

Abstract
Externally added fibroblast growth factor-1 (FGF-1) is capable of crossing cellular membranes to reach the cytosol and the nucleus in a number of cell types. We have monitored the translocation of the growth factor by two methods: phosphorylation of FGF-1, and prenylation of an FGF-1 mutant that contains a C-terminal prenylation signal. Inhibition of endosomal acidification by ammonium chloride or monensin did not block the translocation of FGF-1, whereas bafilomycin A1, a specific inhibitor of vacuolar proton pumps, blocked translocation completely. A combination of ionophores expected to dissipate the vesicular membrane potential (valinomycin plus monensin) also fully inhibited the translocation. The inhibition of translocation by bafilomycin A1 was overcome in the presence of monensin or nigericin, while ouabain blocked translocation under these conditions. The data indicate that translocation of FGF-1 to cytosol occurs from the lumen of intracellular vesicles possessing vacuolar proton pumps, and that a vesicular membrane potential is required. Apparently, activation of vesicular Na+/K+-ATPase by monensin or nigericin generates a membrane potential that can support translocation when the proton pump is blocked.
Keywords: FGF-1/protein translocation/vesicular membrane potential
Introduction
Fibroblast growth factor-1 (FGF-1) is involved in several physiological and pathological processes including angiogenesis and wound healing (Burgess and Maciag, 1989). In cell culture, it can stimulate DNA synthesis and promote cell migration and differentiation. There is evidence that FGF-1 can act both at the cell surface and inside cells (Imamura et al., 1990; Zhan et al., 1992; Więdłocha et al., 1994, 1996). At the surface level, the best understood process is signalling through transmembrane receptors containing an intracellular split tyrosine kinase domain (Mason, 1994), which results in activation of downstream effectors such as the phospholipase Cγ and the MAP kinase pathways.
The growth factor is synthesized as a cytosolic protein (Burgess and Maciag, 1989; Friesel and Maciag, 1995), and it can be secreted in response to stress conditions, such as heat shock (Jackson et al., 1992), by a yet undefined route.
Several reports indicate that exogenous FGF-1 is capable of reaching the cytosol and the nucleus of target cells (Zhan et al., 1992; Imamura et al., 1994; Więdłocha et al., 1994, 1995; Prudovsky et al., 1996), and that this may be required for the mitogenic response, at least in certain cell types. Nuclear transport of exogenous FGF-1 occurs during the G1 period of the cell cycle (Zhan et al., 1993). Evidence for G1 phase-specific nuclear translocation of externally added FGF-2, a protein closely related to FGF-1, has also been presented (Bouche et al., 1987; Baldin et al., 1990). These results imply that the growth factor is able to cross cellular membranes, which could occur from the cell surface or across the membrane of intracellular vesicular compartments. Despite years of intensive research, the mechanism of translocation has remained obscure.
The ability to cross cellular membranes is shared by a number of protein toxins that act on targets in the cytosol. The translocation mechanisms used by diphtheria toxin, anthrax toxin and ricin have been elucidated in great detail. The use of specific drugs that protect cells from intoxication has been instrumental in these studies. Diphtheria and anthrax toxins are translocated to the cytosol from endocytic vesicles upon acidification of the vesicle lumen (Sandvig and Olsnes, 1982; Wesche et al., 1998). Inhibition of the vesicular type proton pump protects cells against these toxins, as do lysosomotropic agents and certain ionophores that neutralize the acidic interior of endosomes. Ricin is translocated from the endoplasmic reticulum and the toxin must be transported retrogradely through the Golgi apparatus to reach this location (Rapak et al., 1997; Wesche et al., 1999). Cells can therefore be protected against ricin by brefeldin A, which disintegrates the Golgi apparatus (Yoshida et al., 1991). In an attempt to elucidate the mechanism of FGF-1 translocation, we have used a similar approach.
While toxins act directly on clearly defined targets in the cytosol, this is not the case with FGF-1. Therefore, a major problem is to demonstrate that the growth factor is really in the cytosol rather than in membrane-bound vesicular compartments. For these reasons, we have previously developed two methods that we consider give reliable data. In one case, we have added a farnesylation signal (CaaX box) onto the C-terminus of the growth factor (Więdłocha et al., 1995). Since farnesyl transferase is found only in the cytosol (Reiss et al., 1990) and in the nucleus (Sinensky et al., 1994), farnesylation of externally added FGF-1 implies that the growth factor has crossed cellular membranes.
The other method relies on the observation that FGF-1 is phosphorylated at a single site by protein kinase C (Klingenberg et al., 1998, 1999). Since protein kinase C has only been found in the cytosol and nucleus (Mellor and Parker, 1998), cells will only be able to phosphorylate growth factor that has crossed cellular membranes.
When testing the effect of drugs on translocation, it is important to exclude the possibility that the tested compounds could interfere in some unsuspected way with the assay system rather than with the translocation process. Here, we have used both assays in parallel, as we consider them to be sufficiently different that it is unlikely that the drugs being tested would interfere with both of them in a similar way.
We have studied the effect of drugs that interfere with the pH of intracellular vesicles and with the electrical potential across vesicular membranes on translocation of FGF-1 into endothelial and fibroblastic cells. The vesicular electrical potential was found to be crucial for translocation of the growth factor.
Results
Effect of neutralization of acidic intracellular compartments on the ability of exogenous FGF-1 to translocate to the cytosol
We tested the effect of various drugs on the translocation of FGF-1 into endothelial cells, which are major targets for the growth factor in vivo. The extent of translocation was monitored by the prenylation assay (Więdłocha et al., 1995). Calf pulmonary artery endothelial (CPAE) cells were serum starved, labelled with [14C]mevalonic acid in the presence of lovastatin, and treated for 6 h with heparin and FGF-1 or FGF-1–CaaX in the absence or presence of various drugs. After lysis, the cellular material was treated with heparin–Sepharose and the extent of prenylation of the growth factor was analysed by SDS–PAGE and fluorography. To provide a better picture of the changes induced in response to some drug treatments, band intensities from several gels were quantitated and expressed as a percentage of the control (no compound added), as summarized in Table I.
Table I. Effect of various compounds on the translocation of FGF-1 as measured by prenylation and phosphorylation assays.
| Compound tested | Translocation (% of control ± SD) | No. of experiments |
|---|---|---|
| Monensin (1 µM) | 102 ± 20 | 10 |
| NH4Cl (20 mM) | 97 ± 24 | 6 |
| Chloroquine (100 µM) | 97 ± 10 | 6 |
| Ouabain (100 µM) | 101 ± 19 | 8 |
| Oligomycin A (1 µM) | 64 ± 14 | 6 |
| Valinomycin (1 µM) | 100 ± 12 | 6 |
| Nigericin (0.1 µM) | 92 ± 14 | 7 |
| Brefeldin A (1 µg/ml) | 85 ± 9 | 6 |
| Nocodazole (33 µM) | 92 ± 5 | 3 |
| Cytochalasin D (10 µg/ml) | 97 ± 11 | 3 |
| Bafilomycin A1 (10 nM) | n.d.a | 15 |
| Concanamycin A (50 nM) | n.d.a | 4 |
an.d., not detectable.
Films were scanned and the band intensities were quantitated. Data are expressed as a percentage of the control (no compound added).
Figure 1A shows that FGF-1–CaaX was efficiently prenylated (lane 2). In contrast, when cells were treated with FGF-1, there was no labelling of the growth factor (lane 1). In the presence of monensin (lane 3) or ammonium chloride (lane 4), prenylation of FGF-1– CaaX was similar to the control. However, in the presence of bafilomycin A1, an inhibitor of the vacuolar proton pump, prenylation of the growth factor was completely blocked (lane 5).

