

Short abstract
The sequencing of genomes provides us with an inventory of the 'molecular parts' in nature, such as protein families and folds, and their functions in living organisms. The genomic occurrence of generalized parts follows a well-known mathematical framework called the power law, with a few parts occurring many times and most occurring only a few times. Cases of power-law behavior, were found, for example in the occurrence of pseudogenes and in levels of gene expression.
Abstract
Background
The sequencing of genomes provides us with an inventory of the 'molecular parts' in nature, such as protein families and folds, and their functions in living organisms. Through the analysis of such inventories, it has been shown that different genomes have very different usage of parts; for example, the common folds in the worm are very different from those in Escherichia coli.
Results
Despite these differences, we find that the genomic occurrence of generalized parts follows a well-known mathematical framework called the power law, with a few parts occurring many times and most occurring only a few times. This observation is true in a wide variety of genomic contexts. Earlier studies found power laws in a few specific cases, such as the occurrence of protein families. Here, we find many further cases of power-law behavior, for example in the occurrence of pseudogenes and in levels of gene expression. We show comprehensively that this behavior applies across many different genomes, for many different types of parts (DNA words, InterPro families, protein superfamilies and folds, pseudogene families and pseudomotifs), and for the many disparate attributes associated with these parts (their functions, interactions and expression levels).
Conclusions
Power-law behavior provides a concise mathematical description of an important biological feature: the sheer dominance of a few members over the overall population. We present this behavior in a unified framework and propose that all these observations are connected to an underlying DNA duplication process as genomes evolved to their current state.
Background
Power-law behaviors have been observed in many different population distributions. Also known as Zipf's law [1], one of the most famous examples is the usage of words in text documents [1]. On grouping words that occur in similar numbers, it was noted that a small selection of words such as 'the' and 'of', are used many times, while most other words are used infrequently. When the size of each group is plotted against its usage, the distribution can be approximated to a power-law function; that is, the number of words (N) with a given occurrence (F) decays according to the equation N = aF-b. This distribution has a linear appearance when plotted on double-logarithmic axes, where -b describes the slope. (Strictly speaking, Zipf's law plots the frequency of occurrence of a word against its rank, where words are ranked according to their occurrence. In addition, the exponent b in the power-law function must be close to 1. Here instead of the rank, we plot the number of words with similar occurrences and do not require an exponent of 1). Some other examples of this behavior include income levels [1], relative sizes of cities [1] and the connectivity of nodes in large networks [2] such as the World Wide Web [3].
In regard to genomic biology, Mantegna et al. [4] discussed the fact that the usage of short base sequences in DNA, or 'DNA words', also follows the power law. They concluded that the behavior applies better to non-coding than to protein-coding sequences and suggested that non-coding DNA resembles a natural language. Further instances cited in genomic biology include the occurrence of protein families or folds [5,6,7,8,9], the connectivity within metabolic pathways [10] and the number of intra- and intermolecular interactions made by proteins [11,12,13].
From the analysis of over 20 of the first genomes sequenced, we show that power-law behavior is prominent throughout genomic biology. As noted above, previous studies have cited the occurrence of power-law behaviors for individual properties. Here we report the behavior for further genomic properties that have not previously been found; in particular, for the occurrence of pseudogene and pseudomotif populations in the intergenic regions of genomes, the number of protein functions associated with a particular fold, and the number of expressed mRNA transcripts within a cell. Furthermore, we bring together all the individual observations within a single framework and demonstrate that the power-law behavior is prevalent across most different genomic properties. Finally, in presenting these data, we discuss the significance of power laws in biology and discuss several models that aim to describe how genomes evolved to their current states to produce this type of behavior.
Results
Genomic occurrence of 'mers, families and folds
We start with the usage of short DNA sequences in genomes; we consider DNA words of size n, termed n-mers, and count the occurrence of distinct words by shifting across the entire genome one base at a time. By grouping the different 'mers by their occurrences, we observe that the occurrence of 6- to 10-mers displays power-law-like behavior. Figure 1a shows the distributions of 6- to 10-mers in the Caenorhabditis elegans genome. The distribution for each 'mer is staggered, which, unsurprisingly, indicates that shorter words have a higher average occurrence in the genome than longer ones. A more unexpected feature of the plot is that the slopes for the different-length words are nearly identical (b = 3.2), indicating that the number of 'mers with given occurrences fall at similar rates regardless of their length (Table 1). Moreover, we find that 'mers in both coding and non-coding regions follow the power-law distribution equally well (see Additional data).
