

Abstract
Yeast mhr1-1 was isolated as a defective mutation in mitochondrial DNA (mtDNA) recombination. About half of mhr1-1 cells lose mtDNA during growth at a higher temperature. Here, we show that mhr1-1 exhibits a defect in the partitioning of nascent mtDNA into buds and is a base-substitution mutation in MHR1 encoding a mitochondrial matrix protein. We found that the Mhr1 protein (Mhr1p) has activity to pair single-stranded DNA and homologous double-stranded DNA to form heteroduplex joints in vitro, and that mhr1-1 causes the loss of this activity, indicating its role in homologous mtDNA recombination. While the majority of the mtDNA in the mother cells consists of head-to-tail concatemers, more than half of the mtDNA in the buds exists as genome-sized monomers. The mhr1-1 δcce1 double mutant cells do not maintain any mtDNA, indicating the strict dependence of mtDNA maintenance on recombination functions. These results suggest a mechanism for mtDNA inheritance similar to that operating in the replication and packaging of phage DNA.
Keywords: concatemer/heteroduplex joints/homologous recombination/rolling circle replication/Saccharomyces cerevisiae
Introduction
Evidence for recombination in the germ line is lacking in mammalian mitochondria because of strict maternal inheritance. Recently, some in vivo evidence for the presence of recombination was obtained from mammalian somatic cultured cells and human heart muscle (Tang et al., 2000; Kajander et al., 2001). Many organisms, including the yeast Saccharomyces cerevisiae, exhibit mitochondrial DNA (mtDNA) recombination during sexual reproduction. Although mtDNA recombination has been characterized in S.cerevisiae (Dujon, 1981), the mechanisms and the functions of mtDNA recombination are not well understood.
Homologous recombination is initiated by double-stranded breaks. The termini of the breaks are processed into single-stranded tails, which interact with homologous double-stranded DNA to form heteroduplex joints (Szostak et al., 1983). Heteroduplex joints that consist of an intermolecular double helix are a general intermediate of homologous DNA recombination (Holliday, 1964). Biochemical studies on the RecA protein (RecAp) of Escherichia coli, a prototype of the RecA/Rad51 family proteins, revealed that the heteroduplex is formed through two successive steps (McEntee et al., 1979; Shibata et al., 1979; Cox and Lehman, 1981; Kahn et al., 1981; Shinohara et al., 1993; Sung, 1994): homologous pairing and branch migration. Homologous pairing is the formation of a core heteroduplex joint from double-stranded DNA and homologous single-stranded DNA, and branch migration is the unidirectional, but slow, extension of the heteroduplex joint. It has been shown that RecAp and the Rad51 protein (Rad51p) must bind ATP to promote homologous pairing, and that ATP hydrolysis is necessary for branch migration (Shibata et al., 2001).
Some homologous recombination systems do not require RecA/Rad51 family proteins. In these systems, homologous pairing is probably carried out by ATP-independent proteins. Examples of proteins that promote the ATP-independent pairing of single-stranded DNA and homologous double-stranded DNA in vitro are the RecT protein (RecTp; Noirot and Kolodner, 1998) and the RecO protein (RecOp; Luisideluca, 1995) of E.coli, and the human Rad52 protein (HsRad52p; Kagawa et al., 2001). In vivo, RecTp promotes RecA-independent plasmid recombination in E.coli (Noirot and Kolodner, 1998), and Rad52p promotes Rad51p-independent mitotic recombination between a pair of small homologous sequences in S.cerevisiae nuclei (Bai and Symington, 1996; Ivanov et al., 1996). Homologous recombination of E.coli phage λ requires the functions of two genes, redα and redβ, but is independent of the host RecAp or its homologs (Enquist and Skalka, 1973). The β protein encoded by redβ is a functional homolog of RecTp (Muyrers et al., 2000).
Mammalian mtDNA exists in a circular form with a genome size (for a review, see Clayton and Smith, 1975), but the major species of S.cerevisiae mtDNA are linear head-to-tail multimers of genomic unit DNA with variable sizes (concatemers; Maleszka et al., 1991; Bendich, 1996). Concatemers can be formed through rolling circle DNA replication or homologous DNA recombination. Rolling circle DNA replication is initiated through recombination-dependent mechanisms in some DNA replication systems, such as bacteriophage λ at the late stage of infection (Enquist and Skalka, 1973), plasmids (Silberstein et al., 1990) and even the chromosome in SOS-induced E.coli cells (Asai et al., 1993). In the later stages of λ infection, the DNA replication switches from a theta (θ) mode to a rolling circle (σ) mode, and this switch requires the proteins encoded by the redα (λ exonuclease) and redβ (β protein) genes required for homologous DNA recombination. In phage T4 of E.coli, concatemers are formed through homologous DNA recombination, which depends on at least two T4-encoded proteins, UvsXp and endonuclease VII, a recombination junction (Holliday junction)-resolving endonuclease encoded by gene 49 (Mosig, 1998).
The S.cerevisiae mhr1-1 mutant is the first mutant defective in wild-type (ρ+) mtDNA recombination and in mtDNA repair (Ling et al., 1995, 2000). The mhr1-1 mutation was mapped to a single locus on a nuclear chromosome. This mutation shows extensive vegetative petite production during cultivation at a higher temperature or after UV irradiation of the cells. About half of such petite cells were found be ρ0 (Ling et al., 1995). This suggests that Mhr1p by itself or an Mhr1p-dependent recombination function plays a role in the maintenance of mtDNA. We have studied the in vivo and in vitro functions of Mhr1p to look for the connections between mtDNA recombination and mtDNA maintenance, and found that Mhr1p has activity to form a heteroduplex joint in vitro. This finding provides a clue towards understanding the basis of the apparently diverse phenotypes of mhr1-1.
Results
The mhr1-1 mutation causes a defect in the partitioning of nascent mtDNA into buds
To characterize the process of mtDNA loss at the restrictive temperature, we investigated the behavior of nascent (newly replicated) mtDNA. The nascent mtDNA of haploid cells was specifically labeled with 5-bromo-2′-deoxyuridine (BrdU) at 30°C by arresting the cells at G1 with mating factor α (α-factor; Nunnari et al., 1997). After the α-factor and BrdU were removed, the cells grew synchronously at 37°C. The labeled nascent mtDNA was bound by a BrdU-labeled DNA-specific antibody and was visualized by confocal scanning laser microscopy. The BrdU-labeled nascent mtDNA was unevenly distributed in the peripheral cytoplasm (Figure 1A). No signal was detected in ρ0 cells (data not shown). The pattern of the nascent mtDNA distribution in the mother cells was very similar between mhr1-1 and MHR1 cells (Figure 1A). In budding (dividing) MHR1 cells, the nascent mtDNA was detected in both the mother cell and the bud in most cases (74%, n = 57), but the majority of budding mhr1-1 cells (77%, n = 64) lacked nascent mtDNA in their buds (Figure 1A). The defect in the partitioning was also shown by a quantitative comparison of the bud mtDNA content between mhr1-1 and MHR1 (Figure 1B), and DAPI staining experiments using the same samples (Supplementary figure 1 available at The EMBO Journal Online).


