

Abstract
DNA double-strand breaks (DSBs) with protein covalently attached to 5′ strand termini are formed by Spo11 to initiate meiotic recombination1,2. The Spo11 protein must be removed for the DSB to be repaired, but the mechanism for removal has been unclear3. We show here that meiotic DSBs in budding yeast are processed by endonucleolytic cleavage that releases Spo11 attached to an oligonucleotide with a free 3′-OH. Surprisingly, two discrete Spo11-oligonucleotide complexes were found in equal amounts, differing with respect to the length of the bound DNA. We propose that these forms arise from different spacings of strand cleavages flanking the DSB, with every DSB processed asymmetrically. Thus, the ends of a single DSB may be biochemically distinct at or before the initial processing step—significantly earlier than previously thought. SPO11-oligonucleotide complexes were identified in extracts of mouse testis, indicating that this mechanism is evolutionarily conserved. Oligonucleotide-topoisomerase II complexes were also present in extracts of vegetative yeast, although not subject to the same genetic control as for generating Spo11-oligonucleotide complexes. Our findings suggest a general mechanism for repair of protein-linked DSBs.
We previously proposed that Spo11 might be removed from DSB ends by either of two mechanisms: direct hydrolysis of the covalent protein-DNA linkage, or single-stranded endonucleolytic cleavage releasing Spo11 covalently attached to a short oligonucleotide (Fig. 1a)1,4. These mechanisms are distinguished by the presence or absence of an oligonucleotide bound to Spo11. We identified this predicted protein-DNA complex using a direct biochemical approach in S. cerevisiae. A strain expressing HA epitope-tagged Spo11 was induced to enter meiosis, denaturing extracts were prepared, then Spo11-HA was immunoprecipitated and treated with 32P-labelled nucleotide and terminal deoxynucleotidyl transferase (TdT), which catalyzes untemplated addition of nucleotides to a free 3′-OH DNA end. A chain terminating nucleotide was used to limit incorporation to a single residue.
Figure 1.
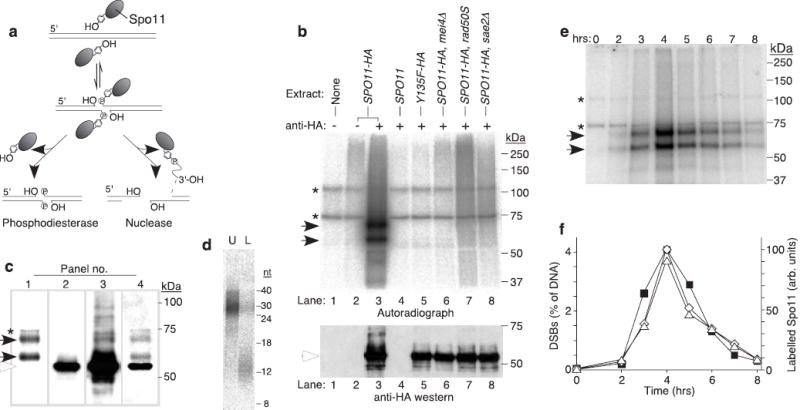
Endonucleolytic processing of covalent Spo11-DSB complexes. a, Alternative mechanisms for Spo11 release1. b, Detection of Spo11-oligonucleotide complexes. Immunoprecipitates from the indicated genotypes with or without antibody were labelled with TdT. Upper panel, autoradiograph; lower panel, anti-HA western. c, Relative mobilities of free and oligonucleotide-associated Spo11-HA. Panel 1, autoradiograph; panels 2–3, low and high exposures of an anti-HA western; panel 4, re-exposure of the blot to film after partial fading of the chemiluminescent signal. d, Sizes of Spo11-associated oligonucleotides from the upper (U) and lower (L) SDS-PAGE bands. Size standards are indicated. e, Time course of appearance of Spo11-oligonucleotide complexes during meiosis. f, Quantification of upper (⋄) and lower (Δ) labelled Spo11 species from e, and the DSB frequency at the HIS4LEU2 recombination hotspot measured in the same culture (▪). Each point is a single measurement. Asterisks, non-specific labelling; closed arrows, Spo11-specific labelled species; open arrows, free Spo11-HA.
