serotype 7), kyushuensis (serotype 11), thomp soni (serotype 12) and konkukian Cry– (serotype 34), B.anthracis Sterne and the B.mycoides type strain.
We investigated whether the absence of complementation in B.thuringiensis 407 Cry– A′Z ΔpapR was due to the peptide sequence of the heterologous PapR polypeptides by sequencing the papR genes from each Bacillus strain tested in the complementation assay. The DNA region including the papR gene was amplified with primers S1 and S2 (Table I). Primers S2 and S3 (Table I) were used for sequencing. The predicted amino acid sequences of all the papR gene products were compared (Figure 7). The N-terminal moiety of the proteins was found to be highly conserved between strains. However, the C-terminal part of the PapR polypeptide was more divergent. As the active peptide contains the last five amino acids of PapR, one or more of these pentapeptide residues may be responsible for the specificity of PlcR activation. The first residue of the C-terminal pentapeptide was found to be a leucine in activating strains, whereas it was a methionine or a valine in non-activating strains.
Table I. Primers.
| Primer name | Sequence | Restriction site |
|---|---|---|
| orf1 | 5′-CCCAAGCTTATCCGAGAACATATGTCATC-3′ | HindIII |
| orf2 | 5′-AAACTGCAGTATGCAATTATGCATATCCAC-3′ | PstI |
| orf3 | 5′-GCTCTAGAGGTACAAGTAGCTGCAG-3′ | XbaI |
| orf4 | 5′-CGGGATCCCTGATTATGGAACTGATG-3′ | BamHI |
| orf5 | 5′-CGGGATCCATGATATATTAAAAGTAAAAAATG-3′ | BamHI |
| orf6 | 5′-GGAAGCTTCACATTCAAGGATTCTTTATTAG-3′ | HindIII |
| PLC1′ | 5′-CCCCAAGCTTAGATCTATAAATATGAGAATAAAGATG-3′ | HindIII |
| PLC2′ | 5′-GGGAATTCAGATCTCACTTTTTCTGTTTTACATC-3′ | EcoRI |
| PO1 | 5′-CCCAAGCTTATAATGGGATGGTGAG-3′ | HindIII |
| PO2 | 5′-CGGGATCCAGGTTGTTTATCTGCTG-3′ | BamHI |
| S1 | 5′-CTATTATTATATGTGAGATGAATTGTATG-3′ | – |
| S2 | 5′-GTAAAGACGTTTGGATGTTACTCC-3′ | – |
| S3 | 5′-CGCAATTGCAAACATTTATGCTGA-3′ | – |
| SP1 | 5′-GGTCTCCCATGCAAGCAGAGAAATTAGG-3′ | BsaI |
| SP2 | 5′-CCGCTCGAGTCTGCTGATTTTATTTACAAGCGC-3′ | XhoI |
| Xyl1 | 5′-CACATGCATGCCATGTCACTATTGCTTCAG-3′ | SphI |
| Xyl7 | 5′-CGGAATTCATATGAGAAGGTGCCATGTCA-3′ | EcoRI |
| Xyl8 | 5′-CGGGATCCTTCGTAAACCACTTTGTTTGC-3′ | BamHI |
| Xyl9 | 5′-CCCAAGCTTCTTTCCCTTCGTAAACCAC-3′ | HindIII |
The restriction sites are underlined.

Fig. 7. Comparison of the PapR peptide sequences from various B.thuringiensis, B.cereus (B c), B.anthracis (B a) and B.mycoides (B m) strains. The number beside each sequence refers to the serotype of the B.thuringiensis strain. 1* and 1# designate strain 407 Cry– and strain berliner 1715, respectively. The sequences were aligned using Megalign (DNASTAR).
The three different synthetic pentapeptides (OS5, LPFEF; OS5-M1, MPFEF; OS5-V1, VPFEF) selected on the basis of the PapR alignment were added, separately, to a B.thuringiensis 407 Cry– A′Z ΔpapR culture, and β-galactosidase activity was assayed (Figure 8A). A pentapeptide (OS5-I1, IPFEF) with an isoleucine was also used as a control. If the OS5-I1 pentapeptide was added to the culture, the lacZ gene was not expressed and no β-galactosidase activity was detected. If the OS5-M1 or the OS5-V1 pentapeptide was added to the culture, then the level of β-galactosidase activity in 407 Cry– A′Z ΔpapR mutant cells 2 h after addition of the peptide was one-quarter to one-sixth that when cells were grown in the presence of OS5.