Fig. 1. The role of Mhr1p in the transmission of nascent mtDNA. (A) Confocal images of BrdU-labeled nascent mtDNA within MHR1 and mhr1-1 cells at 37°C. Nascent (newly synthesized) mtDNA within MHR1 (left column) or mhr1-1 cells (right column), arrested at G1 phase by α-factor, was specifically labeled by BrdU at 30°C. After the removal of BrdU and α-factor, the cells were allowed to divide synchronously at 37°C for 1.5 h in YPD medium. Collected cells were fixed, and the BrdU-labeled mtDNA was detected by using fluorescein-conjugated monoclonal anti-BrdU F(ab′)2 fragments with a confocal scanning laser microscope. Panels in the top two rows are confocal images of BrdU-labeled nascent mtDNA (visible as dots); panels in the bottom two rows are cells under visible light. Bar, 10 µm. (B) Lack of nascent mtDNA in the buds of mhr1-1 cells. Cells were from the same samples used in (A) (top panel: micrographs under visible light). Buds and mother cells were separated, and each part was purified (panels in the second row: micrograph under visible light). Whole-cell lysates of MHR1 and mhr1-1 buds or mother cells (from top to bottom in the panels in the second row from the bottom: ∼0.5, 1 and 1.5 µg in proteins to each dot for bud, ∼0.8, 1.6 and 2.4 µg in protein to each dot for mother cell) were applied to an N+ membrane for immunoblotting using anti-BrdU serum to detect BrdU-labeled nascent mtDNA. The amounts of protein are represented by a mitochondrial protein, porin, in the panels in the bottom row.
The intracellular distribution of the mitochondrial reticulum in budding cells revealed by fluorescent images of mtHSP70 was not altered by mhr1-1, indicating that the partitioning of the mitochondrial reticulum to the growing buds is independent of the MHR1 function (data not shown).
Thus, we conclude that the partitioning of nascent mtDNA to buds is independent of the partitioning of the mitochondrial reticulum into buds and that Mhr1p plays a role in a specific mitochondrial system for the partitioning of mtDNA into buds. This defect can explain the production of ρ0 cells in the mhr1-1 culture at 37°C.
The mhr1-1 mutation is a single base substitution resulting in a single amino acid replacement within Mhr1p
As a step to identify the molecular functions of MHR1, we cloned a 0.94 kbp ClaI–MluI DNA fragment (in pRS416CM) containing a single open reading frame (ORF) that complements the deficiencies of mhr1-1 cells in mtDNA recombination and the maintenance of functional mtDNA at a higher temperature (FL67-1423δura3; Table I). The 678 bp ORF (DDBJ/EMBL/GenBank accession No. AB016430) encodes a protein of 26.9 kDa and is identical to YDR296w. All corresponding ORFs of the three clones independently obtained by PCR of DNA isolated from mhr1-1 cells share a single base pair substitution of A for G at nucleotide 515, causing the substitution of Asp for Gly172. Thus, the ORF is a nuclear gene, MHR1, and the mhr1-1 mutation is the single base substitution.
Table I. Yeast strains.
| Strain | Nuclear genotype | Mitochondrial genotype | Source or reference |
|---|---|---|---|
| IL166-187 | α his1 trp1 can1 MHR1 | ρ+ ω+ ens2 Chl321R | Stock culture of our laboratory |
| FL67 | α his1 trp1 can1 mhr1-1 | ρ+ ω+ ens2 Chl321R | Ling et al. (1995) |
| FL67ρ0 | α his1 trp1 can1 mhr1-1 | ρ0 | Ling et al. (1995) |
| FL67-1423 | α his1 trp1 can1 mhr1-1 | ρ+ ω+ Δens2 Oli2R | Ling et al. (1995) |
| FL67-1423Δura3 | α his1 trp1 Δura3 can1 mhr1-1 | ρ+ ω+ Δens2 Oli2R | ura3 derivative of FL67-1423 |
| FL67-1423Δura3 (pRS416CM) | α his1 trp1 Δura3 can1 mhr1-1 pRS416CM(MHR1, URA3) | ρ+ ω+ Δens2 Oli2R | Transformant of FL67-1423Δura3 |
| FL67-1423Δura3 (pRS416) | α his1 trp1 Δura3 can1 mhr1-1 pRS416(URA3) | ρ+ ω+ Δens2 Oli2R | Transformant of FL67-1423Δura3 |
| FL67-2c | a leu2 trp1 can1 mhr1-1 | ρ+ ω+ ens2 Chl321R | Ling et al. (1995) |
| IL166-6b | a leu2 ura3 trp1 can1 MHR1 | ρ+ ω+ ens2 Chl321R | Ling et al. (1995) |
| IL166-6b-mhr1::LEU2 | a leu2 ura3 trp1 can1 mhr1::LEU2 | ρ– | This study |
| YKN1423 | α leu2 ura3 met3 MHR1 | ρ+ ω+ Δens2 Oli2R | This study |
| FL67-2c-cce1::LEU2ρ0 | a leu2 trp1 can1 mhr1-cce1::LEU2 1 | ρ0 | This study |
| IL166-6b-cce1::LEU2 | a leu2 ura3 trp1 can1 cce1::LEU2MHR1 | ρ+ ω+ ens2 Chl321R | This study |
| YKN1423-cce1::LEU2 | α leu2 ura3 met3 cce1::LEU2 MHR1 | ρ+ ω+ Δens2 Oli2R | This study |
| YKN1423/YES2 | α leu2 ura3 met3 MHR1 YES2(URA3) | ρ+ ω+ Δens2 Oli2R | This study |
| YKN1423/YESMHR1 | α leu2 ura3 met3 MHR1 YESMHR1(MHR1,URA3) | ρ+ ω+ Δens2 Oli2R | This study |
| AFS98 | a ade2 leu2 his3 ura3 trp1 can1::GPD-HSVTK MHR1 | ρ+ | Nunnari et al. (1997) |
| AFS98ρ0 | a ade2 leu2 his3 ura3 trp1 can1 MHR1 | ρ0 | Derived from AFS98 by treatment with ethidium bromide |
| AFS98-4a | a ade2 leu2 his3 ura3 trp1 can1::GPD-HSVTK mhr1-1 | ρ+ | Segregant from FL67ρ0 × AFS98 |
| W303a | a ade2 leu2 his3 ura3 trp1 can1 MHR1 | ρ+ | Nunnari et al. (1997) |
| W303aρ0 | a ade2 leu2 his3 ura3 trp1 can1 MHR1 | ρ0 | Derived from W303a by treatment with ethidium bromide |
| W303a-187 | a ade2 leu2 his3 ura3 trp1 can1 MHR1 | ρ+ ω+ ens2 Chl321R | This study |
| W303α | α ade2 leu2 his3 ura3 trp1 can1 MHR1 | ρ+ | Nunnari et al. (1997) |
| W303α ρ0 | α ade2 leu2 his3 ura3 trp1 can1 MHR1 | ρ0 | Derived from W303α by treatment with ethidium bromide |
| W303α-1423 | α ade2 leu2 his3 ura3 trp1 can1 MHR1 | ρ+ ω+ Δens2 Oli2R | This study |
| W303α/YESMHR1 | α ade2 leu2 ura3 his3 trp1 can1 YESMHR1(MHR1,URA3) | ρ+ ω+ Δens2 Oli2R | This study |
| W303a/YES2 | a ade2 leu2 his3 ura3 trp1 can1 MHR1 YES2(URA3) | ρ+ ω+ ens2 Chl321R | This study |
| W303a/YESMHR1 | a ade2 leu2 his3 ura3 trp1 can1 YESMHR1(MHR1,URA3) | ρ+ ω+ ens2 Chl321R | This study |
| W303aGalMHR1 | a ade2 leu2 his3 ura3 can 1 GalMHR1 | ρ+ ω+ ens2 Chl321R | This study |
| W303aGalCCE1 | a ade2 leu2 his3 ura3 can1 MHR1 GalCCE1 | ρ+ ω+ ens2 Chl321R | This study |
| W303aGalABF2 | a ade2 leu2 his3 ura3 can1 MHR1 GalABF2 | ρ+ ω+ ens2 Chl321R | This study |
| W303αGalCCE1 | α ade2 leu2 ura3 trp1 can1 MHR1GalCCE1 | ρ+ ω+ Δens2 Oli2R | This study |
| W303αGalABF2 | α ade2 leu2 ura3 trp1 can1 MHR1GalABF2 | ρ+ ω+ Δens2 Oli2R | This study |
| W303a/YESMHR1-GFP | a ade2 leu2 his3 ura3 trp1 can1 YESMHR1-GFP(MHR1-GFP,URA3) | ρ+ ω+ ens2 Chl321R | This study |
| pTY24 | a ade2 leu2 ura3 MHR1 | ρ– pMK2 in mitochondria | Thorsness et al. (1990) |
MHR1 encodes a protein (Mhr1p) that is localized in the mitochondrial matrix
PSORT and PSORTII (http://psort.nibb.ac.jp:8800/index.html) analyses predicted that Mhr1p is localized in the mitochondrial matrix. Immunoblotting experiments using rabbit antiserum against recombinant Mhr1p expressed in E.coli showed that a 27 kDa protein is the only protein from the mitochondrial fraction that interacts with this antiserum (Figure 2A) and that the protein is absent from mhr1-null cells (Figure 2A). These results indicate that the 27 kDa protein is encoded by MHR1. We detected Mhr1p in both the inner membrane fraction and the matrix fraction, as is the case for mtHSP70, by examining fractionated mitochondrial components (Figure 2B).