Fig. 1. Effect of drugs that neutralize acidic cellular compartments on the ability of exogenous FGF-1 to become prenylated and phosphorylated in vivo. (A) Serum-starved CPAE cells were pre-incubated with radiolabelled mevalonic acid and lovastatin, and then incubated with 10 U/ml heparin and 100 ng/ml FGF-1 (lane 1) or 100 ng/ml FGF-1–CaaX (lanes 2–5) for 6 h in the absence (lanes 1 and 2) or presence of 1 µM monensin (lane 3), 20 mM NH4Cl (lane 4) or 10 nM bafilomycin A1 (lane 5). After lysis, the material adsorbed onto heparin–Sepharose was analysed by SDS–PAGE and fluorography. The arrow indicates the migration of prenylated FGF-1–CaaX. (B) NIH 3T3 cells were pre-treated as in (A). The incubation was carried out in the absence (lane 1) or presence of 1 µM monensin (lane 2), 20 mM NH4Cl (lane 3), 100 µM chloroquine (lane 4) or 10 nM bafilomycin A1 (lane 5). The lysed cells were analysed as in (A). (C) Serum-starved NIH 3T3 cells were pre-incubated with [33P]phosphate and then incubated with 10 U/ml heparin and 100 ng/ml FGF-1 for 6 h, in the absence (lane 1) or presence of 1 µM monensin (lane 2), 20 mM NH4Cl (lane 3), 100 µM chloroquine (lane 4) or 10 nM bafilomycin A1 (lane 5). After lysis, the material adsorbed onto heparin–Sepharose beads was treated briefly with trypsin and analysed by SDS–PAGE and fluorography. The arrow indicates the migration of phosphorylated FGF-1. (D) HUVE and U2OS cells were incubated with increasing concentrations of diphtheria toxin for 6 h in the absence or presence of 10 nM bafilomycin A1, 1 µM monensin, 20 mM NH4Cl or combination of 10 nM bafilomycin A1 and 1 µM monensin. Then the ability of cells to incorporate [3H]leucine was measured as described previously (Sandvig and Olsnes, 1982).
Similar results were obtained when prenylation experiments were performed in NIH 3T3 cells (Figure 1B). After treatment with monensin (lane 2), NH4Cl (lane 3) or chloroquine (lane 4), the extent of FGF-1–CaaX labelling was similar to the control (lane 1), whereas bafilomycin A1 completely prevented prenylation of the growth factor (lane 5).
The effect of these drugs on translocation of FGF-1 was also monitored by the phosphorylation assay (Klingenberg et al., 1999). NIH 3T3 cells were serum starved, labelled with [33P]phosphate and treated for 6 h with FGF-1 and heparin in the absence or presence of various drugs. After lysis, the cellular material was rotated with heparin– Sepharose and the bound material was subjected to a brief trypsin digestion to reduce background labelling (FGF-1 bound to heparin–Sepharose is very resistant to trypsin, whereas most contaminating proteins are not), and analysed by SDS–PAGE and fluorography.
Treatment of cells with FGF-1 resulted in phosphorylation of the growth factor (Figure 1C, lane 1). The extent of phosphorylation was similar when monensin (lane 2), NH4Cl (lane 3) or chloroquine (lane 4) was present in the medium. However, bafilomycin A1 completely prevented the phosphorylation of FGF-1 (lane 5).
In the concentrations used above, bafilomycin A1, monensin and NH4Cl effectively protected cells against diphtheria toxin in two different cell lines (Figure 1D), indicating that they were efficient in raising the intra vesicular pH. Therefore, it may be concluded that bafilomycin A1 blocks the translocation of exogenous FGF-1 into cells, whereas monensin and lysosomotropic drugs such as NH4Cl and chloroquine do not.
Evidence that translocation of FGF-1 depends on V-type H+-ATPase
At low concentrations, bafilomycin A1 selectively inhibits V-type H+-ATPase (Bowman et al., 1988). The results in Figure 2A show that in NIH 3T3 cells, prenylation of FGF-1–CaaX was fully blocked by 10 nM bafilomycin A1 (lane 6), while partial inhibition was observed at lower concentrations of the drug. Similar concentration dependence was obtained when translocation was monitored by phosphorylation of FGF-1 (Figure 2B). The low concentration of bafilomycin A1 required indicates that the translocation depends on the activity of V-type H+-ATPase.

Fig. 2. Ability of bafilomycin A1 and concanamycin A to prevent prenylation and phosphorylation of exogenous FGF-1 in NIH 3T3 cells. (A) Cells were pre-treated as in Figure 1A. Incubation with 10 U/ml heparin and 100 ng/ml FGF-1 (lane 1) or FGF-1–CaaX (lanes 2–7) was carried out for 6 h in the presence of increasing concentrations of bafilomycin A1. Cellular material was analysed as in Figure 1A. (B) Cells were pre-treated as in Figure 1C and then incubated with 10 U/ml heparin and 100 ng/ml FGF-1 for 6 h in the presence of increasing concentrations of bafilomycin A1. Cellular material was analysed as in Figure 1C. (C) Cells were pre-treated as in Figure 1C and then incubated with 10 U/ml heparin and 100 ng/ml FGF-1 for 6 h in the absence or presence of 50 nM concanamycin A. Cellular material was analysed as in Figure 1C.
The effect of concanamycin A, another inhibitor of V-type H+-ATPase (Drose and Altendorf, 1997), was also tested. The results in Figure 2C show that phosphorylation of the growth factor was completely blocked by 50 nM concanamycin A (lane 2).
We also studied the effect of drugs that inhibit other types of ATPase. Figure 3A shows that, compared with the control (lane 2) treatment, ouabain (lane 3), an inhibitor of the Na+/K+-ATPase, did not affect prenylation of the growth factor in CPAE cells. Oligomycin A (lane 4), which inhibits the F1F0-type ATPase, reduced prenylation by ∼35% (Table I). Also, phosphorylation of FGF-1 in NIH 3T3 cells (Figure 3B, lane 1) was not affected in the presence of ouabain (lane 2), while treatment with oligomycin A reduced it by ∼35% (lane 3) (Table I). This presumably reflects the general toxic effect of oligomycin A in long-term experiments. Thus, the translocation of FGF-1 is highly sensitive to inhibitors of V-type H+-ATPases, but not to inhibitors of E1E2- or F1F0-type ATPases.
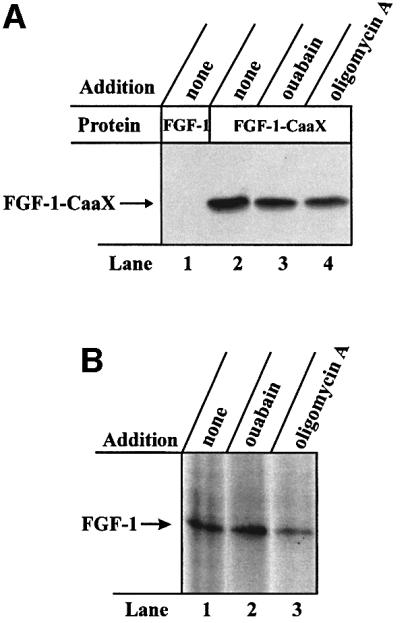
Fig. 3. Effect of inhibitors of various types of ATPase on the ability of exogenous FGF-1 to become prenylated and phosphorylated in vivo. (A) CPAE cells were pre-treated as in Figure 1A. Incubation with 100 ng/ml FGF-1 (lane 1) or FGF-1–CaaX (lanes 2–4) and 10 U/ml heparin was carried out in the absence (lanes 1 and 2) or presence of 200 µM ouabain (lane 3) or 1 µM oligomycin A (lane 4). Cellular material was analysed as in Figure 1A. (B) NIH 3T3 cells were pre-treated as in Figure 1C. Incubation with 100 ng/ml FGF-1 and 10 U/ml heparin was carried out in the absence (lane 1) or presence of 200 µM ouabain (lane 2) or 1 µM oligomycin A (lane 3). After cell lysis, the material was analysed as in Figure 1C.
