

Summary
Mitochondrial aldehyde dehydrogenase (ALDH2) is the major enzyme that oxidizes ethanol-derived acetaldehyde. A nearly inactive form of the enzyme, ALDH2*2, is found in about 40% of the East Asian population. This variant enzyme is defined by a glutamate to lysine substitution at residue 487 located within the oligomerization domain. ALDH2*2 has an increased Km for its coenzyme, NAD+, and a decreased kcat, which lead to low activity in vivo. Here we report the 2.1 Å crystal structure of ALDH2*2. The structure shows a large disordered region located at the dimer interface that includes much of the coenzyme binding cleft and a loop of residues that form the base of the active site. As a consequence of these structural changes, the variant enzyme exhibits rigid-body rotations of its catalytic and coenzyme-binding domains relative to the oligomerization domain. These structural perturbations are the direct result of the inability of lysine 487 to form important stabilizing hydrogen bonds with arginines 264 and 475. Thus, the elevated Km for coenzyme exhibited by this variant likely reflects the energetic penalty for reestablishing this site for productive coenzyme binding, while the structural alterations near the active site are consistent with the lowered Vmax.
Introduction
Mitochondrial aldehyde dehydrogenase, ALDH2, is best known for its role in ethanol metabolism, oxidizing acetaldehyde to acetate (1). More recently, an additional role for ALDH2 has been described as the initiator of nitroglycerin bioactivation (2). About 40% of the East Asian population carries a semi-dominant polymorphism of the ALDH2 gene, ALDH2*2 (3). The associated glutamate to lysine substitution at position 487 causes the enzyme to be nearly inactive in vivo. The resulting phenotype is characterized by an aversive response to ethanol consumption, which may include facial flushing, nausea, and tachycardia, with more severe reactions observed in those individuals who are homozygous for the polymorphism (4). This adverse effect has been linked to a lower frequency of alcoholism among those both hetero- and homozygous for the variant enzyme. Even so, ALDH2*2 has been linked to alcoholic liver disease as well as orapharyngolaryngeal and esophageal cancers in alcoholic patients (5,6).
The kinetic properties of human ALDH2*2 have been determined using protein expressed and purified from E. coli (7). The variant enzyme was found to be active, but exhibits a 200-fold increased Km for NAD+ and a diminished kcat. The enzyme’s Km for NAD+ exceeds the available concentration in the cell by 15 fold, which, combined with the 10-fold lower kcat value, would lead to an approximate 100-fold lower activity in vivo (7).
The three dimensional structure of ALDH2 has been solved and described previously (8,9). The enzyme is a tetramer of four identical subunits each consisting of 500 amino acid residues (Figure 1). This tetramer can be regarded as a dimer of dimers in that the interface between monomers that form a dimer is different and more extensive than the interface between the two dimers that form the tetramer. Each subunit is comprised of three main domains: the catalytic domain, the coenzyme or NAD+-binding domain, and the oligomerization domain (8) (Figure 2). E487 is located within the oligomerization domain, which is important for both dimer and tetramer formation. In wild-type ALDH2, E487 forms hydrogen bonds with R264 of the same subunit and with R475 of the adjacent dimer partner. Consequently, the disruption of these interactions by the presence of K487 was projected to perturb the structure of both its own subunit as well as its dimer partner (8). The interaction across the dimer interface has been thought to be responsible for the dominant effects of this mutation (8).
Figure 1.
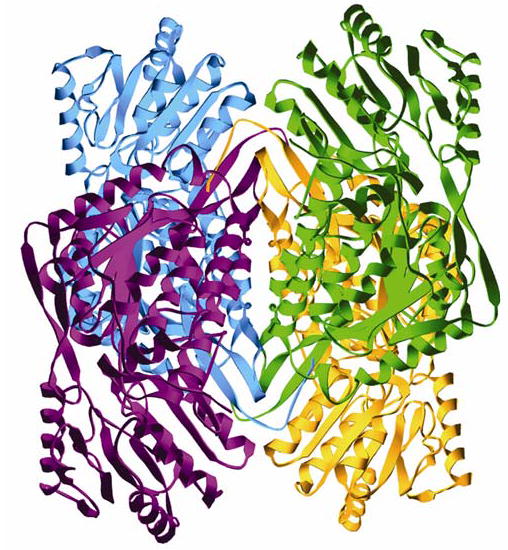
The ALDH2*2 homotetramer. Blue and violet subunits make one dimer pair (subunits A and B, respectively) and gold and green the other (subunits C and D, respectively). The remaining two tetramers in the asymmetric unit are comprised of subunits E–H and I–L.
Figure 2.
