

Abstract
It is important to ensure DNA availability when bacterial cells develop competence. Previous studies in Streptococcus pneumoniae demonstrated that the competence-stimulating peptide (CSP) induced autolysin production and cell lysis of its own non-competent cells, suggesting a possible active mechanism to secure a homologous DNA pool for uptake and recombination. In this study, we found that in Streptococcus mutans CSP induced coordinated expression of competence and mutacin production genes. This mutacin (mutacin IV) is a non-lantibiotic bacteriocin which kills closely related Streptococcal species such as S. gordonii. In mixed cultures of S. mutans and S. gordonii harbouring a shuttle plasmid, plasmid DNA transfer from S. gordonii to S. mutans was observed in a CSP and mutacin IV-dependent manner. Further analysis demonstrated an increased DNA release from S. gordonii upon addition of the partially purified mutacin IV extract. On the basis of these findings, we propose that Streptococcus mutans, which resides in a multispecies oral bio-film, may utilize the competence-induced bacteriocin production to acquire transforming DNA from other species living in the same ecological niche. This hypothesis is also consistent with a well-known phenomenon that a large genomic diversity exists among different S. mutans strains. This diversity may have resulted from extensive horizontal gene transfer.
Introduction
Competence is a physiological state in which bacteria develop a capacity to take up exogenous DNA (Dubnau, 1991a). Competence development is an elaborate process involving multiple protein components and sophisticated regulatory networks (Dubnau, 1991b; Morrison, 1997; Morrison and Lee, 2000; Hamoen et al., 2003; Dagkessamanskaia et al., 2004). It is important to ensure that a DNA pool is available when the cells become competent. In a recent study by Steinmoen et al. it was shown that development of competence in Streptococcus pneumoniae triggers cell lysis and DNA release from a subfraction of the cell population (Steinmoen et al., 2002). Further studies demonstrated that competence-induced cells of S. pneumoniae lyse competence-deficient cells of the same species during cocultivation (Steinmoen et al., 2003). This competence-induced cell lysis has been suggested as a means to provide the competent cells with a donor DNA pool from its own species.
Streptococcus mutans is an oral member of the Streptococcus genus. It lives in a multispecies biofilm commonly known as the dental plaque with over 500 other bacterial species (Moore and Moore, 1994; Kroes et al., 1999; Paster et al., 2001). In addition to having a natural competence system similar to that of S. pneumoniae (Li et al., 2001), S. mutans also produces a plethora of bacteriocins, which are small peptide antibiotics that kill closely related species (Hamada and Ooshima, 1975; Caufield et al., 1985; Baca et al., 1990; Bondi et al., 1991; Baba and Schneewind, 1998). Our laboratory has been interested in understanding the mechanism of bacteriocin gene regulation in S. mutans and its function in interspecies interaction. In this study, we found that the production of one of the bacteriocins, mutacin IV (Qi et al., 2001), was linked to the competence development system in S. mutans. Further analysis of this interesting connection has led to the hypothesis that S. mutans may be able to acquire DNA from other streptococcal species that live in close proximity.
Results
High level mutacin IV gene expression was induced in the pellet of early log phase cells
In previous studies we routinely observed higher mutacin IV production during the transition between late log and early stationary phase (Qi et al., 2001). To further investigate the mechanism of mutacin IV gene regulation, we constructed mutacin IV promoter (nlmA) fusions to the luciferase (luc) and green fluorescent protein (gfp) reporters in strain UA140 (Kreth et al., 2004). To quantify nlmA gene expression as a function of time, an overnight culture of UA140::Ø(nlmAp-luc) was diluted 1:20 into fresh Brain Heart Infusion (BHI) broth and incubated anaerobically as a batch culture. Starting at the early log phase (OD600 ~ 0.1), culture samples were taken at designated time intervals and luciferase activity was measured. As shown in Fig. 1A, after normalization to cell density, the expression level of nlmAp-luc was low throughout log phase (time 0–270 min). However, the expression started to increase at the late log phase (at 330 min) and reached its peak at the transition from the log to the stationary phase (at 390 min). The peak level activity remained for ~60 min and then started to decline. At the peak level, nlmA-luc activity was ~fivefold higher than that at the log phase. A similar pattern was observed when cells were incubated in TH broth (data not shown). These results suggest that mutacin IV gene expression is temporally regulated at the transcriptional level.
Fig. 1.
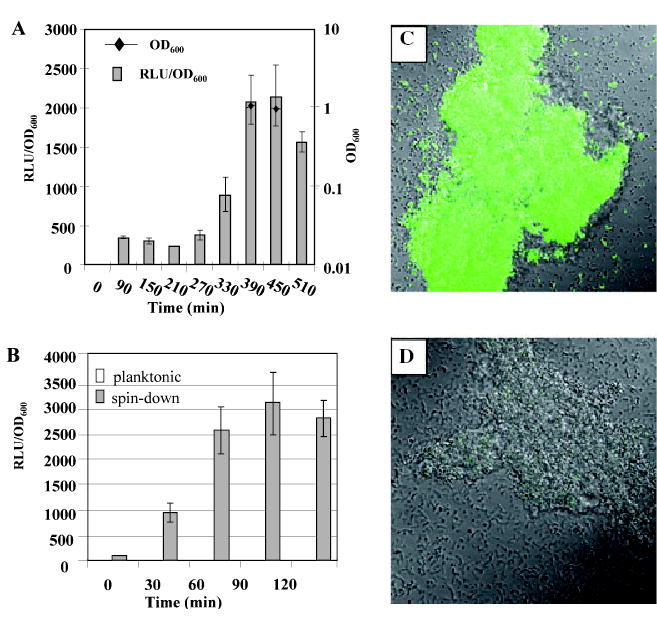
A. Time-course of nlmAp-luc gene expression in strain UA140::Φ(nlmAp-luc). The experiments were repeated two times with variations of less than 20% in between experiments. Presented here is one of the experiments. Values are averages of triplicate samples.