Fig. 8. (A) β-galactosidase activity of the B.thuringiensis 407 Cry– A′Z ΔpapR mutant strain. The cells were grown at 37°C in LB medium and each peptide (OS5, OS5-I1, OS5-M1 and OS5-V1; all 0.5 µM) was added at t1 (1 h after the onset of the stationary phase). The amino acid sequences of the peptides are as follows: OS5, LPFEF; OS5-I1, IPFEF; OS5-M1, MPFEF; OS5-V1, VPFEF. (B) DNase I footprinting analysis of PlcR binding to the plcA promoter region in the presence of various pentapeptides. Lane 1, G+A Maxam and Gilbert reaction; lane 2, no protein; lane 3, 1.2 µg of PlcR (35 pmol); lane 4, 1.2 µg of PlcR and of OS5 10 µg; lane 5, 1.2 µg of PlcR and 10 µg of OS5-I1; lane 6, 1.2 µg of PlcR and 10 µg of OS5-M1; lane 7, 1.2 µg of PlcR and 10 µg of OS5-V1. The region protected by PlcR is indicated by a bracket and corresponds to the PlcR box upstream from the plcA gene.
We investigated whether this difference in activation was due to the specificity of transport or to the specificity between PlcR and the pentapeptides. Binding assays on the plcA promoter region with 50 pmol of PlcR and a range of OS5-M1 or OS5-V1 (from 1000 to 70 pmol) did not result in a detectable protection of the DNA probe (data not shown). A DNase I footprinting assay, using a large excess (15 nmol) of each of the four pentapeptides, was also carried out (Figure 8B). Only OS5 (lane 3) allowed the binding of PlcR to its recognition site. Thus, the specificity of activation depends on the PlcR–PapR complex, and is determined by the first residue of the pentapeptide corresponding to the C-terminal end of PapR.
Discussion
Transcription from the plcA promoter is abolished in the B.thuringiensis 407 Cry– A′Z ΔpapR mutant and this mutant strain presents a non-hemolytic phenotype on sheep blood agar, in contrast to the B.thuringiensis 407 Cry– wild-type strain. Moreover, spores of the ΔpapR and ΔplcR mutants are significantly less virulent than wild-type spores when ingested by insect larvae. Thus, the papR gene is positively involved in PlcR regulon expression. We also showed that the requirement for PapR in PlcR regulon expression extends to in vivo conditions.
Complementation experiments showed that expression of the PlcR regulon was restored in the ΔpapR mutant by a secreted factor present in the wild-type strain culture supernatant but not in the ΔpapR mutant strain culture supernatant. The use of synthetic peptides confirmed that this activating secreted factor resulted from processing of the PapR polypeptide and that it was active as a pentapeptide (LPFEF for the B.thuringiensis 407 Cry– wild-type strain). The effect of PapR on expression of the PlcR regulon is reminiscent of the role of small peptides in the regulation of gene expression in other Gram-positive bacteria such as B.subtilis, Enterococcus faecalis and Staphylococcus aureus (Lazazzera and Grossman, 1998). Unlike the PhrA peptide involved in inactivation of the RapA phosphatase in B.subtilis or the cCF10 peptides regulating plasmid transfer in E.faecalis, in which little of the peptide leaves the cell surface (Perego and Hoch, 1996; Buttaro et al., 2000), the mature PapR peptides are released into the medium.
Previous studies have shown that the oligopeptide permease Opp was required for plcR expression, and thus for PlcR regulon expression (Gominet et al., 2001). This ATP-binding cassette (ABC) transporter is known to be responsible for the uptake of small peptides in bacteria. For example, in B.subtilis, it is involved in the phosphorylation of Spo0A, the sporulation key-factor (Perego and Hoch, 1996; Perego, 1997), and in the development of genetic competence (Lazazzera et al., 1997). Plasmid transfer in E.faecalis is under the control of mating pheromones, which are secreted and then imported into the cell by an oligopeptide permease (Lazazzera, 2001). Alternatively, Opp may function as a receptor in a signaling pathway (Wanner, 1993). Here we show that the pentapeptide (OS5i) corresponding to the C-terminal end of PapR is active within the cell. This implies that the bacteria are able to import this peptide. Once inside the cell, OS5 may bind directly to PlcR or to another target that activates the regulator. DNase I footprinting showed that OS5 was necessary for PlcR binding to its recognition site. However, if the DNA probe was incubated in the presence of PlcR alone, at higher concentrations, the protein bound weakly to the PlcR box (data not shown). These results suggest that OS5 increases the affinity of PlcR for its DNA target, probably by inducing a change in the conformation of the protein. Moreover, binding assays using a range of PapR concentrations show that equivalent amounts of PapR and PlcR are required for an efficient binding. This is the first example of a peptide activating a DNA-binding regulator. Most bacterial regulators are active in a dimer form. The PlcR recognition site is an inverted repeat (TATGNAN4TNCATA), suggesting that the protein binds to its DNA target as a dimer. However, we have not yet demonstrated that PlcR exists as a dimer and have not determined whether OS5 is involved in this possible multimerization.
