

Abstract
Proteins that act at specific DNA sequences bind DNA randomly and then translocate to the target site. The translocation is often ascribed to the protein sliding along the DNA while maintaining continuous contact with it. Proteins also can move on DNA by multiple cycles of dissociation/reassociation within the same chain. To distinguish these pathways, a strategy was developed to analyze protein motion between DNA sites. The strategy reveals whether the protein maintains contact with the DNA as it transfers from one site to another by sliding or whether it loses contact by a dissociation/reassociation step. In reactions at low salt, the test protein stayed on the DNA as it traveled between sites, but only when the sites were <50 bp apart. Transfers of >30 bp at in vivo salt, and over distances of >50 bp at any salt, always included at least one dissociation step. Hence, for this enzyme, 1D sliding operates only over short distances at low salt, and 3D dissociation/reassociation is its main mode of translocation.
Keywords: DNA–protein interaction, recognition sequence, restriction enzyme
Genetic events such as replication, transcription, and restriction depend on proteins acting at specific DNA sites (1–5). The specific sequences constitute a minute fraction of genomic DNA so the protein is unlikely to collide with its target site by random diffusion through bulk solution (6, 7). Moreover, under in vivo conditions, proteins generally bind to specific sites at rates near the diffusion limit for protein–ligand associations, ≈1 × 108 M-1·s-1 (8). However, if a protein has to undergo many separate collisions with the DNA before hitting the target, the association rate could be reduced to far below this limit (9). [Protein–DNA association rates can seemingly exceed the diffusion limit (6, 10), but very rapid rates occur only at low salt and are mainly due to electrostatic factors (8).] Hence, the protein will first collide with a nonspecific site and then transfer to the specific site by an intramolecular process, by facilitated diffusion (6, 8, 9).
Three distinct pathways have been proposed for facilitated diffusion (8–11). In one, called “sliding,” the protein moves along the DNA while remaining in continuous contact with it, by a diffusional process confined to the linear contour of the DNA. The volume of space that the protein needs to search through is thus reduced to the 1D contour (9). However, the random directionality of thermal diffusion gives equal probabilities for forward and reverse steps, so the protein returns repeatedly to its start point (12). Consequently, the rate at which a protein locates its target site is accelerated if the protein slides over relatively short distances, up to ≈10 times the length of the target site, but 1D diffusion over longer distances slows down the rate of target site location (8).
In the second, “hopping/jumping,” the protein moves between sites through 3D space, by dissociating from its initial site before reassociating elsewhere in the same chain (8, 9, 11). After dissociating, the protein will spend some time in the vicinity of the initial site, because of its relatively small diffusion constant (6, 7), so most rebinding events will be short-range “hops” back to or near that site (11, 13). Such hops are distinct from 1D diffusion because even a 1-bp hop breaks the continuous contact between DNA and protein. Occasionally, the protein may “jump” to a new site many bp away from the starting site but that is still in the same chain rather than a different chain. The intramolecular nature of jumping is due to the distances between DNA molecules in solution being much larger than those between two segments of the same chain (8). Hence, a protein can diffuse away from the initial site yet still remain inside the domain of that DNA (14).
In a further scheme, “intersegmental transfer” (8–11), the protein moves from one site to another by transiently binding both at the same time; it then dissociates from one to leave itself on the other. Proteins that bind two sites do so most readily with sites ≈400 bp apart: shorter separations are impeded by the need to bend and/or twist the intervening DNA (15). Intersegmental transfer is thus limited to large steps on DNA by proteins that can bind two sites at the same time. It cannot apply to the system examined here, which involves translocations over short distances by a protein that binds one site at a time.
At present, facilitated diffusion is usually ascribed to 1D diffusion (16, 17). However, much of the experimental data that support sliding can be reconciled to other schemes. For example, the effects of DNA length on the kinetics and/or equilibria of the protein binding to its site are often correlated to 1D diffusion (17–19). However, to distinguish the schemes, it is necessary to vary not only the length of the DNA but also the affinity of the protein for nonspecific DNA (8, 11), the latter normally being implemented by varying the salt concentration (20). Otherwise, the length dependency reveals that the nonspecific DNA is on the path to the specific site, but not the actual pathway (18). Observations of single protein molecules on DNA, by fluorescence or microscopy (21–23), have likewise been reconciled to sliding, although the distinction between the schemes requires a time resolution of <1 ms [because the process may cover 103 bp/s (9)] and a spatial resolution of <1 nm (to see if the protein maintains contact as it takes 1-bp steps on the DNA). No current technique meets both requirements (24).