Fig. 2. Mitochondrial localization of Mhr1p. (A) Presence of Mhr1p in mitochondria. Denatured proteins (3 µg) from the indicated fractions were separated by SDS–PAGE and immunoblotted with rabbit anti-Mhr1p serum as a probe. Lane 1, whole-cell extracts prepared from the mhr1::LEU2 mutant (IL166-6b-mhr1::LEU2); lanes 2 and 4, whole-cell extracts prepared from MHR1 cells (IL166-6b); lane 5, cytoplasmic fraction (without mitochondria) prepared from MHR1 cells; lane 6, mitochondrial fractions prepared from MHR1 cells. (B) Presence of Mhr1p in the mitochondrial matrix. Denatured proteins (4 µg each) prepared from the mitochondrial matrix space (MatSp), inner membrane (IM), intermembrane space (ItMSp) and outer membrane (OM) were subjected to SDS–PAGE and probed with an anti-Mhr1p serum and antisera raised against mitochondrial marker proteins: porin (outer membrane), cytochrome b2 (intermembrane space), the Tim23/Mas6 protein (inner membrane) and mtHSP70/Ens1p (mitochondrial matrix). Of the marker proteins, only HSP70 was detected in more than one fraction, the matrix and inner membrane fractions. (C) Comparison of sizes of Mhr1p expressed in E.coli and that obtained from mitochondria. Purified Mhr1p (without a His tag) expressed in E coli (0.1 µg in lane 1) and a protein fraction prepared from purified mitochondria (1 µg in lane 2) were denatured and subjected to SDS–PAGE, and an immunoblotting analysis was carried out using the anti-Mhr1p serum. (D) Mitochondrial localization of the Mhr1–GFP fusion protein. Cells (W303a) containing the pYESMHR1GFP plasmid were grown in a raffinose medium to an early log phase. Living cells were observed by fluorescence microscopy after staining with DAPI and MitoTracker. Top left, Mhr1–GFP (green); top right, mitochondrial reticulum as indicated by MitoTracker; bottom left, cellular DNA after DAPI staining; bottom right, visible light.
The polypeptides recognized by the anti-Mhr1p serum, in the Mhr1p preparation from E.coli and yeast mitochondria, had the same mobility on SDS–PAGE, and corresponded to a mol. wt of ∼26.7 kDa (Figure 2C). This suggests that no leader sequence with a significant size had been removed from the mitochondrial Mhr1p.
We constructed the gene for an Mhr1–green fluorescent protein (GFP) fusion protein that is fully functional, since its overproduction causes the same peculiar phenotype as that of MHR1 overexpression (F.Ling, unpublished observation). Microscopic observations indicated that the Mhr1–GFP fusion protein expressed in the cells is exclusively associated with mitochondria (Figure 2D).
Mhr1p promotes the pairing of single-stranded DNA and homologous double-stranded DNA in vitro
We purified the overproduced Mhr1p and mhr1-1p from E.coli cells to give a single band on SDS–polyacrylamide gels (Figure 3A). The amino acid sequence of the N-terminus matches the predicted N-terminal 17 amino acid sequence starting with Met of the MHR1 ORF. We did not detect endo- or exonuclease or topoisomerase activities in the Mhr1p preparations (data not shown).
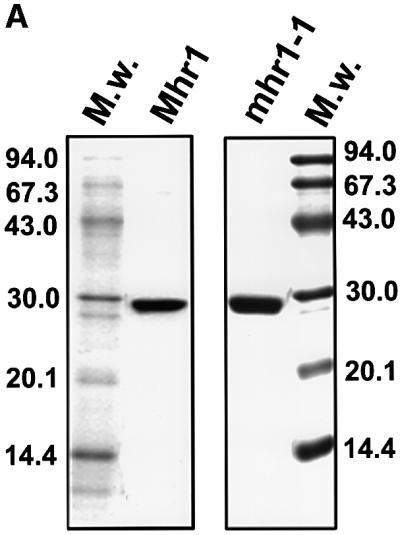


Fig. 3. Pairing of single-stranded DNA with homologous double-stranded DNA promoted by purified Mhr1p, and the absence of this activity in mhr1-1p. (A) SDS–PAGE of purified proteins. Mhr1p (∼5.1 µg) or mhr1-1p (∼5.8 µg) was subjected to 15% SDS–PAGE after purification. Proteins were stained with Coomassie Blue. (B) Mhr1p promotes homologous pairing. The indicated 32P-labeled single-stranded oligo-DNA (pG, SAT-1 containing a sequence from pGsat4; φX, φX-1 containing a sequence from φX174) was incubated with Mhr1p or mhr1-1p for 5 min, and then closed circular double-stranded pGsat4 or φX174 RF I DNA was added, followed by an incubation for 10 min in the presence of 2.0 µM Mhr1p. After the removal of the proteins, the products were analyzed by agarose gel electrophoresis. Homo and hetero indicate homologous and heterologous combinations of double-stranded DNA and single-stranded DNA, respectively. Lanes 1 and 4–8, Mhr1p; lanes 2 and 3, without Mhr1p or RecAp; lanes 13 and 14, mhr1-1p. Lane 1 includes 1.3 mM ATP. Lanes 9–12, E.coli RecA (5 µM) with 13 mM Mg2+ plus (lanes 9 and 10) or minus (lanes 11 and 12) ATP (incubation time, 5 min). Lane 8, after heteroduplex joints were formed from SAT-1 single-stranded oligo-DNA and pGsat4 double-stranded DNA, Mhr1p was inactivated at 75°C for 5 min, and the products were treated at 37°C for 30 min with the restriction endonuclease NheI, which cuts pGsat4 at a single site outside the sequence homologous to SAT-1. (C) Homologous pairing is mediated by Mhr1p, but not by mhr1-1p. The 32P- labeled single-stranded oligo-DNA was SAT-1. After homologous (pGsat4, lanes 1–6 and 9–14) or heterologous (φX174, lanes 7, 8, 15 and 16) double-stranded DNA was added, the reaction mixture was incubated for 10 min. Lanes 2–8, Mhr1p; lanes 10–16, mhr1-1p. Protein amounts: lanes 1 and 9, 0 µM; lanes 2 and 10, 0.49 µM; lanes 3, 7, 11 and 15, 0.98 µM; lanes 4 and 12, 2.0 µM; lanes 5 and 13, 3.9 µM; lanes 6, 8, 14 and 16, 7.8 µM.
