

Abstract
Alcohol (ethanol) abuse is a major societal problem. Although ethanol is a structurally simple, diffusible molecule, its sites of action are surprisingly selective, and the molecular mechanisms underlying specificity in ethanol actions are not understood. The NMDA receptor channel is one of the main targets for ethanol in the brain. We report here that the brain region-specific compartmentalization of Fyn kinase determines NMDA receptor sensitivity to ethanol. We demonstrate that, in the hippocampus but not in the cerebral cortex, Fyn is targeted to the NR2B subunit of the NMDA receptor by the scaffolding protein RACK1. During acute exposure to ethanol, RACK1 is dissociated from the complex, thereby facilitating Fyn-mediated phosphorylation of NR2B, which enhances channel activity, counteracting the inhibitory actions of ethanol. In this way, the selective scaffolding can account for the ethanol-induced acute tolerance of NMDA receptor activity that is detected in the hippocampus but not in the cerebral cortex. The phosphorylation-dependent, region-specific activities of ethanol on the NMDA receptor provide a compelling molecular explanation that accounts for the selective activities of ethanol and may have important implications for elucidating pathways leading to alcohol addiction.
Keywords: ethanol, NMDA, RACK1, Fyn, tyrosine phosphorylation, scaffolding proteins
Introduction
The major excitatory neurotransmitter in the brain, glutamate, binds to the ligand-gated ion receptor channel NMDA (Sucher et al., 1996). The NMDA receptor (NMDAR) is a major target of ethanol. The activities of ethanol on the NMDAR are important contributors to the development of disease states associated with alcohol abuse such as tolerance, dependence, withdrawal, craving, and relapse (for review, see Chandler et al., 1998; Kumari and Ticku, 2000). Acutely, ethanol inhibits NMDAR function (Lovinger et al., 1989), as well as long-term potentiation (LTP) (Morrisett and Swartzwelder, 1993; Givens and McMahon, 1995). Chronic exposure to ethanol causes upregulation of NMDAR subunits (Kumari and Ticku, 2000), specifically the NR2B subunit (Narita et al., 2000), and upregulation of NMDARs are thought to be involved in the expression of withdrawal (Gonzalez et al., 2001).
NMDARs are heteromers composed of an obligatory NR1 subunit and combinations of different NR2 (A-D) subunits (Sucher et al., 1996), and phosphorylation of NMDAR subunits modulates channel function, which contributes to the regulation of postsynaptic responses (Wang and Salter, 1994; Hisatsune et al., 1997; Leonard and Hell, 1997; Zheng et al., 1998; Lu et al., 1999). The NR2 subunits are phosphorylated on tyrosine residue by members of the Src family of protein tyrosine kinases (PTKs), including Fyn (Moon et al., 1994; Ali and Salter, 2001;Takasu et al., 2002). NR2B is tyrosine phosphorylated during acute exposure to ethanol in vivo (Miyakawa et al., 1997), and this phosphorylation is likely to be mediated by Fyn because it is abolished in LacZ-Fyn−/− mice (Miyakawa et al., 1997). Importantly, deletion of the Fyngene resulted in mice that were found to be hypersensitive to the hypnotic effects of ethanol (Miyakawa et al., 1997), suggesting the involvement of Fyn in sensitivity to ethanol.
Important regulators of the phosphorylation state of NMDAR subunits are scaffolding proteins. Scaffolding proteins provide both spatial organization and specificity of signaling cascades (Pawson and Scott, 1997) and play an important role in regulation of the NMDAR channel within the postsynaptic density (PSD) structures (Kennedy, 1997; Sheng and Pak, 1999). Scaffolding proteins in the PSD assemble kinases and phosphatases in close proximity to their substrate (such as the NMDAR subunits), connect the NMDAR to the cytoskeleton, and mediate clustering of the receptors (O'Brien et al., 1998). We recently identified the scaffolding protein RACK1 as part of the NMDAR complex (Yaka et al., 2002). RACK1 directly interacts with the NR2B subunit and with the kinase Fyn. RACK1 decreases Fyn phosphorylation of NR2B and inhibits the function of the NMDAR channel (Yaka et al., 2002). Our results suggest that RACK1 localizes Fyn in close proximity with its substrate (NR2B) and prevents Fyn from phosphorylating NR2B until the appropriate signal occurs. Here we identify ethanol as one such signal by determining the activities of ethanol in the hippocampus and the cerebral cortex on (1) the compartmentalization and function of RACK1 and its association with Fyn and NR2B, (2) the phosphorylation state of the NR2 subunits of NMDAR, and (3) the activity of the NMDAR as a consequence of altered compartmentalization and function of RACK1 and altered phosphorylation state of the NMDAR.
Materials and Methods
Reagents. The polyclonal anti-NR2B, anti-NR2A, anti-actin, and anti-Fyn antibodies and Fyn blocking peptide were purchased from Santa Cruz Biotechnologies (Santa Cruz, CA). The monoclonal anti-phosphotyrosine, anti-RACK1, anti-PSD95, and anti-NR2B antibodies were purchased from Transduction Laboratories (Lexington, KY). The monoclonal anti-hemagglutinin (HA) antibodies were purchased from Roche (Nutley, NJ). The monoclonal anti-NR1 antibodies were purchased fromZymed (South San Francisco, CA). The monoclonal anti-MAP2 (microtubule associated protein-2) and polyclonal anti-NR2B antibodies were purchase from Chemicon (Temecula, CA). The polyclonal anti-(pY1336)NR2B, anti-(pY1252)NR2B, and anti-(pY1472)NR2B antibodies were described previously (Takasu et al., 2002). The Src/Fyn peptide (KVEKIGEGTYGVVYK) was purchased from Upstate Biotechnology (Waltham, MA). Picrotoxin, ifenprodil, and H89 were purchased from Sigma (St. Louis, MO). PP2 was purchased from Calbiochem (La Jolla, CA). 2,3-Dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX) and d-APV were purchased from Tocris (Ellisville, MO).
Recombinant proteins. RACK1 was subcloned into pTAT-HA and expressed in Escherichia coli as described previously (He et al., 2002). Bacteria were homogenized in 20 ml of lysis buffer (8m urea, 200 mm NaCl, and 20 mm HEPES, pH 8.0) containing protease inhibitor mixture (Roche) and phosphatase inhibitor mixture (Sigma), followed by sonication for 2 min. The homogenate was clarified by centrifugation at 4°C for 30 min and purified by using Ni-NTA agarose beads. After incubation at 4°C for 1–2 hr while shaking, the beads were washed three times with >6 vol of lysis buffer containing 20 mm imidizole and then eluted with 500 mm imidizole in lysis buffer. The eluate was dialyzed overnight at 4°C against 10% glycerol in PBS.
Preparation of slice and brain homogenates. Experimental protocols involving the use of vertebrate animals were approved by the Gallo Research Center subcommittee on Research Animal Care and met National Institutes of Health guidelines.
For slice homogenates preparation, transverse hippocampal or coronal cerebral cortical slices (350–400 μm) were prepared from 3- to 4-week-old male Sprague Dawley rats (Simonsen Laboratories, Gilroy, CA). Slices were maintained for 2 hr in artificial CSF (aCSF) that contained the following (in mm): 126 NaCl, 1.2 KCl, 1.2 NaH2PO4, 0.01 MgCl2, 2.4 CaCl2, 18 NaHCO3, and 11 glucose (saturated with 95%O2–5%CO2) at 25°C. After recovery, slices were treated and homogenized in homogenization buffer (HB) [320 mm sucrose, 10 mm Tris-HCl, pH 7.4, 10 mm EDTA, 10 mm EGTA, protease inhibitor mixture (Roche), and phosphatase inhibitor mixture (Sigma)]. Homogenates were centrifuged at 5000 × g. The pellet was resuspended in solubilization buffer (10 mm Tris-HCl, pH 7.4, 10 mm EDTA, 10 mm EGTA, and protease and phosphatase inhibitor mixtures) that includes 1% Triton X-100 for phosphorylation assays or 1% deoxycholate for association assays. The suspension was then centrifuged at 60,000 ×g to yield supernatant (soluble material) and pellet (insoluble material).
For brain homogenates preparation (in vivo), 3- to 4-week-old SVJ/129 male mice (Charles River Laboratories, Wilmington, MA) were intraperitoneally injected with saline or ethanol (3.5 gm/kg) for the time indicated in the figure legends. The cerebral cortex and hippocampus were dissected immediately and frozen in liquid nitrogen. Samples were homogenized and solubilized as described above.