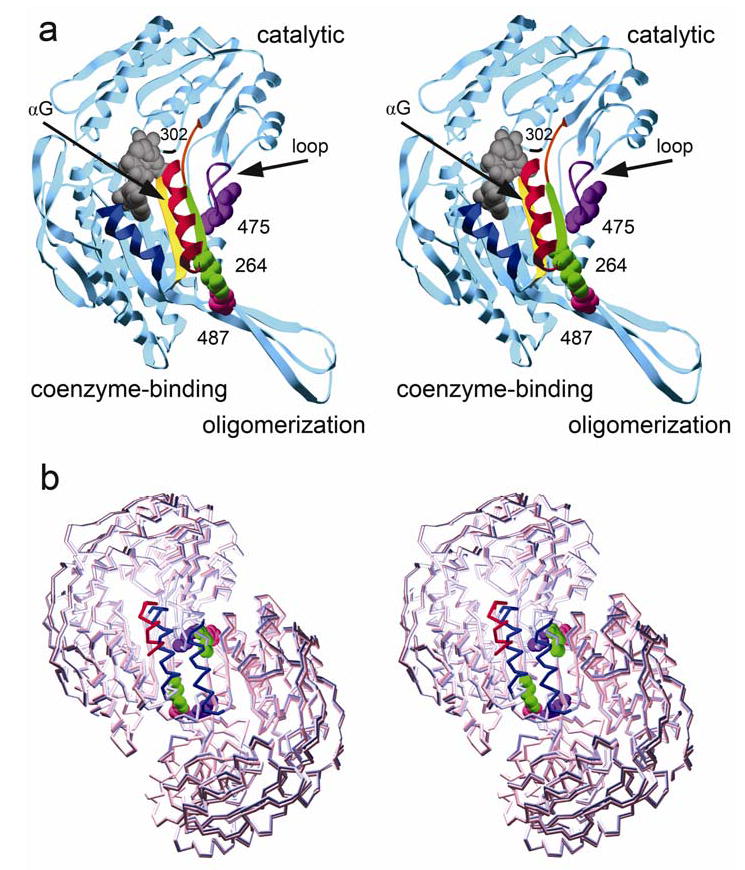
A single subunit of ALDH2: a) Wild-type subunit with its three major domains labeled. The secondary structure features are color coded: navy blue, αF; gold, β10; red, αG; green, β11; orange, loop at residues 269–273; violet, loop at residues 463–478. Residues 264, 487, and 475, shown in space-filled representation, are labeled and colored green, pink, and violet, respectively. The catalytic nucleophile, C302, is labeled and denoted by black ribbon. b) An alignment of ALDH2*2 subunits A and B to wild type. In this dimer representation, the ALDH2*2 αG helix is shown in red, wild type in blue. The helix of ALDH2*2 shifts 3 Å into the NAD+-binding cleft. Residues 264, 487, and 475 from the wild-type structure are colored as in Figure 2a.
In addition to the dominance of ALDH2*2, other examples of intersubunit communication have been observed in ALDH2. Half-of-the-sites reactivity for ALDH2 was first demonstrated in wild-type horse liver ALDH2 where there was a pre-steady-state burst of NADH formation equivalent to 2 moles NADH per ALDH2 tetramer (10). Wild-type rat ALDH2 demonstrated the same pre-steady state burst, and the rate-limiting step was determined to be the deacylation step (7). However, ALDH2*2 does not demonstrate the pre-steady state burst of NADH formation suggesting that for the variant enzyme, the rate-limiting step occurs before NADH formation, possibly involving thiohemiacetal formation (7).
Based on studies conducted with wild-type: ALDH2*2 heterotetramers, the current model of half-of-the sites reactivity is one where only one subunit per dimer pair is active in wild-type ALDH2 (11). Whether activity alternates between active sites or remains fixed in a particular active site is not yet understood. These kinetics studies support a model of dominance where a variant subunit would inactivate its wild-type dimer partner, but would have no effect on the subunits in the dimer pair across the tetramer interface.
Numerous aldehyde dehydrogenase crystal structures have shown that the nicotinamide portion of NAD(P) is flexible and occupies two main conformations (9,12–15). These conformations are likely to play a functional role in catalysis where the extended conformation is primarily occupied by NAD+ and is conducive to hydride transfer, and the other, more contracted conformation is primarily occupied by NADH and is conducive to acyl–enzyme hydrolysis (12). Magnesium is important for stabilizing these two conformations thereby elucidating its documented role in elevating Vmax.
To further study the variant ALDH2 enzyme, we have solved the apoenzyme crystal structure of ALDH2*2 to 2.1 Å resolution. This structure reveals that mutation of E487 to lysine leads to extreme disorder of the dimer interface, including the helix αG that comprises part of the NAD+-binding site. The β-strand containing the general base E268 has shifted away from the active site, and other main-chain and side-chain shifts lead to an altered coenzyme-binding site. Lysine 487 itself, however, is well-ordered, although the residue no longer contributes to favorable intersubunit interactions.
Experimental Procedures
Materials
DTT, DEAE-sepharose, p-hydroxyacetophenone, guanidine-HCL, and ethylene glycol were all purchased from Sigma Chemical Co. (St. Louis, MO). ACES, MgCl2, and propionaldehyde were purchased from Aldrich Chemical Co., Inc. (Milwaukee, WI). Grade I NAD+ was from Roche Diagnostics Corp. (Indianapolis, IN). The Bio-Gel P-6 DG and Bio-Rad Protein Assay reagent were obtained from Bio-Rad Laboratories (Hercules, CA). PEG 6000 was from Hampton Research (Laguna Niguel, CA), and the 24-well sitting drop trays were from Charles Supper Co., Inc (Natick, MA).
Protein Preparation and Crystallization
Human ALDH2 E487K cDNA was expressed in E. coli BL21 (DE3) cells using a pT-7-7 expression system previously described (7,16). The mutant enzyme was isolated by first employing DEAE-sepharose chromatography as previously published (16,17) with a slight modification of the protocol; the pH of the sodium-pyrophosphate buffer was changed to 6.6. Fractions containing ALDH2 were pooled and further purified on a p-hydroxyacetophenone affinity column as described previously (18). The eluate was dialyzed in 4 L 10 mM ACES buffer, 1 mM DTT pH 6.6 for 4 hours. Eluate was then transferred to fresh dialysis buffer for an additional 4 hours. Protein was concentrated in an Amicon stirred cell and then stored at –20°C in a 50% glycerol solution. Before each crystallization tray preparation, protein was removed from –20°C and buffer exchanged on a P6 gel column into 10 mM ACES buffer, 2 mM DTT pH 6.6. Protein was concentrated to 8 mg/ml in a YM30 Amicon Centricon spinning at 2000g. Protein concentration was monitored by the Bio-Rad Protein Assay using bovine serum albumin as the standard.