Prevention of FGF-1 translocation by combinations of ionophores that depolarize the membrane of intracellular vesicles
The above results suggest that the translocation of FGF-1 to the cytosol occurs from intracellular vesicles that possess V-type H+-ATPase, and that dissipation of the pH gradient across the vesicle membrane does not block the translocation. Therefore, the possibility existed that the inside-positive electrical potential across the vesicle membrane (Van Dyke, 1988; Sonawane et al., 2002) generated by V-type proton pumps could be required for translocation. Neutralization of the vesicular pH by monensin or NH4Cl would not be expected to depolarize the membrane, whereas treatment with bafilomycin A1 would prevent the generation of membrane potential by the proton pump.
We tested this possibility by treating cells with the two ionophores monensin and valinomycin. Valinomycin is an electrogenic K+ ionophore, whereas monensin is an electroneutral monovalent cation exchanger. In the presence of active proton pumps, treatment with monensin induces accumulation of K+ ions in the vesicles as a result of exchange with lumenal H+ (Figure 4A). This allows valinomycin to short-circuit the lumen-positive membrane potential.


Fig. 4. Effect of a combination of ionophores expected to depolarize the membrane of intracellular vesicles on the ability of exogenous FGF-1 to become prenylated and phosphorylated in vivo. (A) V-type H+-ATPase (red) pumps H+ from the cytosol into the lumen of the vesicle, lowering lumenal pH and creating a membrane potential (positive-inside). The membrane potential is partly compensated for by the influx of Cl– counter ions (not indicated). Monensin or nigericin (green) exchanges H+ accumulated in the vesicle lumen for K+ present in the cytosol. This raises the lumenal pH but does not dissipate the membrane potential. When both monensin (or nigericin) and valinomycin (blue) are present, there is a net efflux of K+ from the lumen to the cytosol, which results in the dissipation of the membrane potential. (B) NIH 3T3 cells were pre-treated as in Figure 1A. Incubation with 100 ng/ml FGF-1–CaaX and 10 U/ml heparin was carried out in the absence (lane 1) or presence of 1 µM valinomycin (lane 2), 1 µM monensin (lane 3) or combination of both 1 µM valinomycin and 1 µM monensin (lane 4). Cellular material was analysed as in Figure 1A. (C) HUVE cells were pre-treated as in Figure 1A. Incubation with 100 ng/ml FGF-1–CaaX and 10 U/ml heparin was carried out in the absence (lane 1) or presence of 1 µM monensin (lane 2), 1 µM valinomycin (lane 3), 0.1 µM nigericin (lane 5), or combination of both 1 µM valinomycin and either 1 µM monensin (lane 4) or 0.1 µM nigericin (lane 6). After cell lysis, the material was analysed as in Figure 1A. (D) NIH 3T3 cells were pre-treated as in Figure 1C. Incubation with 100 ng/ml FGF-1 and 10 U/ml heparin was carried out in the absence (lane 1) or presence of 1 µM monensin (lane 2), 1 µM valinomycin (lane 3), 0.1 µM nigericin (lane 6), or combination of both 1 µM valinomycin and either 1 µM monensin (lane 4), 20 mM NH4Cl (lane 5) or 0.1 µM nigericin (lane 7). Cellular material was analysed as in Figure 1C. (E) NIH 3T3 cells were pre-treated as in Figure 1C. Incubation with 100 ng/ml FGF-1 and 10 U/ml heparin was carried out in the absence (lane 1) or in the presence of 0.1 µM nigericin in combination with increasing concentrations of valinomycin (lanes 2–5). After cell lysis, the cellular material was treated as in Figure 1C.
The data in Figure 4B demonstrate that there was no difference in the prenylation of FGF-1–CaaX when NIH 3T3 cells were treated in the absence (lane 1) or presence of valinomycin alone (lane 2) or monensin alone (lane 3). However, in the presence of both ionophores, translocation of the growth factor was completely blocked (lane 4).
Human umbilical vein endothelial (HUVE) cells were tested in the same way. Treatment with FGF-1–CaaX resulted in efficient prenylation of the growth factor (Figure 4C, lane 1). In the presence of monensin (lane 2) or valinomycin (lane 3), the labelling was not changed, but when the two ionophores were present together (lane 4), prenylation of FGF-1–CaaX was fully blocked.
In similar experiments, we tested the combination of nigericin and valinomycin. Nigericin is, like monensin, an electroneutral exchanger for monovalent cations, but it has less affinity for Na+ ions than monensin. As shown in Figure 4C lane 5, the addition of nigericin did not affect the prenylation of FGF-1–CaaX. However, the combination of nigericin and valinomycin fully blocked the intracellular labelling of the growth factor (lane 6). Similar results were obtained with CPAE cells (data not shown).
We also performed phosphorylation experiments in NIH 3T3 cells. As compared with the control conditions (Figure 4D, lane 1), neither monensin (lane 2), valino mycin (lane 3) nor nigericin (lane 6) alone affected the phosphorylation of FGF-1. On the other hand, when valinomycin was present in combination with either monensin (lane 4) or nigericin (lane 7), the phosphorylation of the growth factor was fully inhibited. Phosphorylation of FGF-1 was not affected when valino mycin was used in combination with ammonium chloride (lane 5), demonstrating that inhibition of FGF-1 translocation is not due to a simple combinatorial effect of pH gradient dissipation and the presence of valinomycin.
Figure 4E shows that the combination of 0.1 µM nigericin and 1 µM valinomycin was sufficient to completely block FGF-1 phosphorylation in NIH 3T3 cells (lane 5). At lower concentrations of valinomycin (lanes 2–4), the inhibition of the labelling was only partial.
Altogether, the data indicate that combinations of ionophores expected to dissipate the membrane potential block translocation of FGF-1 to cytosol. This suggests that a membrane potential is required for the translocation. Moreover, the requirement of valinomycin to block translocation in the presence of either monensin or nigericin indicates that there are no active K+ channels in the vesicle membrane.
Induction of translocation by activation of vesicular Na+/K+-ATPase
It has been reported that endocytic vesicles contain Na+/K+-ATPase, which can be inhibited by ouabain (Cain et al., 1989; Fuchs et al., 1989). In the absence of an active K+ channel, the Na+/K+-ATPase would soon be inactive due to lack of vesicular K+. We reasoned that treatment with monensin or nigericin would provide the necessary vesicular concentration of K+ (in exchange for Na+) to reactivate the Na+/K+-ATPase. In that case, a membrane potential (inside-positive) could build up across the vesicular membrane and support translocation of FGF-1, even in the presence of bafilomycin A1 (Figure 5A). Treatment of the cells with ouabain should prevent translocation under these conditions.

