

Abstract
The anaphase promoting complex/cyclosome (APC/C), activated by fzy and fzr, degrades cell cycle proteins that carry RXXL or KEN destruction boxes (d-boxes). APC/C substrates regulate sequential events and must be degraded in the correct order during mitosis and G1. We studied how d-boxes determine APC/Cfzy/APC/Cfzr specificity and degradation timing. Cyclin B1 has an RXXL box and is degraded by both APC/Cfzy and APC/Cfzr; fzy has a KEN box and is degraded by APC/Cfzr only. We characterized the degradation of substrates with swapped d-boxes. Cyclin B1 with KEN was degraded by APC/Cfzr only. Fzy with RXXL could be degraded by APC/Cfzy and APC/Cfzr. Interestingly, APC/Cfzy- but not APC/Cfzr-specific degradation is highly dependent on the location of RXXL. We studied degradation of tagged substrates in real time and observed that APC/Cfzr is activated in early G1. These observations demonstrate how d-box specificities of APC/Cfzy and APC/Cfzr, and the successive activation of APC/C by fzy and fzr, establish the temporal degradation pattern. Our observations can explain further why some endogenous RXXL substrates are degraded by APC/Cfzy, while others are restricted to APC/Cfzr.
Keywords: APC/C/cyclosome/destruction box/fizzy/fizzy-related
Introduction
Ubiquitin-mediated degradation plays a major role in ensuring the irreversible nature of the cell cycle. The insight that mitotic cyclins are degraded by this pathway (Glotzer et al., 1991; Hershko et al., 1991) and the subsequent discovery of the anaphase promoting complex/cyclosome (APC/C; King et al., 1995; Sudakin et al., 1995) contributed enormously to the current understanding of how the cell cycle functions. The APC/C is a 1500 kDa complex of a dozen different subunits (Peters et al., 1996) that serves as the ubiquitin ligase (E3) of the ubiquitylation process of various cell cycle proteins that are degraded during mitosis and G1.
More than a dozen different groups of proteins are degraded by the APC/C pathway, including mitotic A and B type cyclins (Sudakin et al., 1995), fzy (Weinstein, 1997; Shirayama et al., 1998), securin (Zou et al., 1999), xkid (Funabiki and Murray, 2000), E2-C (Yamanaka et al., 2000), polo kinase (Shirayama et al., 1998), nek2A (Hames et al., 2001), hsl1 (Burton and Solomon, 2000), cdc6 (Petersen et al., 2000) and geminin (McGarry and Kirschner, 1998). While all these proteins are degraded by the APC/C, they start to be degraded at different time points, such as prometaphase for cyclin A (den Elzen and Pines, 2001; Geley et al., 2001) and nek2A, metaphase for cyclin B1 (Clute and Pines, 1999), securin and xkid, and G1 for cdc6. APC/C substrates carry conserved motifs, so-called destruction boxes (d-boxes), which are required for their degradation. The cyclin B1 d-box (RTALGDIGN) is crudely shared by many of the other APC/C substrates. The arginine (R) and the leucine (L) are conserved in almost all substrates except in pim1, where arginine is replaced by lysine (Leismann et al., 2000), and in cyclin B3, where leucine is replaced by phenylalanine (Lozano et al., 2002). The asparagine (N) at position 9 is conserved in a subset of substrates and is required for the degradation of cyclin B1 in Xenopus extracts (King et al., 1996). Other residues of this RXXL box are much less conserved and it is virtually impossible to identify such a box merely by its sequence. However, the APC/C is evidently able to identify real RXXL boxes because not every protein that carries an RXXL is degraded. Moreover, fine differences in this box can contribute to changes in degradation, as is the case for cyclins A and B (King et al., 1996; Klotzbücher et al., 1996). An important recent advance is the identification of the KEN box as a targeting signal of some APC/C substrates (Pfleger and Kirschner, 2000). The discovery of this motif explained how vertebrate fzy, which lacks an RXXL box, is targeted for degradation by the APC/C. This box also plays a role in the degradation of substrates that do have an RXXL box, such as securin (Zur and Brandeis, 2001), cdc6 (Petersen et al., 2000), clb2 (Hendrickson et al., 2001), hsl1 (Burton and Solomon, 2001) and nek2A (Hames et al., 2001). However, the KEN motif is also abundant in many proteins that are not APC/C substrates.
The APC/C is activated by two WD repeat proteins: fzy/cdc20 and fzr/cdh1/hct1 (Sigrist et al., 1995; Schwab et al., 1997; Sigrist and Lehner, 1997; Visintin et al., 1997; Fang et al., 1998; Kramer et al., 1998; Lorca et al., 1998). In yeast, these two proteins confer some substrate specificity on the APC/C: pds1 is ubiquitylated by APC/Ccdc20, and clb2 by APC/Ccdh1 (Schwab et al., 1997; Visintin et al., 1997). A similar specificity has been suggested in mammalian cells, and it was shown that fzy is ubiquitylated by APC/Cfzr only (Pfleger and Kirschner, 2000). It was recently reported that fzy and fzr directly bind different APC/C substrates in vitro. Fzy is restricted to substrates that have an RXXL box, and fzr binds both RXXL and KEN box substrates (Burton and Solomon, 2001; Hilioti et al., 2001; Pfleger et al., 2001; Schwab et al., 2001).
In order to study the signal specificity of the RXXL and KEN boxes, we inserted artificial motifs into known substrates and studied their degradation in vivo. We show that the degradation of cyclin B1 with a mutated RXXL box can be restored by the insertion of an artificial KEN box close to the N-terminus of the mutated RXXL box. The location of this KEN was critical for its capacity to support degradation. Strikingly, cyclin B1 with a KEN box and a mutated RXXL box is ubiquitylated in vitro and degraded in vivo by APC/Cfzr only. This is in contrast to cyclin B1 with a wild-type RXXL box, which is degraded by both APC/Cfzy and APC/Cfzr.
We also studied the degradation of fzy, which is targeted for degradation by a KEN box and is an APC/Cfzr-specific substrate. Mutation of the KEN box stabilized fzy, and its degradation could be restored by the insertion of an RXXL box. Following the replacement of KEN with RXXL, fzy is targeted by APC/Cfzy, as well as by APC/Cfzr. RXXL inserted anywhere into the N-terminus of fzy could support APC/Cfzr-specific degradation. Degradation by APC/Cfzy was, however, highly dependent on the location of the RXXL, suggesting that flanking sequences or conformation influence the APC/Cfzr/APC/Cfzy specificity of the RXXL box. This could explain why certain RXXL substrates are degraded by APC/Cfzr only while others are degraded by both APC/Cfzr and APC/Cfzy.
We followed the degradation of green fluorescent protein (GFP)-tagged versions of APC/Cfzy and APC/Cfzr- specific substrates in real time. We observed that APC/Cfzy-specific degradation starts upon sister chromatid separation, and that APC/Cfzr-specific degradation starts in early G1.
The results we present here show that d-box type and location determine APC/Cfzy and APC/Cfzr specificity, and that fzy and fzr sequentially activate the APC/C. This specificity could thus form the basis of the ordered degradation of APC/C substrates during the different stages of mitosis and G1.