Complementation experiments between the B.thuringiensis 407 Cry– A′Z ΔpapR mutant strain and various B.thuringiensis strains, as well as B.anthracis, B.mycoides and B.cereus strains, revealed that some, but not all, strains complemented the papR deletion, resulting in the activation of transcription from the plcA promoter in the mutant. This suggested that the activation of PlcR regulon expression by PapR was strain specific. The lack of complementation by the B.anthracis strain was expected because the plcR gene is not functional in this bacterium (Agaisse et al., 1999; Mignot et al., 2001) and, consequently, its papR gene is probably not expressed. However, the B.thuringiensis strains and the B.cereus and B.mycoides strains used in this study generated hemolytic activity similar to that of the B.thuringiensis 407 Cry– strain, suggesting that their respective PapR and PlcR proteins are produced and able to form functional transcriptional activators. Strain-specific expression has been described for the Agr system in S.aureus (Ji et al., 1997). The synthesis of virulence factors in S.aureus is controlled by the agr locus, which encodes a two-component signal transduction system and an autoinducing peptide. This secreted peptide is the activating signal in the two-component system. Staphylococcus strains have been classified into four groups on the basis of cross-inhibition (or activation) activities. The peptide produced by a strain can activate agr expression in strains belonging to the same group, but it inhibits agr expression in strains belonging to another group. In our study, we observed specific activation rather than specific inhibition. Preliminary assays indicated that heterologous pentapeptides (MPFEF and VPFEF) had no antagonist effect on expression of the PlcR regulon in the 407 Cry– [plcA′Z] strain (not shown). Based on the assumption that the specific activation of PlcR regulon expression may be due to PapR, we compared the PapR peptide sequences of the various strains. This analysis revealed that the sequences diverged in terms of the first residue of the C-terminal pentapeptide. Three groups were distinguished on the basis of the peptide sequence alignment. Experiments with the three corresponding synthetic pentapeptides (LPFEF, MPFEF, VPFEF) and the IPFEF pentapeptide demonstrated that the first residue of the peptide must be a leucine if plcA expression in the B.thuringiensis 407 Cry– A′Z ΔpapR mutant is to be activated to the same level as that in the parental strain (B.thuringiensis 407 Cry– [plcA′Z]). The inability of the synthetic peptides IPFEF, MPFEF or VPFEF to restore lacZ expression in B.thuringiensis 407 Cry– A′Z ΔpapR mutant cells, in the way that the LPFEF peptide did, may be due to specificity in transport or to a specificity between PlcR and PapR. DNase I footprinting experiments with the same four synthetic pentapeptides showed that the specificity of activation is probably directly controlled by the interaction between PlcR and PapR. Such differences in the activating capacity of peptides with such similar sequences would mean a very subtle regulation.
Preliminary approach of the molecular basis of PlcR–PapR interaction has been started. A recent study (Perego and Hoch, 2002) showed that the amino acid sequence of PlcR contains structural motifs related to the tetratricopeptide repeats (TPRs). TPRs are structural domains involved in protein–protein interactions. A TPR consists of a degenerate 34-amino-acid sequence folded into two antiparallel α-helices, and TPR motifs are arranged so as to generate an amphipathic channel. TPRs may be involved in TPR–TPR or TPR–non-TPR interactions (Lamb et al., 1995; Blatch and Lassle, 1999). They have been identified, in multiple copies, in a large number of proteins, in both eukaryotic and prokaryotic organisms (Goebl and Yanagida, 1991). TPRs have recently been identified in the Rap proteins of B.subtilis (Perego and Brannigan, 2001). The activity of these phosphatases is inhibited by the Phr pentapeptides, and each Phr peptide interacts specifically with one phosphatase (Perego and Hoch, 1996; Perego, 1997). It has been suggested that TPRs mediate Phr–Rap interactions (Perego and Brannigan, 2001). Further studies, including comparison of the PlcR sequences in various strains belonging to the B.cereus group, are necessary to determine whether TPR motifs play a role in the specificity of the PlcR–PapR interaction.