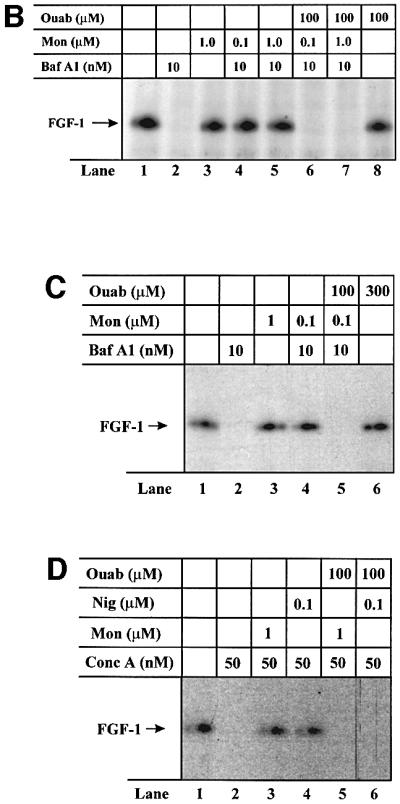
Fig. 5. Ability of monensin and nigericin to overcome the translocation block by an ouabain-sensitive mechanism. (A) In addition to proton pumps (red), endosomes contain also Na+/K+-ATPase (blue), which can contribute to the generation of an inside-positive membrane potential. It is likely that the endosomes soon become deficient in K+, which will lead to inactivation of the Na+/K+-ATPase (dashed arrows). Therefore, treatment with bafilomycin A1, which inactivates proton pumps (dashed arrow), is expected to depolarize the membrane. In the presence of monensin or nigericin (green), K+ is exchanged for Na+ leading to reactivation of the Na+/K+-ATPase (solid arrows) and the generation of a membrane potential even in the presence of bafilomycin A1. Under these conditions, treatment with ouabain inhibits the Na+/K+-ATPase (dashed arrows) leading again to depolarization of the membrane. (B) NIH 3T3 cells were pre-treated as in Figure 1C. Incubation with 50 ng/ml FGF-1 and 10 U/ml heparin was carried out in the absence (lane 1) or presence of 10 nM bafilomycin A1 (lane 2), 1 µM monensin (lane 3), 100 µM ouabain (lane 8), or combination of both 10 nM bafilomycin A1 and either 0.1 µM monensin (lane 4), 1 µM monensin (lane 5), 0.1 µM monensin and 100 µM ouabain (lane 6) or 1 µM monensin and 100 µM ouabain (lane 7). Cellular material was analysed as in Figure 1C. (C) HUVE cells were pre-treated as in Figure 1C. Incubation with 50 ng/ml FGF-1 and 10 U/ml heparin was carried out in the absence (lane 1) or presence of 10 nM bafilomycin A1 (lane 2), 1 µM monensin (lane 3), 300 µM ouabain (lane 6), or combination of 10 nM bafilomycin A1 and either 0.1 µM monensin (lane 4) or 0.1 µM monensin and 100 µM ouabain (lane 5). Cellular material was analysed as in Figure 1C. (D) HUVE cells were pre-treated as in Figure 1C. Incubation with 50 ng/ml FGF-1 and 10 U/ml heparin was carried out in the absence (lane 1) or presence of 50 nM concanamycin A (lane 2), or combination of 50 nM concanamycin A and either 1 µM monensin (lane 3), 0.1 µM nigericin (lane 4), 1 µM monensin and 100 µM ouabain (lane 5) or 0.1 µM nigericin and 100 µM ouabain (lane 6). Cellular material was analysed as in Figure 1C.
The data in Figure 5B show that while treatment of NIH 3T3 cells with bafilomycin A1 alone prevented translocation of FGF-1 (lane 2), bafilomycin A1 in combination with monensin allowed translocation to occur (lanes 4 and 5). On the other hand, when ouabain was added as well, the translocation was blocked (lanes 6 and 7). Neither monensin (lane 3) nor ouabain (lane 8) alone affected the extent of translocation as compared with control conditions (lane 1).
Similar experiments were performed in HUVE cells (Figure 5C). Bafilomycin A1 alone prevented translocation of FGF-1 (lane 2), but treatment with the combination of bafilomycin A1 and monensin allowed translocation to occur (lane 4). In contrast, when ouabain was added as well, translocation was blocked (lane 5). Treatment with ouabain alone did not inhibit translocation (lane 6).
We also tested whether monensin or nigericin could overcome the inhibition of FGF-1 translocation imposed by concanamycin A. In HUVE cells treated with concanamycin A (Figure 5D, lane 2), translocation was fully blocked, but when concanamycin A was used in combination with either monensin (lane 3) or nigericin (lane 4), it was restored. When ouabain was added as well (lanes 5 and 6), translocation was blocked again. Altogether, the data indicate that Na+/K+-ATPase in the vesicle is reactivated by monensin or nigericin to provide sufficient polarization of the vesicular membrane to allow translocation of FGF-1 to occur.
Inability of brefeldin A, nocodazole and cytochalasin D to block FGF-1 translocation
To test whether the translocation involved the Golgi apparatus, which is also acidified by proton pumps (Wu et al., 2001), we treated the cells with brefeldin A, which induces disruption of the Golgi apparatus (Lippincott-Schwartz et al., 1991). Prenylation of FGF-1–CaaX was not blocked by brefeldin A in NIH 3T3 cells (Figure 6A), although ∼15% reduction was observed at the high drug concentration (Table I). This indicates that the Golgi apparatus is not the site of translocation and that retrograde transport across the Golgi apparatus to the endoplasmic reticulum is also not required. Furthermore, treatment of NIH 3T3 cells with cytochalasin D, which disrupts actin microfilaments (Cooper, 1987) or with nocodazole, which induces microtubule depolymerization (Hoebeke et al., 1976), did not affect prenylation of FGF-1–CaaX to a large extent (Figure 6B; Table I) in concentrations of the drugs that efficiently disrupted these cytoskeleton structures (Figure 6C). Therefore, it appears that trafficking of FGF-1-containing vesicles along microtubules and actin microfilaments is not essential for the translocation of the growth factor to the cytosol.
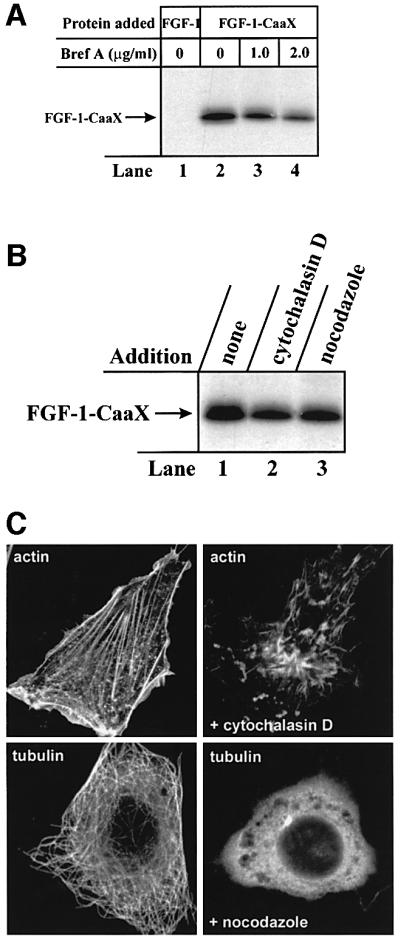
Fig. 6. Inability of brefeldin A, nocodazole and cytochalasin D to block FGF-1–CaaX prenylation in NIH 3T3 cells. (A) Cells were pre-treated as in Figure 1A and then incubated with 10 U/ml heparin and 100 ng/ml FGF-1 (lane 1) or 100 ng/ml FGF-1–CaaX (lanes 2–4) for 6 h in the absence (lanes 1 and 2) or presence of 1 µg/ml brefeldin A (lane 3) or 2 µg/ml brefeldin A (lane 4). Cellular material was analysed as in Figure 1A. (B) Cells were pre-treated as in Figure 1A. Incubation with FGF-1–CaaX and heparin was carried out in the absence (lane 1) or presence of 10 µg/ml cytochalasin D (lane 2) or 33 µM nocodazole (lane 3). Cellular material was analysed as in Figure 1A. (C) Cells were treated with or without 33 µM nocodazole or 10 µg/ml cytochalasin D for 30 min. Microtubuli were visualized with anti-tubulin and rhodamine-conjugated anti-mouse antibody. Actin was visualized with rhodamine-conjugated phalloidin.