Enzyme activity was assayed at 25°C in 100 mM sodium pyrophosphate HCl, pH 9.5 containing 10 mM NAD+ and 200 μM propionaldehyde. NADH production was monitored at 340 nm with a Beckman DU 640 spectrophotometer.
For crystal growth, sitting drop trays were set up at 20°C in a glove box under nitrogen while maintaining a gaseous oxygen concentration less than 2%. Crystals were grown using the sitting drop configuration for vapor diffusion in solutions containing 100 mM ACES, pH 6.2–6.8, 10 mM MgCl2, 100 mM Guanidine HCl, 15–17% w/v PEG 6000, and 4–6 mM DTT. The equilibrium drop volume was 3 μl. Trays were stored at 20°C and crystals were harvested at 3–4 weeks.
Flash freezing of the crystals was accomplished using a rapid two-step procedure to introduce 22% ethylene glycol cryoprotectant. The crystals were immediately flash frozen at 110K in a N2 cryostream.
Data Collection, Processing, and Model Refinement
Data were collected at the Advanced Photon Source (APS) at Argonne National Laboratory (Argonne, IL) on beamline SBC-CAT 19ID. The beamline was equipped with the APS-1 3 x 3 array CCD detector. The X-ray beam wavelength for data collection was 12.4 KeV. Data were indexed, integrated, and scaled with the HKL2000 Program Suite (19).
Phases were solved using the molecular replacement method with the human ALDH2 coordinates as a starting model. Molecular replacement was executed using the program AMoRe as implemented in the CCP4 program package (20,21). The resulting models were refined using the Crystallography and NMR System (CNS) suite (22). For the refinement of the initial apoenzyme model, rigid-body refinement was followed by simulated annealing and minimization that were performed with NCS restraints applied at a weight of 100 Kcal/mol·Å2 for the main-chain atoms and 10 Kcal/mol·Å2 for the side-chain atoms. This was followed by individual restrained isotropic temperature factor refinement. Model rebuilding was performed using the program O (23). To reduce model bias, composite annealed omit maps were calculated. Only energy minimization and temperature factor refinements were utilized in subsequent rounds with residues 246–264 and 466–476 excluded from NCS restraints during minimization. In the final round of refinement, the NCS restraint weights were reduced to 50 Kcal/mol·Å2 for main-chain atoms with no NCS restraints imposed on the side-chain atoms. In this final round, residues 244–271, 424–425, and 463–478 were excluded from the main-chain NCS restraints. Rfree was evaluated using a randomly chosen 5% of the crystallographic data.
Structure Analysis
Hinge axis determination for positional domain differences between ALDH2 and ALDH2*2 were analyzed using the program DynDom as implemented in the CCP4 package (20,24). Tetramer, subunit, and domain alignments were performed using the program LSQKAB as implemented in the CCP4 package (20,25). Root-mean-square deviations (RMSDs) were calculated for the Cα atoms of each alignment. Residues 244–271, 424–425, and 463–478 were omitted for all pairings. To determine the extent of domain reorientation relative to wild type, the cofactor-binding domain was established as the point of reference. This domain was aligned to apoenzyme wild type. The rotated ALDH2*2 subunit was then realigned to wild type using either the catalytic or oligomerization domain. The extent of domain rotation is thus expressed as the amount of rotation necessary to align the domain of interest relative to the common positions of the coenzyme-binding domains.
The extent of secondary structural shifts was determined by aligning the cofactor-binding and catalytic domains together to wild-type enzyme. Therefore, these values are expressed as the amount of shift within the context of these domains and are determined independently of domain shifts.
Residues were considered ordered and were included in the final coordinates if the electron density maps contoured at one standard deviation of the map indicated their presence.
Results
ALDH2*2 crystallized in the triclinic space group with three tetramers in the asymmetric unit. The crystal diffracted to 2.1 Å resolution, and the structure refined to Rfree = 23.8% and Rwork = 20.4%. Like wild-type ALDH2, ALDH2*2 is a tetramer of four identical subunits. Ordered solvent molecules included 2649 water molecules, 21 ethylene glycol molecules, 8 guanidines, and 12 sodium ions. All data collection and refinement statistics are summarized in Table 1.
Table 1.
Data collection and refinement statistics.
| apo ALDH2*2 | |
|---|---|
| resolution (Å) | 50 – 2.10 |
| total/unique observations | 668,406/345,164 |
| completeness (%)a | 97.7 (96.2) |
| < I / σ(I) > | 14.6 (2.3) |
| Rmerge (%) | 5.1 (35.3) |
| Rwork (%) | 20.4 |
| Rfree (%)b | 23.8 |
| Space group | P1 |
| cell dimensions | |
| a, b, c (Å) | 96, 105, 163 |
| α, β, γ (°) | 79, 82, 88 |
| asymmetric unit | 3 tetramers |
| rmsd bonds (Å) | 0.009 |
| rmsd angles (deg.) | 1.36 |
| NCS rmsdc (Å) | 0.136 |
| overall B-factor (Å2) | 40.9 |
| number of solvent molecules | 2690 |
| Protein Data Bank ID code | 1ZUM |
Values for highest resolution shell are in parentheses.
Rfree was determined from a randomly selected 5% of the reflections.
For Cα atoms. Excludes residues 244–271, 424–425, and 463–478.