Subcellular fractionation. Tissue from 3- to 4-week-old male rats was homogenized as described previously (Huttner et al., 1983). Briefly, dounce homogenates of the pellets in ice-cold HB (320 mm sucrose, 10 mm Tris-HCl, pH 7.4, 10 mm EDTA, 10 mmEGTA, and protease and phosphatase inhibitor mixtures) were centrifuged at 1000 × g to remove nuclei and large debris. The supernatant (S1) was centrifuged at 10,000 × g to obtain a crude synaptosomal fraction (P2) and the cytosolic and light membranal fraction (S2). After each centrifugation, the resulting pellet was rinsed briefly with ice-cold HB before subsequent fractionations to avoid possible crossover contamination.
Immunoprecipitation. The supernatant soluble material from slice or brain homogenates were precleared by incubation with protein G agarose. The samples were centrifuged, and protein quantity was determined using BCA reagent. Immunoprecipitation was performed with 5 μg of the appropriate antibody, with ∼0.5 mg of protein diluted in 1× immunoprecipitation buffer [1% Triton X-100, 150 mm NaCl, 10 mm Tris HCl, pH 7.4, 1 mm EDTA, 1 mm EGTA, 0.2 mm sodium orthovanadate, protease inhibitor mixture (Roche), and phosphatase inhibitor mixture (Sigma)]. After overnight incubation at 4°C, protein G agarose was added, and the mixture was incubated at 4°C for 2 hr. The protein G was washed extensively, and pellets were resolved on a 10% SDS-PAGE.
Western blot analysis. Fifty micrograms of protein were resolved on a 10% SDS-PAGE and transferred to a nitrocellulose membrane. Membranes were probed with the appropriate antibodies, and immunoreactivity was detected with enhanced chemiluminescence and processed using the STORM system. Digitized images of the bands corresponding to NMDAR subunits were quantitatively analyzed by densitometry, with NIH Image version 1.61 providing peak areas, and values were expressed as a ratio of phosphorylated to total amount of immunoprecipitated NMDAR subunits or actin as indicated in the figure legends. Statistical analysis was performed using Student'st test for significant differences.
In vitro translation assay. [35S]methionine-labeled proteins were generated in rabbit reticulocyte lysates (TNT kit;Promega, Madison, WI) programmed with cDNAs encoding the cytoplasmic tail of NR2B (amino acids 839–1482), Fyn, and RACK1 as described previously (Yaka et al., 2002). The translation reactions were analyzed by SDS-PAGE and fluorography. Interaction of the proteins was determined by coimmunoprecipitation from the lysates with anti-RACK1 antibodies.
In vitro kinase assay. Fyn (10 U, 0.32 pmol · min−1 · U−1) was incubated in the presence of Src/Fyn substrate peptide (150 μm) without or with increasing concentrations of ethanol in kinase buffer (25 mm Tris-HCl, pH 7.2, 31.5 mm MgCl2, 6.25 mm MnCl2, 10 μCi γ[32P]ATP, and 125 μm cold ATP) at room temperature for 20 min. Aliquots of the reaction were spotted onto P81 paper, Fyn activity was measured using scintillation counting, and specific activity was expressed as pmol of phosphorylation per minute per units of Fyn.
Immunohistochemistry. Three- to 4-week-old male rats were anesthetized and perfused with 4% paraformaldehyde. Brains were removed and sectioned on a vibratome (VT1000S; Leica, Nussloch, Germany). Slices (35 μm) that contained the hippocampus and cerebral cortex were blocked in PBS containing 0.1% Triton X-100 and 10% normal goat serum for 1 hr at room temperature. Blocking solution was aspirated, and sections were then double stained with anti-RACK (1:500) and anti-MAP2 (1:250) antibodies, anti-RACK1 (1:500) and anti-Fyn (1:250) antibodies, and anti-RACK1 (1:500) and anti-NR2B (1:250) antibodies overnight at 4°C. Antibodies were then aspirated, and sections were washed three times in PBS containing 0.1% Triton X-100. Sections were incubated for 2 hr at room temperature with the following secondary antibodies: Texas Red-conjugated antibody (goat anti-mouse IgM for RACK1; 1:250) and FITC-conjugated antibody (goat anti-mouse IgG for MAP2 and goat anti-rabbit IgG for Fyn and NR2B; 1:250). Sections were then washed and mounted on Fisher Superfrost glass slides. Slides were mounted using Vectashield and viewed with aZeiss (Oberkochen, Germany) LSM-1024 laser-scanning microscope. The confocal images were processed using Adobe Photoshop (Adobe Systems, San Jose, CA). Specificity of RACK1 staining was confirmed by preabsorbing anti-RACK1 antibodies with recombinant RACK1 as described previously (Ron et al., 2000). Specificity of Fyn staining was verified by preincubating anti-Fyn antibodies with the blocking peptide (1:5 w/w) for 2 hr at room temperature (data not shown).
Blood ethanol determination. Blood samples of ∼50 μl were obtained from the tail vein of SVJ/129 male mice 15 min after intraperitoneal injection of 3.5 gm/kg ethanol. Blood ethanol concentration was determined using the Sigma Alcohol Diagnostic kit 332.
Electrophysiology. Transverse hippocampal or coronal cerebral cortical slices (350–400 μm) were prepared from 3- to 4-week-old male Sprague Dawley rats (Simonsen Laboratories). Slices were maintained for at least 2 hr in aCSF that contained the following (in mm): 126 NaCl, 1.2 KCl, 1.2 NaH2PO4, and 0.01 for fEPSPs) or 1.2 (for EPSCs) MgCl2, 2.4 CaCl2, 18 NaHCO3, and 11 glucose (saturated with 95%O2–5%CO2 at 25°C). After recovery, slices were submerged and continuously superfused with aCSF at 25°C.
Field recording-field EPSPs (fEPSPs) were recorded from stratum radiatum of CA1 region or layers II–III of the prefrontal cortex with glass microelectrodes filled with 2 m NaCl. To obtain NMDAR-mediated fEPSPs, picrotoxin (100 μm) and NBQX (10 μm) were added to the bath solution. To evoke fEPSPs, Schaffer collateral/commissural (hippocampus) or horizontal (cortex) afferents were stimulated with 0.1 Hz pulses using steel bipolar microelectrodes at intensities adjusted to produce an evoked response that was 50% of the maximum recorded fEPSP for each recording. The maximal rate of change in fEPSP within a time window selected around the rising phase was calculated.
Whole-cell recording–somatic whole-cell voltage-clamp recordings were made from CA1 pyramidal cells using 3–6 MΩ electrodes. The whole-cell solution contained the following (in mm): 117 cesium methansulfonic acid, 2.8 NaCl, 20 HEPES, 0.4 EGTA, 5 TEA-Cl, 2.5 MgATP, and 0.25 MgGTP, pH 7.2–7.4 (285–295 mOsm). To obtain NMDAR-mediated EPSCs, picrotoxin (100 μm) and NBQX (10 μm) were added to the bath solution. Cells were held at +40 mV, and series and input resistances were monitored continuously with a 4 mV depolarizing step, given with every afferent stimulus. The amplitudes of EPSCs were measured using a window at the peak of the event, relative to the baseline taken immediately before the stimulus artifact.
Data were collected using an Axopatch-1D amplifier (Axon Instruments, Foster city, CA), filtered at 2 kHz, and digitized at 5–10 kHz. Bath application of ifenprodil (25 μm) in hippocampal or cortical slices inhibited NMDAR-mediated fEPSPs by nearly 90%, confirming high levels of NR2B-containing NMDARs (data not shown).
Results
Region-specific compartmentalization of Fyn determines the activity of ethanol
We recently found that in the hippocampus, the scaffolding protein RACK1 interacts with the tyrosine kinase Fyn and its substrate, the NR2B subunit of the NMDAR, and prevents Fyn phosphorylation of NR2B. During release of RACK1 from the complex, Fyn phosphorylates the channel, and, as a consequence of the phosphorylation, the activity of the channel is enhanced (Yaka et al., 2002). The NMDAR is one of the main targets for ethanol (Kumari and Ticku, 2000), and Fyn has been linked previously to the activities of ethanol on the NMDAR (Miyakawa et al., 1997). To determine whether the compartmentalization of Fyn to the NR2B subunit contributes to the activities of ethanol, we assessed whether acute exposure to ethanol affected the association between NR2B, RACK1, and Fyn in the hippocampus. Rat hippocampal slices were treated with 25 and 100 mm ethanol, and RACK1 association with Fyn and NR2B was determined. As shown in Figure1A, and as we reported recently (Yaka et al., 2002), in the control hippocampal slices, RACK1 forms a tri-molecular complex with NR2B and Fyn. However, ethanol treatment resulted in the dissociation of the complex in a dose-dependent manner (Fig. 1A). During exposure to ethanol, anti-RACK1 antibodies coimmunoprecipitated less Fyn and NR2B, and anti-NR2B antibodies coimmunoprecipitated less Fyn and RACK1 (Fig.1A and data not shown), and there was no change in the protein level of NR2B or Fyn in the hippocampus in response to acute exposure of the slices to ethanol (Fig.1B).
