

Abstract
The ATPase SecA mediates post-translational translocation of precursor proteins through the SecYEG channel of the bacterial inner membrane. We show that SecA, up to now considered to be a stable dimer, is actually in equilibrium with a small fraction of monomers. In the presence of membranes containing acidic phospholipids or in certain detergents, SecA completely dissociates into monomers. A synthetic signal peptide also affects dissociation into monomers. In addition, conversion into the monomeric state can be achieved by mutating a small number of residues in a dimeric and fully functional SecA fragment. This monomeric SecA fragment still maintains strong binding to SecYEG in the membrane as well as significant in vitro translocation activity. Together, the data suggest that the SecA dimer dissociates during protein translocation. Since SecA contains all characteristic motifs of a certain class of monomeric helicases, and since mutations in residues shared with the helicases abolish its translocation activity, SecA may function in a similar manner.
Keywords: FRET/helicases/monomer/SecA/translocation
Introduction
The post-translational translocation of many proteins across the bacterial inner membrane is mediated by the Sec machinery (Duong et al., 1997; Pohlschröder et al., 1997; Manting and Driessen, 2000). The key component in the membrane is the SecYEG complex, which forms a protein-conducting channel. Another membrane protein complex, the SecDFyajC complex, is associated with it, but is not essential for translocation (Pogliano and Beckwith, 1994; Duong and Wickner, 1997). The cytosolic SecA protein, an ATPase, provides the driving force for polypeptide translocation. SecA probably ‘pushes’ polypeptides through the SecYEG channel. The current evidence suggests that it cycles in an ATP-dependent manner in and out of the channel, and thereby moves successive segments of the polypeptide chain through it (Economou and Wickner, 1994). SecA has several binding partners. It has interaction sites for both the signal sequence and the mature region of the precursor polypeptide (Cunningham and Wickner, 1989). It binds to lipids, particularly acidic phospholipids, an interaction that precedes a stronger association with the SecYEG channel (Lill et al., 1990; Kuster et al., 1991). Finally, SecA binds to the chaperone SecB and to its own mRNA (Hartl et al., 1990; Dolan and Oliver, 1991).
SecA is a dimer in solution, in the absence of binding partners (Akita et al., 1991; Doyle et al., 2000), and some data suggest that it remains a dimer during translocation (Driessen, 1993). On the other hand, a striking sequence similarity between SecA and a certain class of helicases raises the possibility that it may function as a monomer. SecA contains all seven motifs (I, Ia, II–VI) of superfamily I and II helicases (Koonin and Gorbalenya, 1992). These are similar in their three-dimensional structure, and probably function as monomers (Korolev et al., 1997; Kim et al., 1998; Velankar et al., 1999; Soultanas and Wigley, 2000).
The oligomeric state of SecA during translocation is an important issue. As the membrane channel allows polypeptides to slide back and forth (Schiebel et al., 1991; Driessen, 1992; Matlack et al., 1999), SecA must not only bind and move the polypeptide chain, but it must also prevent back-sliding of the chain when it retracts to bind the next polypeptide segment. These considerations suggest that SecA may have two binding sites for the mature region of the polypeptide chain that bind and release the chain in an alternating manner. In the case of the related monomeric helicases, each molecule has two binding sites that bind the nucleic acid in an alternating way (Soultanas and Wigley, 2000).
Here we have analyzed the oligomeric state of SecA under different conditions. On its own, the SecA dimer is in dynamic equilibrium with the monomer, but the equilibrium is on the side of the dimer. In the presence of liposomes containing acidic phospholipids, or certain detergents, SecA fully dissociates into monomers. A syn thetic signal peptide also affects dissociation. A monomeric derivative of a SecA fragment retains strong binding to SecYEG and can promote significant translocation of a substrate polypeptide chain. Mutations in conserved residues corresponding to those found in helicases of superfamilies I and II abolish translocation and ATPase activity of SecA. Therefore, together with our data, this suggests that SecA dissociates into monomers during translocation and that it may employ a mechanism similar to that used by monomeric helicases to move polypeptide chains across the bacterial inner membrane.
Results
SecA dimers are in dynamic equilibrium
We first investigated whether the SecA dimer is in dynamic equilibrium with monomers in the absence of its physiological binding partners. To detect the dimeric state, we employed fluorescence resonance energy transfer (FRET). Spatial proximity of two molecules of SecA is indicated by the transfer of energy from a ‘donor’ fluorophore, attached to one molecule, to an ‘acceptor’ fluorophore, attached to another molecule.
One sample of purified SecA was modified with an iodoacetamido derivative of coumarin, attaching the ‘donor’ fluorophore to cysteine residues, while another sample was modified with an iodoacetamido derivative of fluorescein (‘acceptor’ fluorophore). Approximately 2.0 mol of coumarin and 1.9 mol of fluorescein were bound per mol of SecA. Most of the modification occurred at the extreme C-terminus of SecA, where three of the four cysteines are located (Cys885, Cys887 and Cys896; total length of SecA 901 residues). This was demonstrated by digestion of modified SecA with V8 protease, which removes a 7 kDa C-terminal peptide (Shinkai et al., 1991). We estimate that the C-terminal cysteine cluster carries >90% of the coumarin label, and at least 75% of the fluorescein label (data not shown). It should be noted that the modified SecA proteins have unreduced basal and translocation ATPase activities (data not shown).
To generate mixed heterodimers of SecA containing a donor fluorophore in one subunit and an acceptor fluorophore in the other, the coumarin- and fluorescein-labeled SecA samples were mixed at a 1:1 molar ratio, denatured in urea, and renatured by removal of urea (cf. Matsuyama et al., 1990). Renatured SecA typically had basal and translocation ATPase activities of ∼70% of the untreated protein (data not shown). The heterodimer was excited at a wavelength of 370 nm (absorption maximum of coumarin) and the emitted light was recorded from 420 to 580 nm (Figure 1A). The mixed heterodimers (dark gray trace) show two peaks, one at 466 nm corresponding to the emission maximum of coumarin, the other at 524 nm, corresponding to the emission maximum of fluorescein. This curve has to be compared with those obtained for dimers generated by mixing either coumarin- or fluorescein-labeled SecA with identical amounts of unlabeled SecA (black and light gray traces, respectively). The peak of the dark gray trace at 466 nm is ∼24% lower than for coumarin/unlabeled heterodimers (note that there is no emission of the fluorescein probe at this wavelength), indicating that energy was lost from the donor probe. With a 1:1 molar ratio, a yield of 50% heterodimers was expected, and the actual FRET efficiency (corrected for 100% heterodimers) was 48%. Together, these data show that the two subunits of the SecA dimer are in close spatial proximity.


Fig. 1. Steady-state FRET between coumarin- and fluorescein-labeled SecA molecules. Coumarin- and fluorescein-labeled SecA heterodimers (dark gray traces) are compared with coumarin–SecA/unlabeled SecA (black traces) and fluorescein–SecA/unlabeled SecA (light gray traces) heterodimers. The following additions were made: (A) none; (B) 36 µg of unlabelled SecA; (C) 52 µg of BSA; (D) 24 µg of monomeric N68-SecA; (E) 5 µl of liposomes containing E.coli phospholipids; (F) 5 µl of DOPE/DOPG liposomes; (G) 5 µl of DOPE/DOPC liposomes; (H) 1.5 mg/ml ddm.