B. nlmAp-luc gene expression in planktonic and pelleted cultures over time. Experiments were repeated three times. Presented here are the average values of the three independent experiments. RLU, relative light unit.
C. Confocal micrograph of strain UA140::Φ(nlmAp–gfp) cells in pelleted culture.
D. Confocal micrograph of strain UA140::Φ(nlmAp–gfp) cells in planktonic culture.
To distinguish whether the observed regulation is due to a higher cell density at the early stationary phase, or it is part of a stationary phase phenomenon, we simulated a high cell density environment by centrifuging cells growing in early log phase (OD600 ~ 0.1) when the expression of nlmA was normally low, creating a cell pellet. As a control, a portion of the same culture was kept in planktonic phase. The planktonic culture and the cell pellet were then incubated at 37°C as static cultures, and samples were taken at designated time points to measure luciferase activity. As shown in Fig. 1B, luciferase activity in the centrifuged culture increased ~fivefold after 30 min of incubation compared with cells in planktonic phase. By 60 min after centrifugation, luciferase activity increased nearly 20-fold, reaching a relative light unit (RLU)/OD600 of ~2500. This high activity remained nearly unchanged until the end of the experiment (120 min). In contrast, luciferase activity of the planktonic culture remained low and unchanged through the entire experimental period (120 min). The OD600 of the pelleted culture was ~0.2 after 120 min of incubation while that of the planktonic culture was ~0.4. These results suggest that either high cell density or nutrient limitation or both in the cell pellet may have triggered the high level mutacin IV gene expression.
We also performed experiments to directly visualize the high-level nlmA gene expression in the cell pellet. Strain UA140::Ø(nlmAp–gfp), which carried a nlmAp–gfp fusion, was subjected to a similar pelleting assay. After 2 h of incubation, cells from both planktonic and pelleted cultures were examined under a fluorescence microscope. As shown in Fig. 1C, cells from the pelleted culture displayed bright green fluorescence, even in some single cells. In contrast, cells from the planktonic culture showed only weak fluorescence in cell aggregates (Fig. 1D). These results further confirmed the high-level mutacin IV gene expression in the cell pellet as observed with the luciferase reporter fusion.
The ComDE two-component system regulated mutacin IV gene expression
To explore the molecular mechanism of the high-level mutacin IV gene expression in the cell pellet, we examined the involvement of quorum-sensing systems in mutacin IV gene expression. Two major quorum-sensing systems have been described in S. mutans. The LuxS system is an interspecies quorum-sensing system, which has been shown to be involved in biofilm formation and stress tolerance of S. mutans (Merritt et al., 2003; Wen and Burne, 2004). The ComDE system is a intraspecies quorum-sensing system that has been known to be required for competence development (Li et al., 2001). In addition to controlling competence, the ComDE system was also shown to be involved in biofilm formation of S. mutans (Li et al., 2002; Yoshida and Kuramitsu, 2002). To determine whether mutacin IV gene expression was regulated by either of the two systems, we transferred previously constructed luxS and comD null mutations into the luciferase reporter strain UA140::Φ(nlmAp-luc) and measured luciferase activity in planktonic and pelleted cultures. While the nlmAp-luc gene expression was slightly increased in the ΔluxS background (Fig. 2A) possibly because of inactivation of mutacin I gene expression (Merritt et al. manuscript submitted), it was severely reduced (~10-fold) in the ΔcomD background (Fig. 2A). Furthermore, luciferase activity of the comD mutant in the pelleted culture remained the same as in the planktonic culture, indicating that the comD mutation abolished the high-level mutacin IV gene expression in the cell pellet.
Fig. 2.

nlmAp-luc gene expression in various mutant backgrounds in planktonic and pelleted cultures. Luciferase activity (RLU) was normalized with cell density (OD600) and expressed as a percentage of the wild-type level in planktonic culture, which was arbitrarily assigned as 100%. Values shown here represent the mean of at least two independent experiments performed in triplicate.
To confirm whether the effect of a comD mutation on mutacin IV gene expression was indeed mediated by the same ComDE signalling pathway as for competence gene regulation, we transferred comE, comC and comX mutations into strain UA140::Φ(nlmA-luc). ComE is the cognate response regulator for ComD, ComC is the structural gene for the signaling peptide CSP (Competence Stimulating Peptide), and ComX is an alternative sigma factor. According to the model for competence regulation (Pestova et al., 1996), the binding of CSP to ComD triggers autophosphorylation of ComD, which then transfers the phosphate group to ComE. Phosphorylated ComE activates transcription of ComX, which then enables transcription of the downstream genes for DNA uptake and recombination (Lee and Morrison, 1999; Luo and Morrison, 2003). When nlmAp-luc gene expression was measured in the comC null background, we found that the nlmAp-luc expression was dramatically reduced to the same level as in the comD background in both planktonic and pelleted cultures (Fig. 2A). Interestingly, in the comE background, there was no detectable nlmAp-luc activity in either the planktonic or pelleted cultures. In contrast, in the comX background, nlmAp-luc gene expression was not affected. These data indicate that nlmA is regulated by the ComDE system; however, this regulation is not mediated by ComX.