Figure 1.
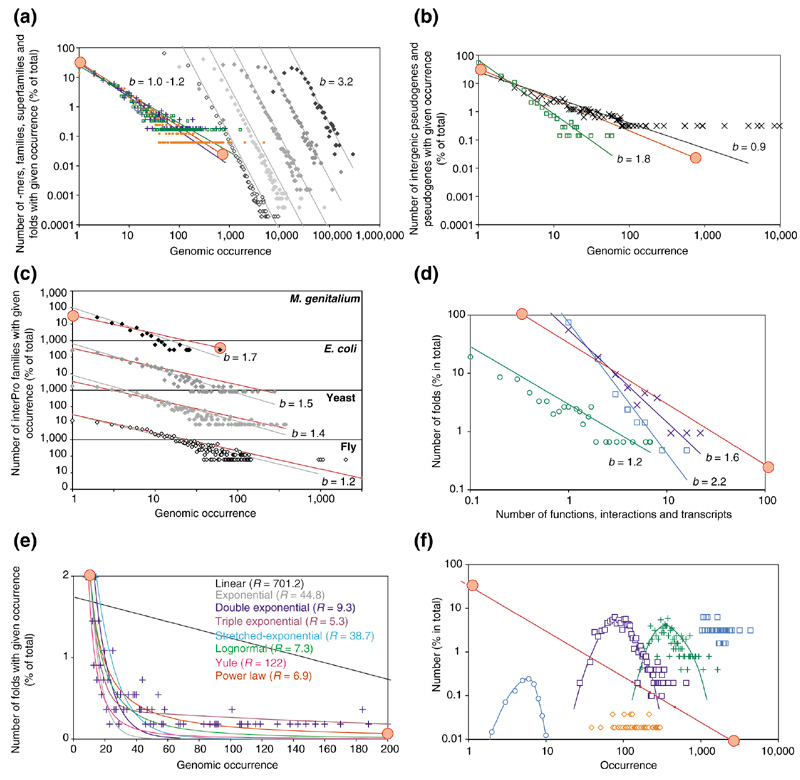
Power-law behaviour is observed for many genomic properties. (a) The occurrence of DNA words, InterPro families and protein folds in the worm genome. Black diamonds, 6-mers; dark-gray diamonds, 7-mers; mid-gray diamonds, 8-mers; light-gray diamonds, 9-mers, open diamonds, 10-mers; red circles, gene families; open green squares, protein superfamilies, blue crosses, protein folds. The solid lines represent the best-fit power-law functions for each distribution. (b) The occurrence of pseudogene families (open green squares) and pseudomotifs (black crosses) in the worm intergenic regions. (c) The occurrence of InterPro families in M. genitalium (black diamonds); E. coli (dark-gray diamonds); S. cerevisiae (mid-gray diamonds); and D. melanogaster (open diamonds). (d) Other properties that follow the power law. Black crosses, the number of assigned functions for each fold; open blue squares, the number of protein-protein interactions each fold makes in the yeast two-hybrid experiment, open green circles, the number of transcripts of each fold during vegetative growth in yeast. (e) Best-fit functions for the occurrence of protein folds in the worm genome (blue crosses): linear (y = a - bx), exponential (y = ae-bx), double-exponential (y = ae-bx + ce-dx), triple exponential (y = ae-bx + ce-dx +fe-gx), stretched-exponential  ,
, 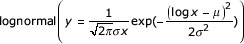 ,
, 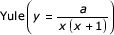 and power-law functions (y = ax-b). The residuals (R) between the functions and genomic data are calculated as Σ (Nfolds(actual) - Nfolds(fitted))2. (f) Properties that do not follow the power law. The occurrence of 3-mers (open blue squares); 4-mers (green crosses); and 5-mers (open dark-blue squares) in the worm genome. Open blue circles, the average composition of asparagine in different folds; open red diamonds, the number of residues involved in protein flexibility in different folds. The slopes (exponent b) are given on the plots. The worm genome was taken from the database at the National Center for Biotechnological Information [41], the family assignments were obtained from the InterPro proteome database[42], and the fold assignments from the Partslist database [20]. Solid red line, best-fit line for worm InterPro families.
