

Abstract
Coronavirus spike (S) proteins are responsible for binding and fusion with target cells and thus play an essential role in virus infection. Recently, we identified a dilysine endoplasmic reticulum (ER) retrieval signal and a tyrosine-based endocytosis signal in the cytoplasmic tail of the S protein of infectious bronchitis virus (IBV). Here, an infectious cDNA clone of IBV was used to address the importance of the S protein trafficking signals to virus infection. We constructed infectious cDNA clones lacking the ER retrieval signal, the endocytosis signal, or both. The virus lacking the ER retrieval signal was viable. However, this virus had a growth defect at late times postinfection and produced larger plaques than IBV. Further analysis confirmed that the mutant S protein trafficked though the secretory pathway faster than wild-type S protein. A more dramatic phenotype was obtained when the endocytosis signal was mutated. Recombinant viruses lacking the endocytosis signal (in combination with a mutated dilysine signal or alone) could not be recovered, even though transient syncytia were formed in transfected cells. Our results suggest that the endocytosis signal of IBV S is essential for productive virus infection.
Assembly and release of virus particles are the final steps of a virus replication cycle. Therefore, studying virus assembly is important for understanding viral diseases and controlling infection. Moreover, studying virus assembly is also useful for understanding the cell biology of protein synthesis and modification, since viruses depend on cellular machinery for their replication. Most well-studied enveloped viruses assemble at the plasma membrane. In these viruses, the membrane-bound envelope proteins are synthesized at the endoplasmic reticulum (ER) and transported through the cellular secretory pathway, where they are modified and accumulate on the cell surface for assembly. However, some enveloped viruses (including flaviviruses, bunyaviruses, and coronaviruses) assemble and bud into the lumen of the ER, the Golgi complex, or the ER-Golgi intermediate compartment (ERGIC) (11, 12, 18, 20). Depending on the host cell type, retroviruses can bud into multivesicular bodies, which are part of the endocytic system (23). Viruses that assemble intracellularly direct their envelope proteins to the assembly site by using normal cellular-protein targeting signals. Virions assembled in intracellular compartments are presumed to exit infected cells by exocytosis.
Coronaviruses have the largest genomes (27 to 31 kb) among known RNA viruses. Coronaviruses belong to the order Nidovirales, members of which produce nested sets of subgenomic RNAs (sgRNAs) by an unusual discontinuous transcription process (29). Interest in coronaviruses has dramatically increased since the recent emergence of a novel coronavirus associated with severe acute respiratory syndrome (SARS) (9, 32). Coronaviruses encode three envelope proteins: small envelope (E), membrane (M), and spike (S). The E protein is a minor component of the virus envelope; however, evidence is mounting for the important role of the E protein in viral budding. E protein interacts with M protein and coexpression of M and E proteins produces virus-like particles that are similar to virions in size and shape (2, 3, 5, 22, 34). Elimination of E protein expression in coronaviruses either completely blocks virus assembly or produces defective virions (13, 25). The M protein is the most abundant envelope protein in virions. M interacts with E, nucleocapsid (N), and S proteins in mouse hepatitis virus (MHV) and other coronaviruses (2, 21, 24). Both the M and E proteins of infectious bronchitis virus (IBV) are targeted near the viral assembly site when expressed independently. The IBV M targeting information resides in its first membrane-spanning domain, and that of IBV E resides in the cytoplasmic tail (3, 17). The coronavirus S protein is a type I transmembrane protein with a large ectodomain, a single transmembrane domain, and a short cytoplasmic tail (8). S proteins are highly glycosylated, with 21 to 28 predicted N-linked glycosylation sites. In some coronaviruses, full-length S protein (S0) is processed to S1 and S2 by a furin-like proteinase(s). The S1 domain interacts with cellular receptors, thus determining the host specificity and tropism. The S2 domain of S is responsible for fusion of the virus envelope with the cell membrane and for fusion of infected cells with neighboring cells late in infection. The S protein is not essential for production of virus-like particles; however, it is essential for the production of infectious virus.
MHV S protein expressed alone reaches the cell surface; however, when coexpressed with the M protein, it is retained near the virus assembly site (24). We recently found that unlike the MHV S protein, the IBV S protein is retained near the virus assembly site when expressed alone (16). In the same study, we showed that the IBV S protein possesses both a dilysine ER retrieval signal and a tyrosine-based endocytosis signal in its cytoplasmic tail that can modulate trafficking of a chimeric reporter protein.
In this study, we investigated how the S trafficking signals in the IBV S cytoplasmic tail affect virus replication during infection. Using an infectious cDNA clone of IBV, we constructed mutants lacking the trafficking signals in the S protein. We show that a recombinant IBV with an S protein lacking the ER retrieval signal has a growth defect at late times postinfection (p.i.), which might be the result of faster trafficking of the mutant S protein through the secretory pathway. Surprisingly, virus lacking the endocytosis signal (alone or in combination with a mutated dilysine signal) could not be rescued. We propose that IBV regulates the level of S at the surface of infected cells using both of the trafficking signals and that the endocytosis signal plays a major role in virus infection, since it is essential for productive virus infection.
MATERIALS AND METHODS
Cells, viruses, and antibodies.
Vero and baby hamster kidney (BHK-21) cells were maintained in Dulbecco's modified Eagle medium (DMEM) containing 5% fetal bovine serum (FBS) supplemented with penicillin G (100 units/ml) and streptomycin (100 μg/ml). HeLa cells were maintained in DMEM containing 10% FBS and antibiotics. The molecularly cloned Beaudette US strain of IBV was maintained as described previously (36). The recombinant vaccinia virus encoding phage T7 RNA polymerase (vTF7-3) has been described previously (6). The rabbit polyclonal anti-IBV E, anti-IBV M, and anti-IBV S tail antibodies used for immunoprecipitation were described previously (3, 16, 17). The mouse monoclonal anti-IBV S antibody (9B1B6) recognizes the luminal domain (35). The mouse anti-vesicular stomatitis virus glycoprotein (VSV G) monoclonal antibody used was described previously (15).
Plasmids, mutagenesis, and isolation of recombinant viruses.
The S2A and S4A mutations were introduced into the E amplicon encoding the structural proteins (36) using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). The codons for lysines in the dilysine ER retrieval signal were changed to those for alanine, followed by replacement of the tyrosine codons in the endocytosis signal with these for alanine using primer sets described previously (16) to create S4A. SYA and S2YA mutations were introduced into the E amplicon by replacing the codons for one or both tyrosines with those for alanine using the method described above. After sequencing, the mutated region was subcloned into the E amplicon to avoid unintentional mutations introduced by PCR. Amplicons A to E or ES2A, ESY, ES2Y, or ES4A were in vitro ligated, and the assembled DNAs were used as templates for in vitro transcription of cRNAs as described previously (36). IBV cRNAs were electroporated with N transcript at 850 V, 25 μF, 950 Ω into BHK-21 cells using an electro-cell manipulator ECM 630 (BTX, San Diego, CA) and cocultured with Vero cells. Transfected cells were harvested approximately 48 h posttransfection, and recombinant viruses were amplified in Vero cells and plaque purified three times. Twelve plaque-purified IBV-S2A clones were sequenced (nucleotides 23540 to 24113) after reverse transcription-PCR. Two independent IBV and IBV-S2A clones were amplified and used for further characterization. Since the two clones behaved identically, the results for one clone for each virus are shown unless otherwise specified.