Addition of synthetic CSP triggered nlmA gene expression
To further confirm the involvement of comDE in mutacin IV gene expression, we chemically synthesized the 21-amino acid CSP peptide. Using the UA140::Φ(nlmAp-luc) reporter strain, we first determined if nlmAp-luc gene expression responded to CSP addition in a dose-dependent manner. CSP was added to the planktonic culture in concentrations ranging from 0.01 to 2 μg ml−1 for 2 h, and luciferase activity was measured. We found that nlmAp-luc gene expression responded to increasing amounts of CSP in a dose-dependent fashion up to 1 μg ml−1 (Fig. 3A). This concentration agrees well with previous reports in which 1 μg ml−1 CSP was used to induce competence or biofilm formation in a comC null mutant (Li et al., 2001; Yoshida and Kuramitsu, 2002). The time of induction was also measured and found to peak at ~3 h (data not shown). Using 1 μg ml−1 CSP and 3 h of induction time, we then measured the response of nlmAp-luc expression in the comE and comC backgrounds in early log phase cells (OD600 ~ 0.1). As shown in Fig. 3B, CSP addition to the wild-type UA140::Φ(nlmAp-luc) strain resulted in ~eightfold higher luciferase activity compared with the control. As expected, a similar response was also observed in the comC background. In contrast, in the comE background, nlmAp-luc expression did not respond to CSP addition. These results further confirmed the involvement of ComE in mutacin IV gene regulation. Addition of CSP to the planktonic culture of strain UA140::Φ(nlmAp–gfp), which was normally not fluorescent (Fig. 1D), also induced fluorescence (Fig. 3C). This was in sharp contrast to strain UA140::Φ(mutAp–gfp) carrying a gfp fusion to the mutacin I promoter mutA (Fig. 3D), which did not become fluorescent upon CSP addition. This demonstrated that CSP-dependent induction of mutacin gene expression was specific to mutacin IV.
Fig. 3.

Response of mutacin IV gene expression to CSP.
A. Response of nlmAp-luc gene expression to different concentrations of CSP.
B. Expression of nlmAp-luc gene expression in the wild-type, comE null, and comC null background in response to CSP. The experiment was repeated three times, and values are averages of the three experiments with triplicate samples of each.
C. Confocal image of nlmAp–gfp gene expression in response to CSP addition.
D. Confocal image of mutpA-gfp gene expression in response to CSP addition.
Mutacin IV gene expression and competence development were temporally coordinated
Results presented above suggested that both mutacin IV production and competence development are regulated by the same ComCDE quorum-sensing system. To determine whether the two events were also coordinated temporally, or their regulation by the same quorum-sensing system was merely coincidental, we measured simultaneously nlmAp-luc expression and transformation efficiency in response to CSP. BHI medium was chosen for this experiment because competence did not develop naturally in this medium (J. Merritt et al. unpubl. obs.). Strain UA140::Φ(nlmAp-luc) cells were grown to OD600 of ~0.05 (early log) in BHI, and CSP was added. At 30 min before each designated time point (30, 60, 120, 180 and 240 min), plasmid pVA838 DNA was added to the cell culture. The culture was first measured for luciferase activity and cell density, and then plated on erythromycin plates to select for transformants. As shown in Fig. 4A, nlmAp-luc gene expression started to increase (~ fivefold) at 30 min after CSP addition, peaked at 3 h, and plateaued at 4 h. Compared with nlmAp-luc gene expression, competence started to increase at 2 h after CSP addition, reached its peak level at 3 h, and dropped to a basal level at 4 h (Fig. 4B, S2). In comparison, luciferase activity in cells without CSP addition remained very low throughout the experiment (Fig. 4A), and no transformants could be detected in these cultures at any time point (Fig. 4B, S1). The 2 h delay in competence after CSP addition was possibly due to the delayed response of comX expression as observed by Aspiras et al. in S. mutans UA159 cells (Aspiras et al., 2004). Taken together, these results demonstrate that both mutacin gene expression and competence development in BHI respond to CSP addition, and that the temporal pattern of mutacin IV gene expression and competence development overlap. This may suggest that mutacin IV production and competence development are coordinated.
Fig. 4.
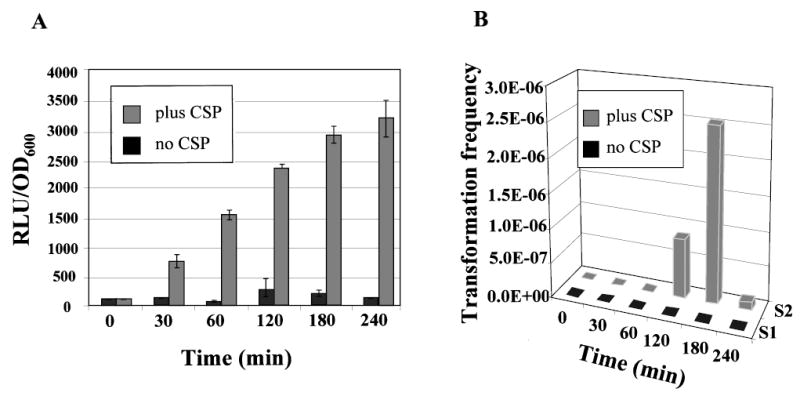
Coordination of competence development and mutacin IV gene expression.
A. Time-course of nlmAp-luc gene expression in response to CSP addition.
B. Transformation frequency of the same culture as (A). S1, no CSP; S2, with CSP. Transformation frequency was calculated as the ratio of transformants versus the number of total viable cells per ml in the transforming culture. The data shown are averages of two independent experiments assayed in triplicate. Although no error bars could be shown in (B) because of the 3-D projection, the variation between experiments is less than 20%.
S. mutans was transformed by plasmid DNA harbored in S. gordonii during co-cultivation
The interesting (and initially surprising) finding of the coordination between mutacin IV production and competence development led to the hypothesis that ComCDE-controlled mutacin IV production may be used to acquire DNA from sensitive streptococcal species living in close proximity during competence of S. mutans in its natural habitat, the dental plaque. To test this hypothesis, we performed a mixed culture transformation assay. We used a member of the mitis group streptococci, S. gordonii, as the potential donor because it naturally coexists with S. mutans in the same niche and is sensitive to mutacin IV (Qi et al., 2001). To monitor DNA transfer, we transformed S. gordonii with the shuttle plasmid pVA838 which carries an erythromycin (Erm) resistance gene (Macrina et al., 1982). To determine the role of mutacin in possible DNA release, we constructed three mutant strains from the parental strain UA140, which produces two mutacins, the lantibiotic mutacin I and the non-lantibiotic mutacin IV (Qi et al., 2001). UA140I−IV+ is defective in the lantibiotic mutacin I production (Qi et al., 2000) but proficient in mutacin IV production; UA140I+IV− is negative in mutacin IV production but positive in mutacin I production; and, UA140I−IV− is defective in both mutacin I and mutacin IV production.