Materials and methods
Bacterial strains and growth conditions
The acrystalliferous strain B.thuringiensis 407 Cry–, which belongs to serotype 1 (Lereclus et al., 1989), was used throughout this study. The B.thuringiensis 407 Cry– [plcA′Z], ΔoppB and ΔplcR mutant strains have been described previously (Salamitou et al., 2000; Gominet et al., 2001). Escherichia coli K-12 strain TG1 [Δ(lac-proAB) supE thi hsdD5 (F′ traD36 proA+ proB+ lacIq lacZΔM15)] (Gibson, 1984) was used as the host strain for plasmid construction and cloning experiments. Plasmid DNA for the electroporation of B.thuringiensis was prepared from E.coli strain SCS110 [rpsL (Strr) thr leu endA thi-1 lacY galK galT ara tonA tsx dam dcm supE44 Δ(lac-proAB) (F′ traD36 proAB lacIqZΔM15)] (Stratagene). Escherichia coli strain BL21λDE3 [F–, ompT hsdSB (rB– mB–) gal dcm (DE3)], containing the pRep4 plasmid (Amrein et al., 1995) was used for protein overproduction. Escherichia coli and B.thuringiensis cells were transformed by electroporation, as described previously (Dower et al., 1988; Lereclus et al., 1989). Escherichia coli strains were grown at 37°C (except for PlcR overproduction) in LB (1% tryptone, 0.5% yeast extract, 0.5% NaCl). Bacillus thuringiensis was grown at 30 or 37°C in LB or in HC, a sporulation-specific medium (Lecadet et al., 1980).
The antibiotic concentrations used for bacterial selection were as follows: 100 µg/ml ampicillin (for E.coli); 2–10 µg/ml erythromycin, 200 µg/ml kanamycin and 10 µg/ml kanamycin (for B.thuringiensis). Bacteria with the Lac+ phenotype were identified on LB plates containing X-gal (80 µg/ml). Columbia medium agar plates (BioMérieux) containing 5% sheep blood were used to evaluate the hemolytic activity of the B.thuringiensis strains. The xylA promoter in B.thuringiensis was induced by including xylose (20 or 1 mM final concentration) in the culture medium.
DNA manipulations
Plasmid DNA was extracted from E.coli by a standard alkaline lysis procedure, using Qiagen kits. Chromosomal DNA was extracted from B.thuringiensis cells harvested in mid-exponential growth phase, and purified as described previously (Msadek et al., 1990). Restriction enzymes and T4 DNA ligase were used as recommended by the manufacturers. The oligonucleotide primers used for PCR amplification (Table I) were synthesized by Genset. PCR was performed with a GeneAmp PCR system 2400 thermal cycler (Perkin-Elmer). Nucleotide sequences were determined by Genome Express (Montreuil, France).
Plasmid construction
pHT315Ωxyl′-orf2, containing the promoterless orf2 gene, the xylR repressor gene and the promoter of the xylA gene, was constructed as follows. The promoter of the xylA gene (pxylA) and the xylR gene, encoding the transcriptional repressor of xylA, was amplified by PCR from chromosomal DNA of the B.subtilis 168 strain, using primers Xyl7 and Xyl8 (Table I). The orf2 gene was amplified by PCR from chromosomal DNA of the B.thuringiensis 407 Cry– strain, using primers orf5 and orf6 (Table I). The two amplified fragments were hydrolyzed with appropriate restriction enzymes and ligated between the HindIII and EcoRI sites of pHT315 (Arantes and Lereclus, 1991), to give pHT315Ωxyl′-orf2.
pHT304.18Ωxyl′-plcR was constructed by inserting the following two fragments between the SphI and BamHI sites of pHT304.18 (Agaisse and Lereclus, 1994): (i) a fragment containing the xylR repressor gene and the promoter of the xylA gene amplified by PCR from chromosomal DNA of the B.subtilis 168 strain with primers Xyl1 and Xyl9 (Table I); (ii) a fragment containing the promoterless plcR gene amplified from chromosomal DNA of the B.thuringiensis 407 Cry– strain with primers PO1 and PO2 (Table I).
The plasmid containing os5i was constructed as follows. Oligonucleotides OLB and OLH (5′-GATCCAAATGTGGGTGATG GAATATGTTACCTTTTGAGTTTTAAA-3′ and 5′-AGCTTTTAAAA CTCAAAAGGTAACATATTCCATCACCCACATTTG-3′, respectively) were synthesized. These oligonucleotides are complementary except for 5′ BamHI and 3′ HindIII sticky ends. Equimolar amounts of the complementary OS5i nucleotides were mixed, heated at 70°C and annealed by slow cooling to room temperature. The resulting double-stranded DNA fragment included the coding sequence of the MLPFEF peptide flanked by 18 bp of sequence upstream from the papR start and a stop codon. The fragment containing the xylR gene and the xylA promoter gene was amplified as described above, using primers Xyl7 and Xyl8 (Table I). The annealed nucleotides and the xylR-pxylA fragment were ligated between the EcoRI and HindIII sites of pHT304 (Arantes and Lereclus, 1991), resulting in pHT304Ωos5i, in which os5i is under the control of the xylose-inducible promoter. The inserts were confirmed by DNA sequencing.