and power-law functions (y = ax-b). The residuals (R) between the functions and genomic data are calculated as Σ (Nfolds(actual) - Nfolds(fitted))2. (f) Properties that do not follow the power law. The occurrence of 3-mers (open blue squares); 4-mers (green crosses); and 5-mers (open dark-blue squares) in the worm genome. Open blue circles, the average composition of asparagine in different folds; open red diamonds, the number of residues involved in protein flexibility in different folds. The slopes (exponent b) are given on the plots. The worm genome was taken from the database at the National Center for Biotechnological Information [41], the family assignments were obtained from the InterPro proteome database[42], and the fold assignments from the Partslist database [20]. Solid red line, best-fit line for worm InterPro families.
Table 1.
List of genomic properties that display power-law behavior and the associated exponent (b) for the best-fitting power-law function
| Exponent b | |||||||||
| Organism | 6-10-mers | Protein families | Protein super-families | Protein folds | Pseudomotifs | Pseudogene families | Functions per protein fold | Interactions per protein fold | Transcripts per protein family |
| Mycoplasma genitalium | 3.7 | 1.7 | 1.6 | 1.9 | - | - | - | - | - |
| Mycoplasma pneumoniae | 3.5 | 1.6 | 1.5 | 1.8 | - | - | - | - | - |
| Rickettsia prowazekii | 3.7 | 1.7 | 1.6 | 1.9 | - | - | - | - | - |
| Chlamydia trachomatis | 3.7 | 1.7 | 1.6 | 1.9 | - | - | - | - | - |
| Treponema pallidum | 3.4 | 1.6 | 1.5 | 1.7 | - | - | - | - | - |
| Chlamydia pneumoniae | 3.6 | 1.6 | 1.5 | 1.7 | - | - | - | - | - |
| Aquifex aeolicus | 3.8 | 1.7 | 1.5 | 1.9 | - | - | - | - | - |
| Helicobacter pylori | 3.5 | 1.6 | 1.5 | 1.7 | - | - | - | - | - |
| Haemophilus influenzae | 3.4 | 1.5 | 1.4 | 1.6 | - | - | - | - | - |
| Methanococcus jannaschii | 3.5 | 1.6 | 1.5 | 1.7 | - | - | - | - | - |
| Methanococcus thermoautotrophicum | 3.8 | 2.0 | 1.8 | 2.2 | - | - | - | - | - |
| Pyrococcus horikoshii | 3.9 | 1.9 | 1.7 | 2.0 | - | - | - | - | - |
| Archaeoglobus fulgidus | 3.8 | 1.8 | 1.6 | 1.9 | - | - | - | - | - |
| Synechocystis sp. | 3.4 | 1.6 | 1.5 | 1.7 | - | - | - | - | - |
| Mycobacterium tuberculosis | 3.4 | 1.5 | 1.4 | 1.6 | - | - | - | - | - |
| Bacillus subtilis | 3.3 | 1.4 | 1.3 | 1.5 | - | - | - | - | - |
| Escherichia coli | 3.2 | 1.5 | 1.4 | 1.6 | - | - | - | - | - |
| Saccharomyces cerevisiae | 3.2 | 1.4 | 1.3 | 1.5 | 0.9 | 1.5 | 1.6 | 2.2 | 1.2 |
| Caenorhabditis elegans | 3.1 | 1.1 | 1.0 | 1.2 | 1.0 | 1.8 | - | - | |
| Drosophila melanogaster | 3.3 | 1.2 | 1.1 | 1.3 | 1.2 | - | - | - | |
| Human chromosomes 21 and 22 | - | - | - | - | 1.0 | 1.9 | - | - | |
Having observed the occurrence of short 'mers, we now shift our focus towards the coding regions of genomes. Most proteins encoded in a genome can be grouped according to their similarity in three-dimensional structure or amino-acid sequence. The most common classifications of proteins are the fold, superfamily and family [14]; each class is a subset of the one before, and groups proteins with increasing similarity. First, proteins are defined to have a common fold if their secondary structural elements occupy the same spatial arrangement and have the same topological connections. Second, proteins are grouped into the same superfamily if they share the same fold, and are deemed to share a common evolutionary origin, owing to a similar protein function, for example. Both the fold and superfamily classes aim to group proteins that are structurally related, but whose similarities cannot necessarily be detected only by their sequences. Finally, proteins are grouped into the same family if their amino-acid sequences are considered similar, most commonly using a measure of percentage sequence identity or an E-value cut-off. Alternatively, they can also be characterized by the presence of a particular sequence 'signature' or 'motif. Here, we have used the fold and superfamily assignments from the SCOP [14] and Superfamily databases [15] and the family classifications from InterPro [16,17].