Conversely, several recent studies have shown that the transfer from nonspecific to specific sites involves multiple dissociation/reassociations (25–27). For instance, the processivity of the EcoRV nuclease, its ability to cleave two recognition sites during one DNA-binding event, decreased as the separation of the sites was increased from 50 to 750 bp in a manner that excluded sliding from being the only route (25). Instead, the data matched best a hopping/jumping scheme but where each reassociation is followed by the protein scanning every sequence for ≈25 bp on either side of the new site. In further experiments on a 3.7-kb plasmid with one recognition site, EcoRV located the site more readily on the supercoiled plasmid than on its relaxed form, which shows that it depends on the 3D rather than the 1D distance to the site (26). The plasmid was converted into a catenane with two interlinked rings of DNA, one of 3.4 kb with only nonspecific sequences and one of 0.3 kb with the recognition site. The transfer from nonspecific to specific site occurred equally well on the catenane, where most of the nonspecific DNA is tethered to, but not contiguous with, the target, as on the plasmid (26). Hence, many proteins seem to translocate along DNA over distances of ≥50 bp primarily by hopping/jumping, but sliding may still play a role in short-range transfers over ≤50 bp. Several theoretical studies also have concluded that the fastest route to a target site in a long DNA chain involves a combination of hopping/jumping steps interspersed by 1D sliding over short distances (8, 12, 28, 29).
An unanswered question about target-site location by proteins on DNA is thus the nature of short-range translocations, over distances of <100 bp. We describe here a strategy that reveals whether the protein maintains continuous contact with the DNA as it translocates from one site to another or whether it loses track of the path of the DNA between the sites one or more times during the transfer: i.e., the fraction of translocation events completed solely by 1D diffusion and the fraction that involves at least one dissociation/reassociation step.
Materials and Methods
Enzymes. The BbvCI restriction enzyme, and its 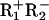 mutant, were purified as described in ref. 30, and their concentrations, in terms of the Mr 64053 R1R2 heterodimer, were determined from A280 readings (31). Other enzymes were from New England Biolabs.
mutant, were purified as described in ref. 30, and their concentrations, in terms of the Mr 64053 R1R2 heterodimer, were determined from A280 readings (31). Other enzymes were from New England Biolabs.
DNA. DNA manipulations were performed according to standard procedures (32) and checked by sequencing (University of Dundee, Dundee, U.K.). A duplex with the recognition sequence for BbvCI (underlined), was made from the oligonucleotides
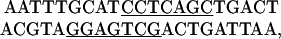 |
(MWG Biotech, Ebersberg, Germany) and ligated to EcoRI-cut pUC19 (33). One recombinant contained a single insert in the above orientation (i.e., with the CCTCAGC sequence in the “top” strand). Further derivatives were constructed from this plasmid by inserting into its polylinker a second duplex with a BbvCI site at the SacI, AvaI, BamHI, or HindIII site. For example, the duplex
 |
was cloned at the HindIII site to yield recombinants with two BbvCI sites 75 bp apart. Some had the insert in the same orientation as above (with CCTCAGC again in the top strand), whereas others had the inverted orientation (with CCTCAGC in the “bottom” strand). The duplexes inserted at the SacI, AvaI, and BamHI sites had the same sequence as the HindIII insert, apart from the “sticky ends,” and gave both repeated and inverted derivatives with two BbvCI sites separated by, respectively, 30, 40, and 45 bp.
For each plasmid, a 301-bp section that spanned the two BbvCI sites was amplified by PCR. The reactions contained 0.1 nM plasmid, 1 μM top and bottom strand primers, 0.37 MBq α32P-dATP (GE Healthcare, Chalfont St. Giles, U.K.), and PCR Master Mix (Promega). The two primers matched, respectively, loci 307–326 and 562–543 in pUC19 (33). The products were recovered with a PCR purification kit (Qiagen, West Sussex, U.K.), and a sample of each was analyzed on agarose gels alongside quantitative GIBCO/BRL markers. Their concentrations were determined by reference to the intensities of the markers (Kodak imagestation 1d software).
Reactions. Solutions at 37°C contained 30 nM 32P-labeled DNA (one of the above PCR fragments) in 100 μl of Reaction Buffer (10 mM Tris·HCl/5 mM MgCl2/1 mM 2-mercaptoethanol/100 μg/ml BSA, pH 7.4), sometimes supplemented with 30, 60, or 150 mM NaCl. An aliquot (5 μl) was taken at zero time and mixed with 10 μl of Stop Mix (26) before adding to the remainder BbvCI (diluted as described in ref. 31) to 0.3 nM. At intervals thereafter, further aliquots were taken, mixed immediately with Stop Mix, and subjected to electrophoresis through 8% polyacrylamide in TBE (32), in water-cooled 20-cm gels at 40 mA for 3 h. Gels were dried, exposed to a Molecular Dynamics phosphorimager, and analyzed in imagequant (Molecular Dynamics) to assess the amounts of each DNA in each sample. The total counts in rectangles encompassing each band were summed and corrected for local background, and the values were converted to DNA concentrations by reference to the number of adenines in the fragment (α32P-dATP was used in the PCR). Initial rates were evaluated from the first 15% of the reaction with grafit (Erithacus Software, Surrey, U.K.).
Nonspecific Binding. Varied concentrations of BbvCI (0–25 μM) were added to a 21-bp duplex (30 nM, 32P-labeled with kinase; ref. 32) in Reaction Buffer (except for CaCl2 instead of MgCl2) with NaCl as indicated. The duplex had the same sequence as those above, except that the BbvCI recognition sequence was replaced by the reverse, 5′-CGACTCC-3′. The fraction of the duplex bound to BbvCI was assessed by gel-retardation (34), and a KD for nonspecific binding was evaluated from the decline in this fraction with increasing [E0] (35).