Fig. 1.

A–C, In the hippocampus, Fyn is compartmentalized to NR2B via RACK1, and ethanol induced the dissociation of the tri-molecular complex. A, Rat hippocampal slices were treated without (control) or with 25 and 100 mm ethanol for 30 min. Immunoprecipitations (IPs) were performed with anti-RACK1 and anti-mouse IgM antibodies as control. Membranes were probed with anti-NR2B, anti-Fyn, and anti-RACK1 antibodies. Histogram depicts the mean ± SD level of Fyn and NR2B dissociation normalized to immunoprecipitated RACK1 (n= 4). B, Rat hippocampal slices were treated as in A, and samples were resolved on SDS-PAGE. Membranes were probed with anti-NR2B and anti-Fyn antibodies (n= 4). C, Three- to 4-week-old SVJ/129 male mice were injected intraperitoneally (IP) with saline or 3.5 gm/kg ethanol. Fifteen minutes later, the hippocampus was dissected, and immunoprecipitation was performed with anti-RACK1 antibodies and probed with anti-NR2B, anti-Fyn, and anti-RACK1 antibodies (n= 3).D, E, In the cerebral cortex, Fyn associated with RACK1 but not with NR2B, and ethanol did not affect the association. D, Rat cortical slices were treated with 100 mm ethanol for 30 min, immunoprecipitated, and probed as in A (n= 3).E, Mice were injected intraperitoneally with saline, and cortical tissues were harvested and analyzed as in C(n= 3).
Next, we determined whether acute administration of ethanol in vivo would induce the dissociation of RACK1 from NR2B and Fyn. To do so, mice were systemically treated with 3.5 gm/kg ethanol by intraperitoneal injection, and, 15 min after injection, blood alcohol concentration was measured, the hippocampus was dissected, and the presence of the complex was assessed. We found that intraperitoneal injection of 3.5 gm/kg ethanol produced blood alcohol concentration of 72 ± 3.3 mm (n = 5), and, at this concentration of ethanol, dissociation of the complex was observed (Fig. 1C).
Unexpectedly, we found that the NR2B–RACK1–Fyn tri-molecular complex did not exist in the cerebral cortex, in slices or in vivo(Fig. 1D,E). RACK1 associated only with Fyn in the cortex, and their interaction was not affected by ethanol (Fig.1D). In summary, in the hippocampus, where RACK1 scaffolds Fyn to the NMDAR, ethanol exposure induced the dissociation of the tri-molecular complex.
RACK1 is compartmentalized to the dendrites in the hippocampus but not in the cortex, and ethanol-induced dissociation of RACK1 from NMDAR complex is mediated via protein kinase A signaling
We hypothesized that the NR2B–RACK1–Fyn complex does not exist in the cortex because of differences in RACK1 compartmentalization in the two brain regions. To test the hypothesis, the localization of RACK1 in hippocampal and cortical slices was determined by immunohistochemistry and confocal microscopy. Double labeling of anti-RACK1 antibodies and the antibodies for the dendritic marker MAP2 revealed that RACK1 is localized to dendrites of CA1 pyramidal neurons as seen by the orange color that resulted from the merged staining of RACK1 (red) and MAP2 (green) (Fig.2A, top panel, left).
Fig. 2.
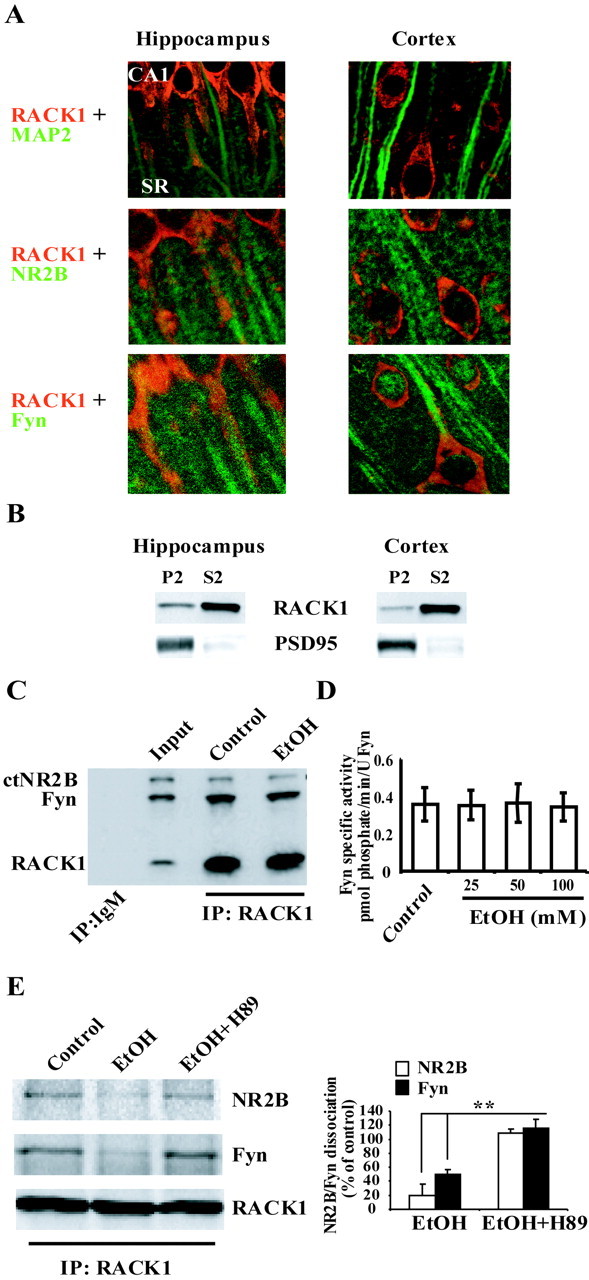
A, B, RACK1 is compartmentalized differently in hippocampus and cerebral cortex.A, Representative sections of rat hippocampal and cortical slices (35 μm) double stained with anti-RACK1 and anti-MAP2 (top panels), anti-NR2B (middle panels), and anti-Fyn (bottom panels) antibodies as described in Materials and Methods. Slices were visualized with laser-scanning confocal microscope at 63× magnification and presented as merged images. Images were taken from the CA1 region of the hippocampus, including the stratum radiatum (SR) and layers II–III of cerebral cortex. B, Subcellular fractionation of rat hippocampal and cortical tissue was performed as described in Materials and Methods. Crude synaptosomal fraction (P2) and cytosolic and light membranes fraction (S2) were resolved on 10% SDS-PAGE, and membranes were probed with anti-RACK1 antibodies. To ensure the integrity of fractionation, membranes were probed with anti-PSD95 antibodies. C, D, Ethanol activities on the NR2B–RACK1–Fyn complex are not direct.C, [35S]Methionine-labeled proteins were generated in rabbit reticulocyte lysates. Translated proteins were incubated in the absence or presence of 100 mm ethanol for 30 min at room temperature. Interaction of the proteins was determined by coimmunoprecipitation from the lysates with anti-RACK1 antibodies. Control immunoprecipitation with anti-IgM and an aliquot of the triple translation reaction ( of reaction volume; Input) are also included; n= 3. D, Fyn kinase (10 U; 0.32 pmol/min/U) was incubated with Src/Fyn substrate peptide (150 μm) and increasing concentration of ethanol (25–100 mm) for 20 min at 20°C. Histogram depicts mean ± SD of Fyn-specific activity of three experiments. E, Ethanol-induced dissociation of RACK1 from NMDA receptor complex is mediated via PKA signaling. Rat hippocampal slices were preincubated with vehicle or with H89 (10 μm) for 30 min at room temperature. Slices were then treated with ethanol (100 mm) for an additional 30 min and homogenized, and immunoprecipitation (IP) was performed with anti-RACK1 antibodies and anti-mouse IgM antibodies as control. Membranes were probed with anti-NR2B, anti-Fyn, and anti-RACK1 antibodies. Histogram depicts the mean ± SD level of Fyn and NR2B dissociation normalized to immunoprecipitated RACK1 (n= 3). **p < 0.01 indicates significantly lower than EtOH plus H89; ttest.
However, in cortical neurons, RACK1 was mainly localized to the cell soma (Fig. 2A, top panel, right). Similar results were obtained when RACK1 distribution was compared in primary hippocampal and cortical cultures (data not shown). Double staining of anti-RACK1 and anti-NR2B antibodies revealed colocalization of the two proteins in CA1 hippocampal dendrites, as seen by the orange color that resulted from the merged staining of RACK1 (red) and NR2B (green) (Fig. 2A, middle panel, left). Colocalization between RACK1 and NR2B was not observed in cortical neurons (Fig. 2A, middle panels). When staining anti-RACK1 together with anti-Fyn antibodies, we found that RACK1 is partly colocalized with Fyn in the cell soma in both hippocampus and cortex; however, dendritic colocalization was observed only in the hippocampus (Fig. 2A, bottom panels). In addition, we performed subcellular fractionation and compared the levels of RACK1 in the crude synaptosomal fraction from hippocampus and cortex. As shown in Figure 2B, RACK1 levels in the crude synaptosomal fraction (P2) was higher in the hippocampus compared with cortex. Together, these results therefore suggest that, in the cortex, RACK1 is not localized in close proximity to NR2B and therefore is not capable of localizing Fyn to the NMDAR.