Four radiolabelled bands were observed between ~60 and 110 kDa (Fig. 1b, lane 3). Two bands (asterisks) were non-specific because they were present when TdT and nucleotide were incubated alone (Fig. 1b, lane 1). Two bands (solid arrows) were specific for Spo11-HA because they were not seen with mock immunoprecipitation (Fig. 1b, lane 2), or untagged Spo11 (Fig.1b, lane 4). Labelling was not observed if DSBs were not formed, namely, when the catalytic tyrosine of Spo11 was mutated to phenylalanine2 (Fig. 1b, lane 5) or in a mei4 mutant4 (Fig. 1b, lane 6). Labelled Spo11 species were also not observed in rad50S or sae2Δ mutants (Fig. 1b, lanes 7–8), in which Spo11 remains covalently attached to DSB ends1,5. Thus, Spo11-oligonucleotide complexes did not arise from non-physiological disruption of covalent Spo11-DSB complexes.
Oligonucleotide-associated Spo11 was compared to the free protein by combined autoradiography and western analysis (Fig. 1c). The predominant western signal (Fig. 1c, panel 2) was from free Spo11-HA (~55 kDa, open arrow). Bands comigrating with the radiolabelled species were not observed after a short western exposure (compare panels 1 and 2, Fig. 1c), indicating that the majority of Spo11 was not DNA-associated. Moreover, Spo11 that has made DSBs in rad50S and sae2Δ mutants remains attached to high molecular weight DNA and is not resolved on SDS-PAGE1, yet free Spo11 levels were not detectably reduced in these mutants relative to strains with no DSBs (Fig. 1b, lower panel). Thus, only a minority of Spo11 protein engages in DSB formation6. Longer western exposure revealed slower migrating bands, one comigrating with the upper radioactive signal (Fig. 1c, panel 3). Free Spo11-HA presumably masked the western signal from the lower radioactively labelled band. After partial fading of the chemiluminescent signal and re-exposure of the blot, free Spo11-HA provided the only signal visible by chemiluminescence, superimposed on the autoradiographic signal (Fig. 1c, panel 4). The labelled species were shifted by +15 and +5 kDa.
Two discrete labelled species were not expected. To determine their nature, they were gel-purified and digested with pronase and the DNA fragments were separated on a sequencing gel (Fig. 1d). The DNA from the upper Spo11 band migrated from ~24–40 nt, whereas DNA from the lower band ran from ~10–15 nt. The smeared migration patterns likely arise from heterogeneities in oligonucleotide lengths, DNA sequence, and the completeness of protease digestion. Estimating true oligonucleotide sizes requires correction for radiolabelled nucleotide incorporation and for any amino acids left after protease digestion7. Also, control experiments indicated that oligonucleotides <10 nt were inefficiently recovered (data not shown), so the lower limit for the smaller oligonucleotides is not accurately known. We estimate that the two complexes contain oligonucleotides of ~21–37 nt and ≤ 12 nt, respectively.
If Spo11-oligonucleotide complexes are products of DSB processing, then they should appear with similar kinetics as resected (Spo11-free) DSBs. In a synchronously sporulating culture, Spo11-oligonucleotide complexes were detected as early as two hours, accumulated to peak levels at four hours, and declined thereafter (Fig. 1e, f). As predicted, the appearance of these complexes closely matched the timing for resected DSBs in the same culture (Fig. 1f). The long and short-oligonucleotide forms were consistently in a 1:1 ratio (long : short = 1.03 ± 0.10, mean ± s.d., n=6 cultures). The ratio did not vary significantly with changes in amount of TdT, labelling time, or dCTP vs. cordycepin as the source of label (data not shown), suggesting that this measurement is a faithful reporter of the relative amounts of the two species.
The Mre11-Rad50-Xrs2 complex is likely the nuclease generating Spo11-oligonucleotide complexes because Mre11 has single-strand endonuclease activity and certain rad50 and mre11 mutants (including nuclease-defective alleles of mre11) cannot remove Spo11 from DSB ends (reviewed in ref. 4). Phosphatase treatment prior to incubation with TdT did not increase labelling (data not shown), indicating that the Spo11-associated oligonucleotides generated in vivo have free 3′-OH termini, consistent with Mre11 nuclease activity. We propose that Mre11 directly cleaves single strands adjacent to Spo11-associated DSBs (see below). It is also possible, however, that Spo11 is released by 3′-5′ exonuclease processing of more distantly placed single-strand breaks. Such a mechanism is consistent with the polarity of weak Mre11 exonuclease activity8, but would require that Mre11 exonuclease work much more efficiently in vivo than in vitro.