By analogy with the earlier 'mers, proteins encoded in a genome can be thought of as longer DNA words. Therefore, by grouping proteins in the classification system above, we can measure the occurrences of collections of sequences of around 1,000-mers in the genome. As we explained above, members of the same superfamily have often diverged beyond detectable sequence similarity and, in the case of folds, may have independently converged to similar structures from unrelated DNA sequences. Nevertheless, the occurrences of families, superfamilies, and folds in the worm approximate to a power-law behavior quite well (Figure 1a). In fact, despite the differing definitions of families, superfamilies and folds, the resulting distributions for each group are very similar. Compared with the 6- to 10-mers, the distributions fall off more gradually (b = 1.0-1.2); this indicates a greater difference in the relative occurrence of the most and least common families.
Returning to the non-coding regions of the genome, we also plotted the occurrence of pseudogene families and pseudomotifs found in intergenic DNA (Figure 1b). Whole pseudogenes were found by searching for matches to SWISS-PROT protein sequences in intergenic DNA, and are usually characterized by frameshift mutations or early stop codons that prevent normal transcription or translation [18,19]. Therefore, they encode non-functional protein sequences. As with functional proteins, pseudogenes were classified into families using InterPro. Pseudomotifs were found by matching PROSITE motifs in intergenic DNA and are thought to be more ancient pseudogenes that have accumulated so many mutations that only small fragments of recognizable motifs remain (Z.Z. and M.G., unpublished observations). These fragments are classified according to the PROSITE classification. As shown in Figure 1b, the occurrences of pseudogenes and the fragments also follow a power law. The distribution for pseudogenes is similar to that for protein families and folds (b = 1.8); this is expected, as pseudogenes in the worm represent a population of DNA sequences that used to encode functional proteins. The distribution of occurrences for pseudomotifs (b = 0.9) has a wide spread and actually bridges those of protein families and 'mers. This is probably because the most frequently occurring PROSITE motifs are only 5-10 amino acids in length, and therefore are similar to 'mers, whereas the less frequently occurring motifs are longer (82 amino-acid residues), and so resemble protein families.
Our findings for the worm genome also apply to at least 20 other prokaryotic and eukaryotic organisms. Figure 1c shows InterPro family distributions in Mycoplasma genitalium, Escherichia coli, Saccharomyces cerevisiae and Drosophila melanogaster; other distributions from many of the recently sequenced genomes are available from our website [20]. Interestingly, smaller genomes (b = 1.0-2.0) tend to have a steeper fall-off than larger genomes; with fewer genes, it would seem natural to expect a narrower distribution in these organisms. Given the prevalence of the power-law behavior, it is likely to be universal to other genomes yet to be analyzed.
Functions, interactions and expression levels
The power law is not only found in the occurrence of words, families, and folds, but also extends to further genomic features of biological macromolecules. As shown in Figure 1d, the distribution fits the number of distinct functions held by a particular protein fold [21,22]. Most folds are only associated with only one or two functions, whereas a few, such as the TIM barrel, have up to 16 (b = 2.2). The behavior also applies to the number of distinct protein-protein interactions made by different folds (b = 1.2) and the number of transcripts for each protein family in yeast in a given cellular state (b = 1.6).
Is the power-law function the best fit?
We have so far demonstrated that disparate types of data display power-law behavior. Not all genomic properties follow a power law, however, and examples include occurrences of 'mers shorter than six bases, the occurrence of particular amino acids in proteins, and the number of residues that are involved in protein flexibility (Figure 1f).
The original publication by Mantegna et al. [4] resulted in a prolonged debate as to whether the power law is actually the best fit for the 'mer distribution [23,24,25,26,27,28], and similar discussions are found for power-law behavior outside biology [29,30,31,32]. Previous publications have only tested the suitability of individual functions. In Figure 1e, however, we examine the best-fit curves of seven alternative functions for protein-fold occurrence in the worm: linear, exponential, double-exponential, triple-exponential, stretched-exponential, lognormal and Yule distributions. The Yule distribution in particular was reported as providing a better fit for the occurrence of 'mers than the power law [27], and the stretched-exponential and lognormal distributions have been cited as providing good fits for non-biological data.