The wild-type (I+IV+) and the mutant strains were mixed with S. gordonii/pVA838 separately, and the mixed cultures were pelleted with and without the addition of 1 μg ml−1CSP. After 4 h of coincubation the cell pellet was plated on Tet/Kan and Erm plates to select for the mutacin defective strains transformed with pVA838, or on Spc and Erm plates to select for wild-type S. mutans UA140 strain transformed with pVA838. Without the addition of CSP (Fig. 5A), the wild-type and the mutacin IV positive strain UA140I−IV+ showed a three- to fivefold higher transformation frequency than the mutacin IV negative strains UA140I+IV− and UA140I−IV−. However, these four strains gave rise to similar transformation frequencies when pure plasmid DNA was used for transformation, which suggested that competence was not affected by any of the mutacin mutations. In addition, monocultures of S. mutans did not show any colonies on Erm plates when no plasmid was added (data not shown), demonstrating that spontaneous Erm resistance did not occur. These results suggested that the S. mutans Erm-resistant colonies obtained from the mixed cultures with S. gordonii/pVA838 likely came from transformants with pVA838 that was probably released from S. gordonii/pVA838. To further confirm this, Erm-resistant S. mutans colonies were randomly selected from the plate, and a colony polymerase chain reaction (PCR) was performed using a pair of primers designed specifically for the erm gene in pVA838 (see Experimental procedures). The analysed 20 colonies all produced positive results (data not shown). Taken together, these data indicated that mutacin IV production was, at least partially, responsible for the increased DNA release from S. gordonii/pVA838, which could then result in increased number of transformants in S. mutans.
Fig. 5.
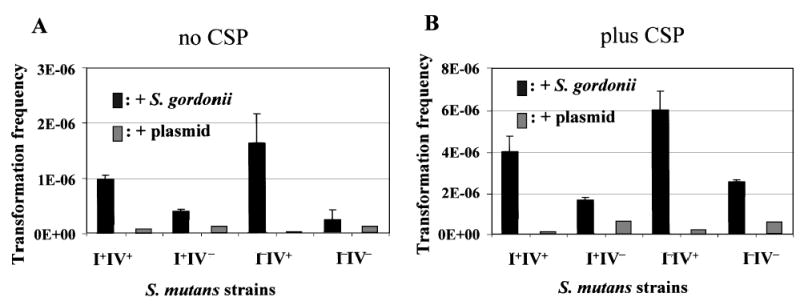
Transformation assay of S. mutans in mixed cultures with S. gordonii/pVA838 in the absence (A) and presence (B) of CSP. Transformation frequency was calculated as in Fig. 4. Data presented here are averages of two independent experiments assayed in triplicate.
Addition of CSP to the mixed culture further increased the transformation frequency in all strains compared to the non-CSP treated cells, but the overall pattern remained similar (Fig. 5B). This is likely due to the fact that CSP addition increased the overall transformability of S. mutans. It is also worth noting that the transformation frequency observed in this experiment was typical for S. mutans transformed with plasmid DNA, while transformation frequency using chromosomal DNA from the same species was usually much higher (Li et al., 2001; Merritt et al., 2005).
Partially purified mutacin IV caused DNA release from S. gordonii
Aiming to provide more direct evidence on mutacin IV-induced DNA release from S. gordonii, we partially purified mutacin IV (referred to as crude extract) from the mutacin I negative, mutacin IV positive strain UA140I−IV+ using established procedures (Qi et al., 2001) (see Experimental procedures). To exclude the possibility that other components in the crude extracts may affect DNA release, crude extract was also obtained from the double mutacin-negative strain UA140I−IV−. Different dilutions of the crude extract from both strains were tested for their activity against S. gordonii in an overlay assay (Fig. 6A). Crude extract from the mutacin IV positive strain UA140I−IV+ displayed inhibitory activity (as shown by a clear zone) even after 64-fold dilution (Fig. 6A, #7), while the crude extract from the mutacin IV negative strain UA140I−IV− showed only weak activity up to a fourfold dilution (Fig. 6A, #3). To test DNA release, equal amounts of the crude extracts were mixed with S. gordonii/pVA838, and the mixtures were incubated at room temperature. As a control, similarly diluted 5 M urea solution (used to resuspend the crude extract) was also mixed with S. gordonii/pVA838. At designated time points aliquots of samples were removed, filtered, and the DNA precipitated with yeast tRNA as carrier. Real-time PCR was used to quantify plasmid pVA838 DNA in the precipitates. As shown in Fig. 6B, 1 min after crude extract addition, four times more plasmid DNA was detected from the culture supernatant treated with UA140I−IV+ crude extract compared with that treated with the control extract. This pattern remained constant throughout the course of the experiment (32 min). Interestingly, compared with the urea control, cultures treated with the control extract (UA140I−IV−) still exhibited higher levels of DNA release from all time points. This may indicate that some minor, uncharacterized bacteriocins were still produced from the double mutacin minus strain, which may have caused the additional DNA release from S. gordonii. The finding that the control extract (UA140I−IV−) also exhibited weak activity against S. gordonii (Fig. 6A) seemed to add further support to this notion.
Fig. 6.

Quantification of DNA release from S. gordonii/pVA838 mediated by partially purified mutacin IV.
A. Inhibitory activity of the partially purified mutacin IV (crude extract) from strain UA140I−IV+ and the control extract from strain UA140I−IV− against S. gordonii. Each spot contained 10 μl of twofold serially diluted extract (e.g. #1, undiluted; #2, twofold diluted; #3, fourfold diluted, etc.).
B. Real-time PCR quantification of DNA release from S. gordonii/pVA838 treated with crude extracts from UA140I−IV+ and UA140I−IV−. The amount of DNA released upon treatment with UA140I−IV+ crude extract at 1 min was arbitrarily assigned as 100%, which was used to normalize all samples. The values shown here represent the mean of at least two independent experiments done in triplicate.