Results
Experimental Strategy. The strategy developed here, to determine whether a protein maintains contact with the DNA as it moves from one site to another, requires a protein that interacts differently with each strand of the DNA. The BbvCI restriction enzyme meets this requirement, as a result of two of its properties (30). First, it recognizes a nonpalindromic site with different 5′-3′ sequences in each strand
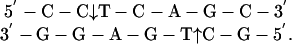 |
The two strands are named after the 5′ dinucleotides, CC and GC, respectively (31). In the presence of Mg2+, it cuts both, as shown. Second, it consists of two different subunits, R1 and R2. Neither subunit has any activity by itself, and the active form is the R1R2 heterodimer. The subunits are each specific for an individual strand: R1 cuts the GC strand and R2 cuts the CC strand (30). Certain mutations in the R1 subunit leave  proteins that cut only the CC strand; conversely,
proteins that cut only the CC strand; conversely, 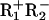 proteins cut only the GC strand. In the wild-type dimer, the R1 subunit cuts the GC strand ≈5 times faster than R2 the CC strand, but both reactions are faster than the subsequent dissociation from the DNA; BbvCI thus releases products cut in both strands (31).
proteins cut only the GC strand. In the wild-type dimer, the R1 subunit cuts the GC strand ≈5 times faster than R2 the CC strand, but both reactions are faster than the subsequent dissociation from the DNA; BbvCI thus releases products cut in both strands (31).
A series of 2.7-kb plasmids were constructed with two BbvCI sites separated by varied distances, from 30 to 75 bp. (Because the products from cutting the plasmids at one or both sites could not be separated from each other by electrophoresis, the reactions were studied on 301-bp fragments with both sites, generated by PCR across the relevant section of each plasmid.) To minimize local sequence effects, both sites had the same flanking sequences for 5 bp around the sites. For each construct, two derivatives were isolated: one with sites oriented in direct (head-to-tail) repeat so that one strand carries both copies of the CC sequence and the other both copies of GC, the targets for R2 and R1, respectively (Fig. 1a); another with sites in inverted (head-to-head) orientation so that the two CC sequences are in opposite strands, and likewise GC (Fig. 1b).
Fig. 1.

Experimental strategy. Shown is a DNA with two BbvCI sites 30 bp apart, in either directly repeated (a) or inverted (b) orientations, as marked by the arrowheads. The points of cleavage in each site are marked by white arrows connected by a jagged line. The distances between the scissile bonds are indicated for both CC → CC and GC → GC separations. In both a and b, the BbvCI restriction endonuclease is shown bound to both sites: its R1 and R2 subunits are shown as blue and green triangles, respectively, with dark blue and dark green hemispheres for their catalytic centers. The subunits are placed on their target strands, R1 on GC and R2 on CC. In both a and b, the minimal motion needed to reposition the enzyme from the left-hand to the right-hand site is shown with red arrow(s): a linear translocation between the directly repeated sites in a and a rotation around and a turnover along the DNA with the inverted sites in b.
For processive action, BbvCI must translocate from one site to the other without leaving the domain of the DNA. With directly repeated sites, this translocation could occur by the protein sliding from one site to the other. If both subunits maintain contact with the DNA, the R2 subunit remains in place to cleave its target phosphodiester bond in the CC strand and likewise the R1 subunit the GC strand (Fig. 1a). However, processive action by BbvCI on a DNA with inverted sites cannot, for the reasons given below, arise solely from sliding and must include a dissociation/reassociation step. Consequently, if the translocation from one site to the other occurs solely by sliding, processivity will be observed with repeated sites but not with inverted sites. If it occurs mainly by sliding, processivity will be observed on both repeated and inverted sites although at a higher level on the repeated sites. However, if the translocation always involves a dissociation step, the extent of processivity on repeated sites will equal that on inverted sites.
If the BbvCI enzyme stays in contact with the DNA as it translocates between inverted sites, both its R1 and its R2 subunits face at the second site not only the “wrong” strand but also the wrong phosphodiester bond (Fig. 1b). To act processively on inverted sites, the protein must rotate around the helical axis of the DNA, to switch the R2 and the R1 subunits to the opposite strands, but it also must turn itself over from left to right along the DNA, to switch the active sites of both subunits onto the target bonds in their respective strands. For example, after BbvCI has cleaved the left-hand site in Fig. 1b and translocated to the right-hand site, a simple rotation of the R2 subunit around the long axis of the DNA will place that subunit onto its cognate CC strand, although in the wrong polarity. Its active site then will be opposite the second phosphodiester bond from the left, the ApG step in the bottom strand. However, to cleave the inverted sequence, the active site of R2 must engage the fifth phosphodiester bond in the bottom strand, the CpT step.