By assaying for the DNA recombination activities of Mhr1p, we found that Mhr1p can promote the pairing of linear single-stranded DNA (a 50mer oligo-DNA) and homologous superhelical closed circular double-stranded DNA to form heteroduplex joints, as follows (Figure 3B and C): the homologous pairing signal is dependent on the homologous sequences of double-stranded DNA and single-stranded DNA, and is so in a sequence independent way, i.e. two unrelated DNAs (pGsat4 and E.coli phage φX174) can be substrates only if the double-stranded DNA and the single-stranded DNA share homology (Figure 3B). The protein-free products of the Mhr1p-promoted reaction migrate at the same velocity on gels as do heteroduplex joints formed by RecAp (McEntee et al., 1979; Shibata et al., 1979). In addition, they dissociate after the release of the topological constraint by cleavage of the double-stranded DNA at a site outside the region of homology (Figure 3B, lane 8; Radding et al., 1977). The extents of the reaction depend on the presence of Mhr1p (Figure 3C) and on the time of incubation with Mhr1p (data not shown), and are similar to those obtained with RecAp (Figure 3B). Homologous pairing reaches an optimum at a medium protein concentration, but seems to be inhibited at a high protein concentration (Figure 3C), as is the case with Rad52p (Kagawa et al., 2001). Unlike RecAp and Rad51p (McEntee et al., 1979; Shibata et al., 1979; Sung, 1994), but like RecTp (Noirot and Kolodner, 1998), RecOp (Luisideluca, 1995) and Rad52p (Kagawa et al., 2001), Mhr1p neither requires nor is stimulated by ATP or any other high energy co-factor to promote homologous pairing (Figure 3B).
The mhr1-1 mutant protein (mhr1-1p) is defective in homologous pairing (Figure 3B and C), i.e. at any con centration tested, or even when 16-fold more mhr1-1p protein was added, no signal for the pairing reaction was detected (Figure 3C). Since homologous pairing is an important step for homologous recombination, and since the single substitution of Asp for Gly172 results in the simultaneous loss of homologous pairing activity in vitro and Mhr1p functions in vivo, including mtDNA recombination, repair, partitioning and maintenance (this study; Ling et al., 1995, 2000), it is very likely that Mhr1p plays a role in the initiation of mtDNA homologous recombination, and that the homologous pairing activity of Mhr1p is also important for its other in vivo functions.
Agarose gel electrophoresis of DNA–protein complexes revealed that Mhr1p was able to bind to single-stranded DNA, as well as linear or closed circular double-stranded DNA (Supplementary figure 2), suggesting that Mhr1p has a structure-independent DNA-binding activity. The mhr1-1 mutation causes an extensive loss of DNA-binding activity, even at 30°C, indicating that the cause of the deficiency in homologous pairing by mhr1-1 is defective DNA binding (Supplementary figure 2).
A genome size monomer is a major species of mtDNA in buds
The majority of mtDNA in S.cerevisiae is found as concatemers with variable sizes consisting of multiple genome units (Maleszka et al., 1991; Bendich, 1996). As expected from the previous studies, most of the mtDNA remained in the well of the gel after pulsed-field gel electrophoresis (PFGE) (Figure 4A; Bendich, 1996). It has been reported that the yeast mtDNA that remains in the well after PFGE is in linear forms larger than the genome size (concatemers) and contains a circular form as a minor group (Bendich, 1996). Compared with the MHR1 cells, the mhr1-1 cells contain a slightly larger fraction of the monomeric form of mtDNA (Supplementary figure 3).
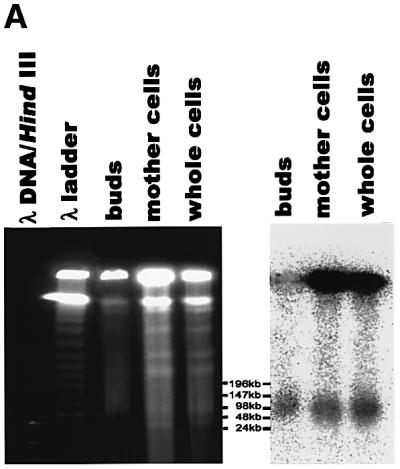

Fig. 4. Genomic-size mtDNA is a major species in the buds of dividing MHR1 cells. (A) PFGE profiles of mtDNA in the bud, the mother cell and the whole cell. MHR1 cells were grown synchronously and were harvested under the conditions described in Figure 1A, except that BrdU was omitted. The buds were separated from the mother cells, and each part was purified, as shown in Figure 1B. Then, the total cellular DNA was prepared and subjected to PFGE. Right panel: the detection of signals from mtDNA, using 32P-labeled mtDNA as a probe. Left panel: the detection of DNA under UV light after ethidium bromide staining. λ ladder DNA (concatemers) and λ DNA digested with HindIII were used as size markers. (B) Standard gel electrophoresis profiles of ρ– mtDNA in the bud, the mother cell and the whole cell. The MHR1 cells (PTY24) with pMK2 plasmid DNA in the mitochondria were grown in YPD medium at 30°C to 2 × 107 cells/ml. Cell growth was arrested at G1 by α-factor for 2 h at 30°C, and the cells then allowed to grow synchronously in YPD medium for 1.5 h at 30°C by the removal of α-factor. The buds were separated from the mother cells, and each part was purified. Whole DNA was isolated and subjected to electrophoresis through a 0.5% agarose gel at 1.0 V/cm in TAE buffer (40 mM Tris–acetate, 10 mM EDTA pH 8.0) at room temperature. Right panel: the detection of signals from mitochondrial pMK2 DNA, using 32P-labeled pUC119 DNA (which shares sequence with pMK2 DNA) as a probe. Left panel: the detection of DNA under UV light after ethidium bromide staining. The size marker was a 5 kb ladder obtained from Bio-Rad. The signals in the area indicated by an asterisk are those from Y-shaped molecules, double Y-shaped molecules and X-shaped molecules. Note that a precise determination of the amounts of mtDNA in the preparations is not possible, and the total amounts of DNA vary among the lanes. Thus, compare the relative amounts between the forms of DNA within each lane. The region indicated as C is concatemers.
A surprising result was obtained when we analyzed the forms of mtDNA in growing buds of MHR1 cells using the same technique. Budding MHR1 cells in culture were synchronized by an α-factor arrest-and-release protocol. The buds and mother cells were separated and purified (Ling et al., 1995), and their DNA was prepared for PFGE. As in case of the whole-cell mtDNA, the majority of the mtDNA isolated from the MHR1 mother cells remained in the well after PFGE (Figure 4A). However, about half of the mtDNA from the buds of MHR1 cells behaves as a monomeric form with a genome size (∼80 kbp). Thus, there is a difference in the size distribution of the mtDNA between mother cells and buds: while concatemers are the major species and monomers are the minor species in mother cells, the monomeric form is the major species in buds.
To confirm the above result with mitochondria in a very different physiological state and to obtain further infor mation about the structures of mtDNA in buds and mother cells, we employed standard gel electrophoresis to avoid a limitation of the analysis by PFGE (circular DNA is trapped in the well; Bendich, 1996) and examined budding cells that had artificial ρ– mitochondrial DNA {pTY24: MHR1 [ρ– maintaining coli plasmid pMK2 DNA (unit size 9.3 kbp) in mitochondria]} (Thorsness and Fox, 1990). We purified buds and mother cells from dividing cells in a culture synchronized by an α-factor arrest-and-release procedure and their DNA was extracted, electrophoresed and analyzed by Southern hybridization using a probe containing the sequence of pMK2 (pUC119). The majority of the mitochondrial plasmid DNA from mother cells and whole cells was detected as a smeared band and species trapped in wells, and these preparations contained nicked circular monomers as a minor species (Figure 4B). In contrast, a nicked circular monomer is the major species of mitochondrial plasmid DNA from the buds, and its signal intensity is of a comparable level to that of concatemers (Figure 4B). Very little DNA from the buds was trapped in the well, whereas more from the mother cells was trapped (Figure 4B). After treatment of the DNA preparations from mother cells with the restriction endonuclease SalI, which cleaves pMK2 DNA once, the smeared bands disappeared, and a linear monomer and significant amounts of fragments with various sizes smaller than the linear monomer appeared (Figure 4B), suggesting that the smeared bands consist of concatemers. Thus, in the ρ– budding cells, the fraction of monomer in the whole mtDNA is much larger in buds than the mother cells, as is the case for ρ cells.