Results
A KEN box can target a cyclin B1 RXXL box mutant for degradation
The N-terminal part of cyclin B1, including its RXXL box (RTALGDIGN), is a portable degradation signal that maintains its degradation specificity when fused to other proteins. We fused the 105 N-terminal residues of cyclin B1 to CAT (chloramphenicol acetyl transferase) and GFP reporters and constitutively expressed them in NIH 3T3 mouse fibroblasts. Enzymatic activity of the B1-CAT fusion protein mimics the levels of endogenous cyclin B1, and is useful for following APC/C-specific proteolysis (Brandeis and Hunt, 1996). Cells were treated with nocodazole, and prometaphase-arrested cells were obtained by shake-off and released into fresh medium. This treatment yields highly synchronous G1 cell populations (Brandeis and Hunt, 1996). Figure 1A shows that when cells expressing a B1-CAT fusion protein were released into G1, CAT activity dropped sharply. CAT activity of cells expressing B1DM, a similar CAT fusion protein with a mutant RXXL box (GTAVGDIGN), remained constant throughout the cell cycle, exactly like that of cells expressing wild-type CAT. We inserted a minimal KEN box motif into B1DM (B1DM-KEN) by mutating two amino acids located five residues N-terminal to the mutated RXXL. We used an already existing lysine (K), to avoid adding another potential ubiquitylation target (Figure 1A). B1DM-KEN was degraded upon release into G1 just like wild-type B1-CAT. Figure 1B shows that analogous fusion proteins with GFP, assayed by immunoblotting, were degraded in a similar fashion. These observations suggest that the minimal KEN box is, like the RXXL box, a portable degradation signal, and that both boxes can target cyclin B1 for degradation in G1.

Fig. 1. A KEN box can target cyclin B1 for degradation in G1. Expression vectors for CAT, B1–CAT, its RXXL box (GTAV) mutant B1DM–CAT, and B1DM-KEN-CAT that has both a GTAV and an artificial KEN, were stably expressed in NIH 3T3 fibroblasts. Prometaphase-arrested cells, obtained by nocodazole treatment and subsequent shake-off, were released into fresh medium, harvested at the indicated time points and assayed for CAT activity (A). Cells stably expressing analogous constructs with a GFP reporter were synchronized in prometaphase and in G1. Degradation of wild-type and mutant B1–GFP reporters was analyzed by immunoblotting with GFP antibodies. Endogenous cyclin B1 was probed in parallel to establish that cells did indeed enter G1. Actin served as a loading control (B). KEN boxes were introduced at the indicated sites of the N-terminus of B1DM-CAT. The degradation of these reporters was assayed upon release into G1, but only KEN36, as used in (A), was degraded in G1 (C).
The short KEN sequence is common in many proteins that are not APC/C substrates, which suggests that it can target proteins to the APC/C only within the correct context of flanking sequences and conformation. To test this idea, we made a series of B1DM-CAT fusion proteins with KEN boxes inserted into various locations. Figure 1C shows that none of these fusion proteins were degraded. These results demonstrate that the sequences or conformation flanking the KEN box determine its capacity to target substrates for degradation.
The B1DM-KEN substrate is degraded by APC/Cfzr
Cyclin B1 is degraded both by APC/Cfzy and by APC/Cfzr. The KEN box has been identified as an APC/Cfzr-specific signal, therefore we wondered whether replacing the RXXL box of cyclin B1 with a KEN box changes its fzy/fzr specificity. We used fzr overexpression to test whether B1DM-KEN can be degraded by the APC/Cfzr. Fzr overexpression triggers unscheduled degradation of APC/Cfzr substrates in cells arrested by nocodazole at the spindle assembly checkpoint (Listovsky et al., 2000; Sorensen et al., 2000). We co-transfected cells with vectors expressing CAT fusion proteins of wild-type B1, B1DM and B1DM-KEN together with a fzr expression vector. Figure 2A shows that fzr overexpression in nocodazole-arrested cells led to the degradation of B1 and B1DM-KEN, but not of B1DM. All three proteins were stable in nocodazole-arrested cells transfected with an empty control vector.
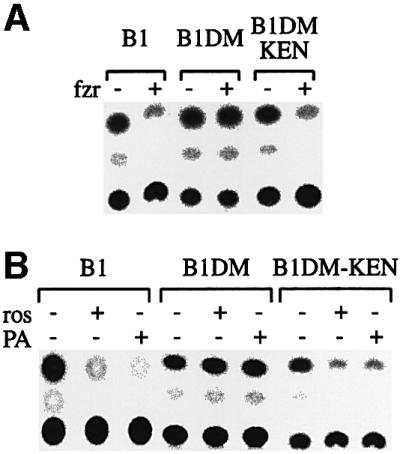
Fig. 2. Cyclin B1DM-KEN can be degraded in prometaphase by fzr overexpression and by cdk1 inhibition. Cells were transiently co-transfected with the indicated reporters together with an empty (–) or a human fzr (+) expression vector in a 1:3 ratio, respectively. After 24 h, cells were treated with nocodazole for 16 h. Prometaphase-arrested cells were obtained by shake-off and assayed for CAT activity (A). Cells stably expressing the indicated reporters were treated with nocodazole for 16 h, and prometaphase cells obtained by shake-off were transferred to fresh medium with nocodazole with or without roscovitin (ros) or purvalanol A (PA). Cells were harvested after 3 h and assayed for CAT activity (B).
APC/Cfzr-specific degradation can also be triggered by inhibition of cdk1 activity in prometaphase-arrested cells (Listovsky et al., 2000). Cells stably expressing fusion proteins were treated with nocodazole, and arrested cells were obtained by shake-off into medium with nocodazole without or with the cdk1 inhibitors roscovitin and purvalanol A. The cells were harvested after 3 h and assayed for CAT activity. Figure 2B shows that B1-KEN was degraded upon cdk1 inactivation with roscovitin or purvanalol A, just like wild-type B1, while B1DM was not affected. These experiments suggest that B1DM-KEN can be degraded in vivo by APC/Cfzr, but they do not preclude the possibility that it is also an APC/Cfzy substrate.
The B1DM-KEN substrate is ubiquitylated exclusively by APC/Cfzr
Cyclin B1 can be ubiquitylated by both APC/Cfzy and APC/Cfzr (Fang et al., 1998; Kramer et al., 1998). Fzy that carries a KEN box is, in contrast, ubiquitylated by APC/Cfzr only (Pfleger and Kirschner, 2000). We examined whether exchanging the cyclin B1 RXXL with a KEN box changed its fzy/fzr specificity in vitro. [35S]methionine-labeled substrates carrying the different mutations were prepared by a coupled in vitro transcription and translation reaction in reticulocyte lysate. The substrates were ubiquitylated by mitotic APC/C, without or with fzy, and by interphase APC/C without or with fzr (see Materials and methods). Figure 3 shows that the wild-type cyclin B1 substrate was, as expected, highly polyubiquitylated by both APC/Cfzy and APC/Cfzr. The B1DM-KEN substrate, which was degraded in vivo (Figure 1C, KEN36), was ubiquitylated in this assay, but by APC/Cfzr only. The non-degradable B1DM and B1DM-KEN13 (Figure 1C) substrates were hardly ubiquitylated at all. This assay shows that the replacement of the functional RXXL box of cyclin B1 with a KEN box converted it, at least in vitro, into an APC/Cfzr-specific substrate.