The rotation around the DNA could conceivably occur while the protein remains bound to it. However, the left-to-right turnaround along the DNA cannot occur while bound to DNA, because it requires the transmutation of protein through DNA, unless the DNA-binding surface of the protein is a flat or a convex plane with no protrusions. If the DNA-binding surface is a flat plane, then it may be possible to turn the DNA around from left to right while keeping it parallel to the plane, but it is physically impossible to turn the DNA around at any angle to the plane. However, protein surfaces are seldom, if ever, smooth planes with no protrusions. If the DNA binds in a concave depression in the protein surface, or if part of the protein protrudes into or around the DNA, then the rotation in the plane of the DNA-binding surface is blocked by steric hindrance. For example, if the DNA binds in a cleft within the protein structure, as is the case with the restriction enzymes bound to specific or nonspecific DNA (5), it may be possible to rotate the protein around the helical axis of the DNA following the screw axis of the helix (23), but it cannot be rotated around any other axis without first taking the DNA completely out of the cleft. Even if the protein just protrudes into the DNA grooves, as is usually the case with DNA-binding proteins (1), the rotation of the DNA around any axis other than its long axis is still blocked. The protein thus can reorient itself relative to the second site only when it is physically separated from the DNA by a sufficient distance to allow for the turnover without encountering a steric block from the protrusions on the surface of the protein and/or the DNA.
After the physical separation of protein from DNA, the protein will reassociate more often with the same DNA than with a different molecule (8, 11). Directly after the dissociation of a DNA–protein complex, the protein may remain near that DNA for some time while sampling all possible orientations relative to the DNA (13). This situation arises because a protein at a particular position in an aqueous solution will, as a result of diffusion, tumble relatively rapidly around that position but only rarely undergo lateral motion to a new position (6–8). Hence, even though the action of BbvCI on a DNA with two inverted sites requires its dissociation from the DNA before cutting the second site, it still has the potential to act processively on such a DNA. Extrapolation of data from the EcoRV nuclease (14, 25) suggests that, after leaving its recognition site on a 301-bp DNA (as here), it may reassociate with the same DNA 10–100 times before escaping to bulk solution.
In the substrates made here, the distances between the BbvCI sites are defined by the number of bp separating the target bonds in the CC sequences rather than the GC sequences. For substrates with repeated sites, the scissile bonds in the CC and the GC strands are the same distance apart (Fig. 1a). For sites in inverted (head-to-head) orientation, the GC → GC separation targeted by the R1 subunit is 6 bp shorter than the CC → CC span for the R2 subunit (Fig. 1b). The GC → GC distance is, however, immaterial, for the following reasons. First, the assays described below measure products with double-strand breaks, and the rate at which wild-type BbvCI cuts both strands is limited by R2 on the CC strand, not by R1 on the GC strand (31). Second, some experiments were done with DNA that had been nicked at both sites in the GC sequence by using the 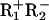 mutant of BbvCI (30). The nicked DNA was tested with wild-type BbvCI, which used its
mutant of BbvCI (30). The nicked DNA was tested with wild-type BbvCI, which used its 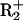 subunit to complete the double-strand break. The levels of processivity on the nicked DNA, which requires only the CC → CC transfer, were similar to those on the intact DNA, which require both CC → CC and GC → GC transfers (data not shown).
subunit to complete the double-strand break. The levels of processivity on the nicked DNA, which requires only the CC → CC transfer, were similar to those on the intact DNA, which require both CC → CC and GC → GC transfers (data not shown).
BbvCI Reactions. On a linear DNA with two sites, the first hit may be a distributive reaction at site 1 alone, to yield fragments A and BC, or at site 2 alone, to give AB and C (Fig. 2a). If these are the only possibilities, the initial rate for forming A (vA) must equal that for BC (vBC); likewise, vAB and vC (19, 25). [In addition, vBC will match vAB if the sites are equally susceptible, but not otherwise (19). Because the sites are both near the middle of the DNA (Fig. 2a) and are flanked by identical sequences, they should be equal. The similarity of vBC and vAB (namely, Fig. 2c) show that they are indeed equal.] After the initial reactions, the species carrying a BbvCI site, BC and AB, can be cleaved in subsequent reactions to yield their component fragments, but these reactions have no effect on the initial rates of product formation. However, instead of distributive reactions at single sites, the first hit may be a processive reaction to yield directly the three final fragments by cutting both before leaving the DNA. If so, the yield of BC will fall below that of A (as in Fig. 2c): likewise, AB relative to C. The proportion of processive reactions, relative to the total number of reactions, can be calculated from
 |
where fP is the processivity factor (expressed here as a %) (19, 25).
Fig. 2.

BbvCI reactions. (a) Shown as ABC is a 301-bp DNA with two BbvCI sites in inverted orientation 75 bp apart. The sites, 1 and 2, have the scissile bonds in the CC sequence at positions 101 and 173, respectively. The initial reaction of BbvCI on this DNA has three possible outcomes: cleavage at site 1 alone to give fragments A (in black) and BC (in blue); at 2 alone for fragments AB (in red) and C (in green); processive cutting at both 1 and 2 to give fragments A, B (in purple), and C. (b and c) BbvCI endonuclease (0.3 nM) was added to the above DNA (32P-labeled, 30 nM) in Reaction Buffer at 37°C. Samples were taken at the indicated times and subjected to electrophoresis through polyacrylamide. b shows a phosphorimager record of the gel, annotated on the right with the mobilities (and sizes) of all of the DNA species from the reaction. For c, the concentrations of the following fragments were evaluated from the record: A, black diamonds; BC, blue circles; AB, red triangles; C, green squares. The dotted lines through each data set are the optimal fits to linear slopes to give, respectively, vA, vBC, vAB, and vC.
