

Abstract
Background
Elastin calcification is a widespread feature of vascular pathology, and circumstantial evidence exists for a correlation between elastin degradation and calcification. We hypothesized that matrix metalloproteinase (MMP)–mediated vascular remodeling plays a significant role in elastin calcification.
Methods and Results
In the present studies, we determined that short-term periadventitial treatment of the rat abdominal aorta with low concentrations of calcium chloride (CaCl2) induced chronic degeneration and calcification of vascular elastic fibers in the absence of aneurysm formation and inflammatory reactions. Furthermore, the rate of progression of calcification depended on the application method and concentration of CaCl2 applied periarterially. Initial calcium deposits, associated mainly with elastic fibers, were persistently accompanied by elastin degradation, disorganization of aortic extracellular matrix, and moderate levels of vascular cell apoptosis. Application of aluminum ions (known inhibitors of elastin degradation) before the CaCl2-mediated injury significantly reduced elastin calcification and abolished both extracellular matrix degradation and apoptosis. We also found that MMP-knockout mice were resistant to CaCl2-mediated aortic injury and did not develop elastin degeneration and calcification.
Conclusion
Collectively, these data strongly indicate a correlation between MMP-mediated elastin degradation and vascular calcification.
Keywords: calcium, metalloproteinases, collagen, arteriosclerosis, apoptosis
Vascular calcification occurs at 2 distinct sites within the vessel wall: the intima and the media.1 Intimal calcification occurs mostly in association with atherosclerosis, subsequent to lipid deposition, macrophage infiltration, and vascular smooth muscle cell proliferation,2 whereas medial calcification can exist independently of atherosclerosis and typically is associated with elastic fibers appearing initially as linear deposits along elastic lamellae.3 Elastin is relatively resistant to proteolysis by all but a limited number of proteinases, such as the matrix metalloproteinases (MMP). The net result of the action of MMPs on elastin is disruption of elastic fiber integrity and subsequent loss of mechanical properties.4
We have shown earlier that pure elastin undergoes extensive degeneration and calcification when implanted subdermally in juvenile rats5 and that local delivery of a synthetic MMP inhibitor significantly reduced elastin calcification in the rat subdermal model.6 Moreover, we have shown that treatment of elastin with aluminum ions before implantation completely abolished elastin calcification by mechanisms that include inhibition of elastolysis by MMPs.7 These results strongly point to a direct correlation between elastin degradation and calcification. To study mechanisms of elastin calcification in a clinically relevant circulatory model, we adapted an arterial injury model that involves a single periarterial application of a calcium chloride (CaCl2) solution.8 As originally described, this short-term, localized insult induced accelerated and extensive aortic calcification in the rabbit carotid artery, accompanied by an intense inflammatory reaction to the calcified deposits, which consecutively induced progressive aneurysm formation. This model was later adapted for the rabbit abdominal aorta9–11 and murine thoracic aorta.12 CaCl2 treated, aneurysmal aortic samples exhibited increased levels of MMPs, indicating that remodeling processes occur in this model.13,14 Moreover, MMP2-and MMP9-knockout mice did not develop aneurysms when subjected to similar CaCl2-mediated aortic injury,15 strongly suggesting that MMPs are essential for onset and progression of aneurysm. Because this model was not purposely used for calcification studies before, we hypothesized that tempering the rate of aortic calcification would allow us to investigate early pathogenic events involved in elastin calcification in the absence of aneurysm formation and inflammatory reactions. We further hypothesized that MMPs play a significant role in elastin degradation and calcification. In the present studies, we show that elastin calcification in this aortic injury model was persistently accompanied by elastin degradation and disorganization of the aortic extracellular matrix. This pathological process was significantly counteracted by pretreatment of the abdominal aorta with aluminum ions before the CaCl2-mediated injury. We also show that MMP-knockout mice were resistant to CaCl2-mediated aortic injury and did not develop elastin degeneration and calcification. Collectively, these data strongly indicate a correlation between elastin degradation and calcification in a clinically relevant aortic injury model.
Methods
Surgical Procedures
In the first study, adult, male Sprague-Dawley rats weighing 250 to 300 g were placed under general anesthesia (2% to 3% isoflurane), and the infrarenal abdominal aorta was treated for 15 minutes with different concentrations of CaCl2 or NaCl (0.1 mol/L, 0.15 mol/L, or 0.2 mol/L, n=4 rats per group) by rubbing (“painting”) of the aorta with a sterile cotton swab. After 7 days, the animals were humanely euthanized by CO2 asphyxiation; the abdominal aorta was excised; and segments from each aorta were processed for histological analysis, immunohistochemistry, and calcium analysis. In a second experiment, rat abdominal aortas were treated for 15 minutes with 0.15 mol/L CaCl2 with the cotton swab method described earlier (12 rats) and separately with a gauze application method (12 rats), which involved placing a rectangle (1.5×0.5 cm) of presoaked, 8-ply, medical-grade sterile cotton gauze on the exposed aorta. After the aorta was treated, the gauze was removed and the abdominal cavity closed followed by subcutaneous sutures and application of staples. Control rats (n=12) were treated with 0.15 mol/L NaCl with the gauze method. After 3 and 7 days, the animals (6 rats per time point) were euthanized, and the aorta processed for histological and mineral analysis as described earlier.