One-step growth curve and titration of released and intracellular virus.
To assess growth kinetics, triplicate wells of Vero cells in six-well plates were infected with virus at a multiplicity of infection (MOI) of 2. The total virus produced was harvested by freezing infected cells every 4 h at −80°C. After three freeze-thaw cycles, the total virus titer was determined by plaque assay on Vero cells. The plaques were stained with crystal violet and counted. For comparison of released and intracellular virus, cells were infected as described above, but the supernatant and cells were harvested separately at 12 and 16 h postinfection. Infectious extracellular virus was harvested from supernatants of infected cells after removing cell debris by centrifugation. Intracellular virus was harvested from infected cells by adding medium, followed by three freeze-thaw cycles. Titers of infectious virus from both supernatant and cells (after freeze-thaw) were determined as described above.
Metabolic labeling, immunoprecipitation, and glycosidase digestion.
Vero cells were infected as described above and, at 7.5 h postinfection, starved for 20 min with methionine- and cysteine-free DMEM at 37°C. The cells were labeled with 100 μCi of [35S]methionine-cysteine (Promix; Amersham Pharmacia Biotech, Piscataway, NJ) per ml in methionine- and cysteine-free DMEM for 20 min at 37°C and then chased in unlabeled DMEM containing 5% FBS for various times as indicated. After a rinse with cold phosphate-buffered saline, the cells were lysed in NP-40-deoxycholate (DOC) buffer (50 mM Tris-HCl [pH 8.0], 62.5 mM EDTA, 0.5% NP-40, and 0.5% deoxycholate) on ice for 3 min. After removal of nuclei and cell debris by centrifugation, the lysates were incubated overnight with 2 μl of rabbit anti-IBV E, -IBV M, or -IBV S antibody. Antibody-antigen complexes were collected by incubation with fixed Staphylococcus aureus (Pansorbin; Calbiochem, San Diego, CA) for 20 min at room temperature. Immunoprecipitates were washed three times in low-DOC RIPA buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% NP-40, 0.5% DOC, and 0.1% sodium dodecyl sulfate [SDS]) and eluted in 20 μl of eluant (50 mM Tris-HCl [pH 6.5] and 1% SDS) by incubation at 100°C for 3 min. After centrifugation to remove the immunoadsorbant, an equal volume of 0.15 M sodium citrate (pH 5.5) containing 0.4 mU of endoglycosidase H (endo H) (New England Biolabs, Beverly, MA) was added to the samples, and they were incubated overnight at 37°C. The samples were then separated on 10% or 15% SDS-polyacrylamide gels, and immunoprecipitated proteins were subjected to quantitation on a Molecular Imager FX phosphorimager (Bio-Rad, Hercules, CA) using Quantity One software (Bio-Rad).
Coimmunoprecipitation of M with S from infected or transiently transfected cells.
M and S protein interaction was assessed in IBV-infected Vero cells and in transiently transfected cells. IBV M and S were expressed transiently in cells infected with a vaccinia virus encoding bacteriophage T7 RNA polymerase using pAR/IBVM and Bluescript-based S or S4A plasmids described previously (3, 16). HeLa cells were infected with vTF7-3 at an MOI of 10 in DMEM for 1 h at 37°C, followed by transfection with a total of 3 μg of M and S plasmids (1:5 ratio) using 6 μl of TransIT-LT1 (Mirus, Madison, WI) as directed by the manufacturer. At 3 h posttransfection (or 8 h postinfection with IBV), the cells were incubated in methionine- and cysteine-free MEM for 15 min, labeled for 30 min with methionine-cysteine, and chased for 30 min before the labeled cells were lysed and immunoprecipitated as described above. To confine the M and S proteins to the ER, HeLa cells infected and transfected as described above were treated with brefeldin A (BFA) at a concentration of 1 μg/ml during the starvation, labeling, and chase.
Northern blot analysis.
Total cellular RNA of IBV- or IBV-S2A-infected Vero cells was extracted using TRIZOL reagent (Invitrogen, Palo Alto, CA) according to the manufacturer's instructions. Approximately 4 μg of total RNA was electrophoresed on a 1% denaturing agarose gel containing 2.2 M formaldehyde. The RNAs were transferred overnight to nylon paper and subjected to Northern hybridization using a random-primed antisense IBV 3′-untranslated-region probe according to standard methods (27).
Indirect immunofluorescence and antibody uptake.
Live-cell antibody uptake was performed by incubating IBV-infected Vero cells grown on coverslips with monoclonal anti-IBV S (35) or mouse anti-VSV G (15) as a negative control (0.5 μg/ml in culture medium) for 15 min at 37°C. After being washed, the cells were fixed in 3% paraformaldehyde in phosphate-buffered saline for 10 min at room temperature and permeabilized with 0.5% Triton X-100 for 3 min. Internal S protein was labeled using a rabbit antibody that recognizes the cytoplasmic tail of IBV. Secondary antibodies were Texas red-conjugated goat anti-mouse immunoglobulin G (IgG) (Jackson Immunoresearch, PA) and Alexa 488-conjugated goat anti-rabbit IgG (Molecular Probes, Eugene, OR). Images were acquired with a Zeiss (Thornwood, NY) Axioscop microscope equipped for epifluorescence with a Sensys charge-coupled-device camera (Photometrics, Tucson, AZ), using IPLab software (Scanalytics, Vienna, VA).
RESULTS
Mutation of the IBV S ER retrieval signal in the IBV infectious clone.
We previously showed that the cytoplasmic tail of IBV S protein has two trafficking signals (16). One signal is a dilysine sequence that retrieves the IBV S protein to the ER and the other is a tyrosine-based endocytosis signal. A reporter protein containing the IBV S cytoplasmic tail is retained in the ERGIC. When the dilysine signal was mutated, the protein was rapidly transported through the Golgi to the plasma membrane and then endocytosed. When both the dilysine and endocytosis signals were mutated, the reporter protein accumulated at the plasma membrane. The implications of these data are that IBV has two mechanisms to prevent accumulation of its S protein at the cell surface. To address the contribution of the IBV S protein trafficking signals to IBV infection, an infectious cDNA clone of IBV described previously (36) was used in this study. The codons for two lysines in the canonical ER retrieval signal of the open reading frame (ORF) encoding S in the E amplicon were replaced with those for alanines by PCR-based site-directed mutagenesis (Fig. 1, S2A). To avoid unintentional mutations during PCR-based mutagenesis, a small fragment containing the mutation was subcloned into the E amplicon after sequencing.
FIG. 1.