Next, we set out to identify a possible mechanism by which ethanol induces the dissociation of RACK1 from the NMDAR complex in the hippocampus. First, we determined whether ethanol affects the binding of RACK1 to NR2B and Fyn using an in vitro translation assay. As shown in Figure 2C, and as reported previously (Yaka et al., 2002), anti-RACK1 antibodies immunoprecipitated in vitro translated RACK1 and coimmunoprecipitated the in vitro translated cytoplasmic tail of NR2B and Fyn. However, ethanol did not alter the interaction (Fig. 2C). In addition, ethanol did not alter Fyn kinase activity in vitro (Fig. 2D), suggesting that ethanol-induced dissociation of the NR2B–RACK1–Fyn complex is not attributable to a direct effect of ethanol.
We found previously that ethanol induces the movement of RACK1 to the nucleus, and ethanol-induced RACK1 nuclear translocation was mediated through a mechanism that requires cAMP-dependent protein kinase (PKA) (Ron et al., 2000; He et al., 2002). Therefore, we hypothesized that ethanol-induced RACK1 dissociation from the tri-molecular complex could be mediated via the PKA pathway. To test this possibility, rat hippocampal slices were preincubated with H89, a selective inhibitor of PKA, followed by ethanol treatment, and the dissociation of the complex was determined. As shown in Figure 2E, inhibition of PKA activity with H89 significantly attenuated ethanol-induced dissociation of NR2B and Fyn from RACK1. These results suggest that PKA signaling is mediating ethanol-induced dissociation of the NR2B–RACK1–Fyn complex.
Ethanol induces phosphorylation of NR2B but not NR2A in the hippocampus but not in the cortex
Previously, we found that the release of RACK1 from Fyn and NR2B enables Fyn to phosphorylate the subunit (Yaka et al., 2002). Therefore, we assessed whether ethanol-induced dissociation of RACK1 from hippocampal NMDAR resulted in an increase in tyrosine phosphorylation of the NR2B subunit. Slices were treated with vehicle or ethanol, the NMDAR was solubilized (Fig.3A), and NR2B phosphorylation was determined. As shown in Figure 3B, ethanol exposure induced an increase in tyrosine phosphorylation of NR2B in hippocampal slices. If ethanol-induced increase in phosphorylation of NR2B is mediated by Fyn, then we predicted that the inhibitor PP2, which is specific for the Src family of PTKs, would inhibit the phosphorylation of the subunit. Indeed, incubation of hippocampal slices with ethanol and PP2 significantly decreased ethanol-induced phosphorylation of NR2B (Fig. 3B). To confirm that these processes occur in vivo, mice were injected intraperitoneally with 3.5 gm/kg ethanol and killed at different time points, and the level of tyrosine phosphorylation of hippocampal NR2B was determined using phospho-specific anti-NR2B antibodies (Takasu et al., 2002). As shown in Figure 3C, increases in the phosphorylation of tyrosine residues Tyr1252 and Tyr1336 were detected 15 min after ethanol injection. Interestingly, there was no change in the major NR2B phosphorylation site identified by Nakazawa et al. (2001), Tyr1472 (Fig. 3C).
Fig. 3.

Acute exposure to ethanol increased tyrosine phosphorylation of the NR2B but not NR2A in the hippocampus.A, Rat hippocampal slices were homogenized and solubilized as described in Materials and Methods, and immunoprecipitations (IPs) were performed with anti-NR2A, anti-NR2B, and anti-NR1 antibodies. Membranes were probed with anti-NR2A, anti-NR2B, and anti-NR1 antibodies. Control hippocampal homogenate was also included (n= 3). B, Rat hippocampal slices were treated with vehicle (DMSO, 1:20,000) or ethanol (100 mm) with or without PP2 (50 nm) for 30 min. Immunoprecipitations were performed with anti-NR2B or anti-NR2A antibodies and probed with anti-NR2B, anti-NR2A, and anti-phosphotyrosine (pY) antibodies. Histogram depicts quantification of the level of tyrosine phosphorylation of NR2B and NR2A. Data are presented as mean ± SD percentage of control (n= 3). **p < 0.01; ttest. C, Mice were injected intraperitoneally with saline or 3.5 gm/kg ethanol. The hippocampus was dissected, homogenized, and resolved by SDS-PAGE. Membranes were probed with the anti-phoshpo-NR2B-specific antibodies (pTyr1252)NR2B, anti-(pTyr1336)NR2B, and anti-(pTyr1472)NR2B and anti-actin antibodies. Histogram depicts quantification of the levels of NR2B phosphorylation normalized to actin and presented as mean ± SD percentage of control (n= 3). D, Rat hippocampal slices were preincubated with PBS or with Tat-RACK1 (1 μm) for 2 hr. Slices were then treated with vehicle or ethanol (100 mm) for 30 min, homogenized, and resolved by SDS-PAGE. To ensure transduction of the Tat-fusion protein, membranes were probed with anti-RACK1 (middle panel) and anti-HA (bottom panel) antibodies. Membranes were probed with anti-(pTyr1252)NR2B and anti-actin antibodies. Histogram depicts quantification of the levels of phosphorylation normalized to actin and presented as mean ± SD percentage of control (n= 3).**p < 0.01; t test.
Next, we predicted that, if ethanol-induced NR2B phosphorylation is mediated via the release of RACK1 from the NMDAR complex, then the transduction of recombinant RACK1 into hippocampal slices would reverse the activities of ethanol by binding to Fyn and/or NR2B and blocking the phosphorylation of NR2B by Fyn. To test this hypothesis, we used the Tat-protein transduction method (Nagahara et al., 1998) to transduce recombinant Tat-RACK1 into hippocampal slices. As shown in Figure 3D (middle and bottom panels), high levels of Tat-RACK1 were transduced into the slices. We measured ethanol-induced phosphorylation in the presence or absence of the fusion protein and found that the transduction of Tat-RACK1 significantly decreased ethanol-induced phosphorylation of both Tyr1252 (Fig. 3D, top panel) and Tyr1336 (data not shown).
As predicted, in the cerebral cortex where Fyn was not localized to the NMDAR (Fig. 1D,E), no change in the phosphorylation state of the NR2B subunit was detected after ethanol treatment (Fig. 4, top panels). In summary, our results suggest that, in the hippocampus, ethanol releases RACK1 from Fyn and NR2B and enables Fyn to phosphorylate the subunit. Importantly, ethanol-induced phosphorylation of the NR2B subunit is brain region specific and is observed in the hippocampus but not in the cortex where RACK1 localized to the NMDAR and presumably is not localizing Fyn near its substrate.
Fig. 4.
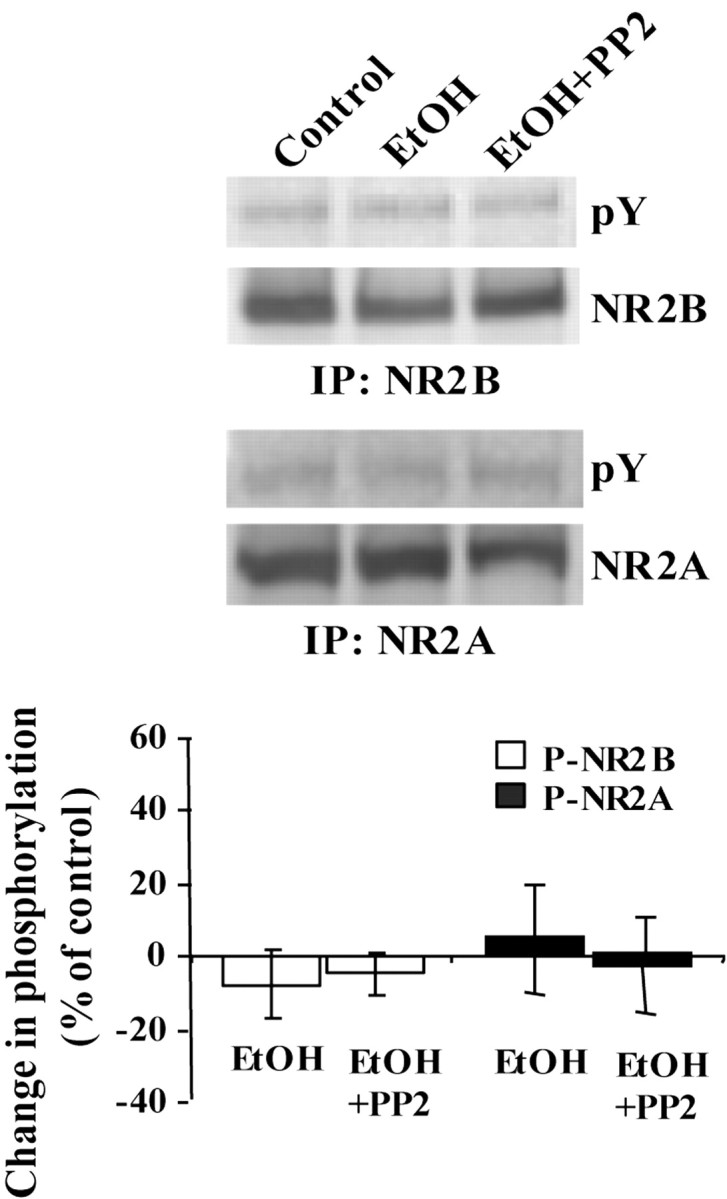
Acute exposure to ethanol does not affect the phosphorylation state of either NR2B and NR2A in the cortex. Rat cortical slices were treated as in Figure 1A and homogenized, and immunoprecipitation (IP) was performed using anti-NR2B or anti-NR2A antibodies. Membranes were probed with anti-NR2B, anti-NR2A, and anti-phosphotyrosine (pY) antibodies. Histogram depicts quantification of the level of phosphorylation. Data are presented as mean ± SD percentage of control (n= 3).