Interactions surrounding K487
The majority of residue K487 is well ordered and clearly present in the electron density maps contoured at one standard deviation (Figure 3a). The loss of hydrogen bonds among residues 487, 475 and 264 is accompanied by changes in side-chain conformation of these residues (Figure 4). No favorable interactions were observed between K487 and R475 in the dimer partner. K487 points away from the dimer interface and forms no new hydrogen bonds. K487 does make van der Waals contacts with the side chain of Y468 (when observed) in the neighboring subunit as well as with the peptide carbonyl oxygen of P158 and the main chain and side chain of V159 within its own subunit. R475 can only be modeled in seven of the twelve subunits. In each of the seven instances, residue 475 occupies the same conformation as wild type with the Nɛ atom within hydrogen bonding distance of the main-chain carbonyl of 488 in the neighboring subunit. The five remaining subunits do not display interpretable electron density for R475. Although not identical, all observed conformations for R264 are rotated away from K487.
Figure 3.
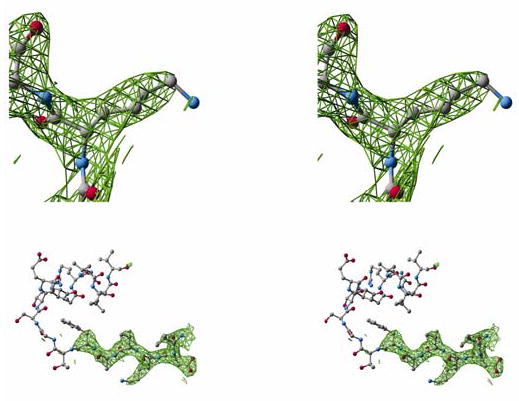
2FO−FC electron density maps contoured at one standard deviation of the map: a) For residue K487, nearly all atoms are well ordered with Nζ and Cɛ being the exceptions in some subunits. b) The β10 strand and αG helix. The electron density maps taper at the C-terminal end of the β10 strand. For eleven of the twelve subunits observed, there is no interpretable density for most of the αG helix.
Figure 4.
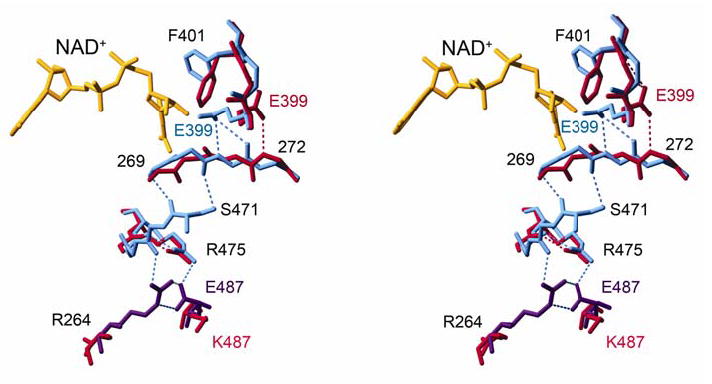
A stereo diagram of side-chain and main-chain shifts from the dimer interface to the NAD+-binding cleft. NAD+-bound wild type (violet, subunit A; blue, subunit B) and apoenzyme ALDH2*2 (red) are aligned.
Disorder at the Interface
The most striking feature of the ALDH2*2 crystal structure is the disordered region surrounding K487 including residues 244–272 and 463–478 (Table 3). Adjacent to 469 in the catalytic domain, residues 424 and 425 are also disordered. Residues 469–473 are disordered in all subunits of the asymmetric unit. In eleven of the twelve subunits within the asymmetric unit, residues 248–262 are not visible in electron density maps contoured at one standard deviation (Figure 3b). These residues comprise the αG helix located at the interface between adjacent subunits within a dimer pair. This helix also comprises part of the adenine binding pocket for NAD+. In subunit A, αG is more ordered with only residues 257–263 missing. Although this helix is observed, it has shifted 3 Å, relative to the wild-type structure (Figure 2b). In the remaining subunits, the general path of αG appears to shift in the same manner as in subunit A; however the residues are insufficiently defined in the electron density maps for confident placement.
Table 3.
Disordered residues.
| Subunit | αG helix | loop | loop |
|---|---|---|---|
| A | 257–263 | 424–425 | 463–478 |
| B | 244–264 | 424–425 | 468–473 |
| C | 246–262 | 424–425 | 468–474 |
| D | 248–263 | 424–425 | 469–476 |
| E | 246–262 | 424–425 | 469–474 |
| F | 246–262 | 424–425 | 468–474 |
| G | 246–262 | 424–425 | 469–474 |
| H | 246–262 | 424–425 | 469–474 |
| I | 246–263 | 424–425 | 469–474 |
| J | 246–263 | 424–425 | 469–475 |
| K | 246–271 | 424–425 | 466–475 |
| L | 245–263 | 424–425 | 466–476 |
Unique to subunit A are the crystal packing contacts between this subunit and the crystallographically related subunit K. The αJ helix of subunit K makes contact with and stabilizes αF of subunit A. This results in a 15% lower average temperature factor for αF as compared to the rest of the subunit. αF is positioned on the opposite side of the NAD+-binding cleft from αG, and its increased order supports αG in this subunit.
Adjacent to αG in both primary and tertiary structure is the β11 strand, which includes R264 and E268. In all subunits of ALDH2*2, the β11 strand’s position is shifted 2 Å in the same direction as αG. The Cα atom of the putative general base, E268, has shifted ~1.5 Å away from the active site, and its side-chain position is ill-defined and not consistent among the subunits. In response to the shifts of both αG and β11, β10 has shifted 0.8 Å in the same direction as αG and β11.