Reactions on 301-bp substrates with two BbvCI sites, in either repeated or inverted orientation, were carried out under steady-state conditions with a 100-fold molar excess of DNA over enzyme (Fig. 2). At this DNA:enzyme ratio, virtually no DNA molecules bind two molecules of enzyme at the same time, so the direct cleavage of the DNA at both sites can only be due to processivity and not to separate events at solitary sites. Samples from the reactions were analyzed by electrophoresis through nondenaturing polyacrylamide under conditions that separate the substrate and all five of the possible reaction products from each other (Fig. 2b). [The DNA with inverted sites 75 bp apart yields a central product (B) of 72 bp that is seen on the gel (Fig. 2b). The substrates with shorter intersite spacing sites gave central products that migrated off the gel.] The DNA was labeled with 32P, and the amounts of the substrate and products in each sample were quantified (Fig. 2c). To ensure that the reaction velocities correspond to initial rates, only data from the first 15% of DNA cleavage were used.
Length and Salt Dependencies. Four pairs of substrates were tested. In one, the two BbvCI sites were separated by 30 bp; in the others, they were separated by 40, 45, and 75 bp. In each pair, one substrate had sites in direct repeat; the other had sites in inverted orientation. All four pairs were examined under four sets of conditions: without NaCl and with 30, 60, or 150 mM NaCl. The concentration of MgCl2 was kept constant at 5 mM; experiments at 10 mM MgCl2 gave much lower fP values (data not shown). In all cases, the initial velocities, vA, vBC, vAB, and vC, were measured as in Fig. 2, and the values were converted to processivity factors (Table 1). For each intersite spacing and reaction condition, the fP value across repeated sites (R) was compared with that on inverted sites (I) by taking the ratio, R/I (Fig. 3).
Table 1. Processivity values.
| Separation, bp
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 30
|
40
|
45
|
75
|
|||||||||
| NaCl, mM | R | I | R/I | R | I | R/I | R | I | R/I | R | I | R/I |
| 0 | 46 ± 2 | 33 ± 4 | 1.39 | 44 ± 3 | 33 ± 1 | 1.33 | 47 ± 3 | 35 ± 4 | 1.34 | 40 ± 2 | 42 ± 2 | 0.95 |
| 30 | 36 ± 2 | 29 ± 3 | 1.24 | 36 ± 1 | 27 ± 3 | 1.33 | 38 ± 2 | 31 ± 1 | 1.23 | 32 ± 6 | 33 ± 2 | 0.97 |
| 60 | 29 ± 3 | 25 ± 5 | 1.16 | 25 ± 1 | 22 ± 1 | 1.14 | 28 ± 8 | 23 ± 3 | 1.21 | 23 ± 6 | 22 ± 1 | 1.05 |
| 150 | 15 ± 1 | 15 ± 1 | 1.0 | 14 ± 1 | 14 ± 3 | 1.0 | 13 ± 3 | 13 ± 2 | 1.0 | 11 ± 2 | 11 ± 1 | 1.0 |
The substrates were 301-bp fragments with two BbvCI sites separated by the number of bp noted, in either repeated or inverted orientations. Reactions contained 0.3 nM BbvCI endonuclease and 30 nM DNA in reaction buffer with NaCl as indicated. Processivity factors on repeated and inverted sites, R and I, were calculated from the initial rates of product formation; the ratios of the values, R/I, are noted in bold. Error margins denote standard deviations from at least four repeats.
Fig. 3.

Processivity ratios. The reactions contained BbvCI and a DNA with two sites in repeated or inverted orientations separated by one of the distances noted below and NaCl as indicated. For each intersite distance and NaCl concentration, the R/I ratio of fP values on repeated sites to inverted sites was calculated as follows: sites 30 bp apart, black circles and line; 40 bp, white triangles; 45 bp, white squares; 75 bp, black diamonds and line.
On the DNA with two sites 30 bp apart, BbvCI displayed in the absence of NaCl a high level of processivity on the substrate with repeated sites but a lower level on the isogenic substrate with inverted sites: the fP on inverted sites was ≈70% of that on repeated sites (Table 1). If the enzyme cuts one site on a linear DNA with two sites and then leaves that site by sliding onto the adjacent DNA with no directional preference, the maximal fP is 50%, because only 50% of the enzyme molecules head toward the intact site (19). In the absence of NaCl, the processivity observed on the DNA with repeated sites 30 bp apart, 46%, is close to the maximum. The levels of processivity on both substrates with sites 30 bp apart fell progressively as the NaCl concentration was raised, but processivity was still detected at 150 mM salt: ≈15% of the reactions were then processive. Strikingly, as the salt was raised, the difference between repeated and inverted sites declined, and no difference in fP values was detected in 150 mM NaCl. The R/I ratio thus fell from a value of 1.4 in the absence of NaCl to 1.0 in the presence of 150 mM NaCl, with intermediate values at 30 and 60 mM NaCl (Fig. 3).