PlcR was overproduced, using pET28.16ΩplcR. This plasmid was constructed by inserting an 880 bp BsaI–XhoI fragment corresponding to the plcR coding sequence between the NcoI and XhoI sites of pET28.16 (A.Chastanet, in preparation), which is derived from pET28a (Novagen). The DNA fragment corresponding to the plcR sequence was generated by PCR, using nucleotides SP1 and SP2 (Table I), thus replacing the TAA stop codon by the XhoI restriction site. This allows the creation of a translational fusion, adding six C-terminal His residues and placing expression of the gene under the control of a T7 promoter.
Construction of the B.thuringiensis 407 Cry–A′Z Δorf2 (or ΔpapR) recombinant strain
The orf2 gene of the B.thuringiensis 407 Cry– [plcA′Z] strain (Gominet et al., 2001) was disrupted by insertion of the aphA3 gene conferring kanamycin resistance. The 5′ and 3′ regions of the orf2 gene were amplified by PCR, using primer pairs orf1, orf2 and orf3, orf4 (Table I). The 5′ end was purified as a HindIII–PstI fragment and the 3′ end as an XbaI–BamHI fragment. The PstI–XbaI fragment containing the aphA3 gene was purified from pDG783 (Trieu-Cuot and Courvalin, 1983). The three fragments were ligated between the HindIII and BamHI sites of the thermosensitive plasmid pRN5101 (Villafane et al., 1987). The ligation mixture was used to transform E.coli cells to ampicillin resistance, and the plasmid isolated from the transformants was verified by restriction mapping. This recombinant plasmid was used to electrotransform B.thuringiensis 407 Cry– [plcA′Z]. Transformants were selected for resistance to erythromycin and kanamycin. The chromosomal wild-type copy of orf2 was replaced with the disrupted copy by homologous recombination, as described previously (Lereclus et al., 1995). The integration of the KanR cassette into the recombinant B.thuringiensis strain was checked by PCR with oligonucleotides flanking the disrupted gene. The resulting strain (407 Cry– [plcA′Z, orf2::aphA3]) was first designated 407 Cry– A′Z Δorf2, then 407 Cry– A′Z ΔpapR.
Use of synthetic peptides
Cells were cultured at 37°C in LB medium until t1. The culture was then fractionated and each synthetic peptide added to one fraction. Incubation was pursued and β-galactosidase activity assayed for each fraction. The peptides were synthesized by Syntem (France).
β-galactosidase assay
The B.thuringiensis cells containing lacZ transcriptional fusions were cultured in LB or HC medium at 30 or 37°C, and β-galactosidase assays were performed as described previously (Msadek et al., 1990). Specific activities are expressed in units of β-galactosidase per milligram of protein (Miller units).
Overproduction and purification of PlcR
We used pET28.16ΩplcR to transform E.coli strain BL21λDE3 carrying pRep4 (Amrein et al., 1995), which bears the groESL operon. This operon encodes chaperone proteins, which help to prevent aggregate formation in the cell. The resulting strain was grown in LB medium until mid-exponential growth phase (OD600 ∼ 0.9); IPTG was added (1 mM) and incubation continued for 4 h at room temperature. The cells were centrifuged at 5000 g for 10 min and resuspended in 1/100 of the culture volume of buffer I (50 mM NaPO4 pH 8, 300 mM NaCl, 20 mM imidazole). The cells were disrupted by sonication, and cell debris was removed by centrifugation at 12 000 g for 20 min. The resulting crude protein extracts were loaded onto a 0.2 ml Ni-NTA–agarose column (Qiagen) previously equilibrated with buffer I. PlcR protein was then eluted with an imidazole gradient (30–500 mM) and analyzed by SDS–PAGE in a 12.5% acrylamide gel, as described previously (Laemmli, 1970). The molecular size reference marker was obtained from Bio-Rad. Protein concentrations were determined with the Bio-Rad protein assay (Bradford, 1976).