Recombination initiation by Spo11 is evolutionarily conserved4. We therefore tested for oligonucleotides bound to mouse SPO11 (mSPO11). An anti-mSPO11 antibody recognized 40.5 and 43.8 kDa polypeptides from adult testis (arrows in Fig. 2a, lane 1), as expected for products of the major splicing isoforms Spo11α and Spo11β (refs. 9,10). Both were absent upon mock immunoprecipitation (Fig. 2a, lane 4) or with extracts from Spo11−/− testes (Fig. 2a, lane 2). TdT-labelling of immunoprecipitates from wild-type testes yielded a smear of 47~59 kDa species (arrows in Fig. 2b; lanes 1 and 4). Spo11−/− testes yielded only background similar to a mock immunoprecipitation (Fig. 2b, compare lanes 2 and 5). The absence of labelled species from Spo11−/− testes could result from spermatocyte death rather than lack of mSPO11 per se11,12. To rule out this possibility, we also examined Dmc1−/− testes, because DSBs are formed and processed in the absence of DMC1, but strand exchange is impaired and spermatocyte apoptosis occurs at the same stage as in Spo11−/− (refs. 13,14). As expected, labelled species were also isolated from Dmc1−/− testes (Fig. 2b, lane 6). Mouse SPO11α was specifically reduced in Dmc1−/− testes (Fig. 2a, lane 3), consistent with RNA expression data12. Nevertheless, mobilities of mSPO11-oligonucleotide complexes were similar in Dmc1−/− and wild type (Fig. 2b, lanes 4 and 6). This result suggests that mSPO11α contributes little to DSB formation, consistent with the relatively late expression of Spo11α (ref. 12).
Figure 2.
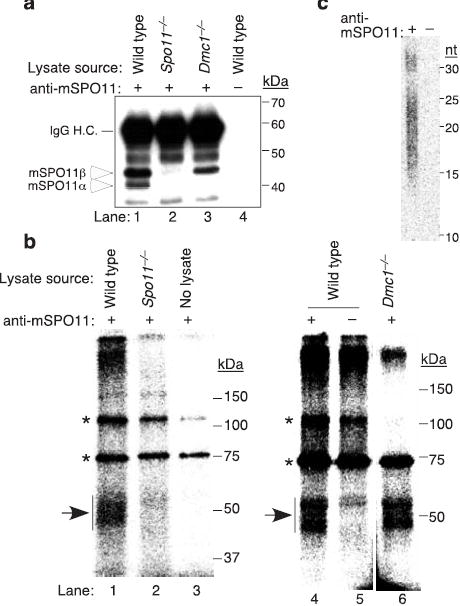
SPO11-oligonucleotide complexes in mouse meiosis. a, Testis extracts immunoprecipitated with or without anti-mSPO11 were probed by western blotting with the same antibody. Each sample represents extract from an equivalent number of testes. IgG H.C., immunoglobulin heavy chain. b, mSPO11-oligonucleotide complexes. mSPO11 was immunoprecipitated from testis extracts and labelled with TdT. Samples were from two testes (lanes 1–2) or six testes (lanes 4–6). Arrows, mSPO11-specific species; asterisks, non-specific bands. c, Sizes of mSPO11-associated oligonucleotides after gel purification and protease digestion. A mock immunoprecipitation lacking the anti-mSPO11 antibody was processed in parallel.
Oligonucleotide lengths from mouse were analyzed as above, except that the protein-DNA complexes were gel-purified en masse because they were not resolved as discrete bands. Two mSPO11-associated oligonucleotide populations were observed, of ~12–26 nt and ~28–34 nt, respectively (Fig. 2c). No signal was obtained from a mock immunoprecipitation processed in parallel (Fig. 2c). The smaller oligonucleotide lengths were more heterogeneous compared to yeast, perhaps accounting for the lack of discrete bands on SDS-PAGE. Our findings reveal that the mechanism for processing Spo11-associated DSBs is evolutionarily conserved.