Increasing the length of DNA between the sites from 30 to 40 or 45 bp, distances that approximate to 1 or 1.5 extra helical turns, had little effect on the fP values (Table 1). As with the 30-bp separation, the DNA molecules with sites 40 or 45 bp apart gave high levels of processivity in the absence of NaCl and progressively reduced levels at higher ionic strengths. Once again, they gave higher fP values on repeated sites than on inverted sites at low salt but very similar values for the two orientations at higher salt concentrations. Their R/I ratios thus declined as the salt was raised, from ≈1.3 in the absence of NaCl to 1.0 in the presence of 150 mM NaCl (Fig. 3).
The two substrates with 75 bp between the sites also were cleaved with high processivity in reactions lacking salt and with progressively lower levels of processivity at elevated ionic strengths (Table 1). However, in marked contrast to the shorter separations, the DNA with repeated sites 75 bp apart gave, at all NaCl concentrations tested, fP values that were virtually identical to those on the equivalent DNA with inverted sites (Table 1). Even in the absence of NaCl, the R/I ratio from the two substrates with sites 75 bp apart was close to 1.0; it had been 1.4 for the DNA with 30 bp between the sites (Fig. 3).
At 30 and 60 mM NaCl, the fP values for repeated sites 75 bp apart were similar to those for inverted sites 40 or 45 bp apart and were smaller than those on repeated sites 40 or 45 bp apart (Table 1). BbvCI thus translocates with similar efficiencies between two inverted sites 40–45 bp apart as it does between either repeated or inverted sites 75 bp apart. Presumably, these processes all proceed by the same pathway, whereas the higher efficiency observed on repeated sites 40–45 bp apart indicates an additional pathway.
The levels of processivity on both repeated and inverted sites declined as the salt was raised (Table 1). To see whether this result was due to salt reducing the affinity of BbvCI for nonspecific DNA, the binding of BbvCI to a 21-bp duplex that lacked the recognition sequence was examined by gel-retardation. The binding buffers were the same as the reaction buffers but with CaCl2 as a noncatalytic cofactor in place of MgCl2. BbvCI bound to the nonspecific duplex with KD values of ≈100 nM in either zero or 30 mM NaCl, ≈200 nM in 60 mM NaCl, and ≈500 nM in 150 mM NaCl (data not shown). The decline in processivity thus can be assigned to the effect of salt on weakening nonspecific binding. The affinities and the salt dependency of nonspecific DNA binding by BbvCI are comparable with other restriction enzymes (34–37).
Discussion
Facilitated Diffusion by BbvCI. The properties of a recently characterized restriction enzyme, BbvCI (30, 31), yielded a general method for analyzing the diffusional translocation of proteins on DNA. Many restriction enzymes are dimers of identical subunits that recognize palindromic sequences (4, 5), but BbvCI is a heterodimer that recognizes a nonpalindromic site. The restriction enzymes that recognize nonpalindromic targets are often monomers (5), and two monomers, each bound to a separate recognition site (38), have to associate to cut the DNA. In contrast, a single molecule of the BbvCI enzyme cleaves both strands of the DNA at an individual site by using different subunits to attack each strand: its R2 subunit cuts the CC strand, and R1 cuts the GC strand (30, 31). Consequently, the degree of processivity of BbvCI on DNA with two sites in repeat orientation, relative to the isogenic DNA with inverted sites (Fig. 1), reveals whether the enzyme stays in contact with the DNA as it moves from one site to another or whether it loses track of the path of the intervening DNA during the transfer.
If the sole pathway for the intramolecular transfer of a protein along DNA is 1D diffusion, then BbvCI may be able to cut the DNA with repeated sites processively, but it can only cut the DNA with inverted sites by distributive reactions at each site, and not by processivity. Conversely, if the protein dissociates from the DNA at least once during the transfer, then BbvCI may still be able to act processively on the two-site DNA, by reassociating back to the same chain. The DNA with inverted sites then must give the same level of processivity as the DNA with repeated sites. A third possibility is that some translocation events involve only sliding steps, whereas others include one or more hopping/jumping step(s). In this case, the fP value for inverted sites will be lower than that on repeated sites, but not zero. The ratios of the fP values on repeated and inverted sites (Fig. 3) thus indicate the contributions of 1D sliding and 3D hopping/jumping to the translocation of the protein along DNA
Under certain circumstances, higher levels of processivity were observed on DNA molecules with two BbvCI sites in repeat orientation than on the isogenic DNA with sites in inverted orientation (Table 1). This result proves that, on a DNA with two sites, the BbvCI endonuclease can cleave one site and then translocate to the other while staying in contact with the DNA throughout the transfer. It has been argued that an enzyme catalyzing processive reactions on a DNA with multiple sites may move along DNA by a different mechanism from that during its motion from its initial random site to its first target site; in particular, that processivity cannot arise from sliding if there is an obligatory dissociation at the end of each catalytic cycle (17). The data presented here show that this scheme is not the case: processivity by BbvCI can involve 1D sliding. However, the differences in the fP values for repeated and inverted sites were observed only with closely spaced sites, up to 45 bp apart, but not at 75 bp, and at low ionic strengths, at 60 mM NaCl, but not at 150 mM. BbvCI thus translocates along DNA by 1D diffusion only over short distances, <75 bp, at low salt concentrations, <150 mM NaCl.