Another NMDAR subunit that is phosphorylated on tyrosine residues is NR2A (Lau and Huganir, 1995); however, RACK1 does not interact with the NR2A subunit (data not shown). Therefore, if ethanol-induced phosphorylation of the NMDAR is indeed mediated via the release of RACK1 and the activation of Fyn, then it should be specific for NR2B. As predicted, no change in the phosphorylation state of NR2A was detected during exposure to ethanol, in either hippocampal or cortical slices (Figs. 3B, 4, bottom panels). In summary, our results suggest that ethanol induction of tyrosine phosphorylation in the hippocampus is indeed specific for the NR2B subunit.
In the hippocampus, ethanol increases NMDAR channel activity and produces a rebound potentiation when ethanol is no longer present
Next, we assessed the physiological consequences of ethanol-induced NR2B tyrosine phosphorylation. Previous studies have shown that acute exposure to ethanol inhibits the activity of the NMDAR channel in hippocampal slice preparation (Lovinger et al., 1989, 1990). However, because tyrosine phosphorylation has been shown to increase the activity of the channel (Kohr and Seeburg, 1996; Yu et al., 1997), we hypothesized that the release of RACK1 that results in tyrosine phosphorylation of NR2B induced by ethanol would, although somewhat paradoxically, increase the activity of the channel. NMDAR-mediated fEPSPs were recorded from stratum radiatum of the CA1 hippocampal region in the presence of ethanol and in the presence or absence of the tyrosine kinase inhibitor PP2. As expected, bath application of 100 mm ethanol resulted in a decrease of NMDAR-mediated fEPSPs (Fig. 5A, black diamonds) (30.3 ± 2.6%; p < 0.006; post hocStudent–Newman–Keuls test). However, application of PP2 (25 nm) together with ethanol resulted in significantly greater inhibition of NMDAR fEPSPs compared with the inhibition induced by ethanol alone (Fig. 5A, white triangles) (47.9 ± 3.3%; p < 0.02; post hoc Student–Newman–Keuls test) or PP2 alone (Fig. 5A, white circles). Interestingly, when ethanol was washed out, the fEPSP slope was increased above baseline [Fig. 5A, black diamonds (127.3 ± 9.7%; p < 0.019; post hocStudent–Newman–Keuls test); Fig.5B (134.0 ± 15.1%; p < 0.01; paired t test)]. We hypothesized that this observed potentiation is attributable to the tyrosine phosphorylation of the receptor induced by ethanol that persisted after washout. To examine this possibility, hippocampal slices were exposed to ethanol (100 mm), but this time ethanol was washed out in the presence of PP2. Adding PP2 to the washout step completely abolished the enhancement in NMDAR-mediated fEPSPs (Fig. 5B) (p < 0.019; Student–Newman–Keuls test), suggesting that, indeed, the increase in NMDAR channel activity is mediated via tyrosine phosphorylation. To confirm that tyrosine phosphorylation is mediating part of the activities of ethanol on the NMDARs, the phosphorylation state of hippocampal NR2B was monitored during the same time course of the electrophysiological experiments. As predicted, the time course for NR2B phosphorylation (Fig. 5C) correlated with the time course of the electrophysiological experiments (Fig.5A,B,D,E). Next, we determined whether inhibition of tyrosine phosphorylation would attenuate the activities of ethanol of hippocampal NMDARs in single-cell recording. We therefore measured NMDAR-mediated whole-cell EPSCs elicited in CA1 pyramidal neurons. As shown in Figure5D, bath application of ethanol inhibited NMDAR EPSCs to the same degree as in field recordings (peak inhibition, 29.3 ± 3.1%) and produced rebound increase after washout (16.9 ± 6.0%). However, when CA1 neurons were intracellularly perfused with PP2 via the patching pipette, ethanol inhibition was significantly greater (44.3 ± 7.9%; p < 0.008; post hoc Student–Newman–Keuls test) compared with the inhibition induced by ethanol alone and remained stable as long as ethanol was present in the bath (Fig. 5E). Moreover, when ethanol was washed out, the rebound increase seen with ethanol alone was no longer observed. Together, these results suggest that, in the hippocampus, ethanol exposure results in two opposing activities on NMDAR function: direct inhibition of NMDAR function as has been reported previously (Lovinger et al., 1989, 1990) and an indirect increase in NMDAR function via the release of RACK1 and the subsequent increase in NR2B phosphorylation.
Fig. 5.
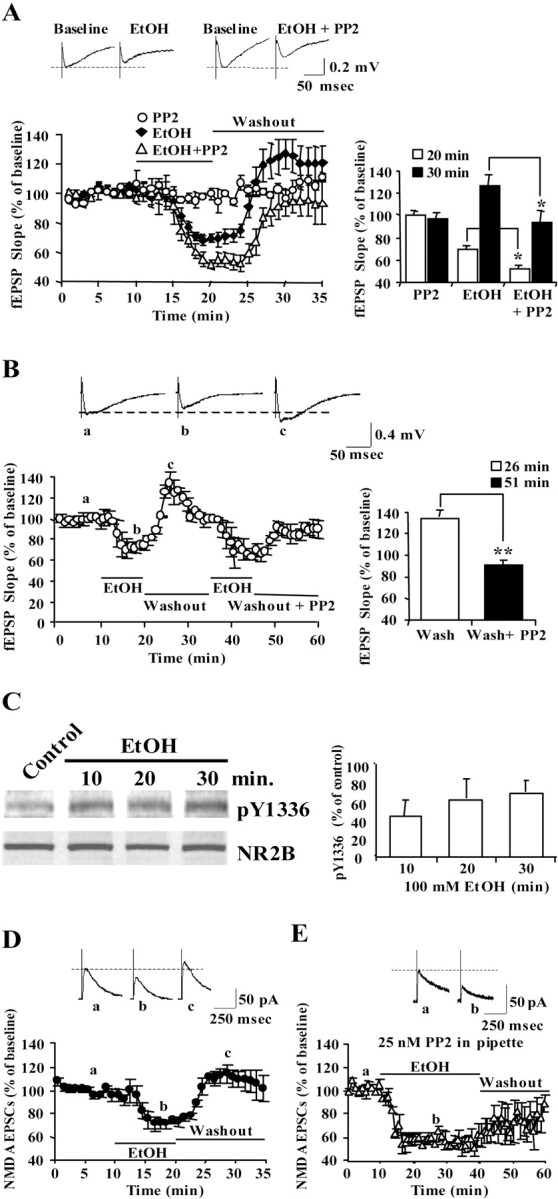
The PTK inhibitor PP2 increases ethanol-induced inhibition of NMDAR-mediated fEPSPs–EPSCs and inhibits ethanol-induced rebound potentiation in hippocampal slices. A, NMDAR-mediated fEPSP slopes were plotted for recordings made from the stratum radiatum in the CA1 region. fEPSPs were measured in the presence of PP2 (25 nm, ○; n = 6 slices), ethanol (100 mm, ♦; n = 10) or ethanol plus PP2 (▵; n = 6), as indicated by the horizontal bar. Data are presented as the mean ± SEM percentage of baseline. The traces in the inset represent NMDA receptor fEPSPs in the CA1 field (average of 12 single sweeps) obtained from the following: left, a slice recorded before (baseline) and during application of ethanol; right, a slice recorded before (baseline) and during coapplication of ethanol and PP2. Histogram on the right shows comparison of fEPSPs at 20 min (peak inhibition) and 30 min (10 min after ethanol washout). Two-way repeated-measures ANOVA showed overall significant interactions between treatments and time points (F(4,16) [infi] = 5.87;p < 0.006). Post hoc analysis showed significant effects of PP2 on ethanol inhibition (20 vs 30 min; *p < 0.02; Student–Newman–Keuls test) and on ethanol-induced rebound potentiation (20 vs 30 min; *p < 0.019; Student–Newman–Keuls test).B, NMDAR-mediated fEPSPs were measured in the CA1 region in the presence of ethanol (100 mm), followed by washout and a subsequent second application of ethanol and washout with PP2 (25 nm), as indicated by the horizontal bars. Data are presented as the mean ± SEM percentage of baseline from six slices. The traces in the inset represent NMDA receptor fEPSPs in the CA1 field (average of 12 single sweeps) obtained from the follow ing: a, a slice recorded before application of ethanol; b, during application of ethanol;c, after ethanol washout. Histogram shows the mean ± SEM fEPSPs slope recorded at 26 min (peak rebound potentiation, 6 min after ethanol washout) or 51 min (6 min after the start of washout with PP2). **p < 0.01; paired ttest. C, Time course of NR2B phosphorylation correlates with recordings. Rat hippocampal slices were incubated without (control) or with (100 mm) ethanol for 10, 20, and 30 min. Slices were homogenized and resolved by SDS-PAGE. Membranes were probed with anti-(pTyr1336)NR2B and anti-NR2B antibodies. Histogram depicts quantification of the levels of phosphorylation normalized to NR2B and are presented as mean ± SD percentage of control (n= 3). D, NMDAR-mediated EPSCs were plotted for recordings made using standard whole-cell voltage clamp from CA1 pyramidal neurons (n = 5). EPSCs were measured in the presence of ethanol (100 mm), followed by washout, as indicated by the horizontal bars. The traces in the inset represent NMDA receptor EPSCs in the CA1 field (average of 12 single sweeps) obtained from the following: a, a neuron recorded before application of ethanol; b, during application of ethanol; c, after ethanol washout.D, NMDAR-mediated EPSCs were recorded in the presence of PP2 (25 nm) in the recording pipette. Ethanol (100 mm) was bath applied as indicated by the horizontal bar, followed by washout (n = 5). The traces in the inset represent NMDA receptor EPSCs in the CA1 field (average of 12 single sweeps) obtained from the following: a, a neuron recorded before application of ethanol; b, during application of ethanol.