Spo11-DSBs are similar to topoisomerase II-DNA complexes formed after inhibition by compounds such as etoposide, in that both consist of phosphotyrosine linkages to 5′ strand termini on both sides of a DSB15,16. We hypothesized that the mechanism to process Spo11-associated DSBs also repairs topoisomerase II-induced DSBs. To test this idea, epitope-tagged topoisomerase II (Top2-HA3) was immunoprecipitated from vegetative yeast cultures and labelled with TdT. A species of appropriate size for a Top2-oligonucleotide complex was observed (Fig. 3a). This species was absent if cells expressed untagged Top2 (Fig. 3a). No difference in mobility of labelled and free Top2-HA could be discerned (Fig. 3b). We estimate that we could detect a shift equivalent to 10 nt or more, suggesting that Top2-associated oligonucleotides are smaller than for Spo11. These Top2 complexes are consistent with products of endonucleolytic processing of a low level of spontaneous topoisomerase-induced DSBs. However, it is also possible that these complexes arise from Top2-mediated cleavage adjacent to a pre-existing nick. Our experiments do not address whether Top2-associated DSBs might also be disjoined by tyrosine-phosphodiester hydrolysis similar to the mechanism of Tdp1 phosphodiesterase17.
Figure 3.
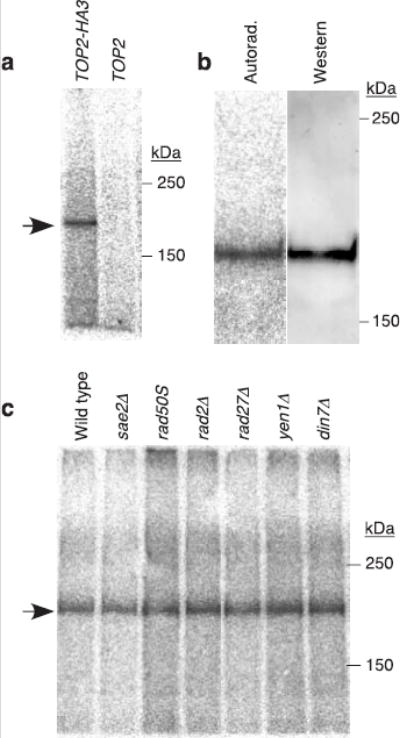
Topoisomerase II-oligonucleotide complexes in non-meiotic yeast cells. a, Extracts were prepared from vegetatively growing yeast strains carrying topoisomerase II with (TOP2-HA3) or without (TOP2) an epitope tag. Samples were immunoprecipitated with anti-HA antibody and labelled with TdT. b, Comparison of electrophoretic mobilities of free and oligonucleotide-associated Top2-HA3. Immunoprecipitated Top2-HA3 was labelled with TdT, separated on 5% SDS-PAGE, and transferred to PVDF membrane. Autoradiograph and anti-HA western of the membrane are shown. c, Formation of Top2-oligonucleotide complexes in nuclease-defective mutants. Arrow, Top2-HA3-specific band.
Mre11 was again a candidate for the nuclease because processing of topoisomerase lesions during bacteriophage T4 replication requires phage homologs of Rad50 and Mre11 (ref. 18), the E. coli homolog of the Rad50-Mre11 complex can remove protein complexes from duplex DNA ends19, and mammalian MRE11 has been proposed to remove the protein covalently attached to the termini of the linear adenovirus genome20. Surprisingly, however, the amount of Top2-oligonucleotide complexes was not altered in rad50S or sae2Δ mutants (Fig. 3c), revealing that the formation of Top2-oligonucleotide complexes is not under the same genetic control as for Spo11. Either Mre11 is not the relevant nuclease, these mutations affect the non-meiotic pathway differently, or Mre11 is redundant with another nuclease(s). We also tested mutations eliminating known or suspected nucleases capable of cleaving 5′ single-stranded DNA at duplex-single-strand junctions (Rad2, Rad27, Yen1, and Din7)21. None affected the spontaneous Top2-oligonucleotide complexes (Fig. 3c). Defining the genetic control for formation of these complexes remains an important challenge.