Outside these limits, its motion from one site to another always involves at least one event where the enzyme loses track of the path of the DNA between the sites, i.e., a dissociation/reassociation step. Buffers containing 150 mM NaCl exclude 1D diffusion over distances of ≥30 bp yet have a lower ionic strength than the cytoplasm of Escherichia coli (39). If the cytoplasm of the bacterium that produces the BbvCI enzyme, Bacillus brevis, has a similar composition to that of E. coli, then it is unlikely that 1D diffusion plays a significant role for BbvCI in vivo. Moreover, elevated fP values from repeated sites, relative to inverted sites, do not necessarily imply no reorientation events during translocations between repeated sites and one or more reorientations with the inverted sites, as proposed here. Instead, the difference in fP values could be due to x reorientation events with repeated sites and x + y events on inverted sites, where x and y are even and odd numbers, respectively. If so, the distance that this protein can travel along DNA by 1D diffusion will be even shorter than the 30-bp limit identified here.
Facilitated Diffusion by Other Systems. This work describes just one DNA-binding protein, the BbvCI restriction endonuclease. Nevertheless, the results obtained with this protein match closely the general conclusions from a number of recent theoretical studies of DNA–protein association rates (8, 28, 29). In particular, at the high DNA concentrations found in vivo, the maximal association rate arises when sliding is limited to relatively short distances, typically ≈10 times longer than the length of the target sequence (8). Hence, the overall conclusions from this study, that proteins find their target sites primarily by hopping/jumping interspersed with sliding over short distances, may be applicable to many DNA-binding proteins. However, the actual path length for sliding, and the effect of salt on this process, are likely to vary from one protein to the next. Some DNA-binding proteins may stay in contact with the DNA for translocations over longer distances than BbvCI, as reported for certain proteins in low salt buffers (10, 18), whereas others may dissociate after shorter distances. The key factor in determining the mean path length for 1D diffusion by a protein on DNA is its affinity for nonspecific DNA (8–11). In this respect, BbvCI is comparable with other restriction enzymes (34–37).
The above strategy for analyzing facilitated diffusion originated from the properties of BbvCI, but it is applicable to any enzyme that interacts differently with each DNA strand. For example, it could be applied to a repair enzyme that removes damaged bases from DNA, by examining its processivity on substrates with two target bases, either one in each strand or two in one strand. Likewise, it could be used on enzymes that act at hemi-methylated sites, by constructing DNA substrates where either some sites are methylated in one strand and others in the opposite strand or where one strand is methylated at all sites and the other fully unmethylated, as is the case in vivo after the semiconservative replication of fully methylated DNA (4, 5).
Acknowledgments
We thank Stuart Bellamy, Lucy Catto, Mark Dillingham, Mark Szczelkun, and John Marko for aid and advice. This work was funded by Wellcome Trust Grant 063111/Z/00.
Author contributions: D.M.G. and S.E.H. designed research and analyzed data; D.M.G. performed research; G.G.W. contributed new reagents/analytic tools; and S.E.H. wrote the paper.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Garvie, C. W. & Wolberger, C. (2001) Mol. Cell 8, 937-946. [DOI] [PubMed] [Google Scholar]
- 2.Stillman, B. (2005) FEBS Lett. 579, 877-884. [DOI] [PubMed] [Google Scholar]
- 3.Browning, D. F. & Busby, S. J. (2004) Nat. Rev. Microbiol. 2, 57-65. [DOI] [PubMed] [Google Scholar]
- 4.Roberts, R. J., Vincze, T., Posfai, J. & Macelis, D. (2005) Nucleic Acids Res. 33, D230-D232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pingoud, A., Fuxreiter, M., Pingoud, V. & Wende, W. (2005) Cell. Mol. Life Sci. 62, 685-707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berg, O. G. & von Hippel, P. H. (1985) Annu. Rev. Biophys. Biophys. Chem. 14, 131-160. [DOI] [PubMed] [Google Scholar]
- 7.Berg, H. C. (1993) Random Walks in Biology (Princeton Univ. Press, Princeton).