A third series of experiments involved a 15-minute exposure of the rat abdominal aorta (n=12) to 0.15 mol/L CaCl2 with the gauze method. A similar number of control rats were exposed to 0.15 mol/L NaCl under identical conditions. The effect of aluminum ions on calcification (n=16) was studied by first applying a 0.0025 mol/L AlCl3 solution (with gauze) for 15 minutes followed by 15 minutes of 0.15 mol/L CaCl2. After 7 days, the animals were humanely euthanized by CO2 asphyxiation; the abdominal aorta was excised; and segments from each aorta were processed for histological analysis, immunohistochemistry, and calcium and desmosine analysis.
For evaluation of endothelial permeability, 2 rats from each group (NaCl, CaCl2, AlCl3) were injected under anesthesia with 0.3 mL IV Evans blue solution (20 mg/mL in phosphate-buffered saline); aortic segments were excised after 40 minutes and used for histology. Age-matched rats, which did not undergo any surgical procedures (n=4), were also used as controls, and their aortic segments were analyzed as described earlier. Aortic diameters were measured by digital photography before and 7 days after application of 0.15 mol/L CaCl2 or NaCl solutions in 8 rats from each group.13 All animals received humane care in compliance with protocols approved by the Clemson University Animal Research Committee as formulated by the NIH (publication No. 86-23, revised 1985).
In a fourth experiment, MMP2-knockout, MMP9-knockout, and wild-type mice (n=10 per group) were anesthetized with 60 mg/kg IP 2,2,2-tribromoethanol (Avertin, Sigma) and underwent laparotomy as described.15 The MMP2-knockout mice were obtained from Dr David Muir, University of Florida, Gainesville, Fla, and the MMP9-knockout mice were a generous gift from Robert Senior of Washington University, St. Louis, Mo. The infrarenal abdominal aorta was treated for 15 minutes with 0.25 mol/L CaCl2 as described earlier. Physiological saline was substituted for CaCl2 in control mice. Ten weeks later, the mice underwent laparotomy and the aorta was embedded in paraffin for histological studies. All experiments were carried out in accordance with the guidelines of the University of Nebraska Medical Center Animal Care Committee for the use and care of laboratory animals. All mice were maintained in the pathogen-free animal facility at the University of Nebraska Medical Center.
Calcium and Desmosine Analyses
Quantitative analysis of calcium content in aortic segments was performed as described previously,16 and desmosine content was analyzed in the same acid hydrolysates by radioimmunoassay.17
Histology, Immunohistochemistry, and Apoptosis Detection
Sections were stained with hematoxylin and eosin (H&E) for general tissue morphology, Dahl’s Alizarin red stain for calcium deposits, Gomori’s trichrome (Poly Scientific) for collagen, and Verhoeff–van Gieson’s stain (VVG) for elastin (Poly Scientific). Sections were also stained for infiltrating macrophages with a mouse anti-rat monocyte/macrophage primary antibody (1:200 dilution, MAB1435, Chemicon) and detection with a mouse IgG Vectastain diaminobenzidine kit (Vector Laboratories) followed by H counterstaining. Paraffin sections from knockout-mice studies (n=3 per group) were stained as described earlier. For cell visualization, sections were also mounted in 4′,6′-diamidino-2-phenylindole (DAPI) mounting medium (Molecular Probes). Apoptotic cells were detected with a Tdt-mediated UTP nick end-label (TUNEL) detection kit (DeadEnd kit, Promega).
Statistical Analysis
Data are reported as mean±SEM. For the first study and aortic diameter measurements, Student’s t test was used to compare means between 2 groups, with P<0.05 as the criterion for statistical significance. Comparison among different groups for other studies was performed with the Kruskal-Wallis test with linear contrasts (α=0.05 for all tests).
Results
Rat Model of Aortic Calcification
Perivascular application of 0.2 mol/L CaCl2 to the infrarenal abdominal aorta induced mural calcification and severe disorganization of the aortic extracellular matrix within 1 week after surgery, as demonstrated by H&E and Alizarin red staining (Figure 1, B and D). Application of an equimolar concentration of NaCl did not induce calcification or any notable alterations in tissue structure (Figure 1, A and C). Moreover, morphological aspects of NaCl-treated aortas were not different from those of control aortas from age-matched rats, which were not subjected to any surgical procedure (not shown), indicating that this injury is calcium dependent and that NaCl application could be used as a proper control. Immunohistochemical staining did not reveal any detectable intramural macrophage infiltration in CaCl2-treated aortas (Figure 1E) or in NaCl controls (not shown). Rat spleen cryosections (Figure 1F) were used as a positive control for macrophage staining (round, brown deposits; arrow). Quantitative calcium analysis also showed that the extent of aortic calcification depended on the CaCl2 concentration applied periadventitially to the abdominal aorta.
Figure 1.