Loop Disorder Linked to Active Site
In wild-type ALDH2, the loop comprised of residues 463–478 interacts with residues 269–272 through two hydrogen bonds—one between the peptide carbonyl oxygen of 472 and the main-chain nitrogen of 269, and the other between the side-chain hydroxyl of S471 and the peptide carbonyl oxygen of residue 270 (Figure 4). Residues 269–272 associate with the active site through a hydrogen bond between the peptide nitrogen of 271 and the side chain of E399. In cofactor-bound wild-type ALDH2, E399 stabilizes the nicotinamide portion of NAD+ by forming hydrogen bonds between its carboxylate and the 2' and 3' hydroxyl oxygens of the nicotinamide ribose (hydride transfer conformation). Yet, in concert with the flexibility of the loop containing residues 463–478 in ALDH2*2, the main chain of residues 269–272 rotates such that the amino nitrogen of 271 no longer stabilizes the side chain of E399 in the active site (Figure 4). E399 assumes a new conformation that is rotated away from the active site and is instead stabilized by the peptide nitrogen of 272. Coincident with this change, the side chain of F401 rotates into the void created by the movement of E399. F401 supports the binding of the NMN portion of the coenzyme in all ALDH2 holoenzyme structures.
The Active Site
The catalytic nucleophile, cysteine 302 is well ordered and retains a wild-type-like conformation. Electron density maps surrounding C302 reveal binding of an unknown molecule. Kinetics experiments have shown that the crystallization precipitant, polyethylene glycol 6000MW, contains aldehydes at an approximate concentration of 0.5 mM (12). Therefore, this unaccounted for density is likely due to unidentified aldehydes arising from the PEG. The resolution of the data is insufficient to fully define the bound molecule, therefore solvent molecules were modeled into the positive difference peaks to partially account for the unknown molecule.
Asparigine 169, important for stabilizing oxyanion formation in the transition state (8), is also well ordered and is in the same conformation as wild-type enzyme. As mentioned above, the side chain of E268 is disordered and not consistent among subunits.
Domain Shifts
Between the cofactor-binding domain and the oligomerization domain lies a hinge axis about which there is a 2.5° rotation in ALDH2*2 relative to wild type, as determined using the program DynDom (20,24). Analysis of relative domain positions within the dimer pair revealed residues that form the hinge, which include the three beta strands of the oligomerization domain, as well as the loop comprised of residues 463–478. Individual domain alignments using the tetramer, dimer, and monomer were also performed (Table 2). These, too, demonstrate that the cofactor-binding and catalytic domains have changed position within the tetramer in a manner which can best be described as a 2.5° rigid-body rotation away from the dimer interfaces (Figure 5).
Table 2.
Root-mean-square deviations of Cα atoms for alignments of ALDH2*2 to apoenzyme wild-type ALDH2 (Å2).
| domain | all domains | oligomerization | coenzyme/catalytic | coenzyme | catalytic |
|---|---|---|---|---|---|
| wt tet1 to 2*2 tet1 | 0.75 | 0.41 | 0.77 | 0.78 | 0.71 |
| wt ab to 2*2 ab | 0.74 | 0.39 | 0.76 | 0.67 | 0.68 |
| wt a to 2*2 a | 0.30 | 0.34 | 0.22 | 0.19 | 0.22 |
| 2*2 a to 2*2 b | 0.17 | 0.15 | 0.14 | 0.13 | 0.10 |
Figure 5.
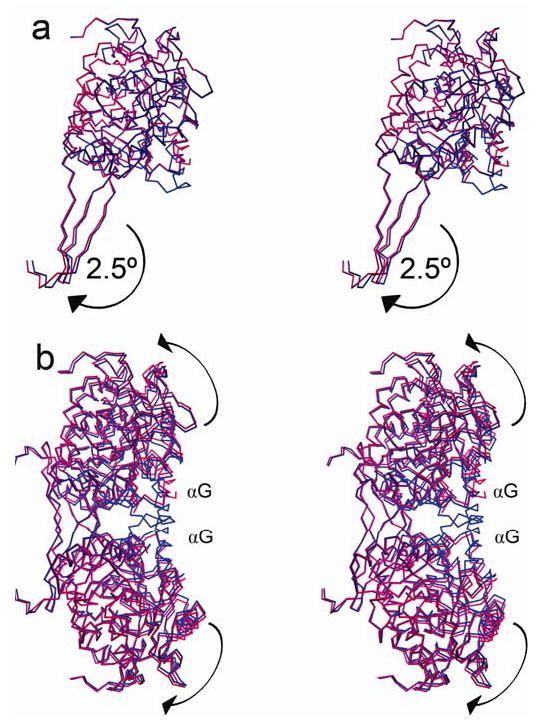
The ALDH2*2 enzyme (red) aligned to wild-type (blue). The view shown is rotated about 90° with respect to Figure 2: a) Monomer view—alignment of the coenzyme-binding and catalytic domains. The 2.5º rotation between the coenzyme-binding and oligomerization domains is illustrated by the shift in the oligomerization domain. b) Dimer view—alignment of the oligomerization domains. The 2.5º rotation causes the coenzyme-binding and catalytic domains to fall away from the interface at the αG helices.
Discussion
The crystal structure studies of ALDH2*2 have revealed that mutation of glutamate to lysine at residue 487 causes a large structural transition within the variant enzyme. While K487 is not itself disordered in the ALDH2*2 crystal structure, its presence at the dimer interface perturbs local secondary structure elements, particularly αG, β10, β11, and the loop consisting of residues 463–478. These structures are either closely or directly involved with NAD+ binding. The disruptions of αG and the aforementioned loop also lead to an unstable dimer interface where the catalytic and coenzyme-binding domains of individual subunits rotate away from the interface. Moreover, the general base, E268 is not stably positioned for catalysis, and the active site is further disrupted at residues 399 and 401 through a cascade of changes that appear to originate in the dimer partner. These shifted main-chain and side-chain positions undoubtedly contribute to the decreased activity observed for this ALDH2 variant and also shed light on the variant’s mechanism of allelic dominance.