DNase I footprinting
DNase I footprinting assays were performed as described previously (Derré et al., 1999). The DNA fragments used for DNase I footprinting were prepared by PCR, using the Pwo polymerase (Roche Diagnostics GmbH) and 20 pmol of each primer, one of which was previously labeled with T4 polynucleotide kinase (New England Biolabs) and [γ-32P]dATP. Purified oligonucleotides PLC1′ and PLC2′ were used as primers. Labeled PCR products were purified with the Qiaquick PCR purification kit (Qiagen). Assays of PlcR binding to DNA were performed as follows, with the addition of BSA (1 µg). Reactions were performed in a 20 µl volume, and concentrations of MgCl2 and CaCl2 were adjusted to 5 and 0.5 mM, respectively, before adding 5 ng of DNase I (Worthington Biochemical). After incubation for 1 min at room temperature, the reaction was stopped by adding 200 µl of stop buffer (0.4 M sodium acetate, 50 µg/ml sonicated calf thymus DNA, 2.5 mM EDTA). DNA fragments were precipitated in ethanol, and an equivalent number of c.p.m. (5 × 104) from each reaction was loaded onto 6% polyacrylamide/7 M urea gels. A + G Maxam and Gilbert reactions (Maxam and Gilbert, 1980) were carried out on the appropriate 32P-labeled DNA fragments and loaded alongside the DNase I footprinting reactions. Gels were dried and analyzed by autoradiography.
Preparation of Cry toxins and bioassay of insecticidal activity
Cry1C toxins were prepared from the asporogenic strain 407 ΔsigK (Bravo et al., 1996) transformed with pHTIC (Sanchis et al., 1996) and last instar G.mellonella were force fed with spore–crystal mixture as described previously (Gominet et al., 2001). Three independent experiments were carried out on 30 insect larvae for each experiment.
Statistical analysis
The strains were compared for the mortalities that they induced in the G.mellonella larvae bioassays using a log–linear model.
Nucleotide and peptide sequence accession numbers
The PapR peptide sequences have been submitted to DDBJ/EMBL/GenBank and have been assigned accession Nos AF465314–AF465329.
Acknowledgments
Acknowledgements
We would like to thank Myriam Gominet for excellent technical help. We also thank Tarek Msadek for helpful discussion, Nathalie Gilois for virulence tests against insects, Michel Gohar for his help with the statistical analysis, Rolland Nageotte for kind assistance with analytical ultracentrifugation and Arnaud Chastanet for providing both pET28.16 plasmid and valuable advice. This work was supported by the Institut Pasteur, the Centre National de Recherche Scientifique and the Institut National de la Recherche Agronomique. L.S. received a PhD grant from the Ministère de la Recherche.
References
- Agaisse H. and Lereclus,D. (1994) Structural and functional analysis of the promoter region involved in full expression of the cryIIIA toxin gene of Bacillus thuringiensis. Mol. Microbiol., 13, 97–107. [DOI] [PubMed] [Google Scholar]
- Agaisse H., Gominet,M., Økstad,O.A., Kolstø,A.B. and Lereclus,D. (1999) PlcR is a pleiotropic regulator of extracellular virulence factor gene expression in Bacillus thuringiensis. Mol. Microbiol., 32, 1043–1053. [DOI] [PubMed] [Google Scholar]
- Agata N., Ohta,M., Mori,M. and Isobe,M. (1995) A novel dodecadepsipeptide, cereulide, is an emetic toxin of Bacillus cereus. FEMS Microbiol. Lett., 129, 17–20. [DOI] [PubMed] [Google Scholar]
- Amrein K.E., Takacs,B., Stieger,M., Molnos,J., Flint,N.A. and Burn,P. (1995) Purification and characterization of recombinant human p50csk protein-tyrosine kinase from an Escherichia coli expression system overproducing the bacterial chaperones GroES and GroEL. Proc. Natl Acad. Sci. USA, 92, 1048–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arantes O. and Lereclus,D. (1991) Construction of cloning vectors for Bacillus thuringiensis. Gene, 108, 115–119. [DOI] [PubMed] [Google Scholar]
- Beecher D.J., Pulido,J.S., Barney,N.P. and Wong,A.C. (1995) Extracellular virulence factors in Bacillus cereus endophthalmitis: methods and implication of involvement of hemolysin BL. Infect. Immun., 63, 632–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatch G.L. and Lassle,M. (1999) The tetratricopeptide repeat: a structural motif mediating protein–protein interactions. BioEssays, 21, 932–939. [DOI] [PubMed] [Google Scholar]
- Bradford M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Bravo A., Agaisse,H., Salamitou,S. and Lereclus,D. (1996) Analysis of cryIAa expression in sigE and sigK mutants of Bacillus thuringiensis. Mol. Gen. Genet., 250, 734–741. [DOI] [PubMed] [Google Scholar]
- Buttaro B.A., Antiporta,M.H. and Dunny,G.M. (2000) Cell-associated pheromone peptide (cCF10) production and pheromone inhibition in Enterococcus faecalis. J. Bacteriol., 182, 4926–4933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derré I., Rapoport,G. and Msadek,T. (1999) CtsR, a novel regulator of stress and heat shock response, controls clp and molecular chaperone gene expression in gram-positive bacteria. Mol. Microbiol., 31, 117–131. [DOI] [PubMed] [Google Scholar]
- Detmers F.J., Lanfermeijer,F.C. and Poolman,B. (2001) Peptides and ATP binding cassette peptide transporters. Res. Microbiol., 152, 245–258. [DOI] [PubMed] [Google Scholar]
- Dower W.J., Miller,J.F. and Ragsdale,C.W. (1988) High efficiency transformation of E.coli by high voltage electroporation. Nucleic Acids Res., 16, 6127–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobniewski F.A. (1993) Bacillus cereus and related species. Clin. Microbiol. Rev., 6, 324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois N.R. and Dean,D.H. (1995) Synergism between Cry1A insecticidal crystal proteins and spores of Bacillus thuringiensis, other bacterial spores and vegetative cells against Lymantria dispar (Lepidoptera: Lymantriidae) larvae. Environ. Entomol., 24, 1741–1747. [Google Scholar]
- Gibson T.J. (1984) Studies on the Epstein–Barr Virus Genome. Cambridge University Press, Cambridge, UK.