In the cortex, ethanol exposure does not result in enhancement of NMDAR channel activity, and rebound potentiation is not observed
Next, we tested the activities of ethanol on NMDAR function in the cortex, where RACK1 was not associated with the channel (Figs.1D,E,2A,B), and ethanol did not induce the phosphorylation of NMDAR subunits (Fig. 4). In the cortex, like the hippocampus, ethanol inhibited NMDAR-mediated fEPSP (Fig.6A). However, unlike in the hippocampus, PP2 had no effect on fEPSPs in the presence of ethanol, and potentiation of NMDAR-mediated fEPSPs after ethanol washout was not observed (Fig.6A). Furthermore, we observed that the peak inhibition of NMDAR-mediated fEPSPs in the presence of ethanol was significantly greater in cortical slices compared with hippocampal slices (Fig. 6B) (47.9 ± 6.4 vs 30.3 ± 2.6%; p < 0.01; ttest). Together, these results suggest that the sensitivity of NMDARs to ethanol varies across brain regions and is determined, at least in part, by the scaffolding of Fyn to the NMDAR via RACK1 and the resulting phosphorylation state of the channel.
Fig. 6.
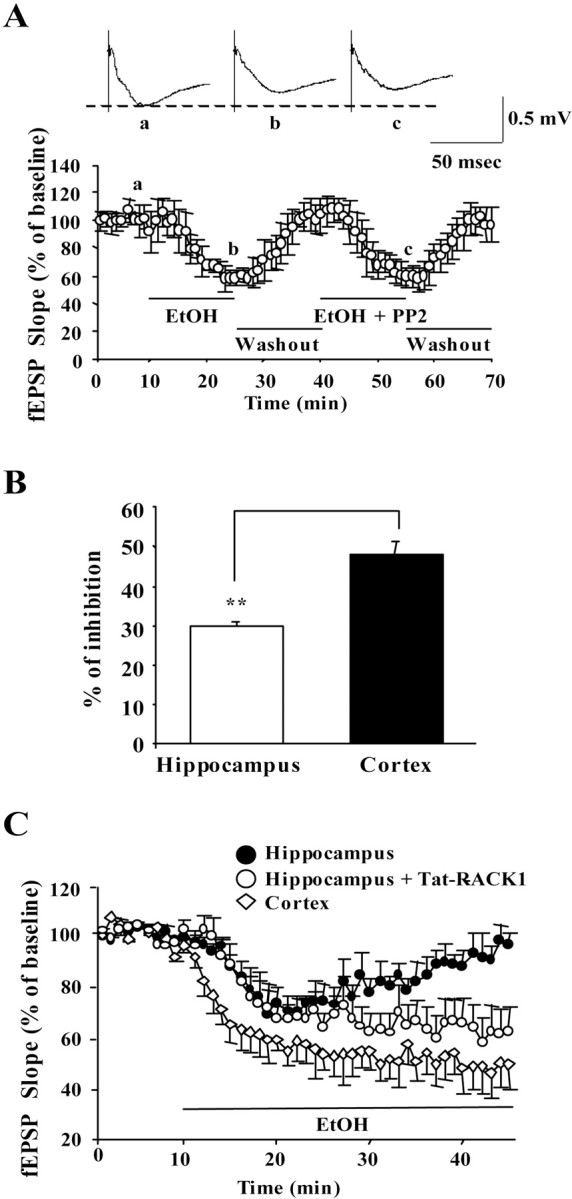
In the cortex, PP2 is inactive, ethanol inhibition is greater compared with the hippocampus, and acute tolerance to ethanol inhibition does not develop. A, NMDAR-mediated fEPSP slopes were plotted for recordings made from layers II–III in the medial prefrontal cortex. fEPSPs were measured in the presence of ethanol (100 mm), followed by washout and subsequent application of ethanol plus PP2, as indicated by the horizontal bar. Data are presented as the mean ± SEM percentage of baseline from seven slices. The traces in the inset represent NMDA receptor fEPSPs in the CA1 field (average of 12 single sweeps) obtained from the following: a, a slice recorded before application of ethanol; b, during application of ethanol;c, during coapplication of ethanol and PP2.B, Histogram depicting the mean ± SEM ratio of fEPSP slopes recorded from hippocampal or cortical slices at the time of maximal effect of ethanol treatment (10 and 15 min, respectively). **p < 0.01; t test.C, NMDAR-mediated fEPSP slopes were measured in the CA1 region in the presence of 100 mm ethanol and in the absence (●; n = 6) or after 2 hr preincubation of hippocampal slices with Tat-RACK1 (1 μm) in the bath solution (○; n = 5). fEPSP slopes were measured in the medial prefrontal cortex in the presence of 100 mmethanol (⋄; n = 5). Horizontal line depicts the period of ethanol application. Data are presented as the mean ± SEM percentage of baseline.
Acute tolerance of NMDAR function is determined by Fyn compartmentalization
Previous studies have demonstrated that ethanol-induced inhibition of NMDAR function is gradually reduced during the period of ethanol exposure (acute tolerance) in the CA1 region of the hippocampus (Grover et al., 1994; Ludvig et al., 2001). Fyn has been implicated in mediating the development of acute tolerance in the hippocampus (Miyakawa et al., 1997). We hypothesized that, if acute tolerance is determined by the scaffolding of Fyn to the NMDAR by RACK1, then the transduction of Tat-RACK1 should inhibit it. As anticipated, when Tat-RACK1 was transduced into hippocampal slices, acute tolerance was completely abolished (Fig. 6C, white circles). Support for the hypothesis also comes from the observation that, when PP2 was intracellularly applied while ethanol was bath applied for 30 min, NMDAR EPSCs remained suppressed and acute tolerance to ethanol inhibition was not observed (Fig. 5E).
We hypothesized that, in the cortex, where RACK1 does not scaffold the kinase to the NMDAR, acute tolerance would be absent. Indeed, in cortical slices, ethanol-induced inhibition of NMDAR-mediated fEPSPs was stable throughout the period of ethanol exposure (Fig.6C, white diamonds). It is unlikely that presynaptic events are mediating the enhancement of NMDAR activities in the presence of ethanol. Ethanol and/or PP2 did not alter hippocampal AMPAR-mediated fEPSPs (data not shown), and PP2 have been shown to induce exocytosis in primary neurons, suggesting that Src PTKs are necessary for the inhibition of presynaptic exocytosis events (Ohnishi et al., 2001).