We measure the fit of each function by calculating the residual between actual protein-fold occurrence and the mathematical functions as follows:
R = 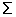 (N(actual) -N(fitted))2
(N(actual) -N(fitted))2
For example, for the fits in Figure 1e we use the following equation:
Rfolds = (Nfolds(worm) - Nfolds(fitted))2
In this calculation, a smaller residual (R) indicates a better fit between the data and the mathematical functions.
The main differences in the fit appear at the tail of the distribution, at high fold occurrences. Although most functions describe the lower end of the distribution well, they do not extend far enough at the upper end of the distribution. The linear and single-exponential curves clearly do not describe the data well. The double-exponential curve provides a reasonable fit for lower genomic occurrences, but diverges from the data at higher values. The same applies for the stretched-exponential and Yule distributions.
Two functions perform well: the triple-exponential and the lognormal distributions. In fact, the triple-exponential displays a smaller residual than the power-law function and one would expect higher-order exponentials to provide increasingly better fits. However, this is at the expense of having more free parameters to fine-tune the shape of the curve. As the fold distribution actually displays a wide spread of values - especially for higher occurrences (Figure 1a,1d) - we conclude that all three mathematical functions describe the data equally well. The same also applies to the other genomic data we discussed earlier. However, given the fit across many different biological distributions, combined with the relative simplicity of the function compared to the higher-order-exponential and lognormal distributions, we suggest that the power law provides the best description of the data.
Discussion
The significance of power-law behavior
Although the power-law behavior has previously been detected in individual biological distributions [4,5,6,7,8,9,10,11,12,13], this is the first time it has been reported for such a wide group of properties associated with genomes. Moreover, here we demonstrate for the first time that power-law distributions are applicable to the occurrence of pseudogenes and pseudomotifs in intergenic regions, the number of functions associated with a protein fold, and the expression levels of different protein families.
At first glance, these observations might appear to be 'biological trivia'. However, power-law behavior actually provides a concise mathematical description of an important biological feature: the sheer dominance of a few members over the overall population. For example, out of the 247 distinct protein folds currently assigned in the worm genome, just 10 account for over half of the 7,805 assigned domains. The top fold, the immunoglobulin-like β-sandwich, accounts for about 829 (10.6%) domains in the genome. For protein superfamilies, 21 out of 606 families account for half of the 15,450 assigned domains, and only 37 of 1,936 InterPro families match half of the 12,589 assigned proteins. Half of all pseudogenes belong to 10 (out of a total of 70) protein families, and just two types of motif make up over half of pseudogenic PROSITE fragments.
Power-law behavior also describes similar underlying observations for the remaining data. For protein-protein interactions, we find that 6 out of 39 protein folds in the yeast genome make up half of the 89 known combinations of interdomain interactions, and for gene-expression levels in yeast, proteins from just 12 out of a total 145 folds comprise half of all the transcripts in the cell at any given state.
Power-law behavior and the underlying evolutionary mechanism
Having discussed the significance of power-law behaviors, this leads us to the question of how genomes achieved these distributions. Given a mathematical description common to many genomes and different genomic properties, it is possible to define evolutionary models that replicate the power-law distributions. Recently, several papers have attempted to answer this question from a theoretical perspective by building mathematical models for evolution.
Mantegna et al. [4] drew analogies with Zipf's original work to suggest that the behavior for 'mers originate from similarities between DNA sequences and natural languages. This suggestion attracted extensive criticism [23,24,25,26,27,28] and the work of Li [33] demonstrated that power-law-like behavior could in fact simply arise from non-equal occurrences of words in random texts. Shorter 'mers (< 6-mers) fail to display power-law behavior because there are insufficient numbers of distinct words to differentiate levels of occurrence. The 4- and 5-mers have larger numbers of distinct sequences and there are hints of power-law behavior in the tails of their distributions (Figure 1d). For 6-mers and above, the reason that words of different lengths have identical slopes is because their distributions are not independent of each other; longer words also contain combinations of the shorter words.