Prevalence of mutacin IV gene in S. mutans clinical isolates
Results presented above demonstrated that there was a link between mutacin IV production and competence development, and that these coordinated events may serve a purpose of acquiring DNA from other streptococcal species. To probe whether this is a rather common phenomenon among S. mutans isolates or just a specific case for some strains like UA140, we used PCR to determine the prevalence of the mutacin IV gene in the S. mutans strain population. Three sets of primers were designed: two for the two mutacins produced by UA140 (mutacin I and mutacin IV) and one for 16S DNA of all S. mutans isolates (see Experimental procedures). The three sets of primers were mixed in a ‘cocktail’ and used to amplify chromosomal DNA from S. mutans strains (e.g. UA140, UA159 and clinical isolates), or from preserved saliva samples from which S. mutans had been detected by using monoclonal antibodies (Shi et al., 1998). Among all S. mutans positive samples (70 total), mutacin IV gene was detected in ~50% of the samples. In a small percentage (~5%) of samples, both mutacin I and IV genes were detected (data not shown). These results demonstrate that at least half of the S. mutans clinical strains harbour the mutacin IV gene in their genomes.
Discussion
In this study we demonstrated that mutacin IV gene expression is dramatically increased in the cell pellet, and that this regulation is mediated by the ComCDE system in a pathway independent of the ComX regulatory pathway (Figs 1 and 2). Further studies suggested that mutacin IV gene expression and competence development may be coordinated temporally in response to CSP (Figs 3 and 4). This raised an interesting question of whether this coordinated mutacin production and competence development could be used as a means for S. mutans to acquire transforming DNA from neighbouring streptococcal species. Mixed culture assays with S. gordonii demonstrated that increased transformation of S. mutans by plasmid DNA originally harboured in S. gordonii was dependent on the presence of the mutacin IV gene, suggesting a role of mutacin IV in DNA release from S. gordonii (Fig. 5). Further investigation using partially purified mutacin IV demonstrated increased DNA release upon addition of mutacin IV crude extract to the S. gordonii culture (Fig. 6). Based on these results, we propose the following model for the coordinated mutacin IV production and competence development (Fig. 7): high cell density or nutrient limitation or both triggers high-level comC gene expression, resulting in high-level production of CSP. CSP binding to the ComDE two-component system activates transcription of a set of genes including the comX gene and nlmA. While expression of the late competence genes such as comY is dependent on ComX, the nlmA gene expression is independent of ComX. The consequence of this complex regulatory pathway (Fig. 7) results in a co-ordinated regulation between competence and bacteriocin production. High levels of mutacin IV production would kill neighbouring streptococcal species such as S. gordonii causing DNA release from these species. Released DNA is then internalized by the ComYA-G complex (Merritt et al., 2005), whose expression is dependent on ComX. It is worth noting that the temporal correlation between nlmA gene expression and competence is not strict; there is a 90 min delay of competence after nlmA gene expression is turned on (Fig. 4). We speculate that this delay is probably due to a signal relay in the competence regulatory pathway to turn on the late competence genes for DNA uptake and recombination, while no such a signal relay is required to turn on the mutacin IV gene expression. This is apparently different from the findings in S. pneumoniae where autolysis and competence strictly correlate (Steinmoen et al., 2002). However, given the reason that mutacin IV has to be produced, secreted, bound to the sensitive cells to kill before donor DNA is released, it would make perfect sense for it to be produced first before the competence system becomes fully functional.
Fig. 7.
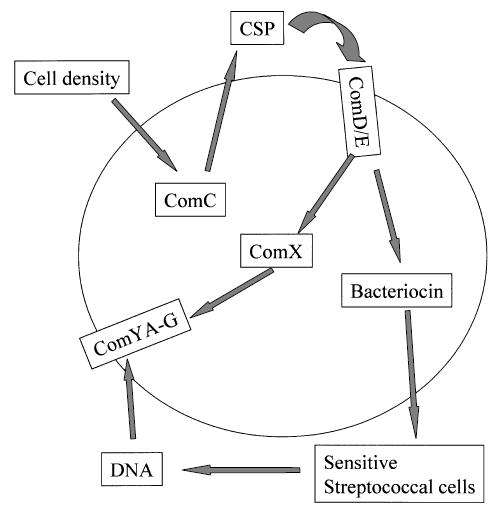
Model for coordinated mutacin IV production and competence in DNA release and uptake from other streptococcal species (see text for details).
Mutacin-mediated interspecies DNA acquisition may not be limited to UA140. Inspection of the clinical isolates of S. mutans revealed that ~50% contain the mutacin IV gene in their chromosome. These mutacin IV genes are likely to be controlled by CSP as their promoter sequences could also be amplified by primers designed using the UA159 sequence (data not shown). In fact, we have tested a number of clinical isolates on a plate assay and showed that all displayed larger inhibition zones against the mitis group streptococci after CSP addition to the plate (J. Kreth et al. unpubl.). Furthermore, mutacin IV may not be the only mutacin controlled by the ComCDE system. In a recent report by Yanezawa and Kuramitsu (Yonezawa and Kuramitsu, 2005), a two-component lantibiotic mutacin called Smb was found to be regulated by CSP in the strain GS-5. As this Smb is active against a group C streptococcus, it would be interesting to see if this mutacin is capable of causing DNA release from the target cells during competence.