Discussion
In the present study, we present evidence for the mechanism by which ethanol induces tyrosine phosphorylation of a specific NMDAR channel subunit, in a specific brain region, and we further show that these changes have important physiological consequences. In the hippocampus, RACK1 is localized in part to the dendrites where RACK1 scaffolds Fyn to the NMDAR at the plasma membrane. Ethanol releases RACK1 from the NMDAR complex, enabling Fyn to phosphorylate the NR2B subunit. These changes lead to phosphorylation-dependent enhancement of NMDAR channel activity during exposure to ethanol, to a rebound potentiation of the channel activity when ethanol is washed out, and to acute desensitization. However, in the cortex, where RACK1 is found mainly in the cell soma but not in the dendrites, Fyn is not associated with the NMDAR. In the cortex, ethanol exposure does not result in changes in the phosphorylation state of the NR2B subunit, and ethanol exposure leads only to the inhibition of the channel activity and not to acute desensitization or rebound potentiation after ethanol washout.
Since the first report by Lovinger et al. (1989) on the ability of ethanol to inhibit NMDAR-mediated currents, the degree of inhibition has been somewhat controversial and varied from study to study and from brain region to brain region. This study provides a molecular mechanism to explain previous results. Our results suggest that, in addition to the direct inhibition of NMDARs, ethanol can also promote an opposing enhancement of activity through the induction of tyrosine phosphorylation of the NR2B subunit. In brain regions where the phosphorylation state of the NR2B subunit is regulated by the compartmentalization of Fyn in close proximity to the NR2B subunit via RACK1, the actions of ethanol on the NMDAR channel are the sum of two opposing activities: an increase of activity attributable to the release of RACK1 and the phosphorylation of NR2B by Fyn, and a decrease in activity attributable to the direct inhibitory activity of ethanol on the NMDAR. The sum of these two opposing activities results in an inhibition that is lower compared with brain regions such as the cortex, where Fyn is not compartmentalized to the NMDAR and NR2B phosphorylation is not observed. Importantly, this molecular mechanism can also account for the acute tolerance observed in hippocampal but not in cortical slices. This specificity of ethanol activity on the NMDAR in some brain regions but not in others is likely to contribute to the behavioral effects of ethanol such as acute sensitivity to the hypnotic effects of ethanol, which has been implicated to be mediated via hippocampal Fyn (Miyakawa et al., 1997).
Our results also suggest that the composition of PSD proteins is different in the cortex compared with the hippocampus. The differences in PSD protein composition between different brain regions may also result in different activities of the same neurotransmitter and receptor systems in different brain regions. Interestingly, several studies allude to such possibilities. For example, Mannaioni et al. (2001) found that, in the hippocampal CA1 region, activation of type 5 metabotropic glutamate receptors (mGluR5) leads to the activation of NMDA receptor channels, whereas in cortical neurons, activation of mGluR1 but not mGluR5 receptors enhances NMDA receptor channel activity (Heidinger et al., 2002). The membranal localization and activity of the mGluR1 and mGluR5 receptors are regulated by the Homer family of scaffolding proteins (Fagni et al., 2002). It is therefore plausible that the differences between the consequences of activation of the same mGluR receptors in the different brain regions may be attributable to differences in Homer expression and/or localization.
What could be the mechanism that accounts for RACK1 dissociation from the NMDAR complex during exposure to ethanol? We found previously that, in cultured cells and in vivo, ethanol induces RACK1 to translocate to the nucleus, via a mechanism that involves the activation of the cAMP–PKA pathway (Ron et al., 2000; He et al., 2002). Here we report that the PKA inhibitor H89 inhibited ethanol-induced dissociation of the NR2B–RACK1–Fyn complex. These results imply that activation of the cAMP–PKA pathway result in both complex dissociation and RACK1 translocation to the nucleus. Although the mechanism to induce dissociation in the presence of ethanol via activation of the cAMP–PKA pathway is unclear, several possibilities are plausible. First, it is possible that exposure to ethanol and activation of the cAMP–PKA pathway result in a putative unidentified posttranslation modification and/or cofactor binding that alters the conformation of RACK1, resulting in the dissociation of RACK1 from the NMDA receptor complex. One obvious possibility is a direct phosphorylation of RACK1 by PKA. However, RACK1 is not a PKA substratein vitro (our unpublished results). Alternatively, exposure to ethanol and activation of the cAMP–PKA pathway induces a posttranslation modification on another protein that now has higher binding affinity to RACK1 compared with NR2B and Fyn. This protein will then replace NR2B and Fyn binding to RACK1, resulting in the dissociation from the complex. We are currently testing these possibilities.
Protein targeting and subcellular localization are very important for processes such as synaptic plasticity but may also contribute (as we show in this paper) to disease states such as alcohol addiction. It is well established that ethanol alters the function of certain kinases (Chandler et al., 1998; Pandey, 1998; Stubbs and Slater, 1999). Our report suggests that these changes may be attributable, at least in part, to the relocalization of a scaffolding protein (RACK1) in the presence of ethanol and that this altered compartmentalization of RACK1 results in profound changes in activity of the NMDARs. Finally, the region-specific regulation of NMDAR function by signaling complexes revealed by the specificity in ethanol activity is likely to have implications for other NMDAR functions. For example, phosphorylation of the cytoplasmic tail of NMDAR subunits is important for the induction of LTP (Lu et al., 1998), and Fyn mice have impaired LTP (Grant et al., 1992). Therefore, the specific scaffolding of Fyn to the NR2B subunit by RACK1 in some brain regions but not others is likely to have implications to other NMDAR-mediated processes such as synaptic plasticity.
Footnotes
This work was supported by the State of California for medical research on alcohol and substance abuse through the University of California, San Francisco (D.R.) and National Institute on Alcohol Abuse and Alcoholism Grant R01AA/MH13438-O1A1 (D.R.).We thank S. Dowdy for supplying the pTAT-HA plasmid and M. Greenberg for the phospho-NR2B antibodies. We thank A. Vagts and D. Nikanjam for the expression and purification of Tat-RACK1 and Viktor Kharazia for technical assistance. We thank our colleagues A. Bonci, M. Moore, F. Hopf, P. Janak, C. Thornton, M. Ungless, N. Vasquez, and J. Whistler for critical reading of this manuscript.
Correspondence should be addressed to Dorit Ron, 5858 Horton Street, Suite 200, Emeryville, CA 94608. E-mail: dorit@itsa.ucsf.edu.