Next we tested whether SecA dimers are in equilibrium with monomers. An excess of unmodified SecA was added to pre-formed heterodimers containing both fluorophores, and the emission spectrum was recorded (Figure 1B). The peak of the dark gray trace at 466 nm was now only slightly lower than that of the black trace (coumarin/unlabeled heterodimers), indicating that FRET was greatly reduced. In contrast, an excess of bovine serum albumin (BSA) or of N68-SecA, an N-terminal fragment of SecA that is monomeric (Karamanou et al., 1999), had no effect on the FRET signal (Figure 1C and D). Thus, unlabeled full-length SecA can replace labeled SecA in the heterodimer, implying dynamic equilibrium between dimers and monomers. In time-course experiments, we determined that the half-time of subunit exchange is ∼5 s (data not shown).
To provide independent evidence for a dimer–monomer equilibrium, we performed pull-down experiments with differently tagged SecA derivatives. FLAG- and His-tagged versions of SecA, both carrying the tag at the C-terminus, were each expressed in Escherichia coli and purified. Equal amounts of these samples were incubated together, and the His-tagged protein was recovered with Ni-NTA beads. The bound material was analyzed by immunoblotting with antibodies to the FLAG and His epitopes (Figure 2). About 30% of the FLAG-tagged SecA was associated with His-tagged SecA (corrected for the recovery of the His-tagged protein), indicating that mixed heterodimers were formed during the incubation. No SecA-FLAG was recovered if untagged SecA, instead of His-tagged SecA, was used (lane 2), or if high concentrations of imidazole were present during the binding reaction (lane 3). Mg2+ and Mg2+ + ATP had only small effects on the co-precipitation (lanes 4 and 5). Together, these results confirm that SecA dimers are in equilibrium with monomers. They are in agreement with a recent report by Woodbury et al. (2002), who used gel filtration and analytical ultracentrifugation to demonstrate a dimer– monomer equilibrium.
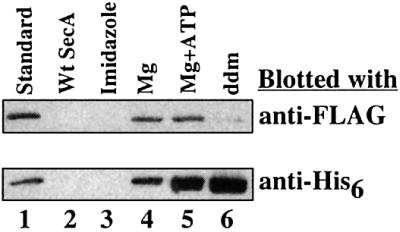
Fig. 2. Exchange of subunits between FLAG- and His6-tagged SecA. In the standard procedure (lane 1), the differently tagged SecA samples were mixed and the His-tagged proteins were recovered with Ni-NTA beads. The bound proteins were separated by SDS–PAGE and analyzed by immunoblotting with anti-FLAG and anti-His antibodies. In lane 2, SecA rather than SecA-His6 was added. For the sample in lane 3, the beads were washed with 140 mM imidazole before analysis of the bound material. In lanes 4 and 5, 2.5 mM MgCl2 and 2.5 mM MgCl2 plus 1.5 mM ATP, respectively, were added. For the sample in lane 6, 2 mg/ml ddm was present during the incubation.
Membranes containing acidic phospholipids cause dissociation of SecA dimers
Since SecA’s first encounter with the membrane probably involves an interaction with lipids, we tested whether liposomes affect the dimer–monomer equilibrium of SecA, again using steady-state FRET. Addition of vesicles made from E.coli phospholipids virtually abolished the FRET signal (Figure 1E); the peak of the dark gray trace of coumarin/fluorescein heterodimers had almost the same height as that of the black trace of coumarin/unlabeled heterodimers. The same result was obtained when vesicles containing SecYEG were employed (data not shown). Liposomes made of a mixture of DOPE and DOPG were equally effective in abolishing the FRET signal (Figure 1F). In contrast, liposomes made of DOPE and DOPC, which lack acidic phospholipids, had little effect on the FRET signal (compare Figure 1G with A). These results suggest that SecA dimers dissociate into monomers upon interaction with a lipid bilayer containing acidic phospholipids.
To provide independent evidence for the effect of lipids, we used a bifunctional cross-linker to probe the oligomeric state of SecA. It should be noted that to approach physiological conditions and to avoid unspecific interactions, we used a low concentration of SecA (1.5 µM) in these experiments. Treatment of SecA with EDAC, a carbodiimide that links neighboring carboxyl and amino groups, led to the appearance of several bands in the 200 kDa region (Figure 3A, lane 3). The same pattern was seen in the presence of ATP, ADP, or ATPγS (lanes 4–6). All bands are likely to correspond to cross-links within the SecA dimer, because in sucrose gradient centrifugation they all sedimented together at the same position as the unmodified SecA dimer (data not shown). The different bands probably originate from cross-links between different positions of the SecA molecules, which may result in different mobilities in SDS gels (Milligan and Koshland, 1988; Plath et al., 1998). When liposomes containing E.coli phospholipids were added before addition of EDAC, all cross-linked products disappeared (Figure 3A, lane 8), again suggesting that lipids affect dissociation of the SecA dimer into monomers. The same effect was seen when the liposomes also contained the SecYEG complex (lane 7), or when liposomes consisting of DOPE and DOPG were used (lane 9). In contrast, liposomes lacking acidic phospholipids did not abolish the appearance of dimer cross-links (lane 10). Other cross-linking reagents like DSS, glutaraldehyde and formaldehyde, which all cross-link amino groups but differ in the size of their spacer arms, gave the same results. Glutaraldehyde and formaldehyde penetrate through membranes, making it unlikely that the phospholipid bilayer prevented cross-linking by shielding reactive groups of SecA. Taken together, both the FRET and cross-linking experiments suggest that SecA dimers dissociate into monomers upon contact with lipid bilayers containing acidic phospholipids.

Fig. 3. Cross-linking of SecA with the bifunctional cross-linker EDAC. (A) The samples were separated by SDS–PAGE and stained with Coomassie Blue. The following additions were made: lane 4, 2 mM ATP; lane 5, 2 mM ADP; lane 6, 2 mM ATPγS; lane 7, 7.5 µl of E.coli liposomes with reconstituted SecYEG; lane 8, 7.5 µl of liposomes made from E.coli phospholipids; lane 9, 7.5 µl of DOPE/DOPG liposomes; lane 10, 7.5 µl of DOPE/DOPC liposomes; lane 11, 3 mg/ml ddm. The control in lane 2 received glycine and sample buffer before EDAC. (B) Cross-linking of [125I]SecA with EDAC. Samples were resolved by SDS–PAGE and analyzed by autoradiography. The following additions were made: lanes 1 and 2, 7.5 µg of SecA; lanes 5–8, 2 µl of E.coli liposomes with reconstituted SecYEG; lane 6, 2 mM ATP; lane 7, 2 mM ADP; lane 8, 2 mM AMP–PNP. Samples 1 and 3 are controls and received glycine and sample buffer before EDAC.
SecA bound to SecYEG is monomeric
We next investigated whether SecA remains a monomer at the next stage of translocation, when it is bound to the SecYEG complex. SecA has a significantly higher affinity for SecYEG than for lipids (Hartl et al., 1990), and at low concentrations it will therefore bind preferentially to SecYEG sites in the membrane. We used a concen tration of iodinated SecA at which at least 40% of the total protein is bound to SecYEG in proteoliposomes. Cross-linking with EDAC was employed to detect dimer formation. SecA at 5 nM was monomeric, even in the absence of any proteoliposomes (Figure 3B, lane 4). When 1.5 µM unlabeled SecA was added, dimer formation was seen (lane 2). These results are consistent with the dissociation constant of 250–500 nM for the SecA dimer determined by gel filtration (Woodbury et al., 2002) and cross-linking experiments (our unpublished data). When cross-linking of [125I]SecA was performed in the presence of proteoliposomes containing SecYEG complex, no dimer formation was seen (lane 5), indicating that SecA remains monomeric when bound to SecYEG. This was also the case in the presence of ATP, ADP or AMP–PNP (Figure 3B, lanes 6–8). We therefore conclude that SecA binds to SecYEG in its monomeric form.