- Goebl M. and Yanagida,M. (1991) The TPR snap helix: a novel protein repeat motif from mitosis to transcription. Trends Biochem. Sci., 16, 173–177. [DOI] [PubMed] [Google Scholar]
- Gominet M., Slamti,L., Gilois,N., Rose,M. and Lereclus,D. (2001) Oligopeptide permease is required for expression of the Bacillus thuringiensis plcR regulon and for virulence. Mol. Microbiol., 40, 963–975. [DOI] [PubMed] [Google Scholar]
- Granum P.E. and Lund,T. (1997) Bacillus cereus and its food poisoning toxins. FEMS Microbiol. Lett., 157, 223–228. [DOI] [PubMed] [Google Scholar]
- Ji G., Beavis,R. and Novick,R.P. (1997) Bacterial interference caused by autoinducing peptide variants. Science, 276, 2027–2030. [DOI] [PubMed] [Google Scholar]
- Johnson D.E. and McGaughey,W.H. (1996) Contribution of Bacillus thuringiensis spores to toxicity of purified Cry proteins towards Indianmeal moth larvae. Curr. Microbiol., 33, 54–59. [DOI] [PubMed] [Google Scholar]
- Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Lamb J.R., Tugendreich,S. and Hieter,P. (1995) Tetratrico peptide repeat interactions: to TPR or not to TPR? Trends Biochem. Sci., 20, 257–259. [DOI] [PubMed] [Google Scholar]
- Lazazzera B.A. (2001) The intracellular function of extracellular signaling peptides. Peptides, 22, 1519–1527. [DOI] [PubMed] [Google Scholar]
- Lazazzera B.A. and Grossman,A.D. (1998) The ins and outs of peptide signaling. Trends Microbiol., 6, 288–294. [DOI] [PubMed] [Google Scholar]
- Lazazzera B.A., Solomon,J.M. and Grossman,A.D. (1997) An exported peptide functions intracellularly to contribute to cell density signaling in B.subtilis. Cell, 89, 917–925. [DOI] [PubMed] [Google Scholar]
- Lecadet M.M., Blondel,M.O. and Ribier,J. (1980) Generalized transduction in Bacillus thuringiensis var. berliner 1715, using bacteriophage CP54 Ber. J. Gen. Microbiol., 121, 203–212. [DOI] [PubMed] [Google Scholar]
- Lereclus D., Arantes,O., Chaufaux,J. and Lecadet,M.-M. (1989) Transformation and expression of a cloned δ-endotoxin gene in Bacillus thuringiensis. FEMS Microbiol. Lett., 60, 211–217. [DOI] [PubMed] [Google Scholar]
- Lereclus D., Agaisse,H., Gominet,M. and Chaufaux,J. (1995) Overproduction of encapsulated insecticidal crystal proteins in a Bacillus thuringiensis spo0A mutant. Bio/Technology, 13, 67–71. [DOI] [PubMed] [Google Scholar]
- Lereclus D., Agaisse,H., Gominet,M., Salamitou,S. and Sanchis,V. (1996) Identification of a Bacillus thuringiensis gene that positively regulates transcription of the phosphatidylinositol-specific phospho lipase C gene at the onset of the stationary phase. J. Bacteriol., 178, 2749–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lereclus D., Agaisse,H., Grandvalet,C., Salamitou,S. and Gominet,M. (2000) Regulation of toxin and virulence gene transcription in Bacillus thuringiensis. Int. J. Med. Microbiol., 290, 295–299. [DOI] [PubMed] [Google Scholar]
- Li R.S., Jarrett,P. and Burges,H.D. (1987) Importance of spores, crystals and δ-endotoxins in the pathogenicity of different varieties of Bacillus thuringiensis in Galleria mellonela and Pieris brassicae. J. Invertebr. Pathol., 50, 277–284. [Google Scholar]
- Maxam A.M. and Gilbert,W. (1980) Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol., 65, 499–560. [DOI] [PubMed] [Google Scholar]
- Mignot T., Mock,M., Robichon,D., Landier,A., Lereclus,D. and Fouet,A. (2001) The incompatibility between the PlcR- and AtxA-controlled regulons may have selected a nonsense mutation in Bacillus anthracis. Mol. Microbiol., 42, 1189–1198. [DOI] [PubMed] [Google Scholar]