Lack of effect of the drugs on FGF-1 binding and FGF-receptor activation
Alteration in the membrane potential is likely to affect the structure of transmembrane proteins. Since binding of the growth factor to specific FGF receptors is required for translocation (Więdłocha et al., 1995), we considered the possibility that the inhibition of FGF-1 translocation could be due to interference with binding of FGF-1 to the receptor or with receptor activation.
The data in Figure 7A demonstrate that treatment with monensin and/or bafilomycin A1 had little or no effect on the apparent affinity and the number of binding sites available at the surface of NIH 3T3 cells.

Fig. 7. Effect of various drugs on the ability of FGF-1 to bind to surface receptors and stimulate phosphorylation of MAP kinase in NIH 3T3 cells. (A) Cells growing on gelatin-coated 12-well plates were incubated for 2 h at 37°C in the presence or absence of 10 nM bafilomycin A1, 1 µM monensin or both. Then the cells were incubated at 4°C for 30 min and increasing amounts of [125I]FGF-1 (29 000 c.p.m./ng) were added. After 4 h incubation at 4°C, the cells were washed, dissolved in 0.1 M KOH and the cell-associated radioactivity was measured according to Munoz et al. (1997) (left panel) and represented as a Scatchard plot (right panel). (B) Serum-starved cells were pre-incubated in the absence (lanes 1 and 2) or presence of 10 nM bafilomycin A1 (lane 3), or combination of 1 µM valinomycin and either 1 µM monensin (lane 4) or 0.1 µM nigericin (lane 5). The cells were left untreated (lane 1) or stimulated for 10 min with 2 ng/ml FGF-1 in the presence of 10 U/ml heparin (lanes 2–5). The cells were subsequently lysed and analysed by western blotting with anti-p44/42 MAP kinase antibodies (lower panel). The membrane was stripped and reprobed with anti-phosphorylated-p44/42 MAP kinase antibodies (upper panel). (C) Serum-starved cells were pre-incubated in the absence (lanes 1–4) or presence (lanes 5–8) of 10 nM bafilomycin A1. Cells were left untreated (lanes 1 and 5) or stimulated for 10 min (lanes 2 and 6), 60 min (lanes 3 and 7) or 180 min (lanes 4 and 8) with 2 ng/ml FGF-1 in the presence of 10 U/ml heparin. The cells were subsequently lysed and analysed as in (B).
We also monitored the extent of MAP kinase phosphorylation, which results from FGF-receptor activation. The results in Figure 7B show that in serum-starved cells, there was only a weak basal level of MAP kinase phosphorylation (upper panel, lane 1). A 10 min treatment with FGF-1 dramatically increased the extent of MAP kinase phosphorylation (lane 2). Neither bafilomycin A1 (lane 3) nor valinomycin in combination with either monensin (lane 4) or nigericin (lane 5) altered the extent of the MAP kinase phosphorylation. Figure 7B (lower panel) shows that in each case a similar amount of MAP kinase was applied to the gel.
We also tested whether the presence of bafilomycin A1 influenced activation of MAP kinase after prolonged exposure to FGF-1. Treatment with FGF-1 for 10, 60 or 180 min induced a similar extent of MAP kinase phosphorylation whether the cells had been pre-incubated with bafilomycin A1 (Figure 7C, upper panel, lanes 5–8) or not (lanes 1–4).
In the experiments demonstrated, we used a low concentration of FGF-1 (2 ng/ml). Since the translocation experiments were carried out with 50–100 ng/ml FGF-1, we repeated the experiments in Figure 7B and C with 50 ng/ml FGF-1, with essentially the same results (data not shown).
Quantitation of the amounts of translocated FGF-1
To get an estimate of how many FGF-1 molecules were translocated into each cell, we cut out the labelled bands from some of the SDS–PAGE gels in the farnesylation experiments and measured the incorporated radioactivity. Calculations were performed as described in Materials and methods. Depending on the cell type, we obtained values of between 2.9 and 11.4 × 105 of FGF-1–CaaX molecules translocated per cell. This corresponds to average intracellular concentrations between 0.18 and 0.58 µM, which represents a 24- to 83-fold up-concentration of the growth factor compared with its concentration in the medium (∼7 nM). It should be noted that in many cases the bands were too weak for the radioactivity to be measured with confidence. The values given above should therefore be considered as maximal values.
Discussion
The main finding in this paper is that translocation of FGF-1 across cellular membranes to reach the cytosol is blocked in the presence of low concentrations of bafilomycin A1. The concentration of bafilomycin A1, which gives complete inhibition of FGF-1 translocation (∼10 nM) corresponds to that needed to block vacuolar proton pumps (V-type H+-ATPases) (Bowman et al., 1988). Much higher concentrations of the drug are required to inhibit E1E2-type ATPases (>10 µM), and even higher to inhibit F1F0-type ATPases (>1 mM). Also, concanamycin A, another inhibitor of V-type H+-ATPases (Drose and Altendorf, 1997), fully blocked the translocation of FGF-1, indicating further that active proton pumps are required for translocation. Since the vacuolar proton pumps are found in endosomes, lysosomes and the Golgi apparatus, the observation indicates that the translocation of FGF-1 occurs from an intracellular membrane-bound compartment.
The proton-motive force, ΔµH, across the membrane of vesicles containing V-type H+-ATPases consists of a proton gradient (inside-acidic) and a membrane potential (inside-positive), which has been estimated at 10–20 mV (Sonawane et al., 2002). Inhibition of vacuolar proton pumps by bafilomycin A1 will result in total collapse of ΔµH. The pH gradient can be selectively dissipated by electroneutral ionophores like monensin and nigericin that exchange Na+ and K+ for protons across membranes or by NH3 and chloroquine, which diffuse across membranes and bind protons. In neither case is the membrane potential expected to change. Our results show that dissipation of the pH gradient alone without changing the membrane potential of the vesicle has no effect on translocation of FGF-1.
Valinomycin is able to transport K+ across membranes according to existing electrochemical gradients. Intra cellular vesicles containing proton pumps are expected to be low in potassium, but if the lumenal H+ are exchanged for K+ by monensin or nigericin, valinomycin will cause leakage of K+ to the cytosol, resulting in the dissipation of the membrane potential. The finding that monensin or nigericin blocked translocation of the growth factor only in combination with valinomycin indicates that the membrane potential is necessary for translocation, whereas the ΔpH is not. The data also indicate that the vesicles do not contain an active K+ channel of their own.
The finding that the inhibition of translocation by bafilomycin A1 could be overcome by monensin or nigericin was interpreted as activation of Na+/K+-ATPase in the vesicular membrane described by other authors (Cain et al., 1989; Fuchs et al., 1989). In fact, treatment with ouabain blocked translocation under these conditions. The Na+/K+-ATPase was found to counteract the acidification of early endosomes, but not of late endosomes, indicating that it is absent or not functional in the late endosomes (Fuchs et al., 1989). We found that in the absence of monensin or nigericin the Na+/K+-ATPase in bafilomycin A1-treated cells was unable to support translocation. This could mean that the translocation occurs from vesicles later than early endosomes.
The ouabain concentrations used here (100–300 µM) are sufficient to completely block the Na+/K+-ATPase in HUVE cells and in CPAE cells, whereas in murine NIH 3T3 cells, the inhibition may only be partial. The α1 subunit of the murine Na+/K+-ATPase expressed in fibroblasts is 1000 times less sensitive to ouabain (ID50 = 10–5–10–4 M) than that from most mammals (Brodsky, 1990; O’Brien et al., 1994). In any event, it is clear that the concentrations of the drug used here are sufficient to block translocation in bafilomycin A1- and monensin-treated NIH 3T3 cells.
The proton-motive force, ΔµH, plays important roles in several systems where proteins are transported across membranes (for a review, see Agarraberes and Dice, 2001). These include transport across the inner membrane of mitochondria, the thylakoid membrane and the inner membrane of bacteria. Particularly interesting is the thylakoid membrane transport in plants and the twin arginine (TAT) system in bacteria as they can transport folded proteins. Also, FGF-1 can be translocated in a folded form (Wesche et al., 2000).