Schematic diagram of IBV-S2A, IBV-S2YA, IBV-SYA, and IBV-S4A mutant virus constructs. The upper open boxes represent each major ORF of the IBV genome, and the closed boxes represent 5′ and 3′ untranslated regions. The lower boxes represent seven amplicons encompassing the entire genome of IBV used to construct recombinant IBVs. Amplicon E was used to eliminate the dilysine ER retrieval sequence located in the C terminus of the IBV S gene by replacing the codons for lysine with those for alanines, producing the S2A amplicon. The S4A amplicon was constructed using the S2A amplicon by additionally replacing the tyrosine codons for the endocytosis signal with those for alanines. The SYA and SY2A amplicons were constructed by replacing the single or double tyrosine codons in the endocytosis signal. Seven DNA fragments, including the E amplicon with or without mutations, were assembled by in vitro ligation and used to transcribe cRNAs. The partial nucleotide sequence and amino acid sequence of the IBV S C terminus are shown. The trafficking signals are shown in boldface. Lines indicate the same nucleotide and amino acid sequences as IBV. Asterisks represent stop codons. The underlined sequence represents the core TRS sequence of IBV subgenomic RNA3.
To investigate the contribution of the IBV S ER retrieval signal to IBV infection, recombinant IBV-S2A virus was produced, along with molecularly cloned IBV (referred to here as IBV) as a control (36). Briefly, in vitro-transcribed cRNA of IBV or IBV-S2A was transfected, along with in vitro-transcribed IBV N transcript, into BHK-21 cells, and the BHK-21 cells were cocultured with Vero cells. At 20 h posttransfection, typical IBV cytopathic effects were observed in cells transfected with cRNAs of IBV and IBV-S2A. Both viruses were harvested at 48 h posttransfection. To further characterize IBV-S2A, the virus was plaque purified three times, and then 12 independent clones were selected and amplified in Vero cells. All 12 clones were confirmed to have alanines instead of lysines at the position of the ER retrieval sequence by reverse transcription-PCR amplification and sequencing (data not shown). All experiments were performed with two independent clones of IBV and IBV-S2A to avoid complication by random mutation of recovered recombinant viruses. Because the two clones of each gave identical results, data from one clone of each virus (IBV-1 and IBV-S2A-1) are presented unless otherwise specified.
IBV-S2A has different growth characteristics than IBV.
One-step growth curves for IBV and IBV-S2A were performed in Vero cells. Cells were infected at an MOI of 2, and the total virus produced was harvested every 4 hours by freezing cells and supernatant together. IBV reached a peak titer by 12 h p.i., and the titer remained high until 24 h p.i., when it declined slightly. Recombinant IBV-S2A also reached a peak titer by 12 h p.i. However, after 12 h p.i., virus production rapidly declined, and at 20 h p.i., total IBV-S2A infectious virus was 10-fold less than that of IBV (Fig. 2).
FIG. 2.
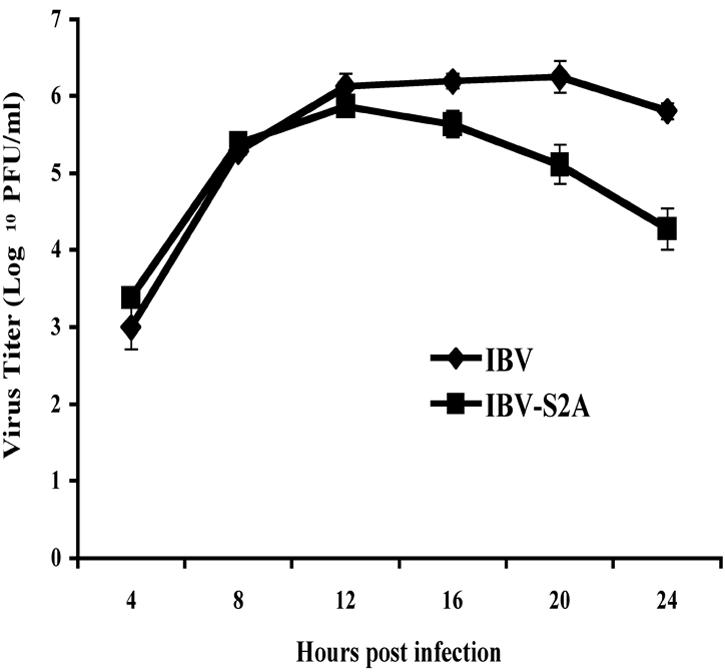
Defective growth of IBV-S2A at late times postinfection. One-step growth curves of IBV and IBV-S2A were performed. Vero cells were infected in triplicate with IBV or IBV-S2A at an MOI of 2. Total virus (supernatant plus cells disrupted by freeze-thaw) was harvested at the time points shown. Titers were measured by plaque assay at each time point. The error bars represent the standard errors of the means.
We also compared the percentage of infectious virus released from cells infected with IBV and IBV-S2A. For this experiment, the supernatant and cells were harvested separately. At 12 h p.i., 54% of the total infectious IBV was present in the supernatant, but only 28% of the total infectious IBV-S2A was released into the supernatant. At 16 h p.i., 59% and 31% of the total infectious IBV and IBV-S2A was present in the supernatant, respectively (Fig. 3). Thus, IBV-S2A-infected Vero cells not only produced less total virus but also released significantly less virus into the supernatant than IBV-infected Vero cells.
FIG. 3.
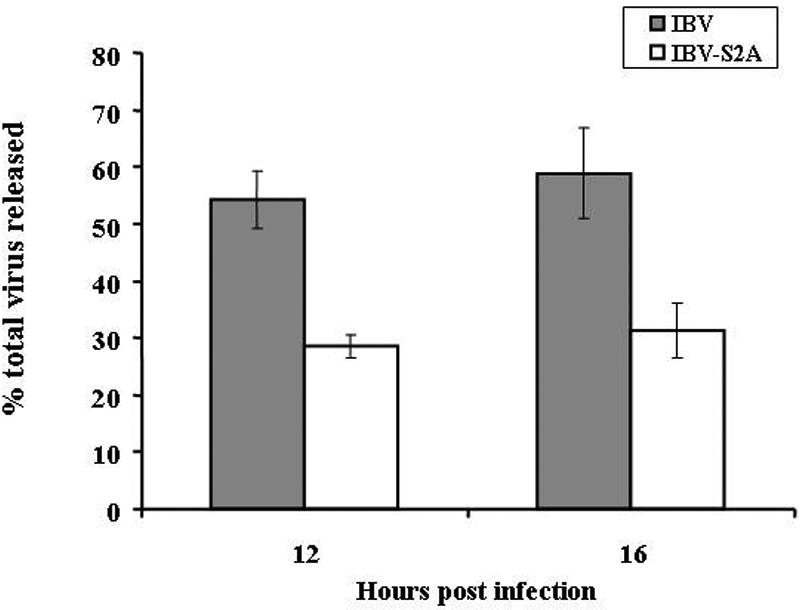
Recombinant IBV-S2A releases less infectious virus than IBV. Vero cells were infected in triplicate with IBV and IBV-S2A at an MOI of 2. The percentage of total virus released was determined after supernatants and cells were harvested separately at 12 and 16 h postinfection and titers were determined as described in Materials and Methods. The percentage of virus released was calculated by dividing the extracellular virus by the total (intracellular plus extracellular) virus. The error bars represent the standard errors of the means.