New details of the meiotic recombination pathway are suggested by characteristics of Spo11-oligonucleotide complexes, especially the presence of different oligonucleotide lengths. The mouse studies indicate that this heterogeneity is a conserved feature. It is possible that an initial longer form is exonucleolytically converted to the shorter form, but a precursor-product relationship seems unlikely from the kinetics in yeast (Fig. 1f). More likely is that endonucleolytic cleavage can occur at either of two spacings from the DSB (Fig. 4). Several scenarios are possible. There might be two DSB classes that are processed symmetrically, with the two classes formed in equal numbers by chance. Alternatively, two cleavage spacings might occur randomly with equal probability, with spacing on one side independent of spacing on the other. In this model, long and short forms would be in equal amounts and there would be three DSB classes: symmetric long, symmetric short, and asymmetric. Finally, the model we favor is that asymmetric cleavage at every DSB gives both a long and a short-oligonucleotide complex, providing a simple explanation for equal amounts of the two forms (Fig. 4).
Figure 4.
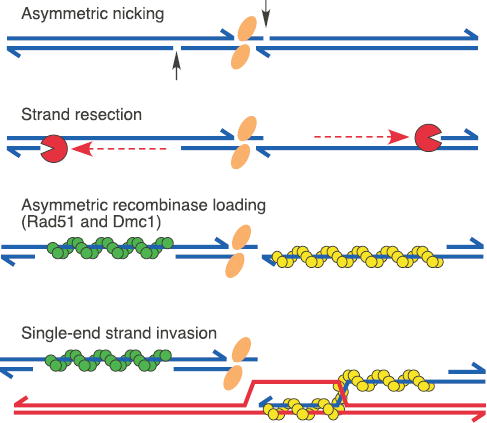
Asymmetric steps in meiotic recombination. A Spo11 dimer (orange elipses) creates a DSB which is processed by asymmetrically spaced nicks. Exonucleolytic resection initiates at these nicks, yielding single-stranded gaps. Rad51 and Dmc1 recombinases (green and yellow, without specifying which) form nucleoprotein filaments on opposite sides of the DSB24,25. Asymmetric strand invasion yields a stable strand exchange intermediate24. Spo11-oligonucleotide complexes, stabilized by base pairing and protein-protein interactions, are proposed to remain associated with DSB ends until a step at or subsequent to strand exchange. See text for further details.
Asymmetrically processed DSBs would have unexpected implications for the mechanism of recombination. Every recombination model posits that the two ends of a DSB behave differently during strand exchange: one end invades the homologous duplex and the second is captured in a separate reaction22,23 (see Fig. 4). Analysis of intermediates in yeast supports this idea24. Dmc1 and Rad51 have been proposed to bind to opposite ends of a DSB, with differences in these recombinases dictating asymmetric behavior of the ends24,25 (Fig. 4). Our findings suggest that DSB ends may already be biochemically distinct at or before DSB processing, raising the possibility that subsequent asymmetric strand exchange is specified earlier than previously thought.
Temporal analysis of Spo11-DSB processing yields another unexpected insight. Kinetics matched closely for appearance and disappearance of both resected DSBs and Spo11-oligonucleotide complexes (Fig. 1f), implying that their lifespans are identical. Matched lifespans were unexpected, because resected DSBs disappear when they give rise to strand exchange intermediates24,26, whereas Spo11-oligonucleotide complexes presumably disappear upon their degradation. It is possible that these distinct processes fortuitously occur with identical timing. However, matched lifespans could also be explained if turnover of Spo11-oligonucleotide complexes is mechanistically linked to turnover of resected DSBs. For example, Spo11-oligonucleotide complexes might remain associated with DSB ends until strand exchange, perhaps contributing further to asymmetry in the recombination reaction (Fig. 4).
Methods
Yeast strains and culture methods
Meiotic cultures were prepared as described5. Vegetative cultures were grown in YPD medium. Strains for meiotic experiments are derivatives of SK1. Tagged SPO11-HA3His6 (referred to for simplicity as SPO11-HA) and spo11-Y135F-HA3His6 were described previously27. Strains for topoisomerase experiments are derivatives of JN362a (ref. 28). Top2 protein was tagged C-terminally with a triple HA repeat by a standard PCR fusion technique29. Other mutations were introduced by one-step replacement and confirmed by Southern blot.