Since the net charge of FGF-1 is positive, it is tempting to speculate that the membrane potential is directly involved as a driving force in FGF-1 translocation, but other possibilities must also be considered. Thus, the membrane potential is likely to influence the structure of the transmembrane part of FGF receptors and it could affect intracellular trafficking of the growth factor– receptor complex (Clague et al., 1994). However, we have found previously that FGF-1 is transported to the recycling endosome compartment and that this is not prevented by bafilomycin A1 (Citores et al., 2001).
FGF-1 translocation does not seem to be dependent on the presence of an intact microtubule and microfilament network. Since vesicle trafficking from early endosomes to lysosomes is thought to be dependent on intact microtubules (Bomsel et al., 1990; Clague et al., 1994), vesicles that traffic along the degradative pathway are probably not involved in FGF-1 translocation. In fact, FGF-1 does not colocalize with lysosome-associated membrane protein 1 (LAMP-1)-positive vesicles to any major extent (Citores et al., 2001). Treatment with brefeldin A, which disrupts the Golgi apparatus, does not block translocation of FGF-1. The moderate reduction in FGF-1 translocation imposed by brefeldin A may reflect the inability to transport newly synthesized FGF receptors to the cell surface in conditions where the Golgi apparatus is no longer functional.
Translocation of FGF-1 to cytosol and nucleus has been observed in several cell types containing endogenous FGF receptors, such as NIH 3T3, CPAE and HUVE cells (Więdłocha et al., 1995; Klingenberg et al., 2000a; this report), and in COS and U2OS cells transfected with FGF receptor 4 (Więdłocha et al., 1996; Klingenberg et al., 2000b). This indicates that the phenomenon is dependent on specific FGF tyrosine kinase receptors and that it is widely distributed. On the other hand, there is a vexing variability in the extent of translocation measured as labelling of the growth factor, particularly in the farnesylation studies. In general, early passages of the cells give better results than later passages. Therefore, it appears that the translocation is a regulated process. It is in accordance with this that treatment of cells with inhibitors of PI3 kinase prevented translocation (Klingenberg et al., 2000a). The farnesylated form of the growth factor is less stable in cells than the unmodified FGF-1 (our unpublished data), similar to translocated farnesylated diphtheria toxin A fragment (Falnes et al., 1995).
In the translocation studies, we used 50–100 ng/ml FGF-1, which is high when compared with the concentration (∼6 ng/ml) giving maximal binding under equilibrium conditions (4 h at 4°C; Więdłocha et al., 1995; Munoz et al., 1997; this study). However, in a dynamic situation at 37°C, higher concentrations of the growth factor are required to obtain maximal internalization. In experiments where we used 10 ng/ml FGF-1, the amount of trans located growth factor was about half of that obtained with 100 ng/ml protein (our unpublished data).
Recent work has demonstrated that several intracellular proteins bind FGF-1. Mortalin (Mizukoshi et al., 1999), S100A13 and synaptotagmin (Carreira et al., 1998; Tarantini et al., 1998), both the α- and the β-subunits of casein kinase 2 (CK2; Skjerpen et al., 2002a), FGF-1 intracellular binding protein (FIBP) (Kolpakova et al., 1998) and p34 (Skjerpen et al., 2002b) were found to bind FGF-1. The dissociation constant for binding of FGF-1 to CK2, FIBP and p34 is similar and in the high nanomolar range (Skjerpen et al., 2002b). Our estimation that intracellular concentrations of translocated FGF-1 can reach up to 0.5 µM suggests that a considerable part of these proteins may form complexes with the growth factor in the living cell.
Materials and methods
Materials
[14C]mevalonolactone (58 mCi/mmol) was from NEN Life Science Products, [33P]phosphoric acid from Hartman Analytic GmbH, heparin–Sepharose from Pharmacia Biotech, protease inhibitor cocktail Complete™ from Roche, lovastatin from Merck, rhodamine–phalloidin from Molecular Probes, phosphatase inhibitor cocktail, anti-tubulin and other chemicals were from Sigma-Aldrich. Rhodamine anti-mouse was from Jackson Immunoresearch. Mouse monoclonal anti-phospho-p44/42 MAP kinase and rabbit polyclonal anti-p44/42 MAP kinase were from Cell Signaling Technology. Endothelial cell culture medium (ECCM) was from Becton Dickinson Labware.
Buffers and media
The I/T medium was Dulbecco’s modified Eagle’s medium (DMEM) containing 2.5 µg/ml insulin and 2.5 µg/ml transferrin. The lysis buffer contained 10 mM Na phosphate pH 7.4, 100 mM NaCl, 1 mM EDTA, 1% Triton X-100 and protease inhibitor cocktail. The P-lysis buffer contained 10 mM Tris–HCl pH 7.4, 50 mM NaCl, 1 mM EDTA, 50 mM NaF, 30 mM Na-pyrophosphate, 100 µM Na-orthovanadate, 1% Triton X-100 and both protease and phosphatase inhibitor cocktails.
Cell cultures
NIH 3T3 cells were propagated under standard conditions (5% CO2, 37°C) in DMEM containing 10% (v/v) newborn calf serum (NCS). CPAE cells were propagated in DMEM containing 20% fetal bovine serum (FBS). HUVE cells were propagated in ECCM containing 10% FBS. Cell cultures were propagated for up to 1 month.
Expression and purification of recombinant proteins
FGF-1 and FGF-1–CaaX were expressed from pTrc-FGF-1 and pTrc-FGF-1–CaaX and purified as described (Więdłocha et al., 1995).
In vivo prenylation
In vivo prenylation was performed essentially as described previously (Więdłocha et al., 1995) with some modifications. Cells were serum starved for 24 h in I/T medium. For the last 3 h, serum starvation was continued in DMEM containing 1% NCS, 0.4 µCi/ml [14C]mevalonic acid and 1 µg/ml lovastatin. The tested compounds were added 30 min before the end of serum starvation. Cells were then treated with 10 U/ml heparin and 100 ng/ml FGF-1 or FGF-1–CaaX and incubation was continued for 6 h more. Cells were washed once in phosphate-buffered saline (PBS), lysed and sonicated. Lysates were adsorbed onto heparin–Sepharose and analysed by SDS–PAGE (Laemmli, 1970) and fluorography.
In vivo phosphorylation
In vivo phosphorylation was performed essentially as described previously (Klingenberg et al., 1999, 2000b), with some modifications. Cells were serum starved for 24–48 h in DMEM (+1% FBS). For the last 3–12 h, serum starvation was continued in phosphate-free DMEM containing 1% FBS and 20 µCi/ml [33P]phosphate. The tested compounds were added 30 min before the end of serum starvation. Then the cells were treated with 10 U/ml heparin and 50–100 ng/ml FGF-1, and incubation was continued for a further 6 h. Cells were washed once with PBS, lysed in P-lysis buffer and sonicated. Lysates were treated with heparin– Sepharose, beads were washed with 0.6 M NaCl and subjected to trypsin digestion (1 µg/ml) for 15 min at room temperature. Samples were analysed by SDS–PAGE and fluorography.
Activation of MAP kinase
Cells were serum starved for 24 h in DMEM + 0.5% FBS. For the last 3 h, serum starvation was continued in the presence of the tested compounds. Then cells were treated with 10 U/ml heparin and 2–50 ng/ml FGF-1 for the indicated period of time, lysed in SDS sample buffer, and analysed by SDS–PAGE and western blotting using anti-p44/p42 MAP kinase antibodies. After stripping, the membrane was re-probed with antibodies against phospho-p44/p42 MAP kinase.