Although random DNA sequences provide a possible explanation for power-law behavior with 'mers, the same is unlikely to apply to protein sequences. Rather, at these levels the distributions probably arise from evolutionary development centered on an underlying process of gene duplication. If we treat gene duplication as a stochastic process, the chance of a given gene being duplicated is proportional to its occurrence in the genome. With each duplication, some genes increase their presence in the genome, enhancing their chance of further duplication. Combined with selective pressure accounting for the functional significance of different protein products, such a process gives prominence to some gene types, or families, over others. Previous studies outside genomic biology have linked such stochastic dynamical processes to power laws [34,35,36,37].
Three evolutionary models proposed by Huynen and van Nimwegen [6], Yanai et al. [38] and Qian et al. [9] successfully replicated the observed biological data using such a duplicative process. All three models rely heavily on gene duplication as the underlying process and, in fact, this process on its own results in an exponential distribution. Each model achieves the power-law distribution by emulating biological processes that perturb the duplicative processes; this is done by including parameters that mimic selective pressure, point mutations or lateral gene transfer.
In the first model, of Huynen and van Nimwegen [6], gene families start the simulation with one member each. Each family is allowed to expand or contract in size through successive multiplications with a random factor, which represents duplication or deletion events depending on its value. In the second model, that of Yanai et al. [38], genomes evolve from a set of precursor genes to a mature size by random gene duplications and gradual accumulation of modifications through point mutations. When an individual family member acquires enough random mutations, it breaks away to form a new family. Finally in the third model, that of Qian et al. [9], genomes evolve from their initial small size using two basic operations: first, duplication of existing genes to expand the size of existing families, and second, the introduction of completely new genes by lateral transfer from other organisms or ab initio creation. Both components are vital for replicating the observed data. In this paper, we have discussed the finding that genomic distributions first take on an exponential appearance and then adopt a power-law behavior after a sufficiently long evolutionary process. In a similar vein, a recent paper by Rzhetsky and Gomez [13] introduced a model in which the underlying DNA duplication mechanism, combined with random production of an inter-protein interaction, successfully simulates the power-law distribution for interaction networks in yeast.
This leads us to speculate on a possible explanation of power-law behaviors for the other properties. For protein functions, folds with high occurrence in the genome also tend to have diverse functions; thus the P-loop-containing NTP hydrolase fold is found 130 times in yeast and has at least six distinct enzymatic functions [20]. Parallel to this, we find that folds with many protein-protein interactions also tend to hold more diverse functions [39]; for example, the NTP hydrolase fold is currently associated with nine interdomain interactions within yeast. Finally, the state of the transcription-regulatory mechanism in a particular cellular state clearly has the most important role in determining gene-expression levels. However, it has previously been shown that some gene families with high occurrences also display high expression levels under diverse experimental conditions [40]. Finally, the occurrence of pseudogene families is related to the occurrence of the protein family from which they originate. In addition, a major method of producing pseudogenes is through reverse transcription of expressed mRNA, and so their occurrences are actually related to expression levels of these families. Given the correlation of all the different genomic properties with the occurrence of gene families in the genome, we can reason that they are connected to an underlying duplicative process.
The main arguments supporting the view that all power-law behaviours are arise from a common duplicative process are that the occurrences of different genomic properties are correlated and the fits between the distributions of biological and simulated data are good. Although these models are based on well-known biological processes, there is, unfortunately, little experimental evidence to directly confirm the validity of these models. However, it is worth noting that most of the properties that do not follow a power law (Figure 1d) are those that are clearly not directly connected with gene duplication.
Additional data files
The CGI script and data that are used to produce the website are available for download and from [20].
Supplementary Material
The CGI script and data used to produce the website
Acknowledgments
Acknowledgements
N.M.L. is supported by the Cancer Bioinformatics Fellowship from the Anna Fuller Fund, and M.G. acknowledges support from the Keck Foundation.
References
- Zipf GK. Human Behavior and the Principle of Least Effort Boston: Addison-Wesley; 1949.