Disorder of residues 463–478 has been observed in other aldehyde dehydrogenase crystal structures. Lamb and Newcomer have reported these residues missing in the electron density maps of cofactor-bound retinal dehydrogenase type II (RalDH2), which shares about 67% sequence identity with ALDH2 (26,27). What is unique to ALDH2*2 is the complete loss of both the loop and the αG helix in the electron density maps.
The positional disorder of the αG helix is likely to be the major contributor to the high Km for NAD+ of ALDH2*2. R264, located at the C-terminal end of the αG helix, forms a hydrogen bond with E487 in wild-type ALDH2, thus helping to anchor the helix. The substitution of lysine for glutamate at 487 leads to the loss of this anchor and the observed flexibility of αG.
No significant differences in main-chain or side-chain positions are found between cofactor-bound and apoenzyme wild-type ALDH2. Therefore, we have compared ALDH2*2 to the coenzyme-bound form of ALDH2 to highlight the potential impact of the structural changes in ALDH2*2 on coenzyme binding. In wild-type enzyme, the adenine ring of NAD+ is nestled between αF and αG and makes Van der Waals contacts with I249 and L252 within αG. The peptide nitrogen and side-chain hydroxyl of S246 at the N-terminal end of αG form hydrogen bonds with the adenosine phosphate. In ALDH2*2, S246 is only observed in subunits A and D, and I249 is only observed in subunit A. In those subunits S246 has shifted 2 Å into the adenine binding site, and I249 has shifted about 2.5 Å toward the adenosine phosphate binding site. These positions would sterically interfere with coenzyme binding. In wild-type ALDH2 the carbonyl oxygen of L269 stabilizes the amide group of the nicotinamide ring, but in ALDH2*2 it has flipped away from the nicotinamide site and toward the dimer interface.
In order for the adenine portion of NAD+ to bind, S246, V249 and L252 must be stably and appropriately positioned, and the αG helix must reorder and shift away from helix αF. For proper positioning of the nicotinamide, the main chain at L269 would need to rotate to position the carbonyl toward the amide, F401 would need to rotate out of the nicotinamide ribose binding site, and E399 must reposition itself as a hydrogen bond acceptor for the ribose oxygen atoms. A substantial loss in binding energy, such as that actually observed, would be required for such secondary structure and side-chain repositioning.
Deacylation is the rate-limiting step for wild-type ALDH2 for most substrates. In contrast, the rate-limiting step for ALDH2*2 has been proposed to occur prior to NADH formation and is perhaps thiohemiacetal formation. However, these studies on ALDH2*2 have been in the presence of saturating concentrations of NAD+. Because the cellular NAD+ concentration in human is only about 500μM (28), it has been further suggested that in vivo, the binding of NAD+ to enzyme may, in fact, be the rate-limiting step of ALDH2*2 oxidation of acetaldehyde (7). This assertion is supported by the presented structural evidence, since at least part of this binding step will also include rate constants associated with the reordering of the binding site for the coenzyme. This effect is most clearly seen where the association rate for NAD+ is decreased 3400 fold, while the dissociation rate is only accelerated 90 fold (7). These data are consistent with most folding transition data where cooperative folding events are seen—while slow to form, once formed, they are marginally stabilized by the cooperative interactions.
NADH
While binding of NAD+ is severely hindered in the ALDH2*2 variant, NADH binding is less affected (7). The Kia for NAD+ in ALDH2*2 is increased 40 fold when compared to wild-type. However, the Kiq of NADH remains relatively unchanged. In ALDH2*2, E399 has changed conformation and is no longer in position to bind both nicotinamide ribose hydroxyls and therefore no longer supports binding for the hydride transfer position of coenzyme, associated with NAD+. The hydrolysis conformation of coenzyme, associated with NADH, relies less on the interaction with E399 and only in this conformation is the nicotinamide ribose further supported by Gln349. Additionally, while a total of three residues support the amide portion of the nicotinamide in the hydrolysis conformation, only one residue supports the same atoms in the hydride transfer conformation. These observations are in accord with the observed Kiq for NADH by suggesting that the nicotinamide of NADH in the hydrolysis conformation would be more stable than NAD+ in the hydride transfer conformation. These data are also consistent with the larger increase in the Km for NAD+, versus the Kia value, since the Kia value is reflective of only a binding event, while the Km value relates both to binding and proper positioning for hydride transfer.
K487 Dominance and kcat
While the disordered αG helix at the interface contributes greatly to the overall decreased activity of ALDH2*2, it does not explain why in saturating concentrations of NAD+ there is still a low kcat. In wild-type ALDH2, both R264 and E487 form hydrogen bonds with the loop at residues 463–478 across the dimer interface. In ALDH2*2 the side chain of R264 is turned away from the dimer interface and its guanidyl group no longer forms hydrogen bonds with the carbonyl group of 469. In fact, R264 no longer engages with the dimer partner at all. As expected, no favorable interactions are observed between K487 and R475 in the dimer partner. The lack of these intersubunit ties likely gives rise to the disorder of the 463–478 loop.
Although the loop at 463–478 does not interact directly with coenzyme, its flexibility initiates a cascade of main-chain and side-chain shifts that disturb the nicotinamide binding pocket in the active site, specifically at residues E399 and F401. This would explain why even with NAD+ bound, ALDH2*2 still exhibits a low kcat; NAD+ may bind, but without these interactions, the nicotinamide portion may not be appropriately constrained for hydride transfer. Mutations of E399 in wild type have been shown to alter the rate-limiting step of ALDH2 and have deleterious effects on activity (29). The mechanism by which a K487 subunit inactivates its dimer partner includes the aforementioned main-chain and side-chain shifts, including E399, the shift of β11, which contains the general base E268, and the disorder of the αG helix. Alternatively, NAD+ binding may reorder these side chains, and the disruptions at the floor of the active site, including E268, may have a more significant effect on the observed kcat.