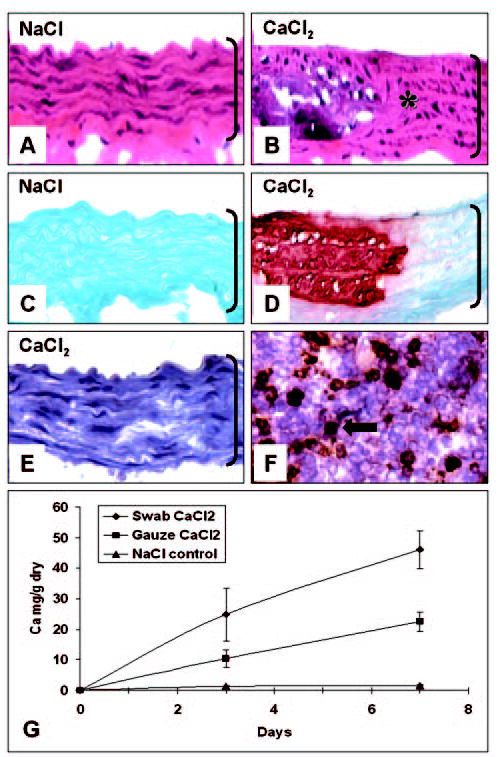
Morphology and kinetics of aortic calcification. H&E stain for general morphology (A, B), Alizarin red for calcium deposits (C, D), and immunohistochemistry for macrophages (E) of abdominal aorta swabbed with 0.2 mol/L NaCl (A, C) or with 0.2 mol/L CaCl2 (B, D, E). Rat spleen cryosections (F) were used as positive control for macrophage staining (brown, round deposits, arrow). Original magnifications were ×400; aortic lumen is on top and brackets designate aorta thickness. Kinetics of aortic calcification (n=6 rats per time point per group) induced by 2 methods of periarterial application of 0.15 mol/L CaCl2 solution is shown in Figure 1G. Abbreviations are as defined in text.
Compared with calcium levels obtained at 7 days with 0.2 mol/L CaCl2 (46±5.2 mg Ca/g dry), application of 0.1 mol/L CaCl2 did not induce aortic calcification (2.35±1.47 mg Ca/g dry; n=4, Student’s t test, P<0.05). A time-course comparative analysis (Figure 1G) between the 2 different application methods (swab versus gauze) demonstrated that aortic calcification with 0.15 mol/L CaCl2 in this model was progressive and that the rate of progression also depended on application method. According to statistical analysis (Kruskal-Wallis test), swab application induced greater calcification compared with gauze for both time points, and there was a significant increase in calcium content from day 3 to day 7 for both swab and gauze methods. Analysis of vascular segments that were not exposed directly to CaCl2 (such as the suprarenal aorta) did not reveal any Alizarin red–positive staining (not shown) and contained minimal amounts of calcium (0.88±0.38 mg Ca/g dry), indicating a localized pathological process.
Comparative measurements of aortic diameter before and after surgery (1.50±0.07 and 1.40±0.08 mm, respectively) revealed that application of 0.15 mol/L CaCl2 did not induce aneurysm formation within 1 week after surgery (Student’s t test, P>0.3, n=8 per group). The original conditions (0.2 to 0.5 mol/L CaCl2) used in this model by others in the past induced arterial calcification very rapidly, and aneurysms formed as a result of heavy inflammatory reactions to the calcific deposits. Therefore, for mechanistic studies described later, we selected to use 0.15 mol/L CaCl2 applied by the gauze method, followed by end-point analysis after 7 days.
Elastin Calcification and Degradation
A salient feature of this model was that the onset of aortic calcification was mainly associated with elastin fibers. Figure 2 depicts typical aspects of early calcium deposits, which were clearly associated with the wavy elastin fibers (Figure 2, A–D). Cell-associated calcification was not noted in any of the samples analyzed as shown by Alizarin red staining for calcium superimposed with DAPI nuclear stain for cells (Figure 2, B, D, F, and H). Progression toward more extensive calcification may involve accretion and fusion of calcium deposits (Figure 2, E and F), ultimately leading to complete encasing of the aorta thickness in calcium deposits (Figure 2G).
Figure 2.
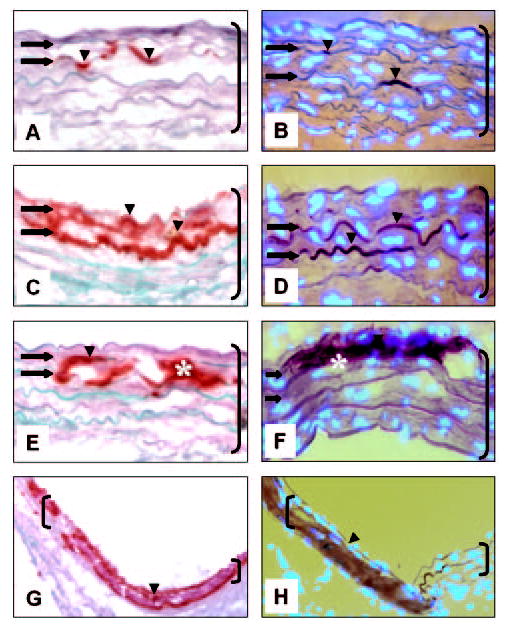
Elastin-associated calcification occurs in rat abdominal aorta injury model. Abdominal aorta collected 7 days after CaCl2 injury was stained with Alizarin red/light green counter-staining (A, C, E, G) and DAPI nuclear stain (bright blue spots) superimposed with Alizarin red without light green counterstaining (B, D, F, H). Arrowheads and * designate calcium deposits; arrows depict elastic lamellae (wavy horizontal fibers). Original magnifications are as follows: A–F, ×400 and G and H, 3200. Brackets designate aorta; sections are oriented with aortic lumen on top. Abbreviations are as defined in text.
A second main trait of calcification in CaCl2-treated aortas was localized disorganization of the aortic extracellular matrix and elastin degradation. Calcified areas were characterized by flattened elastin fibers as demonstrated by VVG stain (Figure 3B) and disorganization of the collagen network (Figure 3E). NaCl-treated aortas exhibited normal histological patterns (Figure 3, A and D), which were indistinguishable from those of control aortas obtained from age-matched rats not subjected to any surgical procedure (not shown). Elastin degradation in calcified aortas was confirmed by quantitative desmosine analysis (Figure 3G), which clearly showed a significant reduction of desmosine content in CaCl2-treated aortas compared with NaCl-treated aortas. Desmosine content showed a strong, negative correlation to calcium content in injured aortas (correlation coefficient of −0.81).