- Barabasi AL, Albert R. Emergence of scaling in random networks. Science. 1999;286:509–512. doi: 10.1126/science.286.5439.509. [DOI] [PubMed] [Google Scholar]
- Albert R, Jeong H, Barabasi AL. Error and attack tolerance of complex networks. Nature. 2000;406:378–382. doi: 10.1038/35019019. [DOI] [PubMed] [Google Scholar]
- Mantegna RN, Buldyrev SV, Goldberger AL, Havlin S, Peng C, Simons M, Stanley HE. Linguistic features of noncoding DNA sequences. Phys Rev Lett. 1994;73:3169–3172. doi: 10.1103/PhysRevLett.73.3169. [DOI] [PubMed] [Google Scholar]
- Gerstein M. A structural census of genomes: comparing bacterial, eukaryotic, and archaeal genomes in terms of protein structure. J Mol Biol. 1997;274:562–576. doi: 10.1006/jmbi.1997.1412. [DOI] [PubMed] [Google Scholar]
- Huynen MA, van Nimwegen E. The frequency distribution of gene family sizes in complete genomes. Mol Biol Evol. 1998;15:583–589. doi: 10.1093/oxfordjournals.molbev.a025959. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Wolf YI, Aravind L. Protein fold recognition using sequence profiles and its application in structural genomics. Adv Protein Chem. 2000;54:245–275. doi: 10.1016/s0065-3233(00)54008-x. [DOI] [PubMed] [Google Scholar]
- Qian J, Stenger B, Wilson CA, Lin J, Jansen R, Teichmann SA, Park J, Krebs WG, Yu H, Alexandrov V, et al. PartsList: a web-based system for dynamically ranking protein folds based on disparate attributes, including whole-genome expression and interaction information. Nucleic Acids Res. 2001;29:1750–1764. doi: 10.1093/nar/29.8.1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Luscombe NM, Gerstein M. Protein family and fold occurrence in genomes: power-law behavior and evolutionary model. J Mol Biol. 2001;313:673–681. doi: 10.1006/jmbi.2001.5079. [DOI] [PubMed] [Google Scholar]
- Jeong H, Tombor B, Albert R, Oltvai ZN, Barabasi AL. The large-scale organization of metabolic networks. Nature. 2000;407:651–654. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- Park J, Lappe M, Teichmann SA. Mapping protein family interactions: intramolecular and intermolecular protein family interaction repertoires in the PDB and yeast. J Mol Biol. 2001;307:929–938. doi: 10.1006/jmbi.2001.4526. [DOI] [PubMed] [Google Scholar]
- Wagner A. The yeast protein interaction network evolves rapidly and contains few redundant duplicate genes. Mol Biol Evol. 2001;18:1283–1292. doi: 10.1093/oxfordjournals.molbev.a003913. [DOI] [PubMed] [Google Scholar]
- Rzhetsky A, Gomez SM. Birth of scale-free molecular networks and the number of distinct DNA and protein domains per genome. Bioinformatics. 2001;17:988–996. doi: 10.1093/bioinformatics/17.10.988. [DOI] [PubMed] [Google Scholar]
- Lo Conte L, Ailey B, Hubbard TJ, Brenner SE, Murzin AG, Chothia C. SCOP: a structural classification of proteins database. Nucleic Acids Res. 2000;28:257–259. doi: 10.1093/nar/28.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough J, Karplus K, Hughey R, Chothia C. Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J Mol Biol. 2001;313:903–919. doi: 10.1006/jmbi.2001.5080. [DOI] [PubMed] [Google Scholar]
- Apweiler R, Attwood TK, Bairoch A, Bateman A, Birney E, Biswas M, Bucher P, Cerutti L, Corpet F, Croning MD, et al. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res. 2001;29:37–40. doi: 10.1093/nar/29.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler R, Biswas M, Fleischmann W, Kanapin A, Karavidopoulou Y, Kersey P, Kriventseva EV, Mittard V, Mulder N, Phan I, Zdobnov E. Proteome Analysis Database: online application of InterPro and CluSTr for the functional classification of proteins in whole genomes. Nucleic Acids Res. 2001;29:44–48. doi: 10.1093/nar/29.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PM, Echols N, Gerstein MB. Digging for dead genes: an analysis of the characteristics of the pseudogene population in the Caenorhabditis elegans genome. Nucleic Acids Res. 2001;29:818–830. doi: 10.1093/nar/29.3.818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PM, Hegyi H, Balisubramanian S, Luscombe NM, Bertone P, Echols N, Johnson T, Gerstein M. Molecular fossils in the human genome: identification and analysis of the pseudogenes in chromosomes 21 and 22. Genome Res. 2002;12:272–280. doi: 10.1101/gr.207102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark Gerstein's lab http://www.partslist.org/powerlaw