The association of bacteriocin production with competence may not be limited to S. mutans. In Bacillus subtilis, it was found that comS, a small peptide required for competence, was embedded within the second open reading frame of the srfA operon-encoding enzymes for the biosynthesis of a surfactin. This organization was suggested as a way for B. subtilis to lyse microorganisms for the release of genetic material during competence (D’Souza et al., 1994; Hamoen et al., 1995). In a recent microarray study in S. pneumoniae, Rimini et al. and Peterson et al. found that some genes encoding bacteriocin-like peptides were induced by CSP, although these findings were not verified by genetic studies (Rimini et al., 2000; Peterson et al., 2004). Although some of the bacteriocin-like peptides (e.g. genes blpA, blpY and blpZ) were later found to be regulated by a different pheromone, BlpR, which does not cross react with the ComDE system (Reichmann and Hakenbeck, 2000; de Saizieu et al., 2000), a recent study by Knutsen et al. reported that an unknown ABC transporter is co-regulated by both the ComCDE and the BlpR system (Knutsen et al., 2004). Whether this unknown ABC transporter is a link between the two quorum-sensing systems is yet to be determined. Nonetheless, findings from this study provide the first genetic evidence for a coordinated production of a bacteriocin and the development of competence as a possible mechanism for DNA acquisition from other species. This mechanism may serve as a basis for acquiring DNA from other species, which may provide the opportunity for horizontal gene transfer in multispecies microbial communities. We speculate that as more bacteriocin and competence systems are characterized in other bacterial species, this mechanism may prove to be a prevalent phenomenon among naturally competent microorganisms that inhabit multispecies environments.
Experimental procedures
Bacterial strains and growth conditions
The mutacin I and mutacin VI positive S. mutans strain UA140 (Qi et al., 2001) was used for this study. The luxS, comC, comD, comE and comX1 mutant strains were constructed by transferring the individual mutations to UA140::Φ(nlmAp-luc) from previously constructed S. mutans strains, JM03 (luxS) (Merritt et al., 2003), JMcC-luc ΔcomC Kanr (Merritt et al., 2005), SMCD1 ComD− Ermr (Li et al., 2001), SMCE1 ComE− Ermr (Li et al., 2001) and SMComX1 ΔcomX1::PcEm Ermr (Li et al., 2002) respectively. Each individual mutation was originally generated by double crossover integration to avoid polar effects on the downstream genes. Strains SMCD1, SMCE1 and SMComX1 were kindly provided by Dr D. Cvitkovitch (University of Toronto, Canada). Transformants were selected on Km (800 μg ml−1 or Erm (15 μg ml−1) plates, and the mutations confirmed by PCR. Plasmid and chromosomal DNA was transformed into S. mutans following previous protocols (Shah and Caufield, 1993). To construct a DNA donor, the mutacin-sensitive Streptococcus gordonii Challis (Kilian et al., 1989; Jenkinson and Easingwood, 1990) was transformed with the shuttle plasmid pVA838 (Macrina et al., 1982), which carries an Erm resistance marker. The presence of the plasmid was confirmed by PCR using a pair of primers specific for the Erm gene. All UA140 variants and S. gordonii were grown in BHI medium (Difco; Sparks, MD) or on BHI agar plates anaerobically (nitrogen 85%, carbon dioxide 5%, hydrogen 10%) at 37°C. E. coli Top10 (Invitrogen; Carlsbad, CA) was used for cloning and plasmid amplification.
DNA manipulations
Restriction enzymes were obtained from Promega (Madison, WIS), Fisher (Pittsburgh, PA) or NEB (Beverly, MA). Ligations were performed using the Quick LigationTM Kit from NEB. PCR products were cloned into the TOPO TA CLONING® Kit from Invitrogen. All plasmids were extracted and purified from E. coli with the QIAGEN (Valencia, CA) Miniprep Kit. DNA extracted from agarose gels (1%) was purified with the QIAGEN QIAquick® Gel Extraction Kit. PCR was performed with a MyCycler™ thermocycler (Bio-Rad; Hercules, CA) using a protocol supplied with the thermocycler. Taq-DNA Polymerase was obtained from Promega and dNTPs from Fisher.
Construction of reporter gene fusion to the mutacin IV promoter
The backbone vector for the construction of all reporter gene fusions was pFW5, which contains a spectinomycin resistance marker (aad9) that works in both Gram-negative and Gram-positive bacteria (Podbielski et al., 1996). To construct the gfp gene fusion, the promoterless gfp–mut2 gene with its own ribosome-binding site was excised from vector pKEN (Cormack et al., 1996) with BamHI and HindIII, and inserted into pFW5 at the BamHI and HindIII sites to generate plasmid pFW5::gfp. To insert the nlmAp in front of the gfp reporter, the promoter region was amplified by PCR from chromosomal DNA of strain UA140 using primers nlmApo-F (5′-taatgcatgctgtagatgttgagcc-3′) and nlmApo-R (5′-ccggatccat tacatcaaattgttcaaatgcc-3′). Primer nlmApo-R had a BamHI site incorporated at its 5′ end (underlined sequence). The PCR product was subcloned into pCR®2.1-Topo® (Invitrogen) to generate pCR2.1::nlmAp. Correct fragment insertion and promoter sequence was confirmed by sequencing. The plasmid pCR2.1::nlmAp was digested with BamHI and EcoRV, and the DNA fragment containing the nlmA promoter was purified. The nlmA promoter region was subsequently inserted upstream of the gfp gene in pFW5::gfp at the BamHI and an upstream StuI site compatible with EcoRV to form plasmid pFW5::Ø(nlmAp–gfp). For the construction of the firefly luciferase (luc) reporter, plasmid pFW5-luc (Elsner et al., 2002) was digested with SalI and BamHI. The nlmA promoter was excised from pCR2.1::nlmAp with BamHI and XhoI, and ligated into the BamHI site and the XhoI compatible SalI site to form plasmid pFW5::Ø(nlmAp-luc). For the construction of the reporter strains UA140::Ø(nlmAp-luc) and UA140::Ø(nlmAp–gfp), plasmids pFW5::Ø(nlmAp-luc) and pFW5::Ø(nlmAp–gfp) were transformed into UA140 and selected with spectinomycin (800 μg ml−1). Integration of each reporter into the chromosome was confirmed by PCR. Confirmed reporter strains had both reporter-gene activity as well as wild-type levels of mutacin production.