Figure 3.
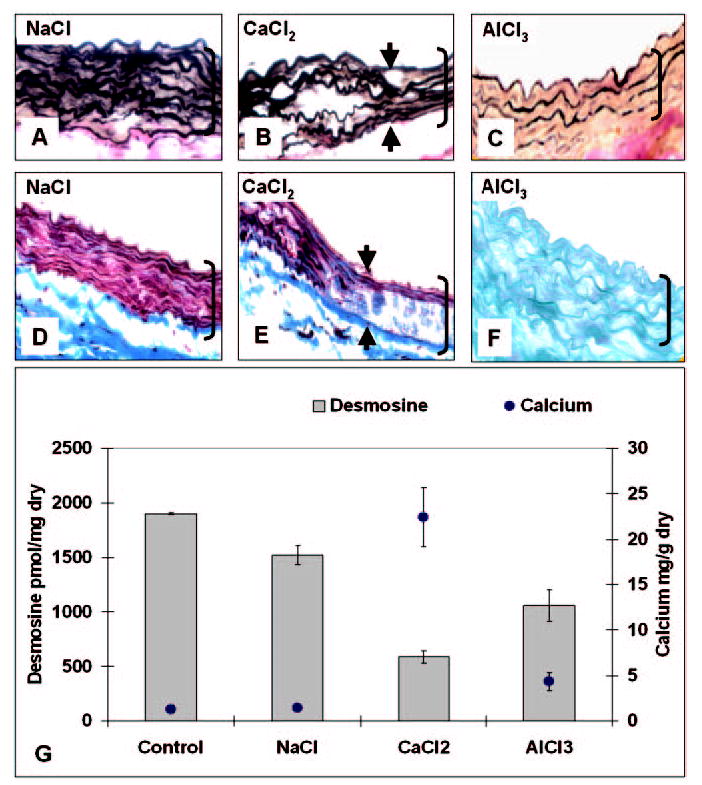
Extracellular matrix degradation accompanies aortic calcification in periadventitial injury model. Abdominal aorta collected after NaCl treatment (A, D), CaCl2 (B, E), and AlCl3 (C, F) stained with VVG (A–C), Masson’s trichrome (D, E), and Alizarin red (F). Arrowheads designate “transition” areas from wavy to flattened elastic fibers. Original magnifications were ×400; brackets designate aorta thickness; and sections are oriented with aortic lumen on top. Desmosine and calcium contents of abdominal aortas for control (no surgery) and for NaCl, CaCl2, and AlCl3 treatments (n=4 per group) are shown in G. Abbreviations are as defined in text.
Cellular Modifications
Although we could not detect direct cell-associated calcification that may have resulted from the CaCl2 injury, some signs of cellular modification were observed. Immunohistochemical analysis by α-smooth muscle cell actin stain did not reveal significant differences between NaCl and CaCl2 treatments (not shown). Moderate levels of apoptosis were identified in CaCl2-injured aortas (Figure 4B) compared with NaCl-treated aortas (Figure 4A), indicating that CaCl2 injury may have induced the apoptotic cascade in aortic cells. Macrophage cell infiltration in the media or intima was not noted in any of the samples analyzed (Figure 1, E and F), indicating that at this early stage, inflammation may not play a major role in aortic calcification.
Figure 4.
Apoptosis of vascular cells in abdominal aorta injury model. Abdominal aorta after treatment with NaCl (A, D, G), CaCl2 (B, E, H), and AlCl3 (C, F, I) were stained with DeadEnd TUNEL for detection of apoptotic cells (A–F) and with DAPI nuclear stain to show cellular distribution (G–I). TUNEL-positive cells are brown (arrows). Negative control for TUNEL assay is shown in E and positive controls (DNase-treated sections) in D and F. Original magnifications were ×400x; brackets designate aorta thickness. L indicates lumen. All other abbreviations are as defined in text.
Endothelial Permeability
Periadventitial CaCl2 injury induced a marked increase in endothelial permeability, as shown by Evans blue experiments (Figure 5, C and G). Evans blue–albumin complexes (red fluorescence) were associated very strongly with the internal (red) and external (red-orange) elastic laminas and, to a lesser extent, with the medial elastic fibers (yellow). Elastic fibers autofluoresce green under UV illumination and a fluorescein isothiocyanate (FITC) filter. NaCl-treated aortas and control, age-matched rats that were not subjected to any surgical intervention when injected to Evans blue exhibited characteristic green elastin autofluorescence and minor orange staining of the internal elastic lamina, possibly reflecting normal albumin transport through the vascular endothelium (Figure 5, A, B, E, and F).
Figure 5.