The different growth kinetics of IBV-S2A might be explained by aberrant or faster trafficking of the S2A protein at late times in the virus replication cycle. This would result in lower virus production because of limited S protein at the virus assembly site. Interestingly, cell fusion was observed in IBV-S2A-infected cells at 8 h p.i., whereas IBV-infected cells began to fuse at 10 h p.i. (data not shown). In subsequent experiments, we repeatedly observed that with identical infection conditions, IBV-S2A-infected cells formed syncytia earlier than IBV-infected cells. IBV-S2A also produced slightly bigger plaques than IBV (Fig. 4). Cell-cell fusion in coronavirus-infected cells is triggered by the interaction of the S protein on the infected cell surface with the virus receptors on adjacent cell surfaces. Therefore, it is likely that the S protein in recombinant IBV-S2A traffics more quickly to the cell surface than wild-type IBV S protein, inducing faster cell fusion and bigger plaques by increased cell-to-cell infection.
FIG. 4.
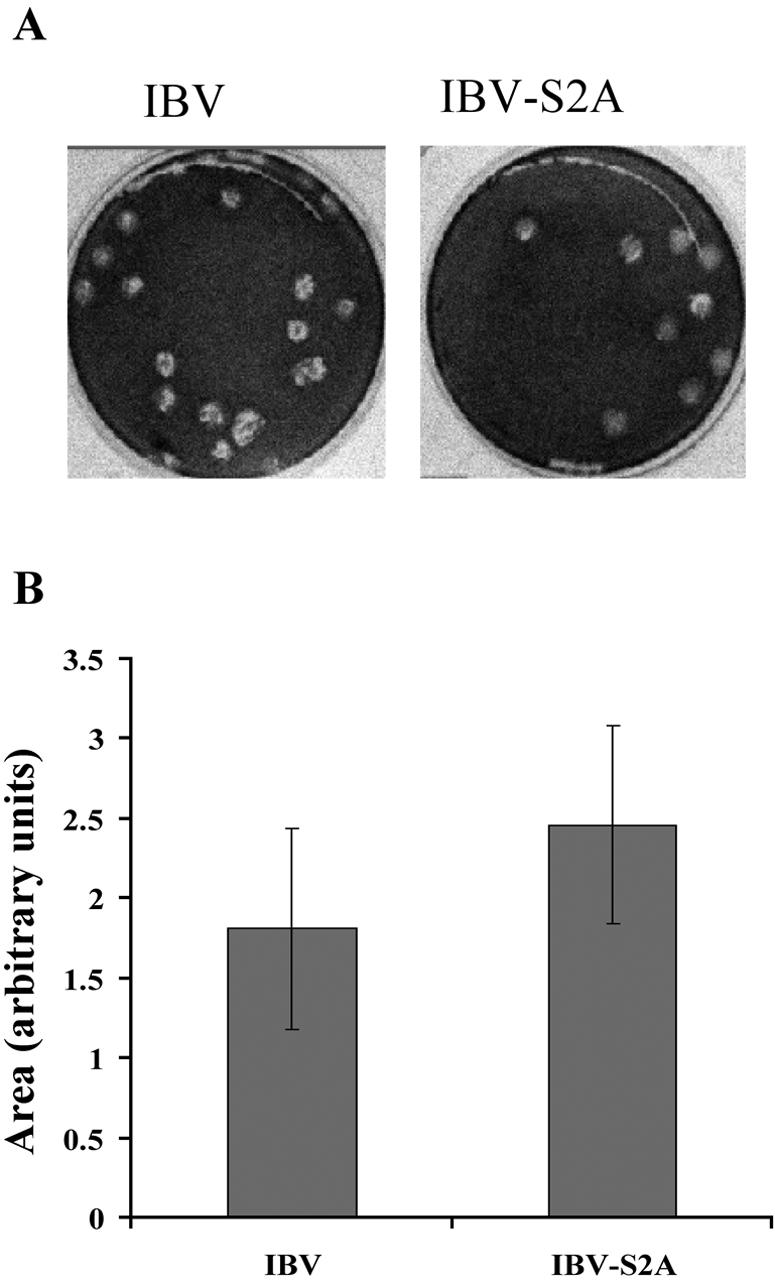
IBV-S2A produces larger plaques than IBV. (A) Vero cells were infected with IBV and IBV-S2A and overlaid with medium containing 0.8% agarose for 3 days. The agarose overlay was removed, and the cells were stained with crystal violet. Plaques produced by IBV are slightly smaller than plaques produced by IBV-S2A. (B) Average areas of plaques produced by IBV and IBV-S2A (IBV, n = 22; IBV-S2A, n = 30) were measured using Quantity One software (version 4.5.2). The error bars represent standard deviations. Student's t test, P = 0.00074.
S2A protein is processed in the Golgi more rapidly than wild-type S protein.
Coronavirus S proteins are highly glycosylated proteins; IBV S has 26 predicted N-linked glycosylation sites. Glycoproteins obtain N-linked oligosaccharides during their synthesis in the ER, and the oligosaccharides are processed as the protein moves though the Golgi stacks. In the medial Golgi, N-linked oligosaccharides gain resistance to digestion with endo H. The rate at which a glycoprotein moves through the Golgi can thus be inferred from the rate at which it acquires endo H resistance. To determine the effect of mutating the ER retrieval signal on IBV S in infected cells, we compared the processing kinetics of S2A to those of S in a pulse-chase labeling experiment. Newly synthesized proteins were labeled with [35S]methionine-cysteine for 20 min at approximately 8 h p.i., and cells were chased in unlabeled medium for up to 90 min. IBV S and S2A proteins were immunoprecipitated from the cell lysates, and immunoprecipitates were digested with endo H. Differences in oligosaccharide processing between S and S2A were observed, with more endo H resistance acquired by S2A during the chase (Fig. 5A and B). Oligosaccharide processing of S2A also appeared to be more extensive than that of S at the chase times analyzed (as shown by the more slowly migrating endo H-resistant species in Fig. 5A). In addition, cleavage of S0 to S1 and S2 was faster for S2A than for S (Fig. 5A). Interestingly, cleavage of IBV S in infected Vero cells was detected after 20 min of labeling and preceded oligosaccharide processing. Our results indicate that IBV S2A traffics through the Golgi faster than wild-type S protein in virus-infected cells.
FIG. 5.

S2A protein traffics through the Golgi faster than wild-type IBV S protein in infected cells. (A) At 8 h p.i., Vero cells infected with IBV and IBV-S2A were pulse-labeled with [35S]methionine-cysteine for 20 min and chased for the times indicated. Lysates were immunoprecipitated with anti-IBV S antibody and treated with endo H prior to sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The arrowhead indicates endo H-sensitive uncleaved S0 protein, the bracket indicates the heterogeneous population of endo H-resistant cleaved S1/S2, and the asterisk indicates endo H-sensitive S1/S2. C, uninfected Vero cells; S, IBV-infected Vero cells; S2A, IBV-S2A-infected Vero cells. (B) The percentage of endo H-resistant S was calculated by dividing the amount of resistant S1/S2 by the total S for each time point. These data are representative of three independent experiments.