- 8.Halford, S. E. & Marko, J. F. (2004) Nucleic Acids Res. 32, 3040-3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.von Hippel, P. H. & Berg, O. G. (1989) J. Biol. Chem. 264, 675-678. [PubMed] [Google Scholar]
- 10.Winter, R. B., Berg, O. G. & von Hippel, P. H. (1981) Biochemistry 20, 6961-6977. [DOI] [PubMed] [Google Scholar]
- 11.Berg, O. G., Winter, R. B. & von Hippel, P. H. (1981) Biochemistry 20, 6929-6948. [DOI] [PubMed] [Google Scholar]
- 12.Gerland, U., Moroz, J. D. & Hwa, T. (2002) Proc. Natl. Acad. Sci. USA 99, 12015-12020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berg, O.G. (1978) Chem. Phys. 31, 47-57. [Google Scholar]
- 14.Baldwin, G. S., Sessions, R. B., Erskine, S. G. & Halford, S. E. (1999) J. Mol. Biol. 288, 87-103. [DOI] [PubMed] [Google Scholar]
- 15.Bellomy, G. R. & Record, M. T., Jr. (1990) Prog. Nucleic Acids Res. Mol. Biol. 30, 81-128. [DOI] [PubMed] [Google Scholar]
- 16.Shimamoto, N. (1999) J. Biol. Chem. 274, 15293-15296. [DOI] [PubMed] [Google Scholar]
- 17.Jeltsch, A. & Urbanke, C. (2004) in Restriction Endonucleases, Nucleic Acids and Molecular Biology, ed. Pingoud, A. (Springer, Heidelberg, Germany), Vol. 14, pp. 95-110. [Google Scholar]
- 18.Jack, W. E., Terry, B. J. & Modrich, P. (1982) Proc. Natl. Acad. Sci. USA 79, 4010-4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terry, B. J., Jack, W. E. & Modrich, P. (1985) J. Biol. Chem. 260, 13130-13137. [PubMed] [Google Scholar]
- 20.Record, M. T., Jr., Zhang, W. & Anderson, C. F. (1998) Adv. Protein Chem. 51, 281-353. [DOI] [PubMed] [Google Scholar]
- 21.Harada, Y., Funatsu, T., Murakami, K., Nonoyama, Y., Ishihama, A. & Yanagida, T. (1999) Biophys. J. 76, 709-715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guthold, M., Zhu, X., Rivetti, C., Yang, G., Thomson, N. H., Kasas, S., Hansma, H. G., Smith, B., Hansma, P. K. & Bustamante, C. (1999) Biophys. J. 77, 2284-2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sakata-Sogawa, K. & Shimamoto, N. (2004) Proc. Natl. Acad. Sci. USA 101, 14731-14735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yildiz, A., Tomishige, M., Vale, R. D. & Selvin, P. R. (2004) Science 303, 676-678. [DOI] [PubMed] [Google Scholar]
- 25.Stanford, N. P., Szczelkun, M. D., Marko, J. F. & Halford, S. E. (2000) EMBO J. 19, 6546-6557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gowers, D. M. & Halford, S. E. (2003) EMBO J. 22, 1410-1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu, W. Y., Thompson, W., Lawrence, C. E. & Derbyshire, K. M. (2001) J. Mol. Biol. 306, 403-416. [DOI] [PubMed] [Google Scholar]
- 28.Coppey, M., Bénichou, O., Voituriez, R. & Moreau, M. (2004) Biophys. J. 87, 1640-1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou, H.-X. (2005) Biophys. J. 88, 1608-1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heiter, D. F., Lunnen, K. D. & Wilson, G. G. (2005) J. Mol. Biol. 348, 631-640. [DOI] [PubMed] [Google Scholar]
- 31.Bellamy, S. R., Milsom, S. E., Scott, D. J., Daniels, L. E., Wilson, G. G. & Halford, S. E. (2005) J. Mol. Biol. 348, 641-653. [DOI] [PubMed] [Google Scholar]
- 32.Sambrook, J. & Russsell, D. W. (2001) Molecular Cloning: A Laboratory Manual (Cold Spring Harbor Lab. Press, Woodbury, NY).
- 33.Yanisch-Perron, C., Vieira, J. & Messing, J. (1985) Gene 33, 103-119. [DOI] [PubMed] [Google Scholar]
- 34.Vipond, I. B. & Halford, S. E. (1995) Biochemistry 34, 1113-1119. [DOI] [PubMed] [Google Scholar]
- 35.Taylor, J. D., Badcoe, I. G., Clarke, A. R. & Halford, S. E. (1991) Biochemistry 30, 8743-8753. [DOI] [PubMed] [Google Scholar]
- 36.Martin, A. M., Horton, N. C., Luseti, S., Reich, N. O. & Perona, J. J. (1999) Biochemistry 38, 8430-8439. [DOI] [PubMed] [Google Scholar]
- 37.Engler, L. E., Sapienza, P., Dorner, L. F., Kucera, R., Schildkraut, I. & Jen-Jacobson, L. (2001) J. Mol. Biol. 307, 619-636. [DOI] [PubMed] [Google Scholar]
- 38.Bath, A. J., Milsom, S. E., Gormley, N. A. & Halford, S. E. (2002) J. Biol. Chem. 277, 4024-4033. [DOI] [PubMed] [Google Scholar]
- 39.Richey, B., Cayley, D. S., Mossing, M. C., Kolka, C., Anderson, C. F., Farrar, T. C. & Record, M. T., Jr. (1987) J. Biol. Chem. 262, 7157-7164. [PubMed] [Google Scholar]