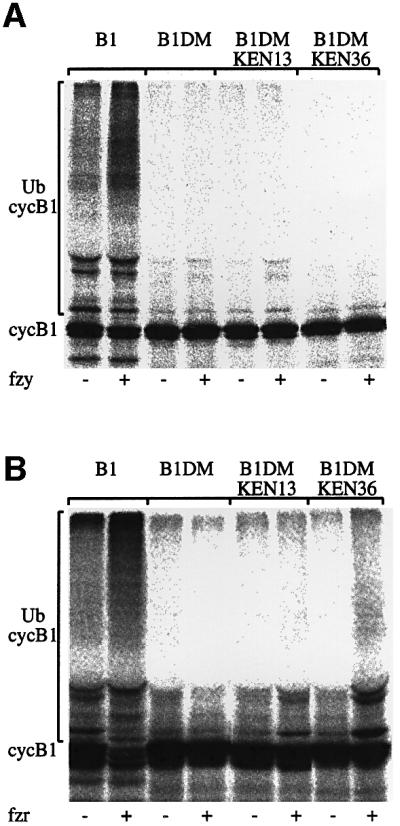
Fig. 3. Cyclin B1 with a mutated RXXL and a KEN box is ubiquitylated in vitro by APC/Cfzr. CAT fusion proteins of cyclin B1, B1DM and B1DM with KEN boxes at positions 13 or 36 only were ubiquitylated in vitro with mitotic APC/C with (+) or without (–) fzy (A) or with interphase APC/C with (+) or without (–) fzr (B).
The B1DM-KEN substrate is degraded in vivo only by APC/Cfzr
The observation that B1DM-KEN can be ubiquitylated in vitro by APC/Cfzr only (Figure 3) suggested that the substitution of RXXL with KEN transformed it into an APC/Cfzr-specific substrate. In order to examine whether B1DM-KEN is degraded by the APC/Cfzy in vivo or not, we had to obtain cells with active APC/Cfzy and inactive APC/Cfzr. APC/Cfzy is normally active only for a short time during mitosis; however, expression of non-degradable kinase-active cyclin B1 arrests cells in telophase (Wheatley et al., 1997) with a highly active APC/Cfzy (Zur and Brandeis, 2001). Fzr can not activate the APC/C in these cells because cdk1–cyclin B1 phosphorylates fzr and prevents its binding to the APC/C (Zachariae et al., 1998; Lukas et al., 1999; Listovsky et al., 2000).
Figure 4A shows round mitotic cells arrested in telophase following transient transfection of the non-degradable catalytically active cyclin B1DM–GFP construct. Cells in telophase would normally be elongated; however, if cytokinesis is prevented by non-degradable cyclin B1, they become round again. Almost all of the round mitotic cells (top right panel) are GFP positives (top left panel). Only <10% of the mitotic cells were GFP negative, and may represent untransfected cells that happened to be in mitosis. Likewise, the vast majority of chromosome spreads prepared from these cells showed cells with fully separated chromatids (Figure 4A, bottom left panel) and <10% showed metaphase chromosomes (Figure 4A, bottom right panel). These cells were collected by gentle shake-off and their APC/C was immunoprecipitated with cdc27 antibodies. Figure 4B shows that only fzy co-precipitated with the APC/C of telophase-arrested cells, as well as with the APC/C of prometaphase-arrested cells. APC/C precipitated from G1 cells was, as reported previously (Kramer et al., 1998; Listovsky et al., 2000), bound only by fzr. Figure 4B also shows that endogenous cyclin B1, a substrate of both APC/Cfzy and APC/Cfzr, was degraded both in telophase and G1 as expected. Fzy, in contrast, which is targeted only by the APC/Cfzr, was stable in telophase-arrested cells and degraded in G1. The fact that no fzr co-precipitated with APC/C immunoprecipitates from telophase cells indicates that these cells were not contaminated with any noticeable amount of G1 cells.

Fig. 4. Cyclin B1 with a mutated RXXL box and a KEN box is degraded in vivo by APC/Cfzr only. Cells were transfected with an expression vector for non-degradable catalytically active cyclin B1–GFP to arrest them in telophase. Almost all the round mitotic cells observed by phase–contrast microscopy are GFP positive (top panels). More than 90% of chromosome spreads prepared from cells arrested by this transfection had fully separated sister chromatids (bottom left panel), compared with only a small fraction that had non-separated metaphase chromosomes (bottom right panel). These results indicate that this protocol yields highly synchronous populations of telophase-arrested cells (A). Cells arrested in telophase by this protocol, and in prometaphase with nocodazole were obtained by gentle shake-off. G1 cells were obtained by growing prometaphase cells obtained by shake-off for 4 h in fresh medium. The APC/C complex was immunoprecipitated with cdc27 antibodies from extracts of prometaphase, telophase and G1 cells and probed with fzy and fzr antibodies. Cell extracts were also directly probed with the indicated antibodies (B). Cells stably expressing the indicated reporters were synchronized as described in (B) and assayed for CAT activity (C). A KEN36 box was introduced into the full-length cyclin B1DM–GFP used in (A). Cells were transfected with this vector and with wild-type and non-degradable cyclin B1–GFP expression vectors on a glass-bottom culture dish. Living transfected cells were photographed after 40 h with fluorescence (GFP and DAPI) and DIC illumination at 630× magnification (D).
Extracts of telophase-arrested cells stably expressing CAT fusion proteins of B1, B1DM, B1DM-KEN and B1-KEN, which has both a KEN and a wild-type RXXL box, were assayed for CAT activity alongside extracts of prometaphase and G1 cells. Figure 4C shows that B1DM-KEN was stable in telophase and degraded in G1. B1 and B1-KEN were, in contrast, degraded both in telophase and G1, and B1DM-CAT was always stable. These results show that B1DM-KEN is stable in the presence of active APC/Cfzy and support the data obtained by in vitro ubiquitylation (Figure 3), which showed that B1DM- KEN is an APC/Cfzr-specific substrate. The fact that B1-KEN was degraded, like wild-type B1, shows that the KEN box is not dominant over the RXXL box.
Fzr phosphorylated by cdk1–cyclin B1 cannot bind the APC/C (Zachariae et al., 1998; Lukas et al., 1999; Listovsky et al., 2000). Degradation of cyclin B1 is therefore required for the activation of the APC/Cfzr. We hypothesized that catalytically active full-length cyclin B1DM-KEN, which can be degraded by APC/Cfzr only, will arrest the cells in telophase, like non-degradable cyclin B1. The top panel of Figure 4D shows that cells transfected with an expression vector for wild-type cyclin B1 do not arrest in mitosis. The middle panel of Figure 4D shows that cells transfected with an expression vector for cyclin B1DM-KEN got arrested in telophase, like cells transfected with non-degradable cyclin B1 shown in the bottom panel. Moreover, chromosomes in spreads of cells arrested by active cyclin B1DM-KEN were fully separated, as seen in the cells expressing non-degradable cyclin B1DM, shown in the bottom left panel of Figure 4A. To confirm that full-length cyclin B1DM-KEN can be degraded by the APC/Cfzr, we treated the arrested cells with cdk1 inhibitors, as described in the legend of Figure 2. The inhibition of cdk1 led to the degradation of cyclin B1DM-KEN, as expected, but had no effect on the levels of cyclin B1DM (data not shown). This observation suggests that APC/Cfzy-mediated degradation of cyclin B1 is indeed essential for mitotic exit.