Yeast protein-oligonucleotide complexes
Cells were harvested from 100 ml of meiotic or 250 ml of vegetative culture, washed with water, and lysed with glass beads in 15% ice-cold trichloroacetic acid (TCA). The TCA slurry was incubated on ice 30 min prior to centrifugation 10 min, 16,000 × g. Pellets were dissolved in 0.5 M Tris-Cl pH 8.1, 2% SDS, 1 mM EDTA, 5% β-mercaptoethanol, boiled 15 min, and centrifuged at 16,000 × g. Soluble protein was diluted 10-fold with IPD buffer (167 mM NaCl, 16.7 mM Tris-Cl pH 8.1, 1.1 mM EDTA, 1.1% Triton X100, 0.01% SDS) and incubated overnight at 4°C with 2 μg monoclonal anti-HA antibody (clone F7; SantaCruz). Antibody complexes were captured with 20-50 μl protein-G-agarose beads (Roche) prewashed in IPD buffer. Immune complexes were collected by centrifugation, washed three times with IPD buffer, twice with 1× NEB4 (50 mM K-acetate, 20 mM Tris-acetate, 10 mM Mg-acetate, 1 mM DTT, pH 7.9), resuspended and incubated 1 h at 37°C in 50 μl 1× NEB4 containing 0.5 mM CoCl2, 50 units TdT (New England Biolabs), and either 10 μCi [α-32P]-cordycepin triphosphate (5000 Ci/mmol) for Spo11 or 50 μCi [α-32P]-dCTP (6000 Ci/mmol) for Top2. Immune complexes were then rinsed twice with IPD buffer and boiled 10 min in 2× Laemmli buffer, then fractionated on SDS-PAGE. Gels were vacuum-dried and radiolabelled species were detected and quantified using film and Fuji phosphor screens and QuantityOne software (BioRad). For western analysis, protein was transferred to PVDF membrane in 10 mM CAPS pH 11, 10% methanol, probed with monoclonal anti-HA conjugated to horseradish peroxidase (clone F7; SantaCruz, 1:4000 dilution for Spo11-HA, 1:500,000 dilution for Top2-HA), and detected using ECL+ reagent (Amersham).
To size Spo11-associated oligonucleotides, radiolabelled complexes were fractionated by 7.5% SDS-PAGE. Wet gels were aligned with the image from a phosphor screen, and the radiolabelled bands separately excised. Complexes were eluted by the crush and soak method, deproteinized 2 h at 37°C with 2 mg/ml Pronase (Roche) in 100 mM Tris-Cl pH 7.5, 0.5% SDS, 50 mM EDTA, 10 mM CaCl2, then extracted twice with phenol/chloroform/isoamyl alcohol (25:24:1), and precipitated with 1 μg tRNA, ammonium acetate to 2.5 M, and 2.5 volumes of ethanol. Precipitate was collected by centrifugation, vacuum dried, resuspended in 80% formamide, 10 mM EDTA, 0.1% xylene cyanol, boiled 2 min, and chilled on ice. Samples were fractionated on an 18 cm long sequencing gel (20% polyacrylamide [19:1], 7 M urea, 1× TBE) at 16 mA for 70 min. Gels were fixed (10% methanol, 7% acetic acid, 5% glycerol) for 15 min, and vacuum dried onto DE81 paper prior to autoradiography.
mSPO11-oligonucleotide complexes
Spo11−/− and Dmc1−/− mice were as previously described11,13. Two to six testes from adult males (≥7 wk old) were used for each experiment. Each testis was decapsulated and lysed in 500 μl 25 mM HEPES-NaOH, pH 7.9, 5 mM EDTA, 1% SDS, 2 mM DTT, 1 mM PMSF, and leupeptin, pepstatin, chymostatin and aprotonin, each at 10 μg/ml. Lysates were heated at 95°C for 10–20 min, chilled on ice, then centrifuged at 100,000 rpm for 15 min in a TLA100.2 rotor. Supernatants were diluted with 4 volumes of 1.25% Triton X-100, 187 mM NaCl, and 18.8 mM Tris-HCl, pH 8.0, then incubated with anti-mSPO11 antibody 129/180 (Kamiya Biomedical Company; 1 μg/testis) at 4°C overnight, followed by addition of 20–50 μl protein A agarose beads (Roche) and incubation for another 4 hr. Beads were washed 3 times with IP buffer (1% Triton X-100, 150 mM NaCl, 15 mM Tris-HCl, pH 8.0, 0.1% SDS), and twice with 1× NEB4. Labelling was at 37°C for 30 min using TdT and either [α-32P] dCTP (Fig. 3b, lanes 4-6; 6000 Ci/mmol) or [α-32P]-cordycepin triphosphate (Fig. 3b, lanes 1–3; 5000 Ci/mmol). Beads were washed twice with IP buffer, boiled in 2× Laemmli sample buffer, and fractionated on 8 % SDS-PAGE.