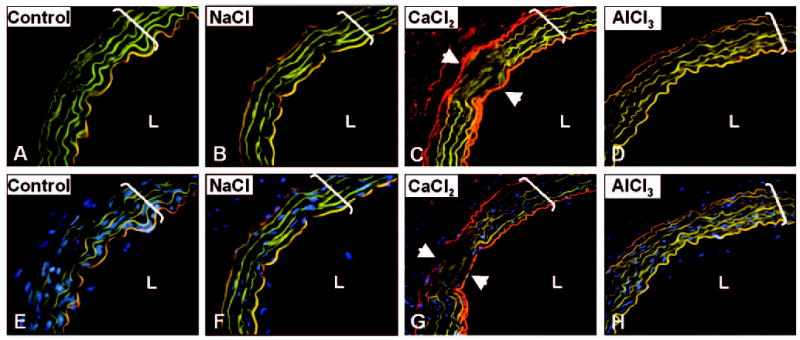
Periadventitial injury induces permeabilization of aortic endothelium to Evans blue. Cryosections collected from controls (A, E), NaCl-treated (B, F), CaCl2-treated (C, G), and AlCl3-treated (D, H) abdominal aortas were analyzed unstained under UV illumination with FITC filter (A–D). Under UV light and FITC filter, elastin fluoresces green, Evans blue appears red, and elastin to which Evans blue was bound appears as yellow–bright orange. Images obtained from DAPI nuclear staining (bright blue spots) were digitally superimposed with those obtained from sections by FITC filter (E–H). Arrowheads designate “transition” areas in calcified aortas (C, G). Original magnifications were ×200; brackets designate aorta thickness. L indicates lumen. All other abbreviations are as defined in text.
Inhibition of Calcification
AlCl3 pretreatment of abdominal aortas before CaCl2 injury reduced calcium levels by >80% (Figure 3G) compared with CaCl2 treatment alone. Calcium levels in the AlCl3 group were not statistically different from NaCl or control groups. Furthermore, AlCl3 pretreatment abolished formation of Alizarin red–stainable calcium deposits (Figure 3F) and largely inhibited elastin degeneration, as revealed by statistically higher desmosine content (Figure 3G) and unaltered fiber morphology (Figure 3C) compared with CaCl2 treatment. Furthermore, AlCl3 pretreatment abolished apoptosis of vascular cells (Figure 4, C, F, and I) and eliminated the CaCl2-mediated increase in endothelial permeability to Evans blue (Figure 5, D and H).
Elastin Calcification in Knockout Mice
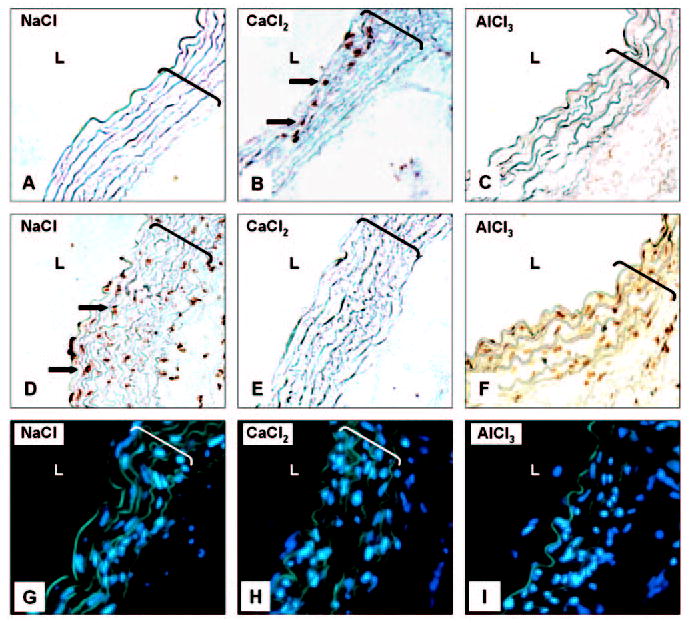
To seek further confirmation for the association of MMP-mediated extracellular matrix degradation with elastin calcification, paraffin sections of murine aortic samples from a previous knockout study were stained for calcification. That study has shown recently that 0.25 mol/L CaCl2 injury to abdominal aortas of MMP2- or to MMP9-knockout mice does not result in aneurysm formation.15 Alizarin red staining of aortas obtained 10 weeks after CaCl2 treatment revealed that CaCl2-treated aortas from both MMP2- and MMP9-knockout mice did not develop calcification (Figure 6, B and E), whereas CaCl2 treatment induced severe calcification in the abdominal aortas of wild-type mice (Figure 6H). Similar to rat experiments, calcification of aortas in wild-type mice was mainly associated with elastin and not with cells (Figure 6I). Control NaCl-treated aortas did not exhibit calcification in any of the experimental groups (Figure 6, A, D, and G). In addition, VVG staining on CaCl2-treated aortas from both strains of knockout mice did not exhibit the characteristic elastin fiber flattening or disorganization (Figure 6, C and F).
Figure 6.