An RXXL box can target fzy for degradation
The results we have presented so far show that replacing the RXXL box of cyclin B1 with a KEN box converted the cyclin into an APC/Cfzr-specific substrate. We wondered whether a reciprocal replacement of the KEN box of fzy with an RXXL box of cyclin B1 would have analogous effects.
Fzy is an APC/Cfzr-specific substrate whose degradation is signaled by a KEN box (Pfleger and Kirschner, 2000). We made a fzy-CAT reporter by fusing the N-terminal 112 residues of human fzy, including the KEN box, to CAT. Figure 5A shows that the fzy-CAT fusion reporter was degraded upon exit from mitosis, and that the Fzy-KM reporter, which has a triple mutation converting KEN to AAA, is not degraded. In order to establish the specificity of the KEN, we made three constructs with conserved replacements of each residue: REN, KDN and KEQ. None of these mutants were degraded (data not shown). Figure 5A shows that fzy-KM-RXXL99, which has an insertion of a cyclin B1 RXXL box close to the mutated KEN box of fzy, is degraded in G1. This degradation depends on the RXXL box because fzy-KM-DM, a similar reporter with a mutated RXXL box, is not degraded. Figure 5B shows similar results obtained with analogous GFP fusion proteins and compares their degradation with that of endogenous fzy.

Fig. 5. The RXXL box can replace the KEN box of fzy and change its fzy/fzr specificity. Degradation of fzy-CAT (fzy) and a mutant with alanine replacement of the KEN box (fzy-KM) was analyzed in cells stably expressing vectors for these fusion reporters. Degradation of fzy-KM either with a cyclin B1 RXXL box (fzy-KM-RXXL99), or with a mutated RXXL box at the same location (fzy-KM-DM) was tested in the same way (A). Degradation of fzy–GFP and its derivatives upon release into G1 was assayed by immunoblotting of cells stably expressing fusion reporters. Mitotic cells were obtained by nocodazole arrest and shake-off, and G1 cells by release of mitotic cells into G1 for 4 h. Endogenous fzy and actin were probed in parallel (B). RXXL boxes were introduced into the indicated locations of the non-degradable fzy-KM-CAT reporter. The CAT activity of these reporters was assayed in prometaphase (M), telophase (T) and G1. This experiment was performed in triplicate (see error bars). All CAT fusion proteins were stable in prometaphase and degraded in G1 by APC/Cfzr. In contrast, only the fzy-KM with an RXXL box introduced into position 78 was degraded as efficiently as cyclin B1 by the APC/Cfzy in telophase. Fzy-KM with RXXL in position 99 was hardly degraded by the APC/Cfzy (C), like wild-type fzy-CAT.
The location of the artificial KEN box we inserted into B1DM-KEN was critical for its capacity to target the substrate for degradation (Figure 1C). We wondered whether the artificial RXXL in fzy is also location dependent. We therefore inserted RXXL boxes into four additional locations of the fzy-CAT fusion protein. Degradation of these substrates by the APC/Cfzr was tested by release of cells into G1, and degradation by the APC/Cfzy was tested by arresting cells with non-degradable cyclin B1. All these substrates were equally degraded by the APC/Cfzr in G1 (Figure 5C). Thus, as far as the APC/Cfzr is concerned, the RXXL did not show any location preference. Interestingly, the degradation of these substrates in telophase-arrested cells varied considerably. A substrate with an RXXL box closest to the mutated KEN (RXXL99) was as stable in telophase cells as wild-type fzy. In contrast, a substrate with an RXXL at a nearby location (RXXL78) was degraded in telophase as efficiently as cyclin B1-CAT (Figure 5C). The other substrates showed intermediate values but were relatively stable. We were surprised by this result and repeated the experiments in triplicate. All experiments yielded very similar results (see error bars), and therefore we are confident that the observed variations are indeed real and not a result of experimental variations. These results suggest that the APC/Cfzy is considerably more sensitive to the location of the RXXL box than the APC/Cfzr.
APC/Cfzr-specific degradation of B1DM-KEN takes place in G1
It is assumed that fzy, which activates the APC/C in metaphase, is replaced in G1 by fzr (Morgan, 1999; Zachariae and Nasmyth, 1999; Peters, 2002). This switch from APC/Cfzy to APC/Cfzr has not been characterized and has never been observed in real time in living cells. We therefore used our B1DM-KEN–GFP fusion construct to follow the onset of APC/Cfzr-specific degradation in living cells. We prepared cell lines stably expressing GFP-tagged reporters of cyclin B1, B1DM, B1DM-KEN, B1-KEN and fzy. Stable lines were used to preclude problems of saturating the degradation machinery, which might be caused by transient transfections. The fact that these reporters are expressed in cells over many cell generations testifies that they do not inhibit proper mitosis.
Figure 6A shows representative cells stably expressing GFP-tagged B1 and B1DM-KEN from different stages of mitosis and early G1. B1–GFP strongly localizes to the chromosomes in metaphase. In anaphase, B1–GFP was no longer observed on chromosomes and the total fluorescence was considerably lower. B1–GFP could hardly be seen at all in cells from later stages of mitosis or early G1. B1DM-KEN–GFP was not only strongly localized to the chromosome in metaphase, but also in anaphase and nuclei of early G1 cells that had already decondensed their chromatin. B1DM-KEN–GFP completely disappeared only after >2 h into G1. Cells shown in this figure represent hundreds of cells with similar phenotypes. The mid-G1 cells shown were followed to this stage from anaphase.

Fig. 6. Live cell imaging of the initiation of APC/Cfzy and APC/Cfzr activity. Cells stably expressing B1–GFP and B1DM-KEN–GFP were photographed at the indicated stages of mitosis and G1 at 630× magnification with GFP and DAPI filter sets, as well as by DIC optics. The mid-G1 cells were followed through mitosis and photographed 3 h after metaphase (A). Cells stably expressing GFP fusion proteins of cyclin B1, B1DM, B1-KEN, B1DM-KEN and fzy were followed through mitosis and photographed every 3 min. Background-subtracted GFP fluorescence was analyzed as described in Materials and methods and plotted as a function of time (B). The stages of mitosis are indicated by arrows.
To quantify the degradation the various fusion proteins, we followed single metaphase cells and photographed them every 3 min. Each time series was quantified and the level of normalized fluorescence was plotted against time. Figure 6B shows quantified data of representative cells from more than a dozen cells of each type (for details, see Materials and methods). Figure 6B shows that degradation of B1–GFP is initiated at the metaphase–anaphase transition and is very rapid. These results are similar to those reported previously for full-length cyclin B1–GFP (Clute and Pines, 1999). Cells expressing B1-KEN were degraded in exactly the same manner. Degradation of B1DM-KEN–GFP, in contrast, started only in early G1 and took ∼2 h to complete. Degradation of the Fzy–GFP, which also depends on a KEN box, was very similar to that of B1DM-KEN. The non-degradable B1DM–GFP was not degraded and remained virtually the same for the duration of the experiment. The relatively constant level of B1DM–GFP shows that the reduction of fluorescence in the cells expressing the other proteins was not due to photobleaching of GFP or to non-specific degradation.