Intersubunit Communication
In wild-type ALDH2, the half-of-the-sites reactivity has been described as a extreme example of negative cooperativity (30). The mechanism of ALDH2*2 dominance has been speculated to be linked to the mechanism of half-of-the-sites reactivity (8,11). Even though the structure presented here does not immediately describe a mode of half-site reactivity, it does provide clues to which residues might be involved in inter–active site communication.
In ALDH2*2, R264 ceases to form hydrogen bonds with the neighboring subunit, contributing to disorder within that dimer partner. In the seven subunits where R475 is observed, it occupies the same conformation as in wild type. This lack of conformational change may suggest that of the two arginines, R264 may have a more dynamic role in intersubunit communication, particularly through indirect alteration of the E399 conformation and subsequent effects on nicotinamide binding.
The changes described by the ALDH2*2 structure affect the interface between monomers in a dimer pair. No major changes at the tetramer interface were observed. This evidence supports the current model for dominance where each dimer pair functions as a unit, and only one active site per dimer pair is functioning (11).
Originally, it was thought that half-of-the-sites reactivity reflected half-of-the-sites NAD+ occupancy, and a number of NADH-binding studies were consistent with stoichiometries of less than four per tetramer (30,31). However, the available holoenzyme ALDH2 structures show full occupancy of coenzyme in all active sites of the tetramer (9,12,15). Although no structural differences have been observed between NAD+-binding domains within a dimer pair, the two distinct conformations of the nicotinamide half of the coenzyme may influence both the activation/inactivation of a particular active site and the observed stoichiometry of NADH binding using fluorescence measurements. Based solely on proximity to bulk solvent, the two observed conformations of the coenzyme when bound to ALDH would be expected to exhibit different fluorescence properties relative to coenzyme in solution and could confound the analysis of stoichiometry. The conformational differences in how the coenzyme is bound during catalysis could be relayed through the αG helices that form the adenosine binding sites in each dimer partner. The large-scale alterations we observe in the ALDH2*2 crystal structure are consistent with a structural connection between changes in the αG helix that forms the adenosine binding cleft in one subunit to changes in the nicotinamide binding site in the dimer partner. However, we have not observed such an activation/inactivation event in wild-type ALDH2 structures, though the molecular averaging inherent in a crystallographic experiment may mask such a difference.
MCAD
Single amino acid substitutions in critical subunit interfaces, as discussed here for ALDH2, are also found to exist in other enzymes. For example, a deficiency in medium chain acyl-CoA dehydrogenase (MCAD), a mitochondrial enzyme involved in the β-oxidation of straight chain fatty acids, is life-threatening (32), especially during periods of fasting in conjunction with viral infection. Although more than twenty variants of the MCAD gene have been reported, one MCAD enzyme mutation, K304E, is present in more than 90% of alleles in patients with MCAD deficiency. This autosomal recessive mutation is responsible for decreased thermal stability and enzyme activity, and may destabilize the active tetramer, once formed. Like E487 in ALDH2, K304 lies at an interface between subunits (33). Although K304 does not make any direct contacts with the neighboring subunit, the residue is located within a helix that makes many contacts across the interface. Perhaps the conversion of lysine to glutamate at 304 within this enzyme perturbs folding at the interface between monomers in a manner similar to glutamate to lysine at 487 in ALDH2.
Conclusion
Mutation of residue 487 from glutamate to lysine in ALDH2 leads to an enzyme that is rendered essentially inactive in vivo. Upon mutation to lysine, residue 487 no longer forms hydrogen bonds with arginine 264 in its own subunit and arginine 475 in the dimer partner. The loss of these interactions leads to disorder of the NAD+-binding site, particularly in the αG helix at the dimer interface. This positional disorder is a major contributor to the high Km observed for NAD+ binding in ALDH2*2, which leads to its inactivity in vivo. A mechanism of dominance is revealed in the altered main-chain and side-chain positions from R264 to across the dimer interface leading eventually to the nicotinamide binding site where E399 is not in position to stabilize the nicotinamide ribose. This alteration may contribute to the low kcat and would predominantly affect the hydride transfer conformation of cofactor since this position more heavily relies on E399 for nicotinamide stability. However, to fully appreciate the dynamics between K487 and E487 subunits within a ALDH2*2/ALDH2 dimer pair, the crystal structures of heterotetramers would prove most valuable.
Acknowledgments
This work has been supported by NIH R01 AA 11982. The authors wish to thank Susan Carlson for her assistance with the purification and crystallization of ALDH2*2, and Norma E.C. Duke for her assistance at beamline 19ID, Advanced Photon Source, Argonne National Laboratories. Use of the Argonne National Laboratory Structural Biology Center beamlines at the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Energy Research, under Contract No. W-31-109-ENG-38.