Detergents also cause dissociation of SecA dimers
Next, we tested whether detergent micelles would also cause dissociation of SecA dimers. Addition of the non-ionic detergent dodecyl-maltoside (ddm) indeed prevented FLAG-tagged SecA from being pulled down along with His-tagged SecA (Figure 2, lane 6). The detergent also abolished the FRET signal seen with coumarin- and fluorescein-labeled heterodimers (Figure 1H), and it prevented the appearance of cross-linked bands when EDAC was added to SecA (Figure 3A, lane 11). All these data indicate that ddm dissociates the SecA dimer into monomers.
To provide further evidence, sucrose gradient centrifugation experiments were performed (Figure 4A and B). Without additions, SecA was found in fractions 17 and 18 at ∼200 kDa, as expected for a dimer (Figure 4A). However, in the presence of ddm, the peak of SecA was found in fractions 13 and 14, close to the position of BSA (Figure 4B). When SecA was first treated with the cross-linker EDAC and then subjected to sucrose gradient centrifugation in the presence of ddm, the cross-linked products migrated in fractions 17 and 18, and the non-cross-linked material in fractions 13 and 14 (data not shown). Together, these experiments demonstrate that a non-ionic detergent induces the conversion of SecA dimers into monomers. It should be noted that dissociation of SecA dimers, as detected by the loss of cross-links induced by EDAC, was also seen with other detergents, such as cymal 6,N,N-dimethyldodecylamine-N-oxide, and 1-myristoyl-2-hydroxy-sn-glycero-3-[phospho-rac-(1-glycerol)] (MLPG), whereas octyl-maltoside, octyl-glucoside, digitonin and polyexyethylene-10-lauryl-ether had no effect (data not shown).

Fig. 4. Analysis of SecA and of SecA derivatives by sucrose density centrifugation. The samples contained: (A) 9 µg of SecA; (B) 9 µg of SecA mixed with 0.15% ddm, run in a gradient containing 0.05% ddm; (C) 17 µg of N95-SecA; (D) 17 µg of N95-SecA-3Ala; (E) 17 µg of N95-SecA-6Ala. All samples also contained 50 µg of BSA. Samples (90 µl) from fractions 1–20 were analyzed by SDS–PAGE and stained with Coomassie Blue.
Interestingly, the dissociation of SecA dimers was not readily apparent in gel filtration experiments (data not shown). In fact, in the presence of MLPG, SecA had an even higher apparent size than in the absence of detergent. Since dissociation by MLPG was clearly evident in cross-linking experiments and sucrose gradient centrifugation, this suggests that in gel filtration experiments, dimer dissociation is obscured by the generation of a SecA–detergent micelle complex.
A synthetic signal peptide causes dimer dissociation
Another early interaction during translocation is the binding of SecA to a signal sequence of a precursor polypeptide chain. We therefore tested the effect of a synthetic signal peptide on the dimer–monomer equi librium. Increasing concentrations of the peptide KRR-LamBwt corresponding to the signal sequence of LamB (Triplett et al., 2001) were added to SecA, and cross-links were induced with EDAC (Figure 5, lanes 3–7). With increasing peptide concentration, the dimer cross-links became progressively weaker (in different experiments half-dissociation was reached at 20–30 µM). The control peptide KRR-LamBΔ78, which lacks a couple of hydrophobic residues and is inactive as a signal sequence in vivo (Triplett et al., 2001), did not abolish the dimer cross-links, even at the highest concentration tested (Figure 5, lanes 8–12). It should be noted that the peptides did not act as detergents when tested with proteoliposomes that contained imported proOmpA; the vesicles remained impermeable to added protease (data not shown). These data suggest that signal sequence binding occurs preferentially to the SecA monomer, thus shifting the dimer– monomer equilibrium towards the latter.

Fig. 5. A signal peptide dissociates SecA dimers. SecA in 60 µl buffer containing 4% DMSO was treated with EDAC in the presence of increasing concentrations of the synthetic wild-type signal peptide KRR-LamBWT or the mutant peptide KRR-LamBΔ78 (Alpha Diagnostic Int.). Samples were analyzed by SDS–PAGE and stained with Coomassie Blue. For a control, glycine and sample buffer were added before EDAC.
Small changes in SecA sequence affect the dimer–monomer equilibrium
Since wild-type SecA is mostly a dimer but is in equilibrium with monomers, we reasoned that small changes in the amino acid sequence of SecA might shift the equilibrium to the monomeric species. Previous work identified a segment from Thr675 to Val831 as important for dimerization (Hirano et al., 1996). This segment contains a sequence of 56 residues (Glu763–Leu818), conserved among SecA proteins from different species. Based on our FRET and cross-linking experiments, we mutated hydrophobic amino acids at the C-terminus of this region. As a starting point, we used a truncated version of SecA (N95-SecA), which is dimeric and as active as full-length SecA protein in both translocation ATPase and preprotein translocation assays (Matsuyama et al., 1990; Shinkai et al., 1991; data not shown). When three hydrophobic residues (Met814, Leu815, Leu818) were changed to alanines, the protein (N95-SecA-3Ala) was found to sediment slightly more slowly than N95-SecA (compare Figure 4C with D, peak fractions 17 and 16, respectively). When analyzed in a native gel, the mutant SecA fragment ran as two bands (Figure 6, lane 3), the upper corresponding to the dimer and the lower to the monomer, whereas the wild-type fragment only showed the upper band (lane 2). Thus, relatively minor changes in SecA sequence significantly shift the dissociation equilibrium towards the monomer.
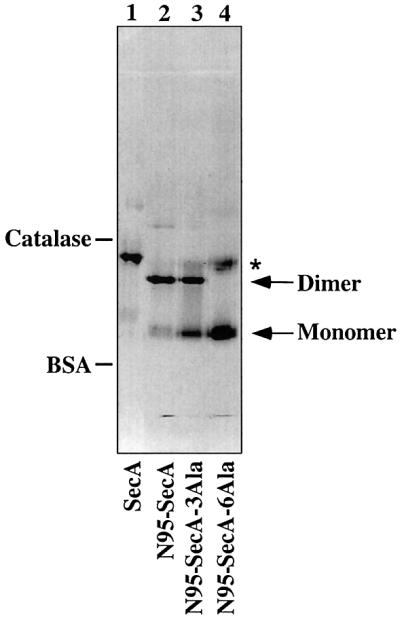
Fig. 6. Native gel electrophoresis of SecA and SecA derivatives. Four micrograms of SecA, 3 µg of N95-SecA, 3 µg of N95-SecA-3Ala or 3 µg of N95-SecA-6Ala were resolved in a native gel. The positions of the size standards catalase (230 kDa) and BSA (68 kDa) are indicated. An asterisk marks the position of misfolded N95-SecA-6Ala.
Complete conversion into the monomeric state was achieved when three additional hydrophobic residues were changed to alanines (Phe808, Met810, Phe811). In sucrose gradient centrifugation, the protein (N95-SecA-6Ala) sedimented at a position slightly faster than BSA (Figure 4E, peak at fraction 11), and in native gels, it ran at the position of the monomer (Figure 6, lane 4). No dimer cross-links were seen upon addition of EDAC (data not shown). A careful estimate of the amounts of N95- SecA-6Ala in each fraction using silver staining revealed that <0.3% of N95-SecA-6Ala exists as dimers (i.e. recovered in fractions 16–19; data not shown). We therefore conclude that mutation of six residues to alanines is sufficient to completely convert the SecA fragment into the monomeric state.