These results show that the exchange of the RXXL with the KEN box, which converted cyclin B1 into an APC/Cfzr substrate, also changed its degradation timing. In addition, this is the first time that the initiation of APC/Cfzr-specific degradation has been observed in real time, suggesting that the APC/Cfzr is activated around the beginning G1.
Discussion
The most important capacity of a regulatory degradation pathway is its capability to identify and degrade its substrates. Thus, elucidating how substrates are identified is a basic requirement for understanding how such a pathway works. Some of the ‘words’ the APC/C understands have been identified; however, the ‘grammar’ of the language of the APC/C is still not fully understood.
APC/C substrates regulate interdependent and sequential events during mitosis, therefore it is paramount that they are degraded in the correct order. It has been proposed that this ordered degradation is achieved by different substrate specificities of fzy and fzr, and by successive APC/C activation, first by fzy and then by fzr (Morgan, 1999; Zachariae and Nasmyth, 1999; Peters, 2002). We made APC/C substrates that differed only at their d-boxes and used them to test this model. A KEN box introduced into cyclin B1 with a mutated RXXL restored its degradation. This artificial KEN box substrate was degraded by APC/Cfzr only. The location of this KEN was critical for degradation. RXXL boxes introduced into fzy with a mutated KEN box restored its degradation. RXXL boxes at different locations could target the substrate for degradation by APC/Cfzr, and one particular RXXL could further target the substrate for degradation by APC/Cfzy. We used GFP-tagged versions of APC/Cfzy- and APC/Cfzr-specific substrates to study degradation in living cells in real time. These studies showed that the APC/Cfzy is switched on at the metaphase–anaphase transition, and that the APC/Cfzr is activated in early G1.
Our conclusions from these observations are that fzy and fzr specificity is determined by the identity of the d-box and its location in the substrate, and that the switch from fzy- to fzr-specific degradation takes place in early G1. These observations support both assumptions of the proposed model (Morgan, 1999), showing that it is indeed capable of establishing the temporal pattern of APC/C-specific proteolysis observed in mitosis.
Fzy and fzr targeting specificity of RXXL and KEN box substrates
Substrate-specific APC/C activation by cdc20 (fzy) and cdh1 (fzr) was observed in budding yeast (Schwab et al., 1997; Visintin et al., 1997), as well as in vertebrates (Pfleger and Kirschner, 2000). Two distinct degradation signals have been identified so far in APC/C substrates: the RXXL box, originally identified in cyclin B1 (Glotzer et al., 1991; King et al., 1996), and the KEN box, originally identified in fzy (Pfleger and Kirschner, 2000).
Several recent reports have shown that the fzy and fzr APC/C activators directly interact with substrates (Burton and Solomon, 2001; Hilioti et al., 2001; Pfleger et al., 2001; Schwab et al., 2001). Mammalian fzr was found to bind both KEN and RXXL box substrates; in contrast, fzy was restricted to the binding of RXXL box substrates (Pfleger et al., 2001). These studies used endogenous substrates and were inconclusive as to whether the KEN box is essential (Schwab et al., 2001) or sufficient (Burton and Solomon, 2001) for fzr specificity.
We used sets of substrates that differed only at their d-boxes in fixed sequence contexts of known APC/C substrates. One set was based on the N-terminal part of cyclin B1 fused to CAT or GFP reporter proteins (Figure 1). Wild-type B1 (RTAL) was universally degraded by both APC/Cfzy and APC/Cfzr. Two amino acid substitutions made a non-degradable B1DM (GTAV). Two additional substitutions made a B1DM-KEN that could be degraded by APC/Cfzr only. The APC/Cfzr specificity of B1DM-KEN was deduced from an in vitro ubiquitylation assay (Figure 3) and from the fact that it was not degraded in cells arrested in telophase with non-degradable cyclin B1 (Figure 4). Such cells contain highly active APC/Cfzy and readily degraded wild-type B1-CAT, as well as endogenous cyclin B1 (Figure 4) and securin (Zur and Brandeis, 2001). Finally, cells transfected with full-length catalytically active cyclin B1DM-KEN arrested in telophase, like cells transfected with the non-degradable cyclin B1. This arrest is the strongest evidence so far that cyclin B1 degradation by the APC/Cfzy is essential for mitotic exit, which could suggest that fzy is essential in multicellular organisms. Cdc20, the budding yeast homologue of fzy, is not essential; however, yeast express the cyclin kinase inhibitor (CKI) p40sic1 that serves as an alternative pathway for inactivating mitotic cyclins (Visintin et al., 1998; Shirayama et al., 1999). In multicellular organisms, no CKI that inhibits cyclin B1 in mitosis has been identified so far.
The second set of substrates we used was based on the N-terminal part of fzy fused to CAT or GFP. Fzy has a KEN box and is degraded by APC/Cfzr only. We mutated this box and obtained a stable fusion protein. RXXL boxes were introduced into five locations of this stable fusion protein and all of them were degraded by APC/Cfzr. One of these substrates was also efficiently degraded by APC/Cfzy (Figure 5C).
The conclusion from the experiments with these artificial substrates is that the RXXL and KEN boxes, together with flanking sequences or conformation, determine whether an APC/C substrate will be targeted by both APC/Cfzy and APC/Cfzr, or by APC/Cfzr only. To what extent can this conclusion describe the degradation of real substrates? Fzy is so far the only protein where it has been shown rigorously that KEN on its own is sufficient for APC/C-specific degradation (Pfleger and Kirschner, 2000); in most other substrates, the KEN box is accompanied by some sort of RXXL box. The dual KEN+RXXL substrates securin, hsl1, clb2 and nek2A are degraded by both APC/Cfzy and APC/Cfzr (Baumer et al., 2000; Yeong et al., 2000; Burton and Solomon, 2001; Hames et al., 2001; Hendrickson et al., 2001; Zur and Brandeis, 2001). This pattern is consistent with our observation that the RXXL box is dominant over the KEN box (Figures 4C and 6B). Interestingly, cdc6 and Aurora-A, which both have RXXL boxes, are degraded by the APC/Cfzr only (Petersen et al., 2000; Castro et al., 2002). Table I shows that the repertoire of artificial substrates we made covers the full range of degradation specificities of endogenous substrates: B1DM-KEN is degraded by the APC/Cfzr, like fzy; B1-KEN is a dual KEN+RXXL substrate degraded by both APC/Cfzy and APC/Cfzr, like securin; fzy-RXXL78 is degraded by both APC/Cfzy and APC/Cfzr, like cyclin B1; and finally, fzy-RXXL99 is degraded like cdc6, Aurora-A and E2-C, by the APC/Cfzr only. Other endogenous substrates that have been characterized so far also fit into one of these groups. We are currently addressing the question of which flanking conformation or sequences are required for activating a putative KEN or determining whether an RXXL can signal targeting by the APC/Cfzy.