Footnotes
The atomic coordinates and structure factors (code 1ZUM) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
References
- 1.Hurley, T. D., Edenberg, H. J., and Li, T. K. (2002) in Pharmacogenomics: The Search for Individualized Therapies (Licinio, J., and Wong, M., eds), pp. 417–441, Culinary and Hospitality Industry Publications Services
- 2.Chen Z, Zhang J, Stamler JS. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:8306–8311. doi: 10.1073/pnas.122225199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seitz HK, Matsuzaki S, Yokoyama A, Homann N, Vakevainen S, Wang XD. Alcoholism: Clinical & Experimental Research. 2001;25:137S–143S. doi: 10.1097/00000374-200105051-00024. [DOI] [PubMed] [Google Scholar]
- 4.Peng GS, Wang MF, Chen CY, Luu SU, Chou HC, Li TK, Yin SJ. Pharmacogenetics. 1999;9:463–476. [PubMed] [Google Scholar]
- 5.Enomoto N, Takase S, Takada N, Takada A. Hepatology. 1991;13:1071–1075. [PubMed] [Google Scholar]
- 6.Yokoyama A, Muramatsu T, Omori T, Matsushita S, Yoshimizu H, Higuchi S, Yokoyama T, Maruyama K, Ishii H. Alcoholism: Clinical & Experimental Research. 1999;23:1705–1710. [PubMed] [Google Scholar]
- 7.Farres J, Wang X, Takahashi K, Cunningham SJ, Wang TT, Weiner H. Journal of Biological Chemistry. 1994;269:13854–13860. [PubMed] [Google Scholar]
- 8.Steinmetz CG, Xie P, Weiner H, Hurley TD. Structure. 1997;5:701–711. doi: 10.1016/s0969-2126(97)00224-4. [DOI] [PubMed] [Google Scholar]
- 9.Ni L, Zhou J, Hurley TD, Weiner H. Protein Science. 1999;8:2784–2790. doi: 10.1110/ps.8.12.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weiner, H., Hu, J. H., and Sanny, C. G. (1976), 3853–3855, 1976 Jul 3810.
- 11.Zhou J, Weiner H. Biochemistry. 2000;39:12019–12024. doi: 10.1021/bi001221k. [DOI] [PubMed] [Google Scholar]
- 12.Perez-Miller S, Hurley TD. Biochemistry. 2003;42:7100–7109. doi: 10.1021/bi034182w. [DOI] [PubMed] [Google Scholar]
- 13.Hammen PK, Allali-Hassani A, Hallenga K, Hurley TD, Weiner H. Biochemistry. 2002;41:7156–7168. doi: 10.1021/bi012197t. [DOI] [PubMed] [Google Scholar]
- 14.Moore S e a. Structure. 1998;6:1541–1551. doi: 10.1016/s0969-2126(98)00152-x. [DOI] [PubMed] [Google Scholar]
- 15.Hurley T, Perez-Miller S, Breen H. Enzymology and Molecular Biology of Carbonyl Metabolism. 2001;130–132:3–14. [Google Scholar]
- 16.Zheng CF, Wang TT, Weiner H. Alcoholism: Clinical & Experimental Research. 1993;17:828–831. doi: 10.1111/j.1530-0277.1993.tb00849.x. [DOI] [PubMed] [Google Scholar]
- 17.Jeng JJ, Weiner H. Archives of Biochemistry & Biophysics. 1991;289:214–222. doi: 10.1016/0003-9861(91)90464-t. [DOI] [PubMed] [Google Scholar]
- 18.Ghenbot G, Weiner H. Protein Expression & Purification. 1992;3:470–478. doi: 10.1016/1046-5928(92)90064-4. [DOI] [PubMed] [Google Scholar]
- 19.Otwinowski, Z., and Minor, W. (1997) in Methods in Enzymology, Volume 276: Macromolecular Crystallography, part A (C.W. Carter, J., and Sweet, R. M., eds), pp. 307–326, Academic Press
- 20.COLLABORATIVE COMPUTATIONAL PROJECT N. Acta Crystallographica Section D-Biological Crystallography. 1994;50:760–763. doi: 10.1107/S1399004714018070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Navaza J. Acta Cryst A. 1994;A50:157–163. [Google Scholar]
- 22.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallographica Section D-Biological Crystallography. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 23.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Acta Cryst A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 24.Hayward S, Berendsen HJC. Proteins. 1998;30:144–154. [PubMed] [Google Scholar]
- 25.Kabsch W. Acta Crystallographica Section A. 1976;32:922–923. [Google Scholar]
- 26.Lamb AL, Newcomer ME. Biochemistry. 1999;38:6003–6011. doi: 10.1021/bi9900471. [DOI] [PubMed] [Google Scholar]
- 27.Bordelon T, Montegudo SK, Pakhomova S, Oldham ML, Newcomer ME. J Biol Chem. 2004;279:43085–43091. doi: 10.1074/jbc.M406139200. [DOI] [PubMed] [Google Scholar]
- 28.Greenbaum AL, Gumaa KA, McLean P. Archives of Biochemistry and Biophysics. 1971;143:617–663. doi: 10.1016/0003-9861(71)90247-5. [DOI] [PubMed] [Google Scholar]
- 29.Ni L, Sheikh S, Weiner H. Journal of Biological Chemistry. 1997;272:18823–18826. doi: 10.1074/jbc.272.30.18823. [DOI] [PubMed] [Google Scholar]
- 30.Ambroziak W, Kosley LL, Pietruszko R. Biochemistry. 1989;28:5367–5373. doi: 10.1021/bi00439a008. [DOI] [PubMed] [Google Scholar]
- 31.Takahashi K, Weiner H, Hu JH. Archives of Biochemistry & Biophysics. 1980;205:571–578. doi: 10.1016/0003-9861(80)90140-x. [DOI] [PubMed] [Google Scholar]
- 32.Bross P, Jespersen C, Jensen TG, Andresen BS, Kristensen MJ, Winter V, Nandy A, Krautle F, Ghisla S, Bolundi L, et al. Journal of Biological Chemistry. 1994;270:10284–10290. doi: 10.1074/jbc.270.17.10284. 11995 Apr 10228. [DOI] [PubMed] [Google Scholar]
- 33.Kim JJ, Wu J. Proc Natl Acad Sci U S A. 1988;85:6677–6681. doi: 10.1073/pnas.85.18.6677. 1988 Sep. [DOI] [PMC free article] [PubMed] [Google Scholar]