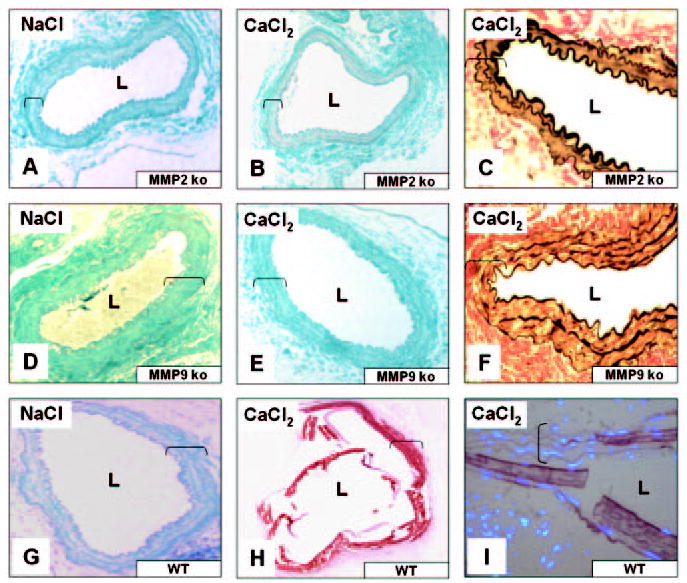
MMPs contribute to elastin-associated calcification in mouse abdominal aorta injury model. MMP2-knockout mice (MMP2 ko), MMP9 ko, and control wild-type (WT) mice were subjected to NaCl (A, D, G) or CaCl2 (B, C, E, F, H, I) abdominal aorta injury. Aortic segments were stained with Alizarin red/light green counterstaining (A, B, D, E, G, H), VVG (C, F), and Alizarin superimposed with DAPI nuclear staining (I). Original magnifications were ×200 (A, B, D, E, G, H) and ×400 (C, F, I); brackets designate aorta thickness. L indicates lumen. All other abbreviations are as defined in text.
Discussion
Medial elastin calcification is a widespread feature of vascular pathology associated with ageing, diabetes, and renal failure and may occur as a result of either proliferative or destructive remodeling. Circumstantial evidence exists for a correlation between elastin degradation and calcification in various pathological situations.18 Although valuable information is available from analysis of human specimens,19 this provides only a snapshot in time and may not always provide insight into pathogenic mechanisms.
Knowledge about the mechanisms of elastin calcification is scarce, mainly owing to the lack of an adequate animal model that closely mimics human pathology. In the current studies, we have shown that perivascular administration of CaCl2 to abdominal aortas induced degradation and calcification of elastic fibers. Our hypothesis stipulated that MMP-mediated degradation of elastin contributed to calcium deposition onto elastic fibers. In support of our hypothesis, we have shown that MMP-knockout mice were resistant to CaCl2-mediated aortic injury and did not develop elastin degeneration and calcification. Furthermore, treatment of aortas with aluminum ions (known to stabilize elastin against the action of MMPs), before CaCl2 injury, significantly reduced elastin degeneration and mitigated elastin-oriented vascular calcification.
In most articles thus far of this injury model, authors have used high (0.2 to 0.5 mol/L) CaCl2 concentrations to induce aneurysm formation that led to extensive aortic calcification within days. Aneurysm formation was mainly the result of an intense inflammatory reaction to the calcified deposits.11,12,14,15 Structurally, most calcium deposits appeared as large, compact masses spanning the entire thickness of the aorta, and rarely could individual elastin fibers be observed. To temper the rate of calcification in this model, we compared several CaCl2 concentrations, as well as techniques of periarterial application, and selected to use a 15-minute gauze application of 0.15 mmol/L CaCl2. Analysis 7 days after surgery allowed us to demonstrate fine details of individual elastic fiber calcification and distinctive morphological aspects of elastin degeneration.
Initial phases of calcification appeared to be related to the formation of small calcium deposits closely associated with elastic fibers, which followed the natural wavy aspect of the aortic lamellae. At later stages, calcium deposits appeared to grow between fibers and connect adjacent structures, which eventually led to flattening of the elastic lamellae. Disorganization of the collagen network and diminution of desmosine content accompanied loss of the natural waviness of the elastic lamellae and paralleled the increase in calcium content of injured aortas. Extensively calcified areas exhibited typical aspects of massive mineral deposits, which apparently acquired the tubular shape of the blood vessel. These morphological and biochemical aspects are reminiscent of human pathology such as Marfan’s syndrome20,21 and Monckeberg’s medial sclerosis22,23 and possibly reflect an enzyme-mediated degenerative process. Elastin has been long recognized as a nucleation site for the calcification process,1 and MMP upregulation has been identified in several vascular degenerative processes,24 but no direct correlation between MMPs and elastin calcification has been established before this study.
Direct involvement of cells in elastin calcification was not evident from our studies. No cell-specific calcification was noted in early phases by Alizarin red staining, and inflammatory cell infiltration in the media was not noted in any of the rat aortic samples analyzed. A moderate number of apoptotic cells were detected in CaCl2-injured rat aortas and none in the NaCl controls, indicating that apoptosis may be associated with tissue injury and degeneration. The involvement of cells in elastin calcification warrants further investigation.
Very little is known about the actual mechanisms involved in this vascular injury model. Of interest is the marked increase in endothelial permeability toward Evans blue, albeit 7 days after CaCl2 injury, suggesting that the putatively injured endothelium did not recuperate its selective permeability during this time interval. Evans blue is known to accurately detect increases in vascular permeability in a variety of vascular segments.25 This increase in permeability may be responsible for the influx of ions (calcium, phosphate), proteins, and other components into the vessel wall.
In an attempt to further understand the mechanisms that underlie elastin-associated calcification, we treated aortas in vivo with aluminum ions before the CaCl2 injury. Aluminum ions have been shown by us to bind strongly and irreversibly to elastin, stabilize it against the action of MMPs, and protect it from degeneration and calcification,7 but we have never attempted to harness this property in a circulatory model of elastin calcification. For the present studies, we chose a very low aluminum concentration (0.0025 mol/L) that is not cytotoxic26 but is sufficient to induce conformational changes in elastin structure that account for inhibition of elastin calcification.16 Periadventitial pretreatment of abdominal aortas with aluminum ions protected the aortic tissue from CaCl2 injury by abolishing elastin degeneration and calcification, preserving endothelial permeability, and reducing apoptosis of vascular cells. The mechanisms of this protective effect are not known, but we speculate that aluminum ions bound to elastin in vivo, and in doing so, prevented it from degradation by MMPs and thus, blocked the cascading sequence of events that may lead to elastin degeneration. Aluminum ion binding to elastin may have also inhibited calcium binding by a direct effect. The effect of aluminum on vascular cell activities also warrants further investigation.