Construction of mutacin defective strains
Streptococcus mutans strain UA140 produces two mutacins, the lantibiotic mutacin I and the non-lantibiotic mutacin IV (Qi et al., 2001). To study the role of each mutacin in causing DNA release from S. gordonii, we constructed three derivative strains defective in either mutacin I (UA140I−IV+), mutacin IV (UA140I+V−), or both (UA140I−IV−). To construct UA140I−IV+, the tetracycline (Tet) resistance gene tetM from Tn916 (Flannagan et al., 1994) was amplified by PCR and cloned into pCR2.1 cloning vector (Invitrogen). A DNA fragment encompassing 1 kb upstream and downstream of mutC (Qi et al., 2000) was amplified by PCR using primers BF1 (5′-TATTTGAAACTCCCAGAAAACTTACAATG-3′) and DR1 (5′-TCCTTTCTTCTTCATACTCTTACC-5′) and cloned into pCR2.1 to form pCRBCD. To delete mutC, an inverse PCR was performed by using two primers BR1 (5′-ggcggcaggc ctTTACTTTGTCTCATTACCCAC-3′) and DF1 (5′-ggcggcag gcctAACTGAACGGAGAAATAATTATGG-3′), both of which had a Stu I restriction site incorporated at their 5′ ends. The tetM gene cassette was released from pCR2.1 by cutting with Stu I restriction enzyme, whose recognition sequence was also incorporated into the primers for amplifying tetM, and inserted into pCRBCD at the same restriction site. The resulting plasmid was digested with Pst I and Sph I and transformed into UA140. The deletion construct was integrated into the chromosome by double cross-over homologous recombination. The transformants were selected on Tet plates (10 μg ml−1). Ten transformants were randomly selected and tested for mutacin I production by the deferred antagonism assay (Hamada and Ooshima, 1975) using S. sobrinus OMZ176 as the indicator, which is sensitive only to mutacin I. The mutacin I negative isolates were further confirmed by PCR. To construct UA140I+V−, a DNA fragment encompassing nlmA and nlmB was PCR amplified by using primers Adh dn1 (5′-CCAGCTAATCCACGTCAGC-3′) and 135 μp1 (5′-CAAGAAAAGATGAGTAAGATGGC-3′), and cloned into pCR2.1 to form pCRnlmAB. An inverse PCR reaction was performed using pCRnlmAB as template and primers nlmA up1 (5′-ggcatcgatTGTGTAGGCACTTGGG GAC-3′) and nlmB dn1 (5′-ggcatcgatGATATAATGGTGGG CAATGTC-3′). Both primers had a Cla I restriction site incorporated at their 5′ ends. The PCR product was digested with Cla I and ligated with a Cla I digested kanamycin (Km) resistance gene cassette (aphIII, Trieu-Cuot and Courvalin, 1983) that was constructed from previous studies (Qi et al., 2004). The plasmid was linearized by Sph I digestion and transformed into UA140. The deletion was confirmed by PCR. As no indicator strain was sensitive only to mutacin IV, the defect in mutacin IV production was further confirmed by mutacin isolation under conditions that mutacin I was not produced (Qi et al., 2001). To construct the double-mutant strain UA140I−IV−, chromosome DNA was isolated from strain UA140I+IV− and transformed into strain UA140I−IV+. The transformed strains were selected on Tet plus Km plates, tested for the lack of mutacin production by using the indicator strain S. sangunis NY101, which was sensitive to both mutacins (Qi et al., 2001). Mutacin defective isolates were further confirmed by PCR.
Luciferase assay
Luciferase assays were performed as previously described (Loimaranta et al., 1998). Briefly, 25 μl of 1 mM D-luciferin (Sigma; St. Louis, MO) suspended in 100 mM citrate buffer, pH 6, was added to 100 μl of the cell culture. To ensure sufficient levels of intracellular ATP pool, cells were recharged with 1% glucose for 10 min prior to luciferin addition. Luciferase activity was measured by using a TD 20/20 luminometer (Turner Biosystems; Sunnyvale, CA). Usually, three parallel cultures were measured at each time point and the average value was taken. Each experiment was repeated at least two times.
Cell pelleting assay
To create an environment mimicking the cell density in the dental plaque (1011 cells g−1wet weight), anaerobically grown log-phase cells were spun down at ~16 000 g for 1.5 min in a microcentrifuge. The cell density in the pellet was found to be close to the cell density in the dental plaque. To study the effect of cell density on mutacin IV gene expression, overnight cultures were diluted 1:20 in fresh BHI and grown at 37°C anaerobically to an OD600 of ~0.2 (early log phase). The culture was divided into 1 ml aliquots in 1.5 ml tubes and half of the samples were spun down. With the supernatant remaining in the tubes, both planktonic and spin-down cultures were further incubated at 37°C anaerobically for a designated amount of time. Both planktonic and pelleted cells were resuspended by vortexing before luciferase activity and OD600 were measured. In general, three parallel cultures for each treatment were used, and the experiment was repeated at least three times.
CSP-dependent transformation assay
To determine the coordination between competence development and mutacin IV (nlmA) gene expression, we simultaneously measured luciferase activity and transformation efficiency in strain UA140::Φ(nlmAp-luc) in response to the CSP. A fresh overnight culture was diluted to OD600 of ~0.025 in BHI and incubated anaerobically until OD600 of ~0.05. The culture was divided into 1 ml aliquots in 1.5 ml tubes with and without the addition of CSP (1 μg ml−1) and incubated anaerobically at 37°C. At 30 min prior to each sampling time, 0.5 μg of pVA838 plasmid DNA was added. Luciferase activity (RLU) and OD600 were measured at each designated time point followed by plating of 700 μl of the samples on Erm plates.