The SalI treatment of DNA from mother cells eliminates the DNA species trapped in the wells and reveals the presence of a species that migrates more slowly than the nicked circular monomer (Figure 4B). This slower moving species was identified to include branched molecules such as X-shaped molecules (Holliday junction), Y-shaped molecules and double Y-shaped molecules, as judged by two-dimensional gel electrophoresis (data not shown). Thus, the mtDNA trapped in the well seems to be a DNA network connected by such junctions. This slower migrating species was not detected in the mtDNA from buds (Figure 4B).
mhr1-1 cce1-null double mutant cells do not maintain any mtDNA
Concatemers are formed through recombination- dependent pathways as obligatory intermediates for the duplication of various phages and plasmids. If concatemers are the obligatory intermediate of mtDNA partioning into buds, then mtDNA maintenance could depend strictly on mtDNA recombination functions. Since some mhr1-1 cells grown at 37°C or cells with disrupted mhr1 still maintain ρ– mtDNA, and we observed residual partitioning of nascent mtDNA in the mhr1-1 cells at 37°C (Figure 1; Ling et al., 1995), we searched for the presence of another recombination-dependent system to maintain mtDNA. CCE1/MGT1 encodes a recombination junction-resolving endonuclease (Cce1p). Although cce1 mutants exhibit a normal level of mtDNA recombination, Cce1p is likely to function in a mtDNA recombination system (Kleff et al., 1992; Ezekiel and Zassenhaus, 1993). In cells with functional ρ+ mtDNA, cce1-null cells show only a mild phenotype for the maintenance of mtDNA (except for hypersuppressiveness, see below; Zweifel and Fangman, 1991; F.Ling, unpublished observation).
We constructed a mhr1-1 cce1::LEU2 (cce1-null) double mutant by disrupting the CCE1 gene within an mhr1-1 mutant. All haploid mhr1-1 cce1-null double mutants were unable to grow in glycerol medium, suggesting that they were ρ– or ρ0. Then, we crossed the mhr1-1 cce1-null double mutant haploids with MHR1 CCE1 haploids to obtain ρ+ diploid cells, which were allowed to sporulate. All of the outgrowing cells on YPD medium at 30°C in the mhr1-1 cce1-null double mutant clones derived from each tetrad were devoid of mtDNA, as shown in Figure 5A. A quantitative Southern hybridization analysis indicated that the signals for mtDNA (normalized to that of the chromosomal gene, URA3) in the mhr1-1 cce1-null clones were much smaller than those in MHR1 cells, mhr1-1 cells or cce1-null cells, and were the background level exhibited by ρ0 cells (Figure 5B). These results indicate that mhr1 cce1-null double mutants are unable to maintain mtDNA (i.e. ρ0 phenotype) and, thus, that the maintenance of mtDNA strictly depends on either of the recombination-related genes, MHR1 or CCE1.


Fig. 5. mtDNA is absent in cells carrying the mhr1-1 cce1-null double mutation. (A) DAPI staining of whole cellular DNA. Cells from a colony on a YPD plate were cultivated in YPD medium at 30°C, and the cells then subjected to DAPI staining. The large bright spot in each cell is DNA in the nucleus, and the small spots are mtDNA. Top left, MHR1 CCE1; top right, mhr1-1 CCE1; bottom left, MHR1 cce1-null; bottom right, mhr1-1 cce1-null double mutant. (B) Quantitative Southern hybridization analysis of the variation in mtDNA per unit number of cells. The amount of mtDNA relative to the nuclear URA3 copy was calculated as described previously (Ling et al., 1995), i.e. signals obtained with a mtDNA probe were normalized by signals obtained with a nuclear URA3 gene probe in a Southern hybridization analysis.
Discussion
Molecular functions of Mhr1p
In this study, we found that Mhr1p has activity to pair linear single-stranded DNA with homologous closed circular double-stranded DNA, as shown in an assay of homologous pairing. This assay (‘D-loop assay’) was used for the first demonstration of homologous pairing by RecA in vitro (Shibata et al., 1979). The in vivo defects in mtDNA recombination and repair, and the in vitro defects in the homologous pairing caused by mhr1-1, suggest that Mhr1p-promoted homologous pairing is important for mtDNA recombination and repair in vivo, as is the case for RecAp.
Unlike with RecAp, the homologous pairing promoted by Mhr1p neither requires nor is stimulated by ATP, and in this sense it resembles the pairing of homologous DNA molecules promoted by RecTp (Noirot and Kolodner, 1998), RecOp (Luisideluca, 1995) and Rad52p (Kagawa et al., 2001). The homologous pairing by ATP-independent proteins may not be surprising, since ATP hydrolysis is not required for homologous pairing by RecAp. ATP-independent proteins might not promote branch migration efficiently by themselves, and instead the core heteroduplex joints formed by homologous pairing are stabilized by the topological stress in the superhelical double-stranded DNA substrates (Holloman et al., 1975). In vivo, double-stranded DNA may experience topological stress, or other proteins, such as DNA helicase, may facilitate branch migration and stabilize heteroduplex joints.
The dependence of mtDNA partitioning on homologous recombination functions
The major species of mtDNA in mother cells are concatemers, which can be either intermediates of the replication/partitioning of mtDNA or byproducts. We found that the fraction of the monomeric form with the single genome size (probably nicked circular DNA) among the mtDNA in buds is much larger than that in either ρ+ or ρ– mother cells. In addition to this finding, the biochemical functions of Mhr1p (ATP-independent pairing of homologous DNA molecules) and Cce1p (resolution of recombination junctions), and the dependence of mtDNA maintenance and partitioning on recombination functions, reveal the resemblance between mtDNA repli cation and partitioning, and E.coli phage DNA replication and maturation. The packaging and maturation of the DNA of E.coli phages such as λ, T4 and T7 require a concatemer as an intermediate, and the concatemer is cleaved into monomers upon packaging into phage particles. The concatemers are formed by rolling circle replication in phage λ and through a crossing-over type of homologous DNA recombination in phage T4. The concatemer formation in phage T4 requires a recombination junction (Holliday junction)-resolving endonuclease as an essential component (Mosig, 1998). In phage λ infection or induction, the phage DNA replicates initially in a theta (θ) mode, and then the replication is switched to a rolling circle (σ) mode. The initiation of rolling circle replication depends on the β protein. The β protein also initiates rolling circle replication of a plasmid DNA that originally replicates in a theta mode (Silberstein et al., 1990). Note that this pathway does not require a recombination junction-resolving enzyme. The β protein is a functional and structural homolog of RecTp and Rad52p (Passy et al., 1999; Muyrers et al., 2000). Mhr1p could functionally be a member of this protein family, because of its ATP-independent homologous pairing activity (this study) and its ability to form a ring structure by itself, as observed by electron microscopy (S.Ikawa, personal communication).
Thus, if we assume that mtDNA is replicated and partitioned by a system analogous to the phage systems, then the phenotype of the mhr1-1 and mhr1-1 cce1-null double mutants described here and previously (Ling et al., 1995, 2000), and the difference in the mtDNA size distribution in mother cells and buds, can be explained by the molecular functions of Mhr1p found in this study. A model based on this assumption is illustrated in Figure 6. In this model, we assume that the concatemer is the essential intermediate for mtDNA partitioning into buds, and transmission occurs with the cleavage of the concatemer into monomers by a mechanism similar to that operating in phage packaging (Figure 6). Like E.coli phages, we further assume that the concatemers are formed through either recombination-dependent rolling circle mtDNA replication or a crossing-over type of recombination (Figure 6). mtDNA may replicate by a θ mode at first, but soon, a recombination pathway is initiated by a double-stranded break (DNA is linearized) followed by processing into single-stranded tails. The Mhr1p-mediated interaction of either tail of the linearized DNA with a homologous circular double-stranded DNA molecule initiates rolling circle replication.