To determine oligonucleotide lengths, mSPO11-DNA complexes from 14 testes were immunoprecipitated, labeled with [α-32P] cordycepin, separated on SDS-PAGE, and transferred to PVDF membrane. The radiolabelled region containing mSPO11-DNA complexes and the corresponding region in the control, in which no antibody was added during the immunoprecipitation, were cut out. Oligonucleotides were eluted from the membrane by Pronase treatment, precipitated, and separated on a sequencing gel as described above.
Acknowledgments
We thank John Nitiss for yeast strains; Susanne Schneider for assistance with strain construction; and Marco Barchi, Francesca Cole, Monica Di Giacomo, and Willie Mark for providing mice. Yeast work was supported by a grant from the NIGMS (to S.K.) and mouse work by a grant from the NICHD (to Maria Jasin). M.J.N. is supported in part by a fellowship from the Human Frontiers Science Program.
Footnotes
Competing interests statement: The authors declare that they have no competing financial interests.
References
- 1.Keeney S, Giroux CN, Kleckner N. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell. 1997;88:375–384. doi: 10.1016/s0092-8674(00)81876-0. [DOI] [PubMed] [Google Scholar]
- 2.Bergerat A, et al. An atypical topoisomerase II from Archaea with implications for meiotic recombination. Nature. 1997;386:414–417. doi: 10.1038/386414a0. [DOI] [PubMed] [Google Scholar]
- 3.Connelly JC, Leach DR. Repair of DNA covalently linked to protein. Mol Cell. 2004;13:307–316. doi: 10.1016/s1097-2765(04)00056-5. [DOI] [PubMed] [Google Scholar]
- 4.Keeney S. Mechanism and control of meiotic recombination initiation. Curr Top Dev Biol. 2001;52:1–53. doi: 10.1016/s0070-2153(01)52008-6. [DOI] [PubMed] [Google Scholar]
- 5.Alani E, Padmore R, Kleckner N. Analysis of wild-type and rad50 mutants of yeast suggests an intimate relationship between meiotic chromosome synapsis and recombination. Cell. 1990;61:419–436. doi: 10.1016/0092-8674(90)90524-i. [DOI] [PubMed] [Google Scholar]
- 6.Diaz RL, Alcid AD, Berger JM, Keeney S. Identification of residues in yeast Spo11p critical for meiotic DNA double-strand break formation. Mol Cell Biol. 2002;22:1106–1115. doi: 10.1128/MCB.22.4.1106-1115.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu J, Wu TC, Lichten M. The location and structure of double-strand DNA breaks induced during yeast meiosis: evidence for a covalently linked DNA-protein intermediate. EMBO J. 1995;14:4599–4608. doi: 10.1002/j.1460-2075.1995.tb00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paull TT, Gellert M. The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol Cell. 1998;1:969–979. doi: 10.1016/s1097-2765(00)80097-0. [DOI] [PubMed] [Google Scholar]
- 9.Romanienko PJ, Camerini-Otero RD. Cloning, characterization, and localization of mouse and human SPO11. Genomics. 1999;61:156–169. doi: 10.1006/geno.1999.5955. [DOI] [PubMed] [Google Scholar]
- 10.Keeney S, et al. A mouse homolog of the Saccharomyces cerevisiae meiotic recombination DNA transesterase Spo11p. Genomics. 1999;61:170–182. doi: 10.1006/geno.1999.5956. [DOI] [PubMed] [Google Scholar]
- 11.Baudat F, Manova K, Yuen JP, Jasin M, Keeney S. Chromosome synapsis defects and sexually dimorphic meiotic progression in mice lacking Spo11. Mol Cell. 2000;6:989–998. doi: 10.1016/s1097-2765(00)00098-8. [DOI] [PubMed] [Google Scholar]
- 12.Romanienko PJ, Camerini-Otero RD. The mouse Spo11 gene is required for meiotic chromosome synapsis. Mol Cell. 2000;6:975–987. doi: 10.1016/s1097-2765(00)00097-6. [DOI] [PubMed] [Google Scholar]