The S protein ER retrieval signal appears to overlap with a critical cis sequence for transcriptional control of subgenomic RNA3.
To complete the characterization of IBV-S2A, we compared the RNA transcription profile of IBV and IBV-S2A by Northern blotting (Fig. 6A). Surprisingly, we found a significant decrease in RNA3 levels (but not the in other subgenomic RNAs) in IBV-S2A-infected cells compared to those infected with IBV. The same results were obtained at earlier times p.i. (8, 10, and 12 h) (data not shown). Subgenomic RNA3 encodes the 3a, 3b, and E proteins. Since the E protein is thought to be essential for efficient virus production, we assessed the amount of E protein produced in infected cells. The reduced RNA3 transcript level in IBV-S2A-infected cells was mirrored by a reduction in IBV E protein (Fig. 6B). Two independent clones of IBV-S2A showed reduced levels of E protein compared to IBV. In contrast to other coronaviruses, most ORFs of IBV overlap by from 1 to 50 nucleotides. The 3′ end of the ORF encoding the S protein overlaps with the transcriptional regulatory sequence (TRS) for RNA3. The mutations in the dilysine signal in IBV S are just downstream of the core TRS (Fig. 1), which could impact the efficiency of the discontinuous transcription process that produces the subgenomic RNAs during infection (30, 37). Thus, the growth defects of IBV-S2A late in infection could be the result of the mutation of the ER retrieval signal, reduced synthesis of E protein, or both.
FIG. 6.

Mutation of the dilysine signal in IBV S affects transcription and translation of the downstream ORF. (A) At 24 h p.i., total cellular RNAs from wild-type IBV- or IBV-S2A-infected Vero cells were extracted and subjected to Northern blotting for subgenomic RNA profile analysis as described in Materials and Methods. Transcription of RNA3 (but not the other subgenomic RNAs) was significantly decreased in IBV-S2A-infected cells compared to that in cells infected with IBV. (B) E protein levels were analyzed by immunoprecipitation from [35S]methionine-cysteine-labeled infected Vero cells. At 9.5 h p.i., IBV- or IBV-S2A-infected Vero cells were labeled for 30 min and lysed, and the lysates were split in two and immunoprecipitated with anti-IBV E or anti-IBV M antibody. The results for two different clones of IBV-S2A (1 and 4) are shown.
Infectious virus cannot be rescued when both targeting signals in IBV S are mutated.
To investigate whether the tyrosine-based endocytosis signal in IBV S protein further impacts virus infection, we constructed the IBV-S4A mutant lacking both the ER retrieval signal and the endocytosis signal (Fig. 1). In vitro-assembled and -transcribed cRNA of IBV-S4A was electroporated into BHK-21 cells, along with IBV N transcript, as described above. At 20 h posttransfection, we could detect several huge syncytia in IBV-S4A-transfected cells (Fig. 7B), while cells transfected with IBV cRNA started to show only small syncytia at the same time point (Fig. 7A). Around 30 h posttransfection, cells transfected with IBV cRNA showed more and larger syncytia (not shown). At the same time point, the huge syncytia seen in cells transfected with IBV-S4A cRNA rounded up and detached from the plate (Fig. 7C). Healthy-looking Vero cells replaced the empty area created by detachment of the huge syncytia. At 48 h postinfection, IBV-transfected cells showed extensive cell death caused by the viral infection while IBV-S4A-transfected cells were overgrown. We passaged IBV-S4A-transfected cell lysates twice; however, we never observed any virus-induced cytopathic effects. We repeated this experiment four times, and the same results (production of huge syncytia followed by their disappearance) were obtained in all cases. Thus, mutation of both targeting signals in the cytoplasmic tail of IBV S was incompatible with recovery of infectious virus.
FIG. 7.

Large transient syncytia formed after electroporation of cRNA with the S4A mutation, but no infectious virus could be recovered. In vitro-transcribed IBV or IBV-S4A cRNAs were electroporated into BHK-21 cells, which were then cocultured with Vero cells. (A) At 20 h posttransfection, normal-size syncytia started to form in IBV cRNA-transfected cells. (B) At 20 h posttransfection of IBV-S4A cRNA, transfected cells showed far fewer but much larger syncytia than IBV cRNA-transfected cells. (C) At 30 h posttransfection of IBV-S4A cRNA, the giant syncytia started to disappear by clumping and detaching from the dish. Uninfected Vero cells expanded to fill the empty areas left by the syncytia. Bars, 100 μm.
The S4A mutation does not disrupt S-M interaction.
The IBV-S4A protein has four amino acid substitutions in its cytoplasmic tail. The cytoplasmic tail of MHV S has been shown to interact with MHV M protein (24). Thus, one explanation for failure to produce recombinant IBV with the S4A mutation is disruption of the interaction between IBV S and IBV M. This possibility was investigated using methods similar to those described for MHV (24). First, to confirm interaction of the IBV M and S proteins, IBV-infected Vero cells were labeled with [35S]methionine-cysteine and chased for 30 min at 8 h p.i. The cells were lysed and immunoprecipitated with an antibody against IBV M. As shown in Fig. 8A, IBV S could be coimmunoprecipitated with anti-M antibody even in the absence of cross-linking. Our antibody against the cytoplasmic tail of IBV S did not coimmunoprecipitate M protein as efficiently as antibody against M coimmunoprecipitated S, perhaps because antibody binding to the S tail disrupted interaction with M. Next, interaction of the S4A protein with IBV M was tested. We transiently expressed IBV M with wild-type IBV S or S4A using a vaccinia virus T7 expression system. Transfected cells were labeled with [35S]methionine-cysteine for 30 min and chased for 30 min. Interaction was assessed by coimmunoprecipitation of S protein with anti-IBV M antibody. Compared to wild-type S, S4A protein interacted with M less efficiently (Fig. 8B). However, this transient-expression system does not reproduce the same timing of protein production as in infected cells. The faster trafficking of S4A protein in this transient system could result in an insufficient amount of time to interact with M protein, which is retained in the Golgi region. To determine if the inefficient interaction of S4A and M was due to a genuine defect in the interaction between these proteins or due to the increased trafficking rate of S4A protein, BFA was used. BFA causes the Golgi complex to collapse into the ER and thus blocks export of proteins from the ER (10). By confining M and S proteins to the ER with BFA treatment, interaction of M and S4A protein comparable to that of M and wild-type S protein was observed (Fig. 8B). Based on these results, it is unlikely that defective interaction of S4A with IBV M explains the inability to rescue IBV-S4A.
FIG. 8.