References
- 1.Ali DW, Salter MW. NMDA receptor regulation by Src kinase signalling in excitatory synaptic transmission and plasticity. Curr Opin Neurobiol. 2001;11:336–342. doi: 10.1016/s0959-4388(00)00216-6. [DOI] [PubMed] [Google Scholar]
- 2.Chandler LJ, Harris RA, Crews FT. Ethanol tolerance and synaptic plasticity. Trends Pharmacol Sci. 1998;19:491–495. doi: 10.1016/s0165-6147(98)01268-1. [DOI] [PubMed] [Google Scholar]
- 3.Fagni L, Worley PF, Ango F. Homer as both a scaffold and transduction molecule. Sci STKE. 2002;2002:RE8. doi: 10.1126/stke.2002.137.re8. [DOI] [PubMed] [Google Scholar]
- 4.Givens B, McMahon K. Ethanol suppresses the induction of long-term potentiation in vivo. Brain Res. 1995;688:27–33. doi: 10.1016/0006-8993(95)00499-g. [DOI] [PubMed] [Google Scholar]
- 5.Gonzalez LP, Veatch LM, Ticku MK, Becker HC. Alcohol withdrawal kindling: mechanisms and implications for treatment. Alcohol Clin Exp Res. 2001;25:197S–201S. doi: 10.1097/00000374-200105051-00032. [DOI] [PubMed] [Google Scholar]
- 6.Grant SG, O'Dell TJ, Karl KA, Stein PL, Soriano P, Kandel ER. Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science. 1992;258:1903–1910. doi: 10.1126/science.1361685. [DOI] [PubMed] [Google Scholar]
- 7.Grover CA, Frye GD, Griffith WH. Acute tolerance to ethanol inhibition of NMDA-mediated EPSPs in the CA1 region of the rat hippocampus. Brain Res. 1994;642:70–76. doi: 10.1016/0006-8993(94)90906-7. [DOI] [PubMed] [Google Scholar]
- 8.He DY, Vagts AJ, Yaka R, Ron D. Ethanol induces gene expression via nuclear compartmentalization of receptor for activated C kinase 1. Mol Pharmacol. 2002;62:272–280. doi: 10.1124/mol.62.2.272. [DOI] [PubMed] [Google Scholar]
- 9.Heidinger V, Manzerra P, Wang XQ, Strasser U, Yu SP, Choi DW, Behrens MM. Metabotropic glutamate receptor 1-induced upregulation of NMDA receptor current: mediation through the Pyk2/Src-family kinase pathway in cortical neurons. J Neurosci. 2002;22:5452–5461. doi: 10.1523/JNEUROSCI.22-13-05452.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hisatsune C, Umemori H, Inoue T, Michikawa T, Kohda K, Mikoshiba K, Yamamoto T. Phosphorylation-dependent regulation of N-methyl-d-aspartate receptors by calmodulin. J Biol Chem. 1997;272:20805–20810. doi: 10.1074/jbc.272.33.20805. [DOI] [PubMed] [Google Scholar]
- 11.Huttner WB, Schiebler W, Greengard P, De Camilli P. Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III: its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol. 1983;96:1374–1388. doi: 10.1083/jcb.96.5.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kennedy MB. The postsynaptic density at glutamatergic synapses. Trends Neurosci. 1997;20:264–268. doi: 10.1016/s0166-2236(96)01033-8. [DOI] [PubMed] [Google Scholar]
- 13.Kohr G, Seeburg PH. Subtype-specific regulation of recombinant NMDA receptor-channels by protein tyrosine kinases of the src family. J Physiol (Lond) 1996;492:445–452. doi: 10.1113/jphysiol.1996.sp021320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumari M, Ticku MK. Regulation of NMDA receptors by ethanol. Prog Drug Res. 2000;54:152–189. [PubMed] [Google Scholar]
- 15.Lau LF, Huganir RL. Differential tyrosine phosphorylation of N-methyl-d-aspartate receptor subunits. J Biol Chem. 1995;270:20036–20041. doi: 10.1074/jbc.270.34.20036. [DOI] [PubMed] [Google Scholar]
- 16.Leonard AS, Hell JW. Cyclic AMP-dependent protein kinase and protein kinase C phosphorylate N-methyl-d-aspartate receptors at different sites. J Biol Chem. 1997;272:12107–12115. doi: 10.1074/jbc.272.18.12107. [DOI] [PubMed] [Google Scholar]
- 17.Lovinger DM, White G, Weight FF. Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science. 1989;243:1721–1724. doi: 10.1126/science.2467382. [DOI] [PubMed] [Google Scholar]
- 18.Lovinger DM, White G, Weight FF. NMDA receptor-mediated synaptic excitation selectively inhibited by ethanol in hippocampal slice from adult rat. J Neurosci. 1990;10:1372–1379. doi: 10.1523/JNEUROSCI.10-04-01372.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu WY, Xiong ZG, Lei S, Orser BA, Dudek E, Browning MD, MacDonald JF. G-protein-coupled receptors act via protein kinase C and Src to regulate NMDA receptors. Nat Neurosci. 1999;2:331–338. doi: 10.1038/7243. [DOI] [PubMed] [Google Scholar]
- 20.Lu YM, Roder JC, Davidow J, Salter MW. Src activation in the induction of long-term potentiation in CA1 hippocampal neurons. Science. 1998;279:1363–1367. doi: 10.1126/science.279.5355.1363. [DOI] [PubMed] [Google Scholar]
- 21.Ludvig N, George MA, Tang HM, Gonzales RA, Bungay PM. Evidence for the ability of hippocampal neurons to develop acute tolerance to ethanol in behaving rats. Brain Res. 2001;900:252–260. doi: 10.1016/s0006-8993(01)02319-8. [DOI] [PubMed] [Google Scholar]
- 22.Mannaioni G, Marino MJ, Valenti O, Traynelis SF, Conn PJ. Metabotropic glutamate receptors 1 and 5 differentially regulate CA1 pyramidal cell function. J Neurosci. 2001;21:5925–5934. doi: 10.1523/JNEUROSCI.21-16-05925.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miyakawa T, Yagi T, Kitazawa H, Yasuda M, Kawai N, Tsuboi K, Niki H. Fyn-kinase as a determinant of ethanol sensitivity: relation to NMDA-receptor function. Science. 1997;278:698–701. doi: 10.1126/science.278.5338.698. [DOI] [PubMed] [Google Scholar]
- 24.Moon IS, Apperson ML, Kennedy MB. The major tyrosine-phosphorylated protein in the postsynaptic density fraction is N-methyl-d-aspartate receptor subunit 2B. Proc Natl Acad Sci USA. 1994;91:3954–3958. doi: 10.1073/pnas.91.9.3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morrisett RA, Swartzwelder HS. Attenuation of hippocampal long-term potentiation by ethanol: a patch-clamp analysis of glutamatergic and GABAergic mechanisms. J Neurosci. 1993;13:2264–2272. doi: 10.1523/JNEUROSCI.13-05-02264.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagahara H, Vocero-Akbani AM, Snyder EL, Ho A, Latham DG, Lissy NA, Becker-Hapak M, Ezhevsky SA, Dowdy SF. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat Med. 1998;4:1449–1452. doi: 10.1038/4042. [DOI] [PubMed] [Google Scholar]
- 27.Nakazawa T, Komai S, Tezuka T, Hisatsune C, Umemori H, Semba K, Mishina M, Manabe T, Yamamoto T. Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-d-aspartate receptor. J Biol Chem. 2001;276:693–699. doi: 10.1074/jbc.M008085200. [DOI] [PubMed] [Google Scholar]
- 28.Narita M, Soma M, Mizoguchi H, Tseng LF, Suzuki T. Implications of the NR2B subunit-containing NMDA receptor localized in mouse limbic forebrain in ethanol dependence. Eur J Pharmacol. 2000;401:191–195. doi: 10.1016/s0014-2999(00)00428-3. [DOI] [PubMed] [Google Scholar]
- 29.O'Brien RJ, Lau LF, Huganir RL. Molecular mechanisms of glutamate receptor clustering at excitatory synapses. Curr Opin Neurobiol. 1998;8:364–369. doi: 10.1016/s0959-4388(98)80062-7. [DOI] [PubMed] [Google Scholar]
- 30.Ohnishi H, Yamamori S, Ono K, Aoyagi K, Kondo S, Takahashi M. A src family tyrosine kinase inhibits neurotransmitter release from neuronal cells. Proc Natl Acad Sci USA. 2001;98:10930–10935. doi: 10.1073/pnas.191368198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pandey SC. Neuronal signaling systems and ethanol dependence. Mol Neurobiol. 1998;17:1–15. doi: 10.1007/BF02802021. [DOI] [PubMed] [Google Scholar]
- 32.Pawson T, Scott JD. Signaling through scaffold, anchoring, and adaptor proteins. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- 33.Ron D, Vagts AJ, Dohrman DP, Yaka R, Jiang Z, Yao L, Crabbe J, Grisel JE, Diamond I. Uncoupling of {beta}IIPKC from its targeting protein RACK1 in response to ethanol in cultured cells and mouse brain. FASEB J. 2000;14:2303–2314. doi: 10.1096/fj.00-0143com. [DOI] [PubMed] [Google Scholar]
- 34.Sheng M, Pak DT. Glutamate receptor anchoring proteins and the molecular organization of excitatory synapses. Ann NY Acad Sci. 1999;868:483–493. doi: 10.1111/j.1749-6632.1999.tb11317.x. [DOI] [PubMed] [Google Scholar]
- 35.Stubbs CD, Slater SJ. Ethanol and protein kinase C. Alcohol Clin Exp Res. 1999;23:1552–1560. [PubMed] [Google Scholar]
- 36.Sucher NJ, Awobuluyi M, Choi YB, Lipton SA. NMDA receptors: from genes to channels. Trends Pharmacol Sci. 1996;17:348–355. [PubMed] [Google Scholar]
- 37.Takasu MA, Dalva MB, Zigmond RE, Greenberg ME. Modulation of NMDA receptor-dependent calcium influx and gene expression through EphB receptors. Science. 2002;295:491–495. doi: 10.1126/science.1065983. [DOI] [PubMed] [Google Scholar]
- 38.Wang YT, Salter MW. Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature. 1994;369:233–235. doi: 10.1038/369233a0. [DOI] [PubMed] [Google Scholar]
- 39.Yaka R, Thornton C, Vagts AJ, Phamluong K, Bonci A, Ron D. NMDA receptor function is regulated by the inhibitory scaffolding protein, RACK1. Proc Natl Acad Sci USA. 2002;99:5710–5715. doi: 10.1073/pnas.062046299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu XM, Askalan R, Keil GJ, II, Salter MW. NMDA channel regulation by channel-associated protein tyrosine kinase Src. Science. 1997;275:674–678. doi: 10.1126/science.275.5300.674. [DOI] [PubMed] [Google Scholar]
- 41.Zheng F, Gingrich MB, Traynelis SF, Conn PJ. Tyrosine kinase potentiates NMDA receptor currents by reducing tonic zinc inhibition. Nat Neurosci. 1998;1:185–191. doi: 10.1038/634. [DOI] [PubMed] [Google Scholar]