Very conclusive evidence to support our data came from analysis of samples obtained from aortas of MMP-knockout mice, which underwent similar CaCl2 injury. The MMP2-knockout mouse expresses only active species of MMP9, whereas the MMP9-knockout mouse expresses active species of MMP2. Both knockout mouse strains were shown to be resistant to CaCl2 periaortic injury, and even 10 weeks after surgery, they did not exhibit signs of aneurysm formation, whereas the wild strain controls developed extensive aneurysms.15 Alizarin red staining revealed a complete lack of calcification in aortas from both MMP2-knockout mice and from MMP9-knockout mice, whereas CaCl2 treatment induced severe calcification in the abdominal aortas of wild-type mice (which express both MMP2 and MMP9 active species). Furthermore, additional histological stains such as VVG, Masson’s, and H&E did not reveal any of the features typical of extracellular matrix disorganization. These results strongly point to a correlation between MMP-mediated matrix degradation, elastin calcification, and aneurysm formation.
It is known that both MMP2 and MMP9 can digest elastin and that MMP2 can activate latent MMP9, but not vice versa.27 We speculate that in this model, most of the damage is possibly caused by MMP9 (obviated in the MMP9-knockout), whereas in the MMP2-knockout, the absence of MMP2 would block the activation of MMP9. Moreover, it is apparent that both MMPs play some role in elastin calcification and are required to work in concert for initiation and progression of elastin calcification and degeneration in this aortic injury model.
We are currently exploring therapeutic approaches for limitations of elastin-associated pathology by investigations into the cellular and molecular mechanisms of vascular cell response to injury and their interactions with extracellular matrix components.
Conclusions
Perivascular administration of low concentrations of CaCl2 induces moderate levels of elastin degradation and calcification within 7 days after surgery, in the absence of aneurysm formation and inflammatory reactions. Calcium deposits are associated mainly with elastic fibers and are accompanied by elastin degradation, disorganization of the aortic extracellular matrix, and moderate levels of vascular cell apoptosis. The presence of both MMP2 and MMP9 is required for elastin degeneration and calcification in the aortic injury model. Vascular remodeling may contribute significantly to calcium deposition onto elastic fibers.
Acknowledgments
This work was supported by National Institutes of Health grant HL61652 (to N.V.).
References
- 1.Proudfoot D, Shanahan CM. Biology of calcification in vascular cells: intima versus media. Herz. 2001;26:245–251. doi: 10.1007/pl00002027. [DOI] [PubMed] [Google Scholar]
- 2.Bobryshev YV, Lord RS, Warren BA. Calcified deposit formation in intimal thickenings of the human aorta. Atherosclerosis. 1995;118:9–21. doi: 10.1016/0021-9150(95)05588-n. [DOI] [PubMed] [Google Scholar]
- 3.Janzen J, Vuong PN. Arterial calcifications: morphological aspects and their pathological implications. Z Kardiol. 2001;90(suppl 3):6–11. doi: 10.1007/s003920170044. [DOI] [PubMed] [Google Scholar]
- 4.Robert L, Robert AM, Jacotot B. Elastin-elastase-atherosclerosis revisited. Atherosclerosis. 1998;140:281–295. doi: 10.1016/s0021-9150(98)00171-3. [DOI] [PubMed] [Google Scholar]
- 5.Bailey MT, Pillarisetti S, Xiao H, Vyavahare NR. Role of elastin in pathologic calcification of xenograft heart valves. J Biomed Mater Res. 2003;66A:93–102. doi: 10.1002/jbm.a.10543. [DOI] [PubMed] [Google Scholar]
- 6.Vyavahare N, Jones PL, Tallapragada S, Levy RJ. Inhibition of matrix metalloproteinase activity attenuates tenascin-C production and calcification of implanted purified elastin in rats. Am J Pathol. 2000;157:885–893. doi: 10.1016/S0002-9440(10)64602-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bailey M, Xiao H, Ogle M, Vyavahare N. Aluminum chloride pretreatment of elastin inhibits elastolysis by matrix metalloproteinases and leads to inhibition of elastin-oriented calcification. Am J Pathol. 2001;159:1981–1986. doi: 10.1016/S0002-9440(10)63048-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gertz SD, Kurgan A, Eisenberg D. Aneurysm of the rabbit common carotid artery induced by periarterial application of calcium chloride in vivo. J Clin Invest. 1988;81:649–656. doi: 10.1172/JCI113368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freestone T, Turner RJ, Coady A, Higman DJ, Greenhalgh RM, Powell JT. Inflammation and matrix metalloproteinases in the enlarging abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 1995;15:1145–1151. doi: 10.1161/01.atv.15.8.1145. [DOI] [PubMed] [Google Scholar]