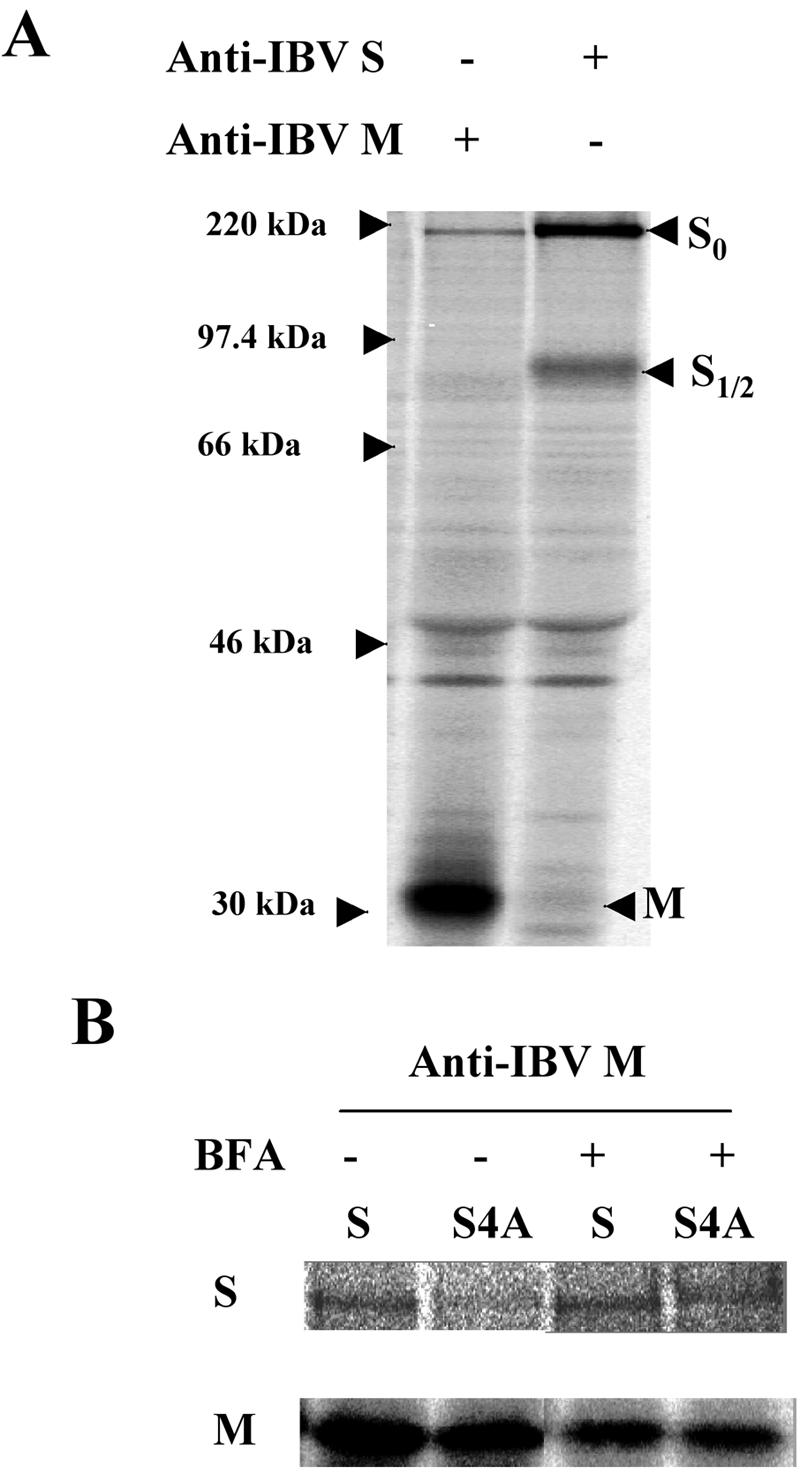
IBV-S4A interacts normally with IBV M. Interaction of S or S4A with IBV M protein was assessed by coimmunoprecipitation. (A) IBV S and M proteins interact in IBV-infected Vero cells. At 8 h p.i., Vero cells infected with IBV were labeled with [35S]methionine-cysteine for 30 min, and the cell lysate was subjected to immunoprecipitation with anti-IBV M or anti-IBV S antibody. (B) S or S4A protein was coexpressed with IBV M protein transiently using vaccinia virus expressing T7 RNA polymerase. After a 30-min pulse-labeling and 30-min chase in the presence or absence of BFA, S or S4A was coimmunoprecipitated with anti-IBV M antibody.
Infectious virus cannot be rescued when only the tyrosine-based endocytosis signal is mutated.
IBV-S4A could not be rescued, even though huge syncytia formed in transfected cells and the S4A protein could interact with IBV M as well as wild-type S protein. We previously showed that the endocytosis signal restricts the amount of IBV S protein on the cell surface, even though the signal itself does not influence trafficking of the S protein from the ER through the Golgi (16). To test the possibility that the endocytosis signal plays an important role in virus replication, we constructed a mutant lacking the two tyrosines in the YYTF endocytosis signal (S2YA) (Fig. 1). To our surprise, we were unable to rescue IBV-S2YA even though we could detect transient, normal-size syncytia approximately 30 h posttransfection (data not shown). These syncytia resolved similarly to those in cells transfected with IBV-S4A cRNA, except that they lasted longer, until approximately 48 h posttransfection. We further constructed a mutant (IBV-SYA) where only the key tyrosine was changed to alanine (Fig. 1). We were unable to rescue the IBV-SYA mutant as well. The latter mutation changed only 2 nucleotides at a site 25 nucleotides upstream of the core TRS for subgenomic RNA3. We believe it is unlikely that the tyrosine-to-alanine mutation interferes with transcription of RNA3, although we could not test this directly because recombinant virus could not be recovered. Thus, we favor the idea that the tyrosine-based endocytosis signal plays an essential role in virus infection.
IBV S is endocytosed from the surfaces of infected cells.
Our results suggested that endocytosis of IBV S plays a critical role in infection. To obtain direct evidence that IBV S is endocytosed in infected cells, we followed trafficking of the protein using exogenously added antibodies. After 15 min at 37°C, the anti-IBV S monoclonal antibody added to the culture medium was internalized into endosome-like puncta, whereas none of a control monoclonal antibody was endocytosed (Fig. 9). Only cells with the highest levels of S expression internalized the antibodies, consistent with the idea that S protein does not reach the plasma membrane until the ER retrieval machinery is saturated. These results suggest that once the IBV S protein reaches the plasma membrane it is rapidly endocytosed.
FIG. 9.

IBV S is rapidly endocytosed in infected cells. Vero cells on coverslips were infected with IBV at a MOI of 0.1 for approximately 18 h. A monoclonal antibody against the IBV S ectodomain (or the VSV G ectodomain as a negative control) was added to the culture medium for 15 min at 37°C. After being thoroughly washed, the cells were fixed, permeabilized, and stained for internal S protein using a rabbit antibody to the S cytoplasmic tail. Exogenously added monoclonal antibodies were detected using Texas red-conjugated goat anti-mouse IgG, and the internal antibody staining was detected with Alexa 488-conjugated goat anti-rabbit IgG. The left panels show internalized antibody, and the right panels show the internal staining of the same field. Note that only the cell with the higher intracellular level of IBV S endocytosed detectable antibody.
DISCUSSION
Contribution of the S protein ER retrieval signal to IBV infection.