Quantitation of translocation of exogenous FGF-1 into cells
Parts of SDS–PAGE gels corresponding to prenylated FGF-1–CaaX were cut out and the incorporated radioactivity was measured. The amount of radiolabelled protein was calculated from the specific activity of the [14C]mevalonic acid by assuming that all translocated FGF-1–CaaX molecules were undergoing prenylation, that three molecules of mevalonic acid were required to form one farnesyl group, and by disregarding the low amounts of unlabelled endogenous mevalonic acid being formed in the presence of lovastatin. The average cell volume (HUVE, 3.8 × 10–12 l; CPAE, 3.9 × 10–12 l; NIH 3T3, 1.1 × 10–12 l) was measured using [14C]urea (Olsnes and Sandvig, 1986). The number of cells used in each experiment (∼3 × 105) was determined in parallel wells.
Acknowledgments
Acknowledgements
The skilful technical assistance of Anne Gro Bredesen and Brit Solvar with the cell cultures is gratefully acknowledged. J.M. and J.W. are post-doctoral Fellows of The Research Council of Norway and The Norwegian Cancer Society, respectively. This work was supported by the Novo Nordisk Foundation, the Blix Fund for the Promotion of Medical Research, Rachel and Otto Kr. Bruun’s Fund and by The Jahre Foundation.
References
- Agarraberes F.A. and Dice,J.F. (2001) Protein translocation across membranes. Biochim. Biophys. Acta, 1513, 1–24. [DOI] [PubMed] [Google Scholar]
- Baldin V., Roman,A.M., Bosc-Bierne,I., Amalric,F. and Bouche,G. (1990) Translocation of bFGF to the nucleus is G1 phase cell cycle specific in bovine aortic endothelial cells. EMBO J., 9, 1511–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomsel M., Parton,R., Kuznetsov,S.A., Schroer,T.A. and Gruenberg,J. (1990) Microtubule- and motor-dependent fusion in vitro between apical and basolateral endocytic vesicles from MDCK cells. Cell, 62, 719–731. [DOI] [PubMed] [Google Scholar]
- Bouche G., Gas,N., Prats,H., Baldin,V., Tauber,J.P., Teissie,J. and Amalric,F. (1987) Basic fibroblast growth factor enters the nucleolus and stimulates the transcription of ribosomal genes in ABAE cells undergoing G0–G1 transition. Proc. Natl Acad. Sci. USA, 84, 6770–6774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman E.J., Siebers,A. and Altendorf,K. (1988) Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc. Natl Acad. Sci. USA, 85, 7972–7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky J.L. (1990) Characterization of the (Na++K+)-ATPase from 3T3-F442A fibroblasts and adipocytes. Isozymes and insulin sensitivity. J. Biol. Chem., 265, 10458–10465. [PubMed] [Google Scholar]
- Burgess W.H. and Maciag,T. (1989) The heparin-binding (fibroblast) growth factor family of proteins. Annu. Rev. Biochem., 58, 575–606. [DOI] [PubMed] [Google Scholar]
- Cain C.C., Sipe,D.M. and Murphy,R.F. (1989) Regulation of endocytic pH by the Na+,K+-ATPase in living cells. Proc. Natl Acad. Sci. USA, 86, 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira C.M., LaVallee,T.M., Tarantini,F., Jackson,A., Lathrop,J.T., Hampton,B., Burgess,W.H. and Maciag,T. (1998) S100A13 is involved in the regulation of fibroblast growth factor-1 and p40 synaptotagmin-1 release in vitro. J. Biol. Chem., 273, 22224–22231. [DOI] [PubMed] [Google Scholar]
- Citores L., Khnykin,D., Sorensen,V., Wesche,J., Klingenberg,O., Więdłocha,A. and Olsnes,S. (2001) Modulation of intracellular transport of acidic fibroblast growth factor by mutations in the cytoplasmic receptor domain. J. Cell Sci., 114, 1677–1689. [DOI] [PubMed] [Google Scholar]
- Clague M.J., Urbe,S., Aniento,F. and Gruenberg,J. (1994) Vacuolar ATPase activity is required for endosomal carrier vesicle formation. J. Biol. Chem., 269, 21–24. [PubMed] [Google Scholar]
- Cooper J.A. (1987) Effects of cytochalasin and phalloidin on actin. J. Cell Biol., 105, 1473–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drose S. and Altendorf,K. (1997) Bafilomycins and concanamycins as inhibitors of V-ATPases and P-ATPases. J. Exp. Biol., 200, 1–8. [DOI] [PubMed] [Google Scholar]
- Falnes P.O., Więdłocha,A., Rapak,A. and Olsnes,S. (1995) Farnesylation of CaaX-tagged diphtheria toxin A-fragment as a measure of transfer to the cytosol. Biochemistry, 34, 11152–11159. [DOI] [PubMed] [Google Scholar]
- Friesel R.E. and Maciag,T. (1995) Molecular mechanisms of angiogenesis: fibroblast growth factor signal transduction. FASEB J., 9, 919–925. [DOI] [PubMed] [Google Scholar]
- Fuchs R., Schmid,S. and Mellman,I. (1989) A possible role for Na+,K+-ATPase in regulating ATP-dependent endosome acidification. Proc. Natl Acad. Sci. USA, 86, 539–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoebeke J., Van Nijen,G. and De Brabander,M. (1976) Interaction of oncodazole (R 17934), a new antitumoral drug, with rat brain tubulin. Biochem. Biophys. Res. Commun., 69, 319–324. [DOI] [PubMed] [Google Scholar]
- Imamura T. et al. (1990) Recovery of mitogenic activity of a growth factor mutant with a nuclear translocation sequence. Science, 249, 1567–1570. [DOI] [PubMed] [Google Scholar]
- Imamura T., Oka,S., Tanahashi,T. and Okita,Y. (1994) Cell cycle-dependent nuclear localization of exogenously added fibroblast growth factor-1 in BALB/c 3T3 and human vascular endothelial cells. Exp. Cell Res., 215, 363–372. [DOI] [PubMed] [Google Scholar]
- Jackson A., Friedman,S., Zhan,X., Engleka,K.A., Forough,R. and Maciag,T. (1992) Heat shock induces the release of fibroblast growth factor 1 from NIH 3T3 cells. Proc. Natl Acad. Sci. USA, 89, 10691–10695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg O., Więdłocha,A., Rapak,A., Munoz,R., Falnes,P. and Olsnes,S. (1998) Inability of the acidic fibroblast growth factor mutant K132E to stimulate DNA synthesis after translocation into cells. J. Biol. Chem., 273, 11164–11172. [DOI] [PubMed] [Google Scholar]
- Klingenberg O., Więdłocha,A. and Olsnes,S. (1999) Effects of mutations of a phosphorylation site in an exposed loop in acidic fibroblast growth factor. J. Biol. Chem., 274, 18081–18086. [DOI] [PubMed] [Google Scholar]
- Klingenberg O., Więdłocha,A., Citores,L. and Olsnes,S. (2000a) Requirement of phosphatidylinositol 3-kinase activity for translocation of exogenous aFGF to the cytosol and nucleus. J. Biol. Chem., 275, 11972–11980. [DOI] [PubMed] [Google Scholar]
- Klingenberg O., Więdłocha,A., Rapak,A., Khnykin,D., Citores,L. and Olsnes,S. (2000b) Requirement for C-terminal end of fibroblast growth factor receptor 4 in translocation of acidic fibroblast growth factor to cytosol and nucleus. J. Cell Sci., 113, 1827–1838. [DOI] [PubMed] [Google Scholar]