Table I. Artificial substrates mimic the degradation specificity and timing of all known endogenous APC/C substrates.
| Synthetic substrate | D-box type | Targeted by | Endogenous substrate | Degradation timing |
|---|---|---|---|---|
| B1DM-KEN | KEN | APC/Cfzr | Fzy | G1 |
| B1-KEN | KEN+RXXL | APC/Cfzy and APC/Cfzr | Securin | Anaphase |
| Fzy-RXXL78 | RXXL | APC/Cfzy and APC/Cfzr | Cyclin B1, Xkid | Anaphase |
| Fzy-RXXL99 | RXXL or KEN+RXXL | APC/Cfzr | E2-C, Cdc6, Aurora-A | G1 |
Sequential activation of the APC/C by fzy and fzr
Multiple experiments in yeast, flies and mammalian cells suggest that the APC/C is first activated in metaphase by fzy, and that fzr activates the APC/C later in mitosis. In Drosophila, fzy is required for degradation of mitotic cyclins in mitosis (Sigrist et al., 1995) and fzr is required for degradation in G1 (Sigrist and Lehner, 1997). Sequential activation of the APC/C by fzy in metaphase and by fzr in G1 is regulated both positively and negatively by cdk1–cyclin B1. Cdk1–cyclin B1, in collaboration with plk1 (Golan et al., 2002), phosphorylates the APC/C, a precondition for its activation by fzy (Shteinberg et al., 1999; Kramer et al., 2000; Rudner and Murray, 2000). At the same time, cdk1–cyclin B1 phosphorylates fzr, thereby preventing it from binding to the APC/C (Zachariae et al., 1998; Lukas et al., 1999; Listovsky et al., 2000). After cdk1–cyclin B1 activity is reduced due to cyclin B1 degradation, fzr is no longer inhibited and can bind to the APC/C and activate it. In contrast to fzy, fzr binds non-phosphorylated interphase APC/C, and it remains associated with it until the end of G1 (Zur and Brandeis, 2001).
We used GFP-tagged versions of the substrates we made to directly observe the onset of APC/C activity in real time. Degradation of APC/Cfzy substrate B1–GFP was initiated at the metaphase–anaphase transition as reported previously (Clute and Pines, 1999). The degradation of APC/Cfzr-specific substrate B1DM-KEN–GFP was initiated only in G1. A recent study has shown that securin with a wild-type KEN and a mutated RXXL box is degraded by APC/Cfzr only (Hagting et al., 2002). The degradation of this APC/Cfzr-specific substrate is initiated in anaphase, somewhat earlier than the degradation of B1DM-KEN that we observed. This small difference might be due to different experimental methods, cell lines or substrates used for monitoring APC/Cfzr activation.
The mechanism that governs the replacement of fzy with fzr is not understood. It is possible that fzy dissociates from the APC/C upon dephosphorylation of the APC/C. Inactivation of APC/Cfzy does not depend on APC/Cfzr activity because it also takes place in embryos that do not express fzr (Lorca et al., 1998) and in cells with a deleted fzr gene (Sudo et al., 2001). Thus, it is not likely that the degradation of fzy by APC/Cfzr plays an important role in this switch from APC/Cfzy to APC/Cfzr.
Finally, the encouraging implication of our results is that the complex issue of substrate recognition by the APC/C and their sequential degradation could, potentially, be explained by a set of relatively simple rules.
Materials and methods
Plasmids
The cyclin B1-CAT fusion reporter consists of the N-terminal 105 amino acids of mouse cyclin B1 fused to bacterial CAT, and expressed by an SV promoter (Brandeis and Hunt, 1996). B1–GFP codes for the same fragment of the cyclin fused to GFP and expressed by an EF1 promoter. Non-degradable full-length mouse cyclin B1–GFP was described previously (Listovsky et al., 2000). To make substrates for ubiquitylation, we cloned B1-CAT and its derivatives into pBluescript downstream of the T7 promoter. All the described mutants were made by using the QuikChange™ site-directed mutagenesis method (Stratagene). Fzy-CAT and fzy–GFP fusion vectors encode the 112 N-terminal residues of human fzy fused to CAT and GFP, respectively. Fzy-RXXL-CAT vectors were made by introducing double-stranded oligos coding for RTALGDIGNK into BssHII (28), BstEII (54), NgoMIV (78), NotI (99) and XbaI (115) sites of fzy-CAT in which KEN was mutated to AAA.
Cell culture, transfections and synchronization
All experiments were performed with NIH 3T3 cells. Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with glutamine, penicillin/streptomycin and 10% fetal calf serum (FCS; Biological Industries, Beit Haemek) and transfected by the CaPO4 co-precipitation method (Ausubel et al., 1994). The various CAT and GFP fusion expression vectors were co-transfected with a G418 resistance vector, and resistant colonies were pooled. Prometaphase-arrested cells were obtained by shake-off of cells that were treated with nocodazole for ∼16 h. G1 cells were obtained by shaking off nocodazole-arrested cells, washing them, and growing them in fresh medium for 4 h, or as indicated. The following drugs were dissolved in DMSO and used at the indicated final concentrations: 2 µM nocodazole (Sigma), 28 µM roscovitin (Biomol) and 30 µM purvalanol A (Alexis).
Antibodies
The following antibodies were used: mouse monoclonal antibodies against cdc27 (AF3), fzr (AR38) and cyclin B1 (152), as well as rabbit anti-GFP, were a gift from J.Gannon and T.Hunt, and goat p55cdc (fzy) and actin were purchased from SCB. Cdc27 antibodies were covalently coupled to protein A–affiprep beads (Bio-Rad) as described previously (Harlow and Lane, 1987). Immunoblots and immunoprecipitation were performed by standard procedures (Harlow and Lane, 1987). CAT assays were performed by standard methods using [14C]chloramphenicol (Amersham) and acetyl CoA (Roche). CAT activity was calculated as the proportion of the mono- and diacetylated chloramphenicol from the total counts of [14C]chloramphenicol.
In vitro ubiquitylation assays
The in vitro ubiquitylation assays were performed as described previously (Yudkovsky et al., 2000). Substrates for ubiquitylation were prepared by in vitro transcription and translation in a coupled reticulocyte lysate system (Promega) in the presence of [35S]methionine. Each reaction contained partially purified APC/C, E1, E2-C, an energy regenerating system, and fzy or fzr. Mitotic APC/C was purified from nocodazole-arrested cells, and interphase APC/C from cells arrested with thymidine, and released for 2 h into S phase.
Time-lapse photography
NIH 3T3 cells stably expressing the various GFP fusion proteins were grown in 60 mm glass-bottom culture dishes (Mattek) in DMEM supplemented with 10% FCS, glutamine and antibiotics. Growth medium was replaced with CO2-independent MEM medium without Phenol Red (Biological Industries) with the same supplements and transferred to a heated stage at 37°C. Cells were photographed at a magnification of ×200 with an Olympus IX70 inverted microscope fitted with a uniblitz shutter (Vincent), to protect the cells from photo damage, and a Magnafire cooled CCD camera (Optronics). This set-up was controlled by Image-Pro 4.5 and Scope-Pro 4.0 (media cybernetics) software. Cells were exposed for 1 s every 3 min. The time series of the region of interest (ROI) with the dividing cell was subtracted with a time series of an adjacent background ROI of identical size. Absolute levels of fluorescence were then normalized and plotted as a function of time using the Time series macro of Image-Pro.