- Hegyi H, Gerstein M. The relationship between protein structure and function: a comprehensive survey with application to the yeast genome. J Mol Biol. 1999;288:147–164. doi: 10.1006/jmbi.1999.2661. [DOI] [PubMed] [Google Scholar]
- Todd AE, Orengo CA, Thornton JM. Evolution of function in protein superfamilies, from a structural perspective. J Mol Biol. 2001;307:1113–1143. doi: 10.1006/jmbi.2001.4513. [DOI] [PubMed] [Google Scholar]
- Israeloff NE, Kagalenko M, Chan K. Can Zipf distinguish language from noise in noncoding DNA? Phys Rev Lett. 1996;76:1976. doi: 10.1103/PhysRevLett.76.1976. [DOI] [PubMed] [Google Scholar]
- Konopka AK, Martindale C. Noncoding DNA, Zipf's law, and language. Science. 1995;268:789. doi: 10.1126/science.7754361. [DOI] [PubMed] [Google Scholar]
- Bonhoeffer S, Herz AV, Boerlijst MC, Nee S, Nowak MA, May RM. No signs of hidden language in noncoding DNA. Phys Rev Lett. 1996;76:1977. doi: 10.1103/PhysRevLett.76.1977. [DOI] [PubMed] [Google Scholar]
- Bonhoeffer S, Herz AV, Boerlijst MC, Nee S, Nowak MA, May RM. Explaining "linguistic features" of noncoding DNA. Science. 1996;271:14–15. [PubMed] [Google Scholar]
- Martindale C, Konopka AK. Oligonucleotide frequencies in DNA follow a Yule distribution. Computer Chem. 1996;20:35–38. doi: 10.1016/0097-8485(95)00091-7. [DOI] [PubMed] [Google Scholar]
- Voss RF. Comment on "Linguistic features of noncoding DNA sequences". Phys Rev Lett. 1996;76:1978. doi: 10.1103/PhysRevLett.76.1978. [DOI] [PubMed] [Google Scholar]
- Perline P. Zipf's law, the central limit theorem, and the random division of the unit interval. Phys Rev E. 1996;54:220–223. doi: 10.1103/physreve.54.220. [DOI] [PubMed] [Google Scholar]
- Laherrere J, Sornette D. Stretched exponential distributions in nature and economy: "fat tails" with characteristic scales. Eur Phys J. 1998;B2:525–539. [Google Scholar]
- Rousseau R. A weak goodness-of-fit test for rank-frequency distributions. In: Macias-Chapula C, editor. In Proc Seventh Conf Int Soc Scientometrics Informetrics. Mexico: Universidad de Colima; 1999. pp. 421–430. [Google Scholar]
- Limpert E, Stahl WA, Abbt M. Lognormal distributions across the sciences: keys and clues. Biosciences. 2001;51:341–352. [Google Scholar]
- Li WT. Random texts exhibit Zipf-like word-frequency distribution. IEEE T Inform Theory. 1992;38:1842–1845. [Google Scholar]
- Yule GU. A mathematical theory of evolution, based on the conclusions of Dr. J.C. Willis, F.R.S. Phil Trans R Soc B. 1924;213:21–87. [Google Scholar]
- Simon HA. On a class of skew distribution functions. Biometrika. 1955;42:425–440. [Google Scholar]
- Kesten H. Random difference equations, and renewal theory for products of random matrices. Acta Math. 1973;131:207–248. [Google Scholar]
- Sornette D, Cont R. Convergent multiplicative processes repelled from zero: power laws and truncated power laws. J Physique. 1997;7:431–444. [Google Scholar]
- Yanai I, Camacho CJ, DeLisi C. Predictions of gene family distributions in microbial genomes: evolution by gene duplication and modification. Phys Rev Lett. 2000;85:2641–2644. doi: 10.1103/PhysRevLett.85.2641. [DOI] [PubMed] [Google Scholar]
- Gerstein M. Integrative database analysis in structural genomics. Nat Struct Biol. 2000;7 Suppl:960–963. doi: 10.1038/80739. [DOI] [PubMed] [Google Scholar]
- Jansen R, Gerstein M. Analysis of the yeast transcriptome with structural and functional categories: characterizing highly expressed proteins. Nucleic Acids Res. 2000;28:1481–1488. doi: 10.1093/nar/28.6.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Center for Biotechnology Information http://www.ncbi.nlm.nih.gov/
- InterPro http://www.ebi.ac.uk/interpro/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The CGI script and data used to produce the website