Fig. 6. Model of the functions of Mhr1p and its role in the transmission of nascent mtDNA. As is the case for phage DNA packaging, concatemers are essential intermediates for partitioning of mtDNA into buds, and are processed into genomic-size monomers (probably a circular form) during the partition. Concatemers are formed by either of two pathways: Mhr1p-initiating rolling circle replication, as in phage λ, or Cce1-dependent crossing over, as in phage T4.
The interaction of both ends of the linearized DNA with a homologous circular double-stranded DNA would initiate a crossing-over type of homologous recombination to generate a dimer or multimer, through the function of Cce1p, as the double-stranded break repair process (Szostak et al., 1983). The crossing over should be promoted by a different homologous pairing protein than Mhr1p. The presence of another mitochondrial homologous pairing protein is suggested by the phenotype of mhr1-1 cells, i.e. mhr1-1 causes a strong deficiency in the gene conversion type of mtDNA recombination, but this mutation exhibits some of the crossing-over type of mtDNA recombination (Ling et al., 1995).
The recombination-dependent rolling circle DNA replication can be initiated by the synthesis of a leading strand from a 3′-OH terminus invading into a double-stranded template DNA and might not require RNA synthesis at the replication origin. On the other hand, S.cerevisiae mtDNA has a conserved sequence, called rep/ori, which shares features with the replication origin of mammalian mtDNA (Van Dyck and Clayton, 1998), and evidence exists for RNA-primed DNA replication in yeast (Graves et al., 1998; MacAlpine et al., 2001), suggesting the contribution of θ mode replication to mtDNA maintenance. The fact that the ρ– type mtDNA is maintained even in the absence of the mitochondrial RNA polymerase indicates that yeast mitochondria also have an RNA priming-independent mtDNA replication system (Van Dyck and Clayton, 1998).
Possible roles of homologous recombination in mammalian mtDNA inheritance
Since the majority of mtDNA exists as a monomeric circle in mammals and in a concatemeric linear form in yeasts and plants, it is possible that the basic mechanisms of mitochondrial genetic inheritance are different between mammals and yeasts or plants (e.g. Lecrenier and Foury, 2000). However, yeast mitochondria and mammalian mitochondria still share various common features, as discussed.
Recent observations suggest a possible mammalian machinery for mtDNA recombination (Introduction). Proteins with an ATP-independent pairing activity on homologous DNA molecules were found in a wide variety of organisms, from prokaryotes (e.g. RecTp, RecOp), yeast (Rad52) and its mitochondria (Mhr1p), to human (HsRad52). It is very likely that mammalian mitochondria have a functional homolog of Mhr1p. Thus, our findings may be applicable beyond yeast and provide insights into the phenomenon of mitochondrial inheritance in other organisms as well.
Materials and methods
Yeast strains and media
Yeast strains are listed in Table I or as described previously (Ling et al., 1995). Raffinose medium consists of 2% raffinose, 0.7% yeast nitrogen base and supplementary amino acids.
Cloning of MHR1 and mhr1-1
A yeast genomic library constructed in the vector YCp50 (Rose et al., 1987) was used to transform FL67-1423δura3 (mhr1-1); Ura+- and temperature-resistant transformants were selected (Ito et al., 1983). The 0.74 kbp mhr1-1 allele was amplified by PCR using total cellular DNA from mhr1-1 cells as the template, with primers containing the sequences from positions –29 to 0 (5′-upstream region) and +683 to +709 (3′-downstream region) of MHR1.
Disruption of MHR1 and CCE1
A 2.8 kbp BglII LEU2 fragment from YEp13 was ligated into the unique NcoI site of the 0.94 kbp MHR1 gene. IL166-6b cells were transformed by the linear DNA fragment containing the disrupted mhr1 (mhr1::LEU2) and LEU2 gene. The Leu+ transformants were selected as IL166-6b-mhr1::LEU2 disruptants.
The DNA fragment starting from position 1 of the CCE1 gene to position 1062 in the 3′-downstream region was amplified by PCR. A 2.2 kbp HpaI LEU2 fragment was ligated to the unique SpeI site of the 1.06 kbp CCE1 gene. The linear DNA fragment containing the cce1::LEU2 (cce1-null) gene was introduced into the MHR1 cells (YKN1423) and the mhr1-1 mutant (FL67-2c), to construct a cce1-null single mutant and a mhr1-1 cce1-null double mutant, respectively. The presence of the disrupted gene in all disruptants was confirmed by a Southern hybridization analysis using an MHR1 or CCE1 probe.
To ensure that mhr1-1 cce1-null double mutant spores with mtDNA were obtained, the diploid formed by mating FL672c-cce1::LEU2 (a mhr1-1 cce1-null trp1 leu2 ρ0) and YKN1423 [α MHR1 CCE1 leu2 met3 ura3 (ρ+)] was allowed to form asci. Each spore was germinated and allowed to form a colony on a YPD plate at 30°C, and then the colonies on the plate were replica plated onto two YPGly plates, with one incubated at 30°C and the other at 37°C. The colonies containing cells carrying mhr1-1 CCE1 formed normal colonies on YPGly plates at 30°C, but not at 37°C. The colonies of cells carrying the cce1-null gene were selected based on the results of a Southern hybridization using a 32P-labeled CCE1 probe on DNA isolated from cells grown on the YPD plates.
Construction of the GFP-tagged Mhr1p
A DNA fragment containing the MHR1 ORF (without the terminator) was introduced into pYES2 at the KpnI–BamHI site. Then, a BamHI–XbaI DNA fragment harboring the GFP gene was excised from the vector pVT100U-GFP2 (provided by J.Nunnari, University of California, Davis) and was introduced into the BamHI–XbaI site (pYESMHR1GFP). Following incubation with 1 µg/ml 4′,6-diamidino-2-phenylindole (DAPI) and 0.5 µg/ml MitoTracker, the unfixed cells with the plasmid were observed with an Olympus IX70 microscope.
Construction of Mhr1p- and mhr1-1p-overproducing plasmids
The MHR1 and mhr1-1 ORFs were ligated into the expression vector pET14b (Novagen) to produce a His6-tagged fusion protein (pET14b MHR1 and pET14 mhr1-1, respectively) and were introduced into E.coli BL21 (DE3) pLyS host cells (Stratagene) by transformation. Proteins expressed in E.coli were detected by immunoblot analysis (Towbin et al., 1979).
Labeling of nascent mtDNA and microscopic observation of the behavior of labeled and unlabeled mtDNA
AFS98 (MHR1) or AFS98-4a (mhr1-1) cells were grown in YPGly medium to log phase at 23°C from 1 × 105 to ∼1 × 107 cells/ml. The nascent mtDNA was labeled with BrdU under cell cycle arrest at G1 by 15 nM α-factor, as described previously (Nunnari et al., 1997), except for the period of incubation with BrdU (30 min). The arrest of cells (75% of the population) at G1 was confirmed by light microscopy.
After the removal of BrdU and α-factor, the cells were incubated at 37°C for 1.5 h with shaking. The cells were collected, fixed with 3.7% formaldehyde and then treated as described previously (Nunnari et al., 1997) for the localization of BrdU-labeled DNA within cells by using fluorescein-conjugated monoclonal anti-BrdU F(ab′)2 fragments (Boehringer Mannheim). Images were collected with an Olympus BX60 microscope equipped with a real-time, spinning-disk confocal scanning unit (Yokogawa CSU10) and were processed on a Macintosh computer using the IPLab Spectrum software (Scanalytics).