We previously showed that the IBV S protein contains two intracellular trafficking signals in its cytoplasmic tail: a dilysine ER retrieval signal and a tyrosine-based endocytosis signal (16). The purpose of this study was to evaluate the contribution of the IBV S protein trafficking signals to virus infection. First, we constructed a recombinant IBV lacking the ER retrieval signal in the S protein. After in vitro assembly and in vitro transcription, followed by transfection of full-length IBV-S2A cRNA, recombinant virus was rescued. IBV-S2A had a growth defect at late times p.i., with a titer 10-fold lower than that of IBV. In infected cells, the S2A protein behaved similarly to the chimeric protein lacking the dilysine signal (16). First, a pulse-chase experiment confirmed that S2A traffics through the Golgi complex faster than wild-type S. Second, we could repeatedly detect earlier syncytium formation in IBV-S2A-infected cells than in cells infected with wild-type virus. Third, IBV-S2A virus produced larger plaques than the wild-type virus. These last two results can be explained by faster trafficking of the S protein to the infected cell surface, thus causing earlier induction of cell-to-cell fusion between the infected cells and neighboring cells. Clearly, the ER retrieval signal in IBV S protein is used in virus infection. The other phenotype of IBV-S2A was a reduced efficiency of release of infectious virus, which was unexpected. Perhaps earlier formation of syncytia induced by faster trafficking of S2A hinders release of virus.
When analyzing subgenomic RNA production by Northern blotting, we made the surprising observation that RNA3 levels were decreased in cells infected with IBV-S2A compared to those infected with IBV. The decrease in RNA3 levels was mirrored by reduced levels of IBV E protein in IBV-S2A-infected cells. Thus, we could not rule out the possibility that the growth defect at late times p.i. was the result of decreased production of E protein. Coronaviruses produce their sgRNAs by a process of discontinuous transcription, producing a nested set of sgRNAs that share the same 5′ leader sequence. Base pairing between the leader sequence and the TRS that precedes each ORF is essential for sgRNA production (33, 37). In contrast to other coronaviruses, most IBV ORFs overlap each other, and the core TRS for RNA3 is just upstream of the sequence that encodes the dilysine signal in the S tail (Fig. 1). We speculate that the mutations in S2A affect the discontinuous transcription process leading to production of RNA3. Sequences flanking the core TRS, particularly those downstream, have been shown to affect sgRNA production in transmissible gastroenteritis virus (30). This region may be important for transcription of RNA3 in IBV, since the core TRS sequence for RNA3 contains a 1-nucleotide mismatch with the leader TRS core. Further study will be required to determine whether the growth defect in IBV-S2A is due solely to the mutation's effect on RNA3 synthesis or if the dilysine signal itself plays an important role in IBV infection.
Contribution of the endocytosis signal in the IBV S cytoplasmic tail to virus infection.
Compared to mutation of the dilysine signal, mutation of the endocytosis signal gave a more dramatic phenotype. Infectious virus could not be recovered even when as few as 2 nucleotides were mutated to change the key tyrosine residue. The mutations of both trafficking signals in the S tail did not block interaction with M; therefore, decreased interaction of S and M is unlikely to explain the failure to recover IBV lacking the tyrosine endocytosis signal. Although we cannot rule out the possibility that the mutation affects RNA3 production or virus replication, we favor the idea that endocytosis of IBV S is essential to productive virus infection. It follows that the dilysine signal in IBV S plays a less important role in virus infection than the endocytosis signal. This idea is supported by the observation that the endocytosis signal is conserved in all IBV strains while the dilysine signal is absent in several due to a stop codon that truncates the S protein. Why would the IBV S protein need an endocytosis signal if it contains a dilysine ER retrieval signal? Even though the dilysine signal is effective at retaining the protein in the ERGIC at low expression levels, it is likely that the ER retrieval machinery is easily saturated. We found this to be true using transient expression of chimeric proteins containing the IBV S tail (16). At late times p.i., when S expression levels are high, it is likely that S escapes to the cell surface, where it can induce cell-cell fusion. We originally hypothesized that the targeting information in IBV S helps to localize the protein to the virus assembly site. However, the data presented here suggest that the IBV S trafficking signals may be more important in regulating the level of IBV S at the cell surface. Syncytia undergo significant internal membrane rearrangements (14), and the distance from the site of virus assembly to the plasma membrane is significantly increased. The production of large syncytia could thus interfere with virus budding or release.
Tyrosine-based sorting signals and implications for the coronavirus replication cycle.
The type of endocytic signal assessed here in the IBV S protein (YXXθ, where X is any amino acid and θ is a bulky hydrophobic residue) has been shown to mediate other sorting events for membrane proteins in addition to endocytosis. This motif can target proteins from the trans-Golgi network directly to endosomes/lysosomes and to the basolateral plasma membrane in polarized epithelial cells (1). Here, we were able to directly demonstrate that IBV S was rapidly endocytosed from the plasma membranes of infected cells. Thus, we favor the interpretation that the YXXθ sequence in IBV functions as a canonical endocytosis signal.
Interestingly, lentivirus Env proteins all contain conserved endocytosis signals (4, 26, 28, 31), and in the case of simian immunodeficiency virus, mutation of the endocytosis signal significantly attenuates infection in vivo (7). Perhaps a common strategy for enveloped viruses that encode fusion proteins that fuse at neutral pH is to regulate the surface levels of these proteins by using endocytosis. Indeed, there is a growing appreciation of the potential role of endocytosis in multiple stages of virus replication cycles (19). Interestingly, the S proteins of group 1 and 3 coronaviruses possess both ER retrieval signals (16) and predicted endocytosis signals in their cytoplasmic domains. SARS S contains a dibasic ER retrieval signal but lacks a predicted endocytosis signal. Group 2 coronaviruses lack both ER retrieval and predicted endocytosis signals. The possibility exists for a novel endocytosis signal in the S proteins from group 2 coronaviruses and SARS. We will continue to explore the requirement for endocytosis of IBV S by introducing endocytosis signals in new places in the cytoplasmic tail of IBV using the infectious clone.
Acknowledgments
This work was supported by Public Health Service grant GM64647 from the National Institute of General Medicine.
We thank the members of the Machamer laboratory for helpful discussions and comments on the manuscript.