Acknowledgments
Acknowledgements
We would like to thank Avram Hershko and Yana Yudkovsky for generous help with the in vitro assays and for many fruitful discussions; Stephan Geley, Julian Gannon and Tim Hunt for providing many of the antibodies used in this study; Dmitri Prilutski and Aryeh Weiss for help with setting up the time-lapse system; and Yosef Gruenbaum for critical reading of the manuscript. Miriam Broit, Michal Macarov, Svetlana Rotlova, Tamar Listovsky and Dalia Sherizen assisted during various stages of this project. This work was funded by grants from the Israeli Academy of Sciences and Humanities (124/98), from the Israel–US Binational Science Fund (1998123) and from the German Israeli Foundation (658/2000).
References
- Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (1994) Current Protocols in Molecular Biology. John Wiley & Sons, New York, NY.
- Baumer M., Braus,G.H. and Irniger,S. (2000) Two different modes of cyclin clb2 proteolysis during mitosis in Saccharomyces cerevisiae. FEBS Lett., 468, 142–148. [DOI] [PubMed] [Google Scholar]
- Brandeis M. and Hunt,T. (1996) The proteolysis of mitotic cyclins in mammalian cells persists from the end of mitosis until the onset of S phase. EMBO J., 15, 5280–5289. [PMC free article] [PubMed] [Google Scholar]
- Burton J.L. and Solomon,M.J. (2000) Hsl1p, a Swe1p inhibitor, is degraded via the anaphase-promoting complex. Mol. Cell. Biol., 20, 4614–4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton J.L. and Solomon,M.J. (2001) D box and KEN box motifs in budding yeast Hsl1p are required for APC-mediated degradation and direct binding to Cdc20p and Cdh1p. Genes Dev., 15, 2381–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro A., Arlot-Bonnemains,Y., Vigneron,S., Labbe,J.C., Prigent,C. and Lorca,T. (2002) APC/Fizzy-related targets Aurora-A kinase for proteolysis. EMBO rep., 3, 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clute P. and Pines,J. (1999) Temporal and spatial control of cyclin B1 destruction in metaphase. Nat. Cell Biol., 1, 82–87. [DOI] [PubMed] [Google Scholar]
- den Elzen N. and Pines,J. (2001) Cyclin A is destroyed in prometaphase and can delay chromosome alignment and anaphase. J. Cell Biol., 153, 121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G., Yu,H. and Kirschner,M.W. (1998) Direct binding of Cdc20 protein family members activates the anaphase-promoting complex in mitosis and G1. Mol. Cell, 2, 163–171. [DOI] [PubMed] [Google Scholar]
- Funabiki H. and Murray,A. (2000) The Xenopus chromokinesin xkid is essential for metaphase chromosome alignment and must be degraded to allow anaphase chromosome movement. Cell, 102, 411–424. [DOI] [PubMed] [Google Scholar]
- Geley S., Kramer,E., Gieffers,C., Gannon,J., Peters,J.M. and Hunt,T. (2001) Anaphase-promoting complex/cyclosome-dependent proteo lysis of human cyclin A starts at the beginning of mitosis and is not subject to the spindle assembly checkpoint. J. Cell Biol., 153, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glotzer M., Murray,A.W. and Kirschner,M.W. (1991) Cyclin is degraded by the ubiquitin pathway. Nature, 349, 132–138. [DOI] [PubMed] [Google Scholar]
- Golan A., Yudkovsky,Y. and Hershko,A. (2002) The cyclin–ubiquitin ligase activity of cyclosome/APC is jointly activated by protein kinases Cdk1–cyclin B and Plk. J. Biol. Chem., 277, 15552–15557. [DOI] [PubMed] [Google Scholar]
- Hagting A., den Elzen,N., Vodermaier,H.C., Waizenegger,I.C., Peters,J.M. and Pines,J. (2002) Human securin proteolysis is controlled by the spindle checkpoint and reveals when the APC/C switches from activation by Cdc20 to Cdh1. J. Cell Biol., 157, 1125–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hames R.S., Wattam,S.L., Yamano,H., Bacchieri,R. and Fry,A.M. (2001) APC/C-mediated destruction of the centrosomal kinase Nek2A occurs in early mitosis and depends upon a cyclin A-type D-box. EMBO J., 20, 7117–7127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow E. and Lane,D. (1987) Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Hendrickson C., Meyn,M.A., Morabito,L. and Holloway,S.L. (2001) The KEN box regulates Clb2 proteolysis in G1 and at the metaphase-to-anaphase transition. Curr. Biol., 11, 1781–1787. [DOI] [PubMed] [Google Scholar]
- Hershko A., Ganoth,D., Pehrson,J., Palazzo,R.E. and Cohen,L.H. (1991) Methylated ubiquitin inhibits cyclin degradation in clam embryo extracts. J. Biol. Chem., 266, 16376–16379. [PubMed] [Google Scholar]
- Hilioti Z., Chung,Y., Mochizuki,Y., Hardy,C.F. and Cohen-Fix,O. (2001) The anaphase inhibitor Pds1 binds to the APC/C-associated protein Cdc20 in a destruction box-dependent manner. Curr. Biol., 11, 1347–1352. [DOI] [PubMed] [Google Scholar]
- King R.W., Peters,J.M., Tugendreich,S., Rolfe,M., Hieter,P. and Kirschner,M.W. (1995) A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell, 81, 279–288. [DOI] [PubMed] [Google Scholar]
- King R.W., Glotzer,M. and Kirschner,M.W. (1996) Mutagenic analysis of the destruction signal of mitotic cyclins and structural characterization of ubiquitinated intermediates. Mol. Biol. Cell, 7, 1343–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotzbücher A., Stewart,E., Harrison,D. and Hunt,T. (1996) The ‘destruction box’ of cyclin A allows B-type cyclins to be ubiquitinated, but not efficiently destroyed. EMBO J., 15, 3053–3064. [PMC free article] [PubMed] [Google Scholar]
- Kramer E.R., Gieffers,C., Holzl,G., Hengstschlager,M. and Peters,J.M. (1998) Activation of the human anaphase-promoting complex by proteins of the CDC20/Fizzy family. Curr. Biol., 8, 1207–1210. [DOI] [PubMed] [Google Scholar]
- Kramer E.R., Scheuringer,N., Podtelejnikov,A.V., Mann,M. and Peters,J.M. (2000) Mitotic regulation of the APC activator proteins CDC20 and CDH1. Mol. Biol. Cell, 11, 1555–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leismann O., Herzig,A., Heidmann,S. and Lehner,C. (2000) Degradation of Drosophila PIM regulates sister chromatid separation during mitosis. Genes Dev., 14, 2192–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listovsky T., Zor,A., Laronne,A. and Brandeis,M. (2000) Cdk1 is essential for mammalian cyclosome/APC regulation. Exp. Cell Res., 255, 184–191. [DOI] [PubMed] [Google Scholar]
- Lorca T., Castro,A., Martinez,A., Vigneron,S., Morin,N., Sigrist,S., Lehner,C., Doree,M. and Labbe,J. (1998) Fizzy is required for activation of the APC/cyclosome in Xenopus egg extracts. EMBO J., 17, 3565–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano J.C., Perret,E., Schatt,P., Arnould,C., Peaucellier,G. and Picard,A. (2002) Molecular cloning, gene localization and structure of human cyclin B3. Biochem. Biophys. Res. Commun., 291, 406–413. [DOI] [PubMed] [Google Scholar]