For the detection of whole cellular DNA, the fixed cells were stained with 1 µg/ml DAPI and observed with an Olympus IX70 microscope.
The separation of buds and mother cells, and the quantitation of nascent mtDNA
To compare the amount of BrdU-labeled DNA in mhr1-1 and MHR1 buds and mother cells, and to examine the mtDNA structures in buds and mother cells, the buds were separated from the mother cells using a previously published procedure (Ling et al., 1995), except that the sonication of zymolyase-treated cells was omitted. Total DNA from mhr1-1 and MHR1 mother cells was prepared from the separated buds and mother cells, which had been adjusted previously to have a fixed amount of the mitochondrial protein porin.
For the quantitation, the same volume of DNA solution from each sample was applied to an N+ membrane (Amersham), and the DNA was fixed to the membrane by UV cross-linking. BrdU-labeled DNA was detected through immunoblotting using anti-BrdU serum (Sigma) as described previously (Towbin et al., 1979).
Total bud DNA and total mother cell DNA for PFGE were prepared as described previously (Jacobs et al., 1996).
Subfractionation of mitochondria
Mitochondria were prepared from IL166-6b cells by the fractionation of whole MHR1 cell extracts, according to a previously published procedure (Daum et al., 1982). Mitochondrial intermembrane space and matrix fractions were prepared (Daum et al., 1982). Submitochondrial fractions, including outer and inner membranes, were further separated on linear sucrose gradients (Pon et al., 1989). Immunoblotting was carried out as described previously (Towbin et al., 1979).
Purification of Mhr1p and mhr1-1p
Escherichia coli strains carrying pET14bMHR1 or pET14 mhr1-1 were cultured in LB medium containing ampicillin at 37°C. An initial culture of 5 × 106 cells/ml was grown to 1.5 × 108 cells/ml and then cultured at 16°C for another 4 h in the presence of 1 mM isopropyl-β-d(–)-thiogalactopyranoside (IPTG). Mhr1p and mhr1-1p were purified using the HisTrap™ system (Pharmacia Biotech). The His-tag sequence was removed by the use of a Cleavage Capture kit (Novagen). According to the procedure described above, ∼0.8–1.0 mg of each purified protein was obtained from a 300 ml culture.
Assay for homologous pairing (‘D-loop’ assay)
The standard reaction mixture (21 µl) for the assay consists of 15.6 mM Tris–HCl pH 7.5, 1.8 mM dithiothreitol, 1 mM MgCl2 and 88 µg of bovine serum albumin per milliliter. After the 32P-labeled single-stranded DNA (1.0 µM in nucleotides) was incubated with Mhr1p in the reaction mixture (20 µl) for 5 min at 37°C, 15 µM (in nucleotides) double-stranded DNA was added, and the incubation was continued at 37°C for the indicated time. For the reaction with RecAp, 1.3 mM ATP and MgCl2 (final 13 mM) were added together with double-stranded DNA. After the proteins were removed, the products were examined by agarose gel electrophoresis. After electrophoresis, the gel was dried and exposed to an imaging plate, which was analyzed with a Fuji BAS2000 image analyzer.
The preparation of superhelical double-stranded pGsat4 and φX174 RF I (replicative form I) DNA, with care to avoid denaturation, the preparation of single-stranded DNA (50mer oligonucleotides; SAT1 and φX-1), the 5′-end 32P-labeling of the single-stranded DNA and the details of the assay have been described previously (Kagawa et al., 2001). SAT1 and φX-1 contain sequences from pGsat4 and φX174, respectively.
PFGE
PFGE was performed using the CHEF-DR III (Bio-Rad) apparatus in 0.5% low melting agarose (FMC) in 0.5× TBE buffer (45 mM Tris, 45 mM boric acid, 1 mM EDTA pH 8.0) at 4°C, with a pulse time of 50 s for 20 h and 100 s for 2 h. Total cellular DNA for PFGE was prepared according to a previously published procedure (Jacobs et al., 1996).
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
The authors wish to express their sincere thanks to Kyohei Umebayashi, Chieko Saito and Akihiko Nakano (RIKEN) for confocal microscopic techniques, Hirohisa Masuda and Saeko Takada for their technical guidance in immunofluorescence microscopy, Ms Shelly Meeusen and Jodi Nunnari (University of California, Davis) for providing the vector DNA and their help in the strain construction, M.D.Rose (Princeton University) for providing us the yeast genomic library, Mr Masao Chijimatsu (RIKEN) for the N-terminal sequence analysis of the proteins, Hitoshi Kurumizaka (RIKEN) and Shukuko Ikawa (this laboratory) for advice regarding the D-loop assay, C.M.Radding (Yale Medical School) for comments, Shukuko Ikawa for her communication about the ring structures formed by Mhr1p prior to publication, and S.Nishikawa and Toshiya Endo (Nagoya University, Japan) for providing antisera. This study was supported partly by a grant for the ‘Bioarchitect Research program’ of RIKEN to F.L. and T.S., and by a grant for CREST from JST to T.S.
References
- Asai T., Sommer,S., Bailone,A. and Kogoma,T. (1993) Homologous recombination-dependent initiation of DNA replication from DNA damage-inducible origins in Escherichia coli. EMBO J., 12, 3287–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y. and Symington,L.S. (1996) A rad52 homolog is required for RAD51-independent mitotic recombination in Saccharomyces cerevisiae. Genes Dev., 10, 2025–2037. [DOI] [PubMed] [Google Scholar]
- Bendich A.J. (1996) Structural analysis of mitochondrial DNA molecules from fungi and plants using moving pictures and pulsed-field gel electrophoresis. J. Mol. Biol., 255, 564–588. [DOI] [PubMed] [Google Scholar]
- Clayton D.A. and Smith,C.A. (1975) Complex mitochondrial DNA. Int. Rev. Exp. Pathol., 14, 1–67. [PubMed] [Google Scholar]
- Cox M.M. and Lehman,I.R. (1981) recA protein of Escherichia coli promotes branch migration, a kinetically distinct phase of DNA strand exchange. Proc. Natl Acad. Sci. USA, 78, 3433–3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum G., Bohni,P.C. and Schatz,G. (1982) Import of proteins into mitochondria. Cytochrome b2 and cytochrome c peroxidase are located in the intermembrane space of yeast mitochondria. J. Biol. Chem., 257, 13028–13033. [PubMed] [Google Scholar]
- Dujon B. (1981) Mitochondrial genetics and function. In Strathern,J.N., Jones,E.W. and Broach,J.R. (eds), The Molecular Biology of the Yeast Saccharomyces: Life Cycle and Inheritance. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 505–635.