Monomeric SecA retains strong interaction with SecYEG and significant translocation activity
We tested whether the monomeric SecA derivative could still bind to SecYEG. [125I]SecA (50 nM) was incubated with proteoliposomes containing reconstituted SecYEG in the presence of increasing concentrations of N95-SecA-6Ala, SecA or N95-SecA, and the bound radioactivity was determined after centrifugation of the vesicles (Figure 7A). Half-maximal inhibition of [125I]SecA binding was achieved at 470 and 410 nM SecA and N95-SecA, respectively, while 760 nM were required for N95-SecA-6Ala. Thus, the monomeric SecA derivative retains a strong interaction with SecYEG.

Fig. 7. (A) A monomeric derivative of SecA retains strong interaction with SecYEG. Binding of [125I]SecA (50 nM) to proteoliposomes containing reconstituted SecYEG was performed in the presence of 0–4 µM SecA, 0–4.5 µM N95-SecA or 0–4.5 µM N95-SecA-6Ala. Non-specific binding, which was 16% of total binding, was subtracted. The specific binding of [125I]SecA in the absence of competitor was taken as 100%. (B) A monomeric derivative of SecA retains significant translocation activity. Translocation of [35S-Met]proOmpA into reconstituted proteoliposomes was tested with full-length SecA and a monomeric SecA fragment (N95-SecA-6Ala). The reconstituted proteoliposomes contained either wild-type SecYEG complex (rec.YEG) or a mutant complex containing the prlA4 mutation in SecY [rec.Y(prlA4)EG]. Where indicated, ATP was present during the reaction. All samples were treated with proteinase K. Where indicated, Triton X-100 was present during proteolysis. Lane 1 shows 15% of input [35S-Met]proOmpA. (C) Kinetics of translocation of [35S-Met]proOmpA into proteoliposomes containing reconstituted SecY(PrlA4)EG was followed in the presence of either SecA or N95-SecA-6Ala at 25°C. Protease-protected proOmpA were quantitated using a phosphoimager.
Next we tested the monomeric derivative for its ability to translocate proOmpA in vitro. Proteoliposomes containing purified SecYEG were mixed with purified SecA, ATP and in vitro synthesized [35S]methionine-labeled proOmpA. Translocation of proOmpA into the vesicles was tested after proteinase K treatment by SDS–PAGE and autoradiography. Compared with wild-type SecA (or N95-SecA; data not shown), monomeric N95-SecA-6Ala had 4.5% activity (Figure 7B, compare lane 7 with 2). Although low, the activity is real as no translocation was observed in the absence of ATP (lane 8). Interestingly, when the monomeric SecA derivative was tested with proteoliposomes containing a mutant SecYEG complex, harboring a signal sequence suppressor mutation in SecY (prlA4 mutation in SecY), translocation was ∼20% of that of wild-type SecA (Figure 7B, compare lanes 9 and 2). The mutant SecYEG complex was as active as the wild-type complex in the presence of wild-type SecA (lane 5 versus 2). No protease-protected proOmpA was seen in the absence of ATP or of N95-SecA-6Ala (Figure 7B, lanes 10 and 12), or when detergent was present during protease treatment (lane 11). The initial rate of proOmpA translocation seen with N95-SecA-6Ala was ∼7% of that with wild-type SecA, and translocation leveled off at a later time point (Figure 7C). Taken together, these data show that the monomeric SecA derivative shows significant translocation activity with SecY(PrlA4)EG complex.
Mutations in residues conserved in monomeric helicases abolish SecA activity
Since SecA can act as a monomer and shares significant sequence similarity with a class of monomeric helicases, we wished to obtain further evidence for a related mechanism of their function. We mutated Arg574 in motif VI, which is absolutely conserved in helicases of superfamilies I and II, as well as in all SecAs, and Thr504 in motif V, which is conserved in many SecAs. Mutation of the residue equivalent to Arg574 abolished all helicase activity of the UvrD helicase (Hall et al., 1998). Both the arginine and threonine residues are not found in other classes of ATPases and therefore provide a good test for the relatedness of SecA with this particular class of helicases. We found that SecA(R574A) and SecA(T504A) have the same basal ATPase activity as wild-type SecA but, in contrast to the latter, show little stimulation upon addition of proOmpA and proteoliposomes containing SecYEG (Figure 8). Both mutants also had very low in vitro translocation activity (data not shown). Even a conserved replacement of Arg574 for lysine abolished almost all stimulated ATPase and translocation activity (data not shown). These data show that two critical residues in monomeric helicases are also essential for the activity of SecA.

Fig. 8. Residues conserved between SecA and monomeric helicases are important for SecA translocation ATPase activity. Wild-type SecA, SecA(Thr504Ala) and SecA(Arg574Ala) were tested for ATPase activity in the presence of proteoliposomes containing reconstituted SecYEG (+rec.YEG) or in the absence of liposomes (–rec.YEG). All samples also contained proOmpA.
Discussion
Our results demonstrate that the SecA dimer, on its own, is in dynamic equilibrium with monomers. SecA has been thought to be a stable dimer (Akita et al., 1991; Driessen, 1993; Doyle et al., 2000), but our data show that subunit exchange can readily occur, indicating that a small pool of monomers exists. These data are in agreement with a recent study by Woodbury et al. (2002). We have estimated from immunoblots that the total concentration of SecA in E.coli cells is ∼5 µM (data not shown). Taking into account that ∼50% of total SecA is on the membrane (Cabelli et al., 1991) and that the dissociation constant is ∼0.3–0.5 µM (Woodbury et al., 2002), it seems that cytosolic SecA is mostly in the dimeric state in vivo. The interface between the SecA monomers and the dimer does not seem to be extensive because the equilibrium can be shifted towards the monomeric state by relatively small changes in the SecA primary sequence. An important region for dimerization appears to be a hydrophobic segment (Phe808–Leu818) that is conserved among SecAs from all species. A relatively moderate reduction of the hydrophobicity of only three residues is sufficient to significantly affect the dimer–monomer equilibrium, and six changes completely shift the equilibrium to the monomer. Although the mutated region appears to be important for monomer association, it is perhaps not the only one; at high concentrations, an N-terminal fragment of SecA lacking this region can form tetramers whose functional significance is unknown (Dempsey et al., 2002).
We provide strong evidence that the SecA dimer dissociates upon contact with phospholipid bilayers containing acidic phospholipids. One piece of evidence is that, in the presence of vesicles, SecA monomers can no longer be cross-linked with bifunctional reagents. Of course, on its own, this result could also be explained by a conformational change in SecA, rather than dissociation into monomers. However, all tested cross-linkers gave the same result and the FRET experiments support dissociation. The energy transfer between fluorophores in different subunits is essentially abolished upon contact with the lipid bilayer, suggesting that their distance must have increased to at least 70 Å (assuming that 50% FRET corresponds to a distance of ∼50 Å; see Rychlik et al., 1983; Snyder and Hammes, 1984). Our experiments with detergents support complete dissociation, rather than a drastic conformational change of the dimer. The effect of detergents is consistent with the idea that the subunits of the SecA dimer are held together by hydrophobic interactions. Interestingly, dissociation of SecA by vesicles requires a net negative charge in the head group of phospholipids, while some uncharged detergents have the same effect. One possibility is that a bilayer lacking acidic phospholipids may not permit SecA to cross the barrier posed by the head groups of phospholipids to reach the hydrophobic interior of the membrane; negative charges may induce disorder in the bilayer, which could facilitate contact of the hydrophobic segments of SecA with the hydrocarbon chains of phospholipids. Alternatively, interactions of SecA with both the hydrophobic hydrocarbon chains and negatively charged head groups may facilitate dissociation of the dimer.