- Lukas C., Sorensen,C.S., Kramer,E., Santoni-Rugiu,E., Lindeneg,C., Peters,J.M., Bartek,J. and Lukas,J. (1999) Accumulation of cyclin B1 requires E2F and cyclin-A-dependent rearrangement of the anaphase-promoting complex. Nature, 401, 815–818. [DOI] [PubMed] [Google Scholar]
- McGarry T.J. and Kirschner,M.W. (1998) Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell, 93, 1043–1053. [DOI] [PubMed] [Google Scholar]
- Morgan D. (1999) Regulation of the APC and the exit from mitosis. Nat. Cell Biol., 1, E47–E53. [DOI] [PubMed] [Google Scholar]
- Peters J.M. (2002) The anaphase-promoting complex. Proteolysis in mitosis and beyond. Mol. Cell, 9, 931–943. [DOI] [PubMed] [Google Scholar]
- Peters J.M., King,R.W., Hoog,C. and Kirschner,M.W. (1996) Identification of BIME as a subunit of the anaphase-promoting complex. Science, 274, 1199–1201. [DOI] [PubMed] [Google Scholar]
- Petersen B.O. et al. (2000) Cell cycle- and cell growth-regulated proteolysis of mammalian CDC6 is dependent on APC-CDH1. Genes Dev., 14, 2330–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfleger C.M. and Kirschner,M.W. (2000) The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev., 14, 655–665. [PMC free article] [PubMed] [Google Scholar]
- Pfleger C.M., Lee,E. and Kirschner,M.W. (2001) Substrate recognition by the Cdc20 and Cdh1 components of the anaphase-promoting complex. Genes Dev., 15, 2396–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudner A.D. and Murray,A.W. (2000) Phosphorylation by Cdc28 activates the Cdc20-dependent activity of the anaphase-promoting complex. J. Cell Biol., 149, 1377–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab M., Lutum,A. and Seufert,W. (1997) Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell, 90, 683–693. [DOI] [PubMed] [Google Scholar]
- Schwab M., Neutzner,M., Mocker,D. and Seufert,W. (2001) Yeast Hct1 recognizes the mitotic cyclin Clb2 and other substrates of the ubiquitin ligase APC. EMBO J., 20, 5165–5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayama M., Zachariae,W., Ciosk,R. and Nasmyth,K. (1998) The Polo-like kinase Cdc5p and the WD-repeat protein Cdc20p/fizzy are regulators and substrates of the anaphase promoting complex in Saccharomyces cerevisiae. EMBO J., 17, 1336–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayama M., Toth,A., Galova,M. and Nasmyth,K. (1999) APC(Cdc20) promotes exit from mitosis by destroying the anaphase inhibitor Pds1 and cyclin Clb5. Nature, 402, 203–207. [DOI] [PubMed] [Google Scholar]
- Shteinberg M., Protopopov,Y., Ganoth,D., Listovsky,T., Brandeis,M. and Hershko,A. (1999) Phosphorylation of the cyclosome is required for its stimulation by Fizzy/Cdc20. Biochem. Biophys. Res. Commun., 260, 193–198. [DOI] [PubMed] [Google Scholar]
- Sigrist S.J. and Lehner,C.F. (1997) Drosophila fizzy-related down-regulates mitotic cyclins and is required for cell proliferation arrest and entry into endocycles. Cell, 90, 671–681. [DOI] [PubMed] [Google Scholar]
- Sigrist S.J., Jacobs,H., Stratmann,R. and Lehner,C.F. (1995) Exit from mitosis is regulated by Drosophila fizzy and the sequential destruction of cyclins A, B and B3. EMBO J., 14, 4827–4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen C.S., Lukas,C., Kramer,E.R., Peters,J.M., Bartek,J. and Lukas,J. (2000) Nonperiodic activity of the human anaphase-promoting complex-Cdh1 ubiquitin ligase results in continuous DNA synthesis uncoupled from mitosis. Mol. Cell. Biol., 20, 7613–7623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakin V., Ganoth,D., Dahan,A., Heller,H., Hershko,J., Luca,F.C., Ruderman,J.V. and Hershko,A. (1995) The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol. Biol. Cell, 6, 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudo T., Ota,Y., Kotani,S., Nakao,M., Takami,Y., Takeda,S. and Saya,H. (2001) Activation of Cdh1-dependent APC is required for G1 cell cycle arrest and DNA damage-induced G2 checkpoint in vertebrate cells. EMBO J., 20, 6499–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin R., Prinz,S. and Amon,A. (1997) CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science, 278, 460–463. [DOI] [PubMed] [Google Scholar]
- Visintin R., Craig,K., Hwang,E.S., Prinz,S., Tyers,M. and Amon,A. (1998) The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol. Cell, 2, 709–718. [DOI] [PubMed] [Google Scholar]
- Weinstein J. (1997) Cell cycle-regulated expression, phosphorylation and degradation of p55Cdc. A mammalian homolog of cdc20/fizzy/slp1. J. Biol. Chem., 272, 28501–28511. [DOI] [PubMed] [Google Scholar]
- Wheatley S.P., Hinchcliffe,E.H., Glotzer,M., Hyman,A.A., Sluder,G. and Wang,Y. (1997) CDK1 inactivation regulates anaphase spindle dynamics and cytokinesis in vivo. J. Cell Biol., 138, 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka A., Hatakeyama,S., Kominami,K., Kitagawa,M., Matsumoto,M. and Nakayama,K. (2000) Cell cycle-dependent expression of mammalian E2-C regulated by the anaphase-promoting complex/cyclosome. Mol. Biol. Cell, 11, 2821–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeong F.M., Lim,H.H., Padmashree,C.G. and Surana,U. (2000) Exit from mitosis in budding yeast: biphasic inactivation of the Cdc28–Clb2 mitotic kinase and the role of Cdc20. Mol. Cell, 5, 501–511. [DOI] [PubMed] [Google Scholar]
- Yudkovsky Y., Shteinberg,M., Listovsky,T., Brandeis,M. and Hershko,A. (2000) Phosphorylation of Cdc20/fizzy negatively regulates the mammalian cyclosome/APC in the mitotic checkpoint. Biochem. Biophys. Res. Commun., 271, 299–304. [DOI] [PubMed] [Google Scholar]
- Zachariae W. and Nasmyth,K. (1999) Whose end is destruction: cell division and the anaphase-promoting complex. Genes Dev., 13, 2039–2058. [DOI] [PubMed] [Google Scholar]
- Zachariae W., Schwab,M., Nasmyth,K. and Seufert,W. (1998) Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science, 282, 1721–1724. [DOI] [PubMed] [Google Scholar]
- Zou H., McGarry,T.J., Bernal,T. and Kirschner,M.W. (1999) Identification of a vertebrate sister-chromatid separation inhibitor involved in transformation and tumorigenesis. Science, 285, 418–422. [DOI] [PubMed] [Google Scholar]
- Zur A. and Brandeis,M. (2001) Securin degradation is mediated by fzy and by fzr and is required for complete chromatid separation but not for cytokinesis. EMBO J., 20, 792–801. [DOI] [PMC free article] [PubMed] [Google Scholar]