REFERENCES
- 1.Bonifacino, J. S., and L. M. Traub. 2003. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72:395-447. [DOI] [PubMed] [Google Scholar]
- 2.Corse, E., and C. E. Machamer. 2003. The cytoplasmic tails of infectious bronchitis virus E and M proteins mediate their interaction. Virology 312:25-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corse, E., and C. E. Machamer. 2000. Infectious bronchitis virus E protein is targeted to the Golgi complex and directs release of virus-like particles. J. Virol. 74:4319-4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Danis, C., J. Deschambeault, S. Do Carmo, E. A. Cohen, E. Rassart, and G. Lemay. 2004. The tyrosine-based YXXO targeting motif of murine leukemia virus envelope glycoprotein affects pathogenesis. Virology 324:173-183. [DOI] [PubMed] [Google Scholar]
- 5.de Haan, C. A., L. Kuo, P. S. Masters, H. Vennema, and P. J. Rottier. 1998. Coronavirus particle assembly: primary structure requirements of the membrane protein. J. Virol. 72:6838-6850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuerst, T. R., E. G. Niles, F. W. Studier, and B. Moss. 1986. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. USA 83:8122-8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fultz, P. N., P. J. Vance, M. J. Endres, B. Tao, J. D. Dvorin, I. C. Davis, J. D. Lifson, D. C. Montefiori, M. Marsh, M. H. Malim, and J. A. Hoxie. 2001. In vivo attenuation of simian immunodeficiency virus by disruption of a tyrosine-dependent sorting signal in the envelope glycoprotein cytoplasmic tail. J. Virol. 75:278-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gallagher, T. M., and M. J. Buchmeier. 2001. Coronavirus spike proteins in viral entry and pathogenesis. Virology 279:371-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holmes, K. V., and L. Enjuanes. 2003. Virology. The SARS coronavirus: a postgenomic era. Science 300:1377-1378. [DOI] [PubMed] [Google Scholar]
- 10.Klausner, R. D., J. G. Donaldson, and J. Lippincott-Schwartz. 1992. Brefeldin A: insights into the control of membrane traffic and organelle structure. J. Cell Biol. 116:1071-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klumperman, J., J. Locker, A. Meijer, M. Horzinek, H. Geuze, and P. Rottier. 1994. Coronavirus M proteins accumulate in the Golgi complex beyond the site of virion budding. J. Virol. 68:6523-6534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krijnse-Locker, J., M. Ericsson, P. Rottier, and G. Griffiths. 1994. Characterization of the budding compartment of mouse hepatitis virus: evidence that transport from the RER to the Golgi complex requires only one vesicular transport step. J. Cell Biol. 124:55-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuo, L., and P. S. Masters. 2003. The small envelope protein E is not essential for murine coronavirus replication. J. Virol. 77:4597-4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lai, M. M., and D. Cavanagh. 1997. The molecular biology of coronaviruses. Adv. Virus Res. 48:1-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lefrancois, L., and D. S. Lyles. 1982. The interaction of antibody with the major surface glycoprotein of vesicular stomatitis virus. II. Monoclonal antibodies of nonneutralizing and cross-reactive epitopes of Indiana and New Jersey serotypes. Virology 121:168-174. [DOI] [PubMed] [Google Scholar]
- 16.Lontok, E., E. Corse, and C. E. Machamer. 2004. Intracellular targeting signals contribute to localization of coronavirus spike proteins near the virus assembly site. J. Virol. 78:5913-5922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Machamer, C. E., and J. K. Rose. 1987. A specific transmembrane domain of a coronavirus E1 glycoprotein is required for its retention in the Golgi region. J. Cell Biol. 105:1205-1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mackenzie, J. M., and E. G. Westaway. 2001. Assembly and maturation of the flavivirus Kunjin virus appear to occur in the rough endoplasmic reticulum and along the secretory pathway, respectively. J. Virol. 75:10787-10799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marsh, M., and A. Pelchen-Matthews. 2000. Endocytosis in viral replication. Traffic 1:525-532. [DOI] [PubMed] [Google Scholar]
- 20.Matsuoka, Y., S. Y. Chen, and R. W. Compans. 1991. Bunyavirus protein transport and assembly. Curr. Top. Microbiol. Immunol. 169:161-179. [DOI] [PubMed] [Google Scholar]
- 21.Narayanan, K., A. Maeda, J. Maeda, and S. Makino. 2000. Characterization of the coronavirus M protein and nucleocapsid interaction in infected cells. J. Virol. 74:8127-8134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen, V. P., and B. G. Hogue. 1997. Protein interactions during coronavirus assembly. J. Virol. 71:9278-9284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ono, A., and E. O. Freed. 2004. Cell-type-dependent targeting of human immunodeficiency virus type 1 assembly to the plasma membrane and the multivesicular body. J. Virol. 78:1552-1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Opstelten, D. J., M. J. Raamsman, K. Wolfs, M. C. Horzinek, and P. J. Rottier. 1995. Envelope glycoprotein interactions in coronavirus assembly. J. Cell Biol. 131:339-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ortego, J., D. Escors, H. Laude, and L. Enjuanes. 2002. Generation of a replication-competent, propagation-deficient virus vector based on the transmissible gastroenteritis coronavirus genome. J. Virol. 76:11518-11529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rowell, J. F., P. E. Stanhope, and R. F. Siliciano. 1995. Endocytosis of endogenously synthesized HIV-1 envelope protein. Mechanism and role in processing for association with class II MHC. J. Immunol. 155:473-488. [PubMed] [Google Scholar]
- 27.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Plainview, N.Y.
- 28.Sauter, M. M., A. Pelchen-Matthews, R. Bron, M. Marsh, C. C. LaBranche, P. J. Vance, J. Romano, B. S. Haggarty, T. K. Hart, W. M. Lee, and J. A. Hoxie. 1996. An internalization signal in the simian immunodeficiency virus transmembrane protein cytoplasmic domain modulates expression of envelope glycoproteins on the cell surface. J. Cell Biol. 132:795-811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sawicki, S. G., and D. L. Sawicki. 2005. Coronavirus transcription: a perspective. Curr. Top. Microbiol. Immunol. 287:31-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sola, I., J. L. Moreno, S. Zuniga, S. Alonso, and L. Enjuanes. 2005. Role of nucleotides immediately flanking the transcription-regulating sequence core in coronavirus subgenomic mRNA synthesis. J. Virol. 79:2506-2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song, C., S. R. Dubay, and E. Hunter. 2003. A tyrosine motif in the cytoplasmic domain of Mason-Pfizer monkey virus is essential for the incorporation of glycoprotein into virions. J. Virol. 77:5192-5200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stadler, K., V. Masignani, M. Eickmann, S. Becker, S. Abrignani, H. D. Klenk, and R. Rappuoli. 2003. SARS—beginning to understand a new virus. Nat. Rev. Microbiol. 1:209-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Marle, G., J. C. Dobbe, A. P. Gultyaev, W. Luytjes, W. J. Spaan, and E. J. Snijder. 1999. Arterivirus discontinuous mRNA transcription is guided by base pairing between sense and antisense transcription-regulating sequences. Proc. Natl. Acad. Sci. USA 96:12056-12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vennema, H., G. Godeke, J. Rossen, W. Voorhout, M. Horzinek, D. Opstelten, and P. Rottier. 1996. Nucleocapsid-independent assembly of coronavirus-like particles by co-expression of viral envelope protein genes. EMBO J. 15:2020-2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang, L., R. L. Parr, D. J. King, and E. W. Collisson. 1995. A highly conserved epitope on the spike protein of infectious bronchitis virus. Arch. Virol. 140:2201-2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Youn, S., J. L. Leibowitz, and E. W. Collisson. 2005. In vitro assembled, recombinant infectious bronchitis viruses demonstrate that the 5a open reading frame is not essential for replication. Virology 332:206-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zuniga, S., I. Sola, S. Alonso, and L. Enjuanes. 2004. Sequence motifs involved in the regulation of discontinuous coronavirus subgenomic RNA synthesis. J. Virol. 78:980-994. [DOI] [PMC free article] [PubMed] [Google Scholar]