- 13.Pittman DL, et al. Meiotic prophase arrest with failure of chromosome synapsis in mice deficient for Dmc1, a germline-specific RecA homolog. Mol Cell. 1998;1:697–705. doi: 10.1016/s1097-2765(00)80069-6. [DOI] [PubMed] [Google Scholar]
- 14.Barchi, M. et al. Surveillance of different recombination defects in mouse spermatocytes yields distinct responses despite elimination at an identical developmental stage. Mol. Cell. Biol., in press (2005). [DOI] [PMC free article] [PubMed]
- 15.Walker JV, Nitiss JL. DNA topoisomerase II as a target for cancer chemotherapy. Cancer Invest. 2002;20:570–589. doi: 10.1081/cnv-120002156. [DOI] [PubMed] [Google Scholar]
- 16.Fortune JM, Osheroff N. Topoisomerase II as a target for anticancer drugs: when enzymes stop being nice. Prog Nucleic Acid Res Mol Biol. 2000;64:221–253. doi: 10.1016/s0079-6603(00)64006-0. [DOI] [PubMed] [Google Scholar]
- 17.Pouliot JJ, Yao KC, Robertson CA, Nash HA. Yeast gene for a Tyr-DNA phosphodiesterase that repairs topoisomerase I complexes. Science. 1999;286:552–555. doi: 10.1126/science.286.5439.552. [DOI] [PubMed] [Google Scholar]
- 18.Stohr BA, Kreuzer KN. Repair of topoisomerase-mediated DNA damage in bacteriophage T4. Genetics. 2001;158:19–28. doi: 10.1093/genetics/158.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Connelly JC, de Leau ES, Leach DR. Nucleolytic processing of a protein-bound DNA end by the E. coli SbcCD (MR) complex. DNA Repair (Amst) 2003;2:795–807. doi: 10.1016/s1568-7864(03)00063-6. [DOI] [PubMed] [Google Scholar]
- 20.Stracker TH, Carson CT, Weitzman MD. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature. 2002;418:348–352. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- 21.Johnson RE, Kovvali GK, Prakash L, Prakash S. Role of yeast Rth1 nuclease and its homologs in mutation avoidance, DNA repair, and DNA replication. Curr Genet. 1998;34:21–29. doi: 10.1007/s002940050362. [DOI] [PubMed] [Google Scholar]
- 22.Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW. The double-strand-break repair model for recombination. Cell. 1983;33:25–35. doi: 10.1016/0092-8674(83)90331-8. [DOI] [PubMed] [Google Scholar]
- 23.Paques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hunter N, Kleckner N. The single-end invasion: an asymmetric intermediate at the double- strand break to double-holliday junction transition of meiotic recombination. Cell. 2001;106:59–70. doi: 10.1016/s0092-8674(01)00430-5. [DOI] [PubMed] [Google Scholar]
- 25.Shinohara M, Gasior SL, Bishop DK, Shinohara A. Tid1/Rdh54 promotes colocalization of Rad51 and Dmc1 during meiotic recombination. Proc Natl Acad Sci USA. 2000;97:10814–10819. doi: 10.1073/pnas.97.20.10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwacha A, Kleckner N. Identification of joint molecules that form frequently between homologs but rarely between sister chromatids during yeast meiosis. Cell. 1994;76:51–63. doi: 10.1016/0092-8674(94)90172-4. [DOI] [PubMed] [Google Scholar]
- 27.Henderson KA, Keeney S. Tying synaptonemal complex initiation to the formation and programmed repair of DNA double-strand breaks. Proc Natl Acad Sci USA. 2004;101:4519–4524. doi: 10.1073/pnas.0400843101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nitiss JL, et al. Amsacrine and etoposide hypersensitivity of yeast cells overexpressing DNA topoisomerase II. Cancer Res. 1992;52:4467–4472. [PubMed] [Google Scholar]
- 29.Longtine MS, et al. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]