Our observation that SecA dimer dissociation requires acidic phospholipids in the bilayer is consistent with previous results showing a similar requirement for overall protein translocation. In vivo, the export of PhoE and OmpA is severely impaired in pgsA3 cells, which have no phosphatidylglycerol phosphate synthase activity and, consequently, very low levels of phosphatidylglycerol and cardiolipin (de Vrije et al., 1988). Conversely, overexpression of pgsA, which increases the content of acidic phospholipids by 10%, suppresses the cold-sensitive growth phenotypes of SecAcsR11 and ΔSecG mutants (Suzuki et al., 1999). Acidic phospholipids are also required for in vitro translocation of precursor proteins and for SecA translocation ATPase activity (Lill et al., 1990; Kuster et al., 1991; Suzuki et al., 1999; van der Does et al., 2000). Finally, acidic phospholipids are required for membrane interaction of SecA and for a resulting conformational change in SecA (Lill et al., 1990; Shinkai et al., 1991; Ulbrandt et al., 1992). Together with our results, it seems that the dissociation of SecA dimers into monomers induced by acidic phospholipids is a critical step during protein translocation. It is possible that acidic phospholipids have additional roles in translocation.
We have found that a synthetic signal peptide also affects the dissociation of the SecA dimer, suggesting that the signal sequence of a substrate is recognized by the monomer with a higher affinity than the dimer. Our results are in agreement with a report in which a biotinylated synthetic signal peptide has a much lower affinity for dimeric full-length SecA than for a monomeric N-terminal fragment (Triplett et al., 2001). Since the binding site for signal sequences is expected to be hydrophobic, it seems possible that this site would be at least partially buried in the dimer.
The interaction of SecA with phospholipids and the signal sequence are early events during protein translocation. The resulting complex at the membrane appears to be a precursor to a tighter interaction with the SecYEG complex, forming the actual translocation complex. We have provided evidence that SecA remains monomeric even at this next stage of translocation; SecA does not re-dimerize when it binds to SecYEG, and a monomeric SecA derivative retains a strong interaction with SecYEG as well as significant translocation activity. The translocation activity reached 4.5% of that of the wild-type protein when tested with wild-type SecYEG, and 20% with SecYEG from the prlA4 mutant. It should be noted that the prlA4 mutant has an increased affinity for SecA and a relaxed conformation that facilitates translocation in other instances as well (van der Wolk et al., 1998; Duong and Wickner, 1999). According to sucrose gradient centrifugation experiments, <0.3% of the monomeric SecA derivative is present as dimers, making it unlikely that residual dimers account for the translocation activity. The monomeric SecA derivative is clearly less active than the wild-type protein, suggesting that the mutations introduced have additional effects, besides causing dissociation into monomers.
A previous report proposed that SecA functions as a dimer throughout translocation (Driessen, 1993). One piece of evidence came from steady-state FRET experiments similar to those described in the present paper. In the earlier experiments, the steady-state FRET signal in heterodimers carrying coumarin and fluorescein probes was unrealistically high (the ∼45% FRET signal observed with a 1:1 molar ratio corresponds to ∼90% efficiency in the heterodimer). In addition, the control for FRET be tween the donor and acceptor fluorophores in a heterodimer was the loss of the signal upon protease treatment. In our hands, proteolysis leads to drastic changes in coumarin fluorescence intensity, even when it is the only fluorophore present, presumably because the environment of the probe is changed (data not shown). Finally, the earlier experiments, in which no effect of membrane addition was seen on the FRET signal, were conducted differently from ours; the membranes were re-isolated by sedimentation together with bound SecA and resuspended prior to FRET measurements. Perhaps, a significant percentage of SecA dissociated from the lipids, re-dimerized and gave rise to the FRET signal. It should be noted that we have performed extensive controls for the FRET experiments and that our FRET data are confirmed by results obtained with other techniques. Driessen (1993) also reported that a heterodimer of SecA, in which one of the subunits is inactivated by treatment with azido-ATP and UV irradiation, has a lower activity than expected from the percentage of inactive molecules, suggesting that the two subunits function together. These data raise the possibility that oligomerization of SecA may be required at some stage during the translocation process.
SecA contains all seven motifs that are characteristic for helicases of superfamilies I and II. Our data provide further evidence for the relatedness of these proteins. Mutation of Arg574 in motif VI, which is conserved among all SecAs and helicases, essentially abolishes translocation ATPase and protein translocation activities. The X-ray structure of the PcrA helicase indicates that this arginine serves as an ‘arginine finger’; it is present in domain 2A and moves close to the γ phosphate of ATP bound to domain 1A. We would therefore infer that the equivalent residue in SecA has a similar role. Motif VI contains another arginine residue (Arg577), which is completely conserved among all SecAs and helicases of superfamilies I and II. Although the function of this residue is unknown, its mutation also abolishes the activities of SecA (Sianidis et al., 2001), further supporting the relatedness of the proteins. We have found that Thr504 in motif V is also important. This residue may be involved in transferring conformational changes caused by nucleotide binding or hydrolysis to the substrate-binding site(s). Consistent with this idea, mutations G508S and G510A in motif V greatly increase the basal ATPase activity of SecA (Schmidt et al., 2001; Sianidis et al., 2001). Interestingly, mutation of the residue in between (Arg509) abolishes translocation ATPase activity (Mitchell and Oliver, 1993), as is the case with Thr504. Although many ATPases contain Walker A and B motifs (contained in motifs I and II), motifs V and VI are characteristic for SecA and the related helicases. Most of the evidence indicates that helicases of superfamilies I and II function as monomers, and our results now indicate that SecA is similar in this respect as well. It should be noted that SecA has RNA helicase activity (Park et al., 1997), but whether this is of relevance for translocation is unclear. Schmidt et al. (2001) made several mutants that had reduced RNA helicase but maintained significant translocation activity; most of the mutations were in residues not conserved between SecA and helicases.
Monomeric helicases are thought to use an ‘inchworm’ mechanism to move along single-stranded nucleic acid (Soultanas and Wigley, 2000). The seven characteristic motifs that are conserved in SecA are located in two separate domains, each with a RecA-like fold, which can bind ATP at their interface. One domain binds the nucleic acid strand tightly and the other more weakly. Upon ATP binding, the domains move towards each other, and swap binding affinities. Following ATP hydrolysis, the two domains move apart, now with the previously weakly binding domain holding tightly onto the nucleic acid strand. Since SecA contains all seven helicase motifs, and since it is monomeric at least during some stages of its functional cycle, an ‘inchworm’ mechanism may also underlie protein translocation. SecA may undergo similar conformational changes as the monomeric helicases, although in this case, SecA would be bound to the SecYEG channel, and the polypeptide chain would move into and through the channel.
If SecA functions as a monomer, why does it start out as a dimer? Perhaps, the hydrophobic surfaces required for lipid and signal sequence interactions pose a problem for monomeric SecA in the cytosol. We have indeed found that SecA tends to aggregate at low concentrations (25 nM) when a larger fraction exists as monomers. In addition, the monomeric SecA fragment readily forms inclusion bodies in E.coli (see Materials and methods). Dimerization might also function as a safeguard against premature activation of the SecA ATPase activity. Finally, it is still possible that the dimeric state of SecA is required at some point during the translocation cycle. Further experiments are required to fully understand the transitions between the oligomeric states of SecA during translocation.