- Kolpakova E., Więdłocha,A., Stenmark,H., Klingenberg,O., Falnes,P.O. and Olsnes,S. (1998) Cloning of an intracellular protein that binds selectively to mitogenic acidic fibroblast growth factor. Biochem. J., 336, 213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J., Yuan,L., Tipper,C., Amherdt,M., Orci,L. and Klausner,R.D. (1991) Brefeldin A’s effects on endosomes, lysosomes, and the TGN suggest a general mechanism for regulating organelle structure and membrane traffic. Cell, 67, 601–616. [DOI] [PubMed] [Google Scholar]
- Mason I.J. (1994) The ins and outs of fibroblast growth factors. Cell, 78, 547–552. [DOI] [PubMed] [Google Scholar]
- Mellor H. and Parker,P.J. (1998) The extended protein kinase C superfamily. Biochem. J., 332, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizukoshi E., Suzuki,M., Loupatov,A., Uruno,T., Hayashi,H., Misono,T., Kaul,S.C., Wadhwa,R. and Imamura,T. (1999) Fibroblast growth factor-1 interacts with the glucose-regulated protein GRP75/mortalin. Biochem. J., 343, 461–466. [PMC free article] [PubMed] [Google Scholar]
- Munoz R., Klingenberg,O., Więdłocha,A., Rapak,A., Falnes,P.O. and Olsnes,S. (1997) Effect of mutation of cytoplasmic receptor domain and of genistein on transport of acidic fibroblast growth factor into cells. Oncogene, 15, 525–536. [DOI] [PubMed] [Google Scholar]
- O’Brien W.J., Lingrel,J.B., and Wallick,E.T. (1994) Ouabain binding kinetics of the rat α two and α three isoforms of the sodium-potassium adenosine triphosphate. Arch. Biochem. Biophys., 310, 32–39. [DOI] [PubMed] [Google Scholar]
- Olsnes S. and Sandvig,K. (1986) Interactions between diphtheria toxin entry and anion transport in Vero cells. I. Anion antiport in Vero cells. J. Biol. Chem., 261, 1542–1552. [PubMed] [Google Scholar]
- Prudovsky I.A., Savion,N., LaVallee,T.M. and Maciag,T. (1996) The nuclear trafficking of extracellular fibroblast growth factor (FGF)-1 correlates with the perinuclear association of the FGF receptor-1α isoforms but not the FGF receptor-1β isoforms. J. Biol. Chem., 271, 14198–14205. [DOI] [PubMed] [Google Scholar]
- Rapak A., Falnes,P.O. and Olsnes,S. (1997) Retrograde transport of mutant ricin to the endoplasmic reticulum with subsequent translocation to cytosol. Proc. Natl Acad. Sci. USA, 94, 3783–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss Y., Goldstein,J.L., Seabra,M.C., Casey,P.J. and Brown,M.S. (1990) Inhibition of purified p21ras farnesyl: protein transferase by Cys-AAX tetrapeptides. Cell, 62, 81–88. [DOI] [PubMed] [Google Scholar]
- Sandvig K. and Olsnes,S. (1982) Entry of the toxic proteins abrin, modeccin, ricin, and diphtheria toxin into cells. II. Effect of pH, metabolic inhibitors, and ionophores and evidence for toxin penetration from endocytotic vesicles. J. Biol. Chem., 257, 7504–7513. [PubMed] [Google Scholar]
- Sinensky M., Fantle,K., Trujillo,M., McLain,T., Kupfer,A. and Dalton,M. (1994) The processing pathway of prelamin A. J. Cell Sci., 107, 61–67. [DOI] [PubMed] [Google Scholar]
- Skjerpen C.S., Nilsen,T., Wesche,J. and Olsnes,S. (2002a) Binding of FGF-1 variants to protein kinase CK2 correlates with mitogenicity. EMBO J., 21, 4058–4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skjerpen C.S., Wesche,J. and Olsnes,S. (2002b) Identification of ribosome-binding protein p34 as an intracellular protein that binds acidic fibroblast growth factor. J. Biol. Chem., 277, 23864–23871. [DOI] [PubMed] [Google Scholar]
- Sonawane N.D., Thiagarajah,J.R., and Verkman,A.S. (2002) Chloride concentration in endosomes measured using a ratioable fluorescent Cl- indicator: evidence for chloride accumulation during acidification. J. Biol. Chem., 277, 5506–5513. [DOI] [PubMed] [Google Scholar]
- Tarantini F., LaVallee,T., Jackson,A., Gamble,S., Carreira,C.M., Garfinkel,S., Burgess,W.H. and Maciag,T. (1998) The extravesicular domain of synaptotagmin-1 is released with the latent fibroblast growth factor-1 homodimer in response to heat shock. J. Biol. Chem., 273, 22209–22216. [DOI] [PubMed] [Google Scholar]
- Van Dyke R.W. (1988) Proton pump-generated electrochemical gradients in rat liver multivesicular bodies. Quantitation and effects of chloride. J. Biol. Chem., 263, 2603–2611. [PubMed] [Google Scholar]
- Wesche J., Elliott,J.L., Falnes,P.O., Olsnes,S. and Collier,R.J. (1998) Characterization of membrane translocation by anthrax protective antigen. Biochemistry, 37, 15737–15746. [DOI] [PubMed] [Google Scholar]
- Wesche J., Rapak,A. and Olsnes,S. (1999) Dependence of ricin toxicity on translocation of the toxin A-chain from the endoplasmic reticulum to the cytosol. J. Biol. Chem., 274, 34443–34449. [DOI] [PubMed] [Google Scholar]
- Wesche J., Więdłocha,A., Falnes,P.O., Choe,S. and Olsnes,S. (2000) Externally added aFGF mutants do not require extensive unfolding for transport to the cytosol and the nucleus in NIH 3T3 cells. Biochemistry, 39, 15091–15100. [DOI] [PubMed] [Google Scholar]
- Więdłocha A., Falnes,P.O., Madshus,I.H., Sandvig,K. and Olsnes,S. (1994) Dual mode of signal transduction by externally added acidic fibroblast growth factor. Cell, 76, 1039–1051. [DOI] [PubMed] [Google Scholar]
- Więdłocha A., Falnes,P.O., Rapak,A., Klingenberg,O., Munoz,R. and Olsnes,S. (1995) Translocation of cytosol of exogenous, CAAX-tagged acidic fibroblast growth factor. J. Biol. Chem., 270, 30680–30685. [DOI] [PubMed] [Google Scholar]
- Więdłocha A., Falnes,P.O., Rapak,A., Munoz,R., Klingenberg,O. and Olsnes,S. (1996) Stimulation of proliferation of a human osteosarcoma cell line by exogenous acidic fibroblast growth factor requires both activation of receptor tyrosine kinase and growth factor internalization. Mol. Cell. Biol., 16, 270–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M.M., Grabe,M., Adams,S., Tsien,R.Y., Moore,H.P. and Machen,T.E. (2001) Mechanisms of pH regulation in the regulated secretory pathway. J. Biol. Chem., 276, 33027–33035. [DOI] [PubMed] [Google Scholar]
- Yoshida T., Chen,C.C., Zhang,M.S. and Wu,H.C. (1991) Disruption of the Golgi apparatus by brefeldin A inhibits the cytotoxicity of ricin, modeccin, and Pseudomonas toxin. Exp. Cell Res., 192, 389–395. [DOI] [PubMed] [Google Scholar]
- Zhan X., Hu,X., Friedman,S. and Maciag,T. (1992) Analysis of endogenous and exogenous nuclear translocation of fibroblast growth factor-1 in NIH 3T3 cells. Biochem. Biophys. Res. Commun., 188, 982–991. [DOI] [PubMed] [Google Scholar]
- Zhan X., Hu,X., Friesel,R. and Maciag,T. (1993) Long term growth factor exposure and differential tyrosine phosphorylation are required for DNA synthesis in BALB/c 3T3 cells. J. Biol. Chem., 268, 9611–9620. [PubMed] [Google Scholar]