- 10.Freestone T, Turner RJ, Higman DJ, Lever MJ, Powell JT. Influence of hypercholesterolemia and adventitial inflammation on the development of aortic aneurysm in rabbits. Arterioscler Thromb Vasc Biol. 1997;17:10–17. doi: 10.1161/01.atv.17.1.10. [DOI] [PubMed] [Google Scholar]
- 11.Tambiah J, Franklin IJ, Trendell-Smith N, Peston D, Powell JT. Provocation of experimental aortic inflammation and dilatation by inflammatory mediators and Chlamydia pneumoniae. Br J Surg. 2001;88:935–940. doi: 10.1046/j.0007-1323.2001.01801.x. [DOI] [PubMed] [Google Scholar]
- 12.Ikonomidis JS, Gibson WC, Gardner J, Sweterlitsch S, Thompson RP, Mukherjee R, Spinale FG. A murine model of thoracic aortic aneurysms. J Surg Res. 2003;115:157–163. doi: 10.1016/s0022-4804(03)00193-8. [DOI] [PubMed] [Google Scholar]
- 13.Chiou AC, Chiu B, Pearce WH. Murine aortic aneurysm produced by periarterial application of calcium chloride. J Surg Res. 2001;99:371–376. doi: 10.1006/jsre.2001.6207. [DOI] [PubMed] [Google Scholar]
- 14.Tambiah J, Powell JT. Chlamydia pneumoniae antigens facilitate experimental aortic dilatation: prevention with azithromycin. J Vasc Surg. 2002;36:1011–1117. doi: 10.1067/mva.2002.128928. [DOI] [PubMed] [Google Scholar]
- 15.Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002;110:625–632. doi: 10.1172/JCI15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vyavahare N, Ogle M, Schoen FJ, Levy RJ. Elastin calcification and its prevention with aluminum chloride pretreatment. Am J Pathol. 1999;155:973–982. doi: 10.1016/S0002-9440(10)65197-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Starcher BC. Determination of the elastin content of tissues by measuring desmosine and isodesmosine. Anal Biochem. 1977;79:11–15. doi: 10.1016/0003-2697(77)90372-4. [DOI] [PubMed] [Google Scholar]
- 18.Robert L, Jacob MP, Fulop T. Elastin in blood vessels. Ciba Found Symp. 1995;192:286–299. doi: 10.1002/9780470514771.ch15. [DOI] [PubMed] [Google Scholar]
- 19.Tintut Y, Demer LL. Recent advances in multifactorial regulation of vascular calcification. Curr Opin Lipidol. 2001;12:555–560. doi: 10.1097/00041433-200110000-00012. [DOI] [PubMed] [Google Scholar]
- 20.Bunton TE, Biery NJ, Myers L, Gayraud B, Ramirez F, Dietz HC. Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ Res. 2001;88:37–43. doi: 10.1161/01.res.88.1.37. [DOI] [PubMed] [Google Scholar]
- 21.Pereira L, Andrikopoulos K, Tian J, Lee SY, Keene DR, Ono R, Reinhardt DP, Sakai LY, Biery NJ, Bunton T, Dietz HC, Ramirez F. Targeting of the gene encoding fibrillin-1 recapitulates the vascular aspect of Marfan syndrome. Nat Genet. 1997;17:218–222. doi: 10.1038/ng1097-218. [DOI] [PubMed] [Google Scholar]
- 22.Shioi A, Taniwaki H, Jono S, Okuno Y, Koyama H, Mori K, Nishizawa Y. Monckeberg’s medial sclerosis and inorganic phosphate in uremia. Am J Kidney Dis. 2001;38(suppl 1):S47–S49. doi: 10.1053/ajkd.2001.27396. [DOI] [PubMed] [Google Scholar]
- 23.Shanahan CM, Cary NR, Salisbury JR, Proudfoot D, Weissberg PL, Edmonds ME. Medial localization of mineralization-regulating proteins in association with Monckeberg’s sclerosis: evidence for smooth muscle cell-mediated vascular calcification. Circulation. 1999;100:2168–2176. doi: 10.1161/01.cir.100.21.2168. [DOI] [PubMed] [Google Scholar]
- 24.Kieffer P, Giummelly P, Schjoth B, Carteaux JP, Villemot JP, Hornebeck W, Atkinson J. Activation of metalloproteinase-2, loss of matrix scleroprotein content and coronary artery calcification. Atherosclerosis. 2001;157:251–254. doi: 10.1016/s0021-9150(00)00704-8. [DOI] [PubMed] [Google Scholar]
- 25.Uwatoku T, Shimokawa H, Abe K, Matsumoto Y, Hattori T, Oi K, Matsuda T, Kataoka K, Takeshita A. Application of nanoparticle technology for the prevention of restenosis after balloon injury in rats. Circ Res. 2003;92:e62–e69. doi: 10.1161/01.RES.0000069021.56380.E2. [DOI] [PubMed] [Google Scholar]
- 26.Jones DL, Kochian LV. Aluminum interaction with plasma membrane lipids and enzyme metal binding sites and its potential role in Al cytotoxicity. FEBS Lett. 1997;400:51–57. doi: 10.1016/s0014-5793(96)01319-1. [DOI] [PubMed] [Google Scholar]
- 27.Brassart B, Randoux A, Hornebeck W, Emonard H. Regulation of matrix metalloproteinase-2 (gelatinase A, MMP-2), membrane-type matrix metalloproteinase-1 (MT1-MMP) and tissue inhibitor of metalloproteinases-2 (TIMP-2) expression by elastin-derived peptides in human HT-1080 fibrosarcoma cell line. Clin Exp Metastasis. 1998;16:489–500. doi: 10.1023/a:1006550503612. [DOI] [PubMed] [Google Scholar]