Materials and methods
Overexpression and purification of proteins
All mutants and derivatives of SecA are derived from plasmid pT7SecA2 expressing wild-type SecA (Rajapandi et al., 1991). N68-SecA has an amber stop codon replacing Leu610. SecA-His6 and SecA-FLAG have the last six residues of SecA replaced with a His6 tag or the sequence DYKDED, respectively. N95-SecA ends at Val831, followed by a His6 tag. N95-SecA-3Ala was derived from N95-SecA by altering Met814, Leu815 and Leu818 to alanines, and N95-SecA-6Ala was derived from N95-SecA-3Ala by mutating Phe808, Met810 and Phe811 to alanines as well. SecA and its derivatives were expressed in BL21(λDE3) grown on LB/Amp and induced with IPTG at mid-log phase. After 1.5–3.5 h, cells were pelleted, resuspended in 50 mM Tris–HCl pH 7.5, 50 mM KCl, 1 mM dithiothreitol (DTT) and 1 mM PMSF, and ruptured using a French Press. Membranes and insoluble material were removed by centrifugation (44 000 r.p.m. for 1 h at 4°C). Supernatants were applied onto a Reactive Blue 4 column (Sigma) connected to FPLC and further purified on a Q-Sepharose column. N68-SecA and N95-SecA-6Ala were recovered in inclusion bodies, which were solubilized with 50 mM Tris–HCl pH 7.5 and 6 M guanidinium–HCl. N68-SecA was renatured over a Sephacryl-100 column pre-equilibrated with renaturation buffer (50 mM Tris–HCl pH 7.5, 0.1 M KCl, 5 mM MgCl2, 2 mM 2-mercaptoethanol, 1 mM ATP, 10% glycerol). His-tagged SecA derivatives were purified using Ni-NTA–agarose followed by chromatography on a ResourceQ column. N95-SecA-6Ala was renatured while bound to Ni-NTA–agarose. Purified proteins were concentrated and stored in 25 mM Tris–HCl pH 7.5, 50 mM KCl, 1 mM DTT and 10% glycerol at –70°C. SecYE(His6)G and SecY(prlA4)E(His6)G were expressed in BL21(λDE3)c43 cells carrying pBAD22 and purified as described previously (Collinson et al., 2001). ProOmpA was expressed in W3100 cells harboring pTRC-Omp9 and purified from inclusion bodies as described previously (Crooke et al., 1988).
Liposomes
Stocks (20 mg/ml) of E.coli polar lipid extract [Avanti Polar Lipids (APL) #100600] were prepared in 50 mM K–HEPES pH 7.5, 10% glycerol, 10 mM DTT and 2% deoxyBigCHAP (Calbiochem) as described previously (Görlich and Rapoport, 1993). Liposomes without or with reconstituted SecYEG or SecY(prlA4)EG were prepared as described elsewhere (Collinson et al., 2001). Liposomes made of a 7:3 mixture of DOPE (APL #850725) and DOPC (APL #850375) or of a 7:3 mixture of DOPE and DOPG (APL #840475) were prepared as described previously (van der Does et al., 2000), and stored in 50 mM K–HEPES pH 7.5, 1 mM DTT and 10% glycerol at –70°C.
ATPase and translocation assays
ATPase assays were performed by measuring the hydrolysis of [γ-32P]ATP as described previously (Misselwitz et al., 1998; Collinson et al., 2001). ProOmpA mRNA was transcribed from pSP65-proOmpA using RiboMax (Promega), and [35S-Met]proOmpA was synthesized using rabbit reticulocyte lysate (Promega). Translocation reactions were carried out for 20 min at 33°C in 50 µl of 50 mM K–HEPES pH 7.5, 50 mM KCl, 4 mM MgCl2, 1 mM DTT and 0.5 mg/ml BSA. Complete reactions contained 30 pmol of SecA or N95-A-6Ala, 4 µl of proteoliposomes containing reconstituted SecYEG or SecY(prlA4)EG and [35S-Met]proOmpA plus 150 ng of proOmpA. Samples were treated with proteinase K, precipitated with TCA and resolved by SDS–PAGE. Gels were analyzed by autoradiography.
Iodination of SecA and liposome binding assays
SecA was labeled with Na[125I] using Iodogen (Pierce) as described by Economou and Wickner (1994). [125I]SecA essentially retained full translocation ATPase activity. Binding assays were performed according to Hartl et al. (1990); 100 µl of translocation buffer contained 50 nM [125I]SecA (80 000 c.p.m.), 2 µl of proteoliposomes with reconstituted SecYEG and increasing concentrations of the competing protein. Samples were incubated for 15 min at 22°C before separation of bound from unbound [125I]SecA (75 000 r.p.m. 15 min at 4°C). Supernatants were withdrawn and pellets were dissolved in 150 µl of 0.1% SDS. All samples were then counted in a gamma counter.
Labeling of SecA with fluorescein or coumarin
SecA (10 mg/ml) in 250 µl of 25 mM Tris–HCl pH 7.5, 50 mM KCl, 10% glycerol, 5 mM EDTA and 11 mM DTT was incubated for 30 min at 37°C. The buffer was changed to 50 mM K–HEPES pH 7.5 and 5 mM EDTA by passage though a desalting column, and SecA was then incubated at 22°C in the dark with 120 nmol of 5-IAF (Molecular Probes; I-3) or with 250 nmol of DACIA (Molecular Probes; D-10252) for 11 or 18 h, respectively. Supernatants were dialyzed against 500 ml of 50 mM K–HEPES pH 7.5 with one change of buffer and then passed through a desalting column pre-equilibrated with 50 mM K–HEPES pH 7.5 and 10% glycerol. The protein concentration of modified SecA was determined using the Bio-Rad protein assay and BSA as a standard. The absorbancies of 5-IAF-modified SecA (at 498 nm) and DACIA-modified SecA (at 393 nm) were determined in the presence of 6 M guanidinium. The values obtained, together with the extinction coefficients of 5-IAF (ε = 75 500 cm–1 M–1) and DACIA (ε = 24 100 cm–1 M–1), were used to calculated the level of modification.
Steady-state FRET
SecA–coumarin was mixed with either SecA–fluorescein or unlabeled SecA (each 300 µg plus 300 µg). Similarly, unlabeled SecA was mixed with either SecA–fluorescein or unlabeled SecA. All four mixtures were denatured with 8 M urea in 50 mM K–HEPES pH 7.5 and renatured by passage through a desalting column pre-equilibrated with renaturation buffer. Steady-state FRET experiments were conducted at 25°C on an SLM AMINCO Series 2 spectrofluorimeter (AMINCO). Samples contained 9 µg of total SecA protein in 50 mM K–HEPES pH 7.5, 50 mM KCl, 1 mM DTT, 2 mM Na-EDTA and 2 mM Na-EGTA. Additional components were added as indicated. In a single scan, a sample was excited at 370 nm (slit width 4 nm), and emitted light was scanned three times from 420 to 580 nm (slit width 4 nm, scan rate 0.75 nm/s). The readout was averaged automatically. Each FRET experiment consisted of four separate scans. In every experiment, the background scan (‘unmodified SecA/unmodified SecA’) was subtracted. The scans were subsequently normalized taking the peak fluorescence of ‘SecA–coumarin/unmodified SecA’ as a reference.