- Miller J.M., Hair,J.G., Hebert,M., Hebert,L., Roberts,F.J.,Jr and Weyant,R.S. (1997) Fulminating bacteremia and pneumonia due to Bacillus cereus. J. Clin. Microbiol., 35, 504–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Msadek T., Kunst,F., Henner,D., Klier,A., Rapoport,G. and Dedonder,R. (1990) Signal transduction pathway controlling synthesis of a class of degradative enzymes in Bacillus subtilis: expression of the regulatory genes and analysis of mutations in degS and degU. J. Bacteriol., 172, 824–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen H., Engelbrecht,J., Brunak,S. and von Heijne,G. (1997) Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng., 10, 1–6. [DOI] [PubMed] [Google Scholar]
- Økstad O.A., Gominet,M., Purnelle,B., Rose,M., Lereclus,D. and Kolstø,A.-B. (1999) Sequence analysis of three Bacillus cereus loci under PlcR virulence gene regulator control. Microbiology, 145, 3129–3138. [DOI] [PubMed] [Google Scholar]
- Perego M. (1997) A peptide export–import control circuit modulating bacterial development regulates protein phosphatases of the phosphorelay. Proc. Natl Acad. Sci. USA, 94, 8612–8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perego M. and Brannigan,J.A. (2001) Pentapeptide regulation of aspartyl-phosphate phosphatases. Peptides, 22, 1541–1547. [DOI] [PubMed] [Google Scholar]
- Perego M. and Hoch,J.A. (1996) Cell–cell communication regulates the effects of protein aspartate phosphatases on the phosphorelay controlling development in Bacillus subtilis. Proc. Natl Acad. Sci. USA, 93, 1549–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perego M. and Hoch,J.A. (2002) Two-component systems, phosphorelays and regulation of their activities by phosphatases. In Sonenshein,A.L., Losick,R.M. and Hoch,J.A. (eds), Bacillus subtilis and Its Relatives: From Genes to Cells. ASM Press, Washington, DC, pp. 473–481.
- Salamitou S., Ramisse,F., Brehélin,M., Bourguet,D., Gilois,N., Gominet,M., Hernandez,E. and Lereclus,D. (2000) The plcR regulon is involved in the opportunistic properties of Bacillus thuringiensis and Bacillus cereus in mice and insects. Microbiology, 146, 2825–2832. [DOI] [PubMed] [Google Scholar]
- Sanchis V., Agaisse,H., Chaufaux,J. and Lereclus,D. (1996) Construction of new insecticidal Bacillus thuringiensis recombinant strains by using the sporulation non-dependent expression system of cryIIIA and a site specific recombination vector. J. Biotechnol., 48, 81–96. [DOI] [PubMed] [Google Scholar]
- Schnepf E., Crickmore,N., Van Rie,J., Lereclus,D., Baum,J., Feitelson,J., Zeigler,D.R. and Dean,D.H. (1998) Bacillus thuringiensis and its pesticidal crystal proteins. Microbiol. Mol. Biol. Rev., 62, 775–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trieu-Cuot P. and Courvalin,P. (1983) Nucleotide sequence of the Streptococcus faecalis plasmid gene encoding the 3′5′′-aminoglycoside phosphotransferase type III. Gene, 23, 331–341. [DOI] [PubMed] [Google Scholar]
- Villafane R., Bechhofer,D.H., Narayanan,C.S. and Dubnau,D. (1987) Replication control genes of plasmid pE194. J. Bacteriol., 169, 4822–4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanner B.L. (1993) Gene regulation by phosphate in enteric bacteria. J. Cell. Biochem., 51, 47–54. [DOI] [PubMed] [Google Scholar]
- Zhang M.-Y., Lövgren,A., Low,M.G. and Landén,R. (1993) Characterization of an avirulent pleitropic mutant of the insect pathogen Bacillus thuringiensis: reduced expression of flagellin and phospholipases. Infect. Immun., 61, 4947–4954. [DOI] [PMC free article] [PubMed] [Google Scholar]