- Enquist L.W. and Skalka,A. (1973) Replication of bacteriophage λ DNA dependent on the function of host and viral genes. I. Interaction of red, gam and rec. J. Mol. Biol., 75, 185–212. [DOI] [PubMed] [Google Scholar]
- Ezekiel U.R. and Zassenhaus,H.P. (1993) Localization of a cruciform cutting endonuclease to yeast mitochondria. Mol. Gen. Genet., 240, 414–418. [DOI] [PubMed] [Google Scholar]
- Graves T., Dante,M., Eisenhour,L. and Christianson,T.W. (1998) Precise mapping and characterization of the RNA primers of DNA replication for a yeast hypersuppressive petite by in vitro capping with guanylyltransferase. Nucleic Acids Res., 26, 1309–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday R. (1964) A mechanism for gene conversion in fungi. Genetic Res. (Camb.), 5, 282–304. [DOI] [PubMed] [Google Scholar]
- Holloman W.K., Wiegand,R., Hoessli,C. and Radding,C.M. (1975) Uptake of homologous single-stranded fragments by superhelical DNA: a possible mechanism for initiation of genetic recombination. Proc. Natl Acad. Sci. USA, 72, 2394–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H., Fukuda,Y., Murata,K. and Kimura,A. (1983) Transformation of intact yeast cells treated with alkali cations. J. Bacteriol., 153, 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov E.L., Sugawara,N., Fishman-Lobell,J. and Haber,J.E. (1996) Genetic requirements for the single-strand annealing pathway of double-strand break repair in Saccharomyces cerevisiae. Genetics, 142, 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs M.A., Payne,S.R. and Bendich,A.J. (1996) Moving pictures and pulsed-field gel electrophoresis show only linear mitochondrial DNA molecules from yeasts with linear-mapping and circular-mapping mitochondrial genomes. Curr. Genet., 30, 3–11. [DOI] [PubMed] [Google Scholar]
- Kagawa W., Kurumizaka,H., Ikawa,S., Yokoyama,S. and Shibata,T. (2001) Homologous pairing promoted by the human Rad52 protein. J. Biol. Chem., 276, 35201–35208. [DOI] [PubMed] [Google Scholar]
- Kahn R., Cunningham,R.P., DasGupta,C. and Radding,C.M. (1981) Polarity of heteroduplex formation promoted by Escherichia coli recA protein. Proc. Natl Acad. Sci. USA, 78, 4786–4790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajander O.A., Karhunen,P.J., Holt,I.J. and Jacobs,H.T. (2001) Prominent mitochondrial DNA recombination intermediates in human heart muscle. EMBO rep., 2, 1007–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleff S., Kemper,B. and Sternberg,R. (1992) Identification and characterization of yeast mutants and the gene for a cruciform cutting endonuclease. EMBO J., 11, 699–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecrenier N. and Foury,F. (2000) New features of mitochondrial DNA replication system in yeast and man. Gene, 246, 37–48. [DOI] [PubMed] [Google Scholar]
- Ling F., Makishima,F., Morishima,N. and Shibata,T. (1995) A nuclear mutation defective in mitochondrial recombination in yeast. EMBO J., 14, 4090–4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling F., Morioka,H., Ohtsuka,E. and Shibata,T. (2000) A role for MHR1, a gene required for mitochondrial genetic recombination, in the repair of damage spontaneously introduced in yeast mtDNA. Nucleic Acids Res., 28, 4956–4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luisideluca C. (1995) Homologous pairing of single-stranded DNA and superhelical double-stranded DNA catalyzed by RecO protein from Escherichia coli. J. Bacteriol., 177, 566–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAlpine D.M., Kolesar,J., Okamoto,K., Butow,R.A. and Perlman,P.S. (2001) Replication and preferential inheritance of hypersuppressive petite mitochondrial DNA. EMBO J., 20, 1807–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maleszka R., Skelly,P.J. and Clark-Walker,G.D. (1991) Rolling circle replication of DNA in yeast mitochondria. EMBO J., 10, 3923–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEntee K., Weinstock,G.M. and Lehman,I.R. (1979) Initiation of general recombination catalyzed in vitro by the recA protein of Escherichia coli. Proc. Natl Acad. Sci. USA, 76, 2615–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosig G. (1998) Recombination and recombination-dependent DNA replication in bacteriophage T4. Annu. Rev. Genet., 32, 379–413. [DOI] [PubMed] [Google Scholar]
- Muyrers J.P., Zhang,Y., Buchholz,F. and Stewart,A.F. (2000) RecE/RecT and Redα/Redβ initiate double-stranded break repair by specifically interacting with their respective partners. Genes Dev., 14, 1971–1982. [PMC free article] [PubMed] [Google Scholar]
- Noirot P. and Kolodner,R.D. (1998) DNA strand invasion promoted by Escherichia coli RecT protein. J. Biol. Chem., 273, 12274–12280. [DOI] [PubMed] [Google Scholar]
- Nunnari J., Marshall,W.F., Straight,A., Murray,A., Sedat,J.W. and Walter,P. (1997) Mitochondrial transmission during mating in Saccharomyces cerevisiae is determined by mitochondrial fusion and fission and the intramitochondrial segregation of mitochondrial DNA. Mol. Biol. Cell, 8, 1233–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passy S.I., Yu,X., Li,Z., Radding,C.M. and Egelman,E.H. (1999) Rings and filaments of β protein from bacteriophage λ suggest a superfamily of recombination proteins. Proc. Natl Acad. Sci. USA, 96, 4279–4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pon L., Moll,T., Vestweber,D., Marshallsay,B. and Schatz,G. (1989) Protein import into mitochondria: ATP-dependent protein translocation activity in a submitochondrial fraction enriched in membrane contact sites and specific proteins. J. Cell Biol., 109, 2603–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radding C.M., Beattie,K.L., Holloman,W.K. and Wiegand,R.C. (1977) Uptake of homologous single-stranded fragments by superhelical DNA. IV. Branch migration. J. Mol. Biol., 116, 825–839. [DOI] [PubMed] [Google Scholar]
- Rose M.D., Novick,P., Thomas,J.H., Botstein,D. and Fink,G.R. (1987) A Saccharomyces cerevisiae genomic plasmid bank based on a centromere-containing shuttle vector. Gene, 60, 237–243. [DOI] [PubMed] [Google Scholar]
- Shibata T., DasGupta,C., Cunningham,R.P. and Radding,C.M. (1979) Purified Escherichia coli recA protein catalyzes homologous pairing of superhelical DNA and single-stranded fragments. Proc. Natl Acad. Sci. USA, 76, 1638–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata T., Nishinaka,T., Mikawa,T., Aihara,H., Kurumizaka,H., Yokoyama,S. and Ito,Y. (2001) Homologous genetic recombination as an intrinsic dynamic property of a DNA structure induced by RecA/Rad51-family proteins: a possible advantage of DNA over RNA as genomic material. Proc. Natl Acad. Sci. USA, 98, 8425–8432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara A., Ogawa,H., Matsuda,Y., Ushio,N., Ikeo,K. and Ogawa,T. (1993) Cloning of human, mouse and fission yeast recombination genes homologous to RAD51 and recA. Nat. Genet., 4, 239–243. [DOI] [PubMed] [Google Scholar]
- Silberstein Z., Maor,S., Berger,I. and Cohen,A. (1990) λ Red-mediated synthesis of plasmid linear multimers in Escherichia coli K12. Mol. Gen. Genet., 223, 496–507. [DOI] [PubMed] [Google Scholar]
- Sung P. (1994) Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science, 265, 1241–1243. [DOI] [PubMed] [Google Scholar]
- Szostak J.W., Orr-Weaver,T.L., Rothstein,R.J. and Stahl,F.W. (1983) The double-strand-break repair model for recombination. Cell, 33, 25–35. [DOI] [PubMed] [Google Scholar]
- Tang Y., Manfredi,G., Hirano,M. and Schon,E.A. (2000) Maintenance of human rearranged mitochondrial DNAs in long-term cultured transmitochondrial cell lines. Mol. Biol. Cell, 11, 2349–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsness P.E. and Fox,T.D. (1990) Escape of DNA from mitochondria to the nucleus in Saccharomyces cerevisiae. Nature, 346, 376–379. [DOI] [PubMed] [Google Scholar]
- Towbin H., Staehelin,T. and Gordon,J. (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl Acad. Sci. USA, 76, 4350–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyck E. and Clayton,D.A. (1998) Transcription-dependent DNA transactions in the mitochondrial genome of a yeast hypersuppressive petite mutant. Mol. Cell. Biol., 18, 2976–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifel S.G. and Fangman,W.L. (1991) A nuclear mutation reversing a biased transmission of yeast mitochondrial DNA. Genetics, 128, 241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]