Exchange of subunits between SecA-FLAG and SecA-His6
SecA-FLAG and SecA-His6 (2 µg each) were mixed in 80 µl of buffer (50 mM K–HEPES pH 7.5, 50 mM KCl, 1 mM 2-mercaptoethanol). Mixtures contained additional components as indicated. After 20 min incubation at 22°C, 20 µl of 80 mM imidazole in buffer were added followed by 10 µl of BSA-pre-coated Ni-NTA–agarose beads (Qiagen). After incubation for 40 min at 22°C, the beads were washed with 150 µl of 25 mM imidazole in buffer for 10 min at 4°C. Protein bound to the beads was eluted with 80 µl of 2× sample buffer for 15 min at 40°C and resolved on a 7.5% SDS gel. Proteins were blotted onto nitrocellulose and incubated with either anti-His6 (Santa Cruz Biotechnology; cs-803) or anti-FLAG M2 (Sigma; F-3165) antibodies. Anti-rabbit IgG or anti-mouse IgG (both coupled to peroxidase) were used as secondary antibodies.
Cross-linking
SecA (7.5 µg) or [125I]SecA (26 ng) in 50 µl of 50 mM K–HEPES pH 7.5, 50 mM KCl, 2.5 mM MgCl2 and 1 mM DTT was treated with 20 mM EDAC (Sigma; E-1769) at 22°C. Additional components were present as indicated. Reactions were terminated after 15 min with 16 µl of 2 M glycine plus 24 µl of 5× sample buffer. Samples were separated on 5% SDS gels.
Sucrose gradient centrifugation and native gels
Protein samples in 400 µl of buffer (50 mM K–HEPES pH 7.5, 50 mM KCl, 2 mM MgCl2, 1 mM DTT) were layered on top of 12 ml 5–12% sucrose gradients in the same buffer. Gradients were centrifuged at 40 000 r.p.m. (SW40) for 16 h at 22°C. Twenty-four fractions of 520 µl were collected from the top. Colorless native gels were run as described previously (Schagger et al., 1994).
Acknowledgments
Acknowledgements
We thank Don Oliver for the plasmid pT7SecA2 and the strain BL21.19 (λDE3) as well as for advice regarding purification of SecA. We thank Bill Wickner for strain W3100 expressing proOmpA, Ian Collinson for purified SecYEG, Tom Ellenberger for suggesting mutations in the helicase motifs of SecA, and Alfred Goldberg for allowing us to use his Bio-Rad FPLC. We thank Andrew Osborne and Stuart Lecker for critical reading of the manuscript. The work was supported by a grant from the NIH to T.A.R., who is a Howard Hughes Medical Institute Investigator.
References
- Akita M., Shinkai,A., Matsuyama,S.I. and Mizushima,S. (1991) SecA, an essential component of the secretory machinery of Escherichia coli, exists as homodimer. Biochem. Biophys. Res. Commun., 174, 211–216. [DOI] [PubMed] [Google Scholar]
- Cabelli R.J., Dolan,K.M., Qian,L. and Oliver,D.B. (1991) Characterization of membrane-associated and soluble states of SecA protein from wild-type and SecA51(TS) mutant strains of Escherichia coli. J. Biol. Chem., 266, 24420–24427. [PubMed] [Google Scholar]
- Collinson I., Breyton,C., Duong,F., Tziatzios,C., Schubert,D., Or,E., Rapoport,T.A. and Kuhlbrandt,W. (2001) Projection structure and oligomeric properties of a bacterial core protein translocase. EMBO J., 20, 2462–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooke E., Gurthrie,B., Lecker,S., Lill,R. and Wickner,W. (1988) ProOmpA is stabilized for membrane translocation by either purified E.coli trigger factor or canine signal recognition particle. Cell, 54, 1003–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham K. and Wickner,W. (1989) Specifc recognition of the leader region of precursor proteins is required for the activation of translocation ATPase of Escherichia coli. Proc. Natl Acad. Sci. USA, 86, 8630–8634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vrije T., de Swart,R.L., Dowhan,W., Tommassen,J. and de Kruijff,B. (1988) Phosphatidylglycerol is involved in protein translocation across Escherichia coli inner membrane. Nature, 334, 173–175. [DOI] [PubMed] [Google Scholar]
- Dempsey B.R., Economou,A., Dunn,S.D. and Shilton,B.H. (2002) The ATPase domain of SecA can form a tetramer in solution. J. Mol. Biol., 315, 831–843. [DOI] [PubMed] [Google Scholar]
- Dolan K.M. and Oliver,D.M. (1991) Characterization of Escherichia coli SecA protein binding to a site on its mRNA involved in autoregulation. J. Biol. Chem., 266, 23329–23333. [PubMed] [Google Scholar]
- Doyle S.M., Braswell,E.H. and Teschke,C.M. (2000) SecA folds via a dimeric intermediate. Biochemistry, 39, 11667–11676. [DOI] [PubMed] [Google Scholar]
- Driessen A.J.M. (1992) Precursor protein translocation by the Escherichia coli translocase is directed by the protonmotive force. EMBO J., 11, 847–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driessen A.J.M. (1993) SecA, the peripheral subunit of the Escherichia coli precursor protein translocase, is functional as a dimer. Biochemistry, 32, 13190–13197. [DOI] [PubMed] [Google Scholar]
- Duong F. and Wickner,W. (1997) Distinct catalyic roles of the SecYE, SecG and SecDFyajC subunits of preprotein translocase holoenzyme. EMBO J., 16, 2756–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong F. and Wickner,W. (1999) The PrlA and PrlG phenotypes are caused by a loosened association among the translocase SecYEG subunits. EMBO J., 18, 3263–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong F., Eichler,J., Price,A., Leonard,M.R. and Wickner,W. (1997) Biogenesis of the Gram-negative bacterial envelope. Cell, 91, 567–573. [DOI] [PubMed] [Google Scholar]
- Economou A. and Wickner,W. (1994) SecA promotes preprotein translocation by undergoing ATP-driven cycles of membrane insertion and deinsertion. Cell, 78, 835–843. [DOI] [PubMed] [Google Scholar]
- Görlich D. and Rapoport,T.A. (1993) Protein translocation into proteoliposomes reconstituted from purified components of the endoplasmic reticulum membrane. Cell, 75, 615–630. [DOI] [PubMed] [Google Scholar]
- Hall M.C., Ozsoy,Z.A. and Matson,S.W. (1998) Site-directed mutations in motif VI of Escherichia coli DNA helicase II result in multiple biochemical defects: evidence for the involvement of motif VI in the coupling of ATPase and DNA binding activities via conformational changes. J. Mol. Biol., 277, 257–271. [DOI] [PubMed] [Google Scholar]
- Hartl F.U., Lecker,S., Schiebel,E., Hendrick,J.P. and Wickner,W. (1990) The binding cascade of SecB to SecA to SecY/E mediates preprotein targeting to the E.coli plasma membrane. Cell, 63, 269–279. [DOI] [PubMed] [Google Scholar]
- Hirano M., Matsuyama,S.I. and Tokuda,H. (1996) The carboxyl-terminal region is essential for SecA dimerization. Biochem. Biophys. Res. Commun., 229, 90–95. [DOI] [PubMed] [Google Scholar]
- Karamanou S., Vrontou,E., Sianidis,G., Baud,C., Roos,T., Kuhn,A., Politou,A.S. and Economou,A. (1999) A molecular switch in SecA protein couples ATP hydrolysis to protein translocation. Mol. Microbiol., 34, 1133–1145. [DOI] [PubMed] [Google Scholar]
- Kim J.L., Morgenstern,K.A., Griffith,J.P., Dwyer,M.D., Thomson,J.A., Murcko,M.A., Lin,C. and Caron,P.R. (1998) Hepatitis C virus NS3 helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure, 6, 89–100. [DOI] [PubMed] [Google Scholar]
- Koonin E.V. and Gorbalenya,A.E. (1992) Autogenous translation regulation by Escherichia coli ATPase SecA may be mediated by an intrinsic RNA helicase activity of this protein. FEBS Lett., 298, 6–8. [DOI] [PubMed] [Google Scholar]
- Korolev S., Hsieh,J., Gauss,G.H., Lohman,T.N. and Waksman,G. (1997) Major domain swiveling revealed by the crystal structures of complexes of E.coli Rep helicase bound to single-stranded DNA and ADP. Cell, 90, 635–647. [DOI] [PubMed] [Google Scholar]
- Kuster R., Dowhan,W. and de Kruijff,B. (1991) Negatively charged phospholipids restore prePhoE translocation across phosphatidylglycerol-depleted Escherichia coli inner membranes. J. Biol. Chem., 266, 8659–8662. [PubMed] [Google Scholar]
- Lill R., Dowhan,W. and Wickner,W. (1990) The ATPase activity of SecA is regulated by acidic phospholipids, SecY and the leader and mature domains of precursor proteins. Cell, 60, 271–280. [DOI] [PubMed] [Google Scholar]
- Manting E.H. and Driessen,A.J. (2000) Escherichia coli translocase: the unravelling of a molecular machine. Mol. Microbiol., 37, 226–238. [DOI] [PubMed] [Google Scholar]
- Matlack K.E.S., Misselwitz,B., Plath,K. and Rapoport,T.A. (1999) BiP acts as a molecular ratchet in posttranslational protein transport across the ER membrane. Cell, 97, 553–564. [DOI] [PubMed] [Google Scholar]
- Matsuyama S.I., Kimura,E. and Mizushima,S. (1990) Complementation of two overlapping fragments of SecA, a protein translocation ATPase of Escherichia coli, allows ATP binding to its amino-terminal region. J. Biol. Chem., 265, 8760–8765. [PubMed] [Google Scholar]
- Milligan D.L. and Koshland,D.E.,Jr (1988) Site-directed cross-linking. J. Biol. Chem., 263, 6268–6275. [PubMed] [Google Scholar]
- Misselwitz B., Staeck,O. and Rapoport,T.A. (1998) J proteins catalytically activate Hsp70 molecules to trap a wide range of peptide sequences. Mol. Cell, 2, 593–603. [DOI] [PubMed] [Google Scholar]
- Mitchell C. and Oliver,D. (1993) Two distinct ATP-binding domains are needed to promote protein export by Escherichia coli SecA ATPase. Mol. Microbiol., 10, 483–497. [DOI] [PubMed] [Google Scholar]
- Park S.K., Kim,D.W., Choe,J. and Kim,H. (1997) RNA helicase activity of Escherichia coli SecA protein. Biochem. Biophys. Res. Commun., 235, 593–597. [DOI] [PubMed] [Google Scholar]
- Plath K., Mothes,W., Wilkinson,B.M., Stirling,C.J. and Rapoport,T.A. (1998) Signal sequence recognition in posttranslational protein transport across the yeast ER membrane. Cell, 94, 795–807. [DOI] [PubMed] [Google Scholar]
- Pogliano J.A. and Beckwith,J. (1994) SecD and SecF facilitate protein export in Escherichia coli. EMBO J., 13, 554–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohlschröder M., Prinz,W.A., Hartmann,E. and Beckwith,J. (1997) Protein translocation in the three domains of life: variation on a theme. Cell, 91, 563–566. [DOI] [PubMed] [Google Scholar]
- Rajapandi T., Dolan,K.M. and Oliver,D.M. (1991) The first gene in the Escherichia coli secA operon, gene X, encodes a nonessential secretory protein. J. Bacteriol., 173, 7092–7097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychlik W., Odom,O.W. and Hardesty,B. (1983) Localization of the elongation factor Tu binding site on Escherichia coli ribosomes. Biochemistry, 22, 85–93. [DOI] [PubMed] [Google Scholar]
- Schagger H., Cramer,W.A. and von Jagow,G. (1994) Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal. Biochem., 217, 220–230. [DOI] [PubMed] [Google Scholar]
- Schiebel E., Driessen,A.J.M., Hartl,F.U. and Wickner,W. (1991) δµH+ and ATP function at different steps of the catalytic cycle of preprotein translocase. Cell, 64, 927–939. [DOI] [PubMed] [Google Scholar]
- Schmidt M.O., Brosh,R.M.,Jr and Oliver,D.B. (2001) Escherichia coli SecA helicase activity is not required in vivo for efficient protein translocation or autogenous regulation. J. Biol. Chem., 276, 37076–37085. [DOI] [PubMed] [Google Scholar]
- Shinkai A., Mei,L.H., Tokuda,H. and Mizushima,S. (1991) The conformation of SecA, as revealed by its protease sensitivity, is altered upon interaction with ATP, presecretory proteins, everted membrane vesicles and phospholipids. J. Biol. Chem., 266, 5827–5833. [PubMed] [Google Scholar]
- Sianidis G., Karamanou,S., Vrontou,E., Boulias,K., Repanas,K., Kyrpides,N., Politou,A.S. and Economou,A. (2001) Cross-talk between catalytic and regulatory elements in a DEAD motor domain is essential for SecA function. EMBO J., 20, 961–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder B. and Hammes,G.G. (1984) Structural mapping of chloroplast coupling factor. Biochemistry, 23, 5787–5795. [DOI] [PubMed] [Google Scholar]
- Soultanas P. and Wigley,D.B. (2000) DNA helicases: ‘inching forward’. Curr. Opin. Struct. Biol., 10, 124–128. [DOI] [PubMed] [Google Scholar]
- Suzuki H., Nishiyama,K.I. and Tokuda,H. (1999) Increases in acidic phospholipid contents specifically restore protein translocation in a cold-sensitive secA or secG null mutant. J. Biol. Chem., 274, 31020–31024. [DOI] [PubMed] [Google Scholar]
- Triplett T.L., Sgrignoli,A.R., Gao,F.B., Yang,Y.B., Tai,P.C. and Geirasch,L.M. (2001) Functional signal peptides bind a soluble N-terminal fragment of SecA and inhibit its ATPase activity. J. Biol. Chem., 276, 19648–19655. [DOI] [PubMed] [Google Scholar]
- Ulbrandt N.D., London,E. and Oliver,D. (1992) Deep penentration of a portion of Escherichia coli SecA protein into model membranes is promoted by anionic phospholipids and by partial unfolding. J. Biol. Chem., 267, 15184–15192. [PubMed] [Google Scholar]
- van der Does C., Swaving,J., van Klompenburg,W. and Driessen,A.J.M. (2000) Non-bilayer lipids stimulate the activity of the reconstituted bacterial protein translocase. J. Biol. Chem., 275, 2472–2478. [DOI] [PubMed] [Google Scholar]
- van der Wolk J.P.W., Fekkes,P., Boorsma,A., Huie,J.L., Silhavey,T.J. and Driessen,A.J.M. (1998) PrlA4 prevents the rejection of signal sequence defective preproteins by stabilizing the SecA–SecY interaction during the initiation of translocation. EMBO J., 17, 3631–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velankar S.S., Soultanas,P., Dillingham,M.S., Subramanya,H.S. and Wigley,D.B. (1999) Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell, 97, 75–84. [DOI] [PubMed] [Google Scholar]
- Woodbury R.L., Hardy,S.J.S. and Randall,L.L. (2002) Complex behavior in solution of homodimeric SecA. Protein Sci., 11, 875–882. [DOI] [PMC free article] [PubMed] [Google Scholar]