Interspecies transformation assay
To determine the possible S. mutans competence-induced DNA release from S. gordonii, the shuttle vector pVA838 (ErmR) was transformed into S. gordonii Challis. S. mutans UA140::Φ(nlmAp-luc) (wild-type) and mutacin-negative derivative strains and S. gordonii/pVA838 were grown overnight in BHI. The overnight cultures were centrifuged and washed twice to eliminate possible released plasmid DNA during stationary phase, and the cell pellets were resuspended in fresh BHI to an OD600 of ~0.1. Both cultures were allowed to grow to OD600 of 0.2 and mixed in a 5:1 ratio of S. mutans: S. gordonii/pVA838. The mixed culture was pelleted and incubated at 37°C for 4 h. The cell pellet was plated on BHI plates containing both Spc and Erm to detect transformants. Colonies on this plate could come from S. mutans transformed with the shuttle plasmid, or S. gordonii transformed with chromosomal DNA from S. mutans. Different colony morphologies would allow us to distinguish the two species. As a control, 0.5 μg of the shuttle plasmid DNA isolated from E. coli was added to S. mutans single species cultures to normalize variations in transformation efficiency of different isogenic strains.
Mutacin IV induced DNA release assay by real-time PCR
To test if mutacin IV is responsible for the release of plasmid DNA from S. gordonii cells, mutacin IV was partially purified (referred to as crude extract) as described previously (Qi et al., 2000). To measure mutacin IV-induced DNA release, an overnight culture of S. gordonii/pVA838 was washed twice in BHI and adjusted to OD600 of 0.4 in TE containing 10 mM EDTA to inhibit DNase activity. To 1.75 ml of the cell suspension, 250 μl of crude extract from either UA140I−IV+ or UA140I−IV− was added. As a negative control, 250 μl of 5 M urea was added (crude mutacin extract was suspended in 5 M urea). The cells were incubated at 37°C and 600 μl of the cell suspension was removed after 1, 16 and 32 min. The suspension was centrifuged at 16 000 g for 5 min and 500 μl of supernatant was immediately removed, filter-sterilized, and stored on ice until further processing. The DNA was precipitated by ethanol precipitation using yeast tRNA as carriers and used as template for real-time PCR. A pair of erm-cassette specific primers was used to detect the presence of the shuttle plasmid, and real-time PCR was performed as previously described (Merritt et al., 2005).
Confocal Laser Scanning Microscopy
For microscopy of planktonic, pelleted and CSP-treated cells, an overnight culture in BHI was diluted 1:20 and incubated at 37°C anaerobically to OD600 of ~0.2. The cultures were treated with CSP or centrifuged as described in previous sections. Before microscopy, the cells were washed with PBS and incubated for 10 min at room temperature to promote folding of the gfp fluorophore (Hansen et al., 2001). Images were obtained with a 40 × 1.4 Plan-Neofluar oil objective.
Detection of mutacin IV in S. mutans clinical isolates
To see the prevalence of the mutacin genes in S. mutans isolates, three sets of primers were designed. M1F (5′-ATAG TAAAGTGGGTAGTTTC-3′) and M1R (5′-GCCTAATGG TTTTCTGTAC-3′) would amplify a 270 bp fragment from the mutacin I gene (Qi et al., 2000), Orf1 (5′-ATGGATACACAGG CATTTGAAC-3′) and Orf2 (5′-TTTAGTGTGGAAAAACTACA GATCC-3′) would amplify a 450 bp fragment from the mutacin IV gene (Qi et al., 2001), and SmF205 (5′-TTGATTGAAA GATGCAAGC-3′) and BacR335 (5′-GCTGCCTCCCGTA GGTG-3′) would amplify a 130 bp fragment of the S. mutans 16S DNA. Chromosomal DNA was isolated by using the bead beater. After testing the specificity and sensitivity of each pair of primers with S. mutans UA140, which produces both mutacin I and IV (Qi et al., 2001), and closely related species such as S. sobrinus OMZ176 and S. sanguinis NY101, a primer cocktail was made, in which all primers (25 μM each) were mixed in equal portions. For PCR, Taq polymerase was used under standard conditions with the primer cocktail at a final concentration of 1 μM. PCR was performed for one cycle at 94°C/3 min, 50°/1 min, and 72°C/30 s, and 25 cycles of 94°C/1 min, 50°/1 min and 72°C/30 s.
Chemical synthesis of CSP
The mature peptide of CSP (SGSLSTFFRLFNRSFTQALGK) was synthesized by automated 9-fluorenylmethoxy carbonyl chemistry (Applied Biosystems 431 A Peptide Synthesizer) and confirmed with reverse-phase high-performance liquid chromatography and mass spectrometry profiles. Purified CSP was dissolved in dionized water, aliquoted (5 mg ml−1) and stored at −20°C.
Acknowledgments
We thank R. Eckert for synthesis of the CSP peptide, Dr Cvitkovitch for providing the comD, comE and comX mutant constructs. This work was supported in part by NIH grants U01-DE15018 to W.S., R01-DE014757 to F.Q., NIDCR T32 Training grant DE007296 to J.M., and a Delta Dental grant WDS78956 to W.S.
References
- Aspiras MB, Ellen RP, Cvitkovitch DG. ComX activity of Streptococcus mutans growing in biofilms. FEMS Microbiol Lett. 2004;238:167–174. doi: 10.1016/j.femsle.2004.07.032. [DOI] [PubMed] [Google Scholar]
- Baba T, Schneewind O. Instruments of microbial warfare: bacteriocin synthesis, toxicity and immunity. Trends Microbiol. 1998;6:66–71. doi: 10.1016/S0966-842X(97)01196-7. [DOI] [PubMed] [Google Scholar]
- Baca P, Liebana J, Piedrola G. Epidemiological application of a new bacteriocin typing scheme for Streptococcus mutans. Community Dent Oral Epidemiol. 1990;18:194–196. doi: 10.1111/j.1600-0528.1990.tb00055.x. [DOI] [PubMed] [Google Scholar]
- Bondi M, Neglia RG, Messi P, Manicardi G, Fabio U. Streptococcus mutans: classification in bacteriocin-types. Microbiologica. 1991;14:223–228. [PubMed] [Google Scholar]
- Caufield PW, Childers NK, Allen DN, Hansen JB. Distinct bacteriocin groups correlate with different groups of Streptococcus mutans plasmids. Infect Immun. 1985;48:51–56. doi: 10.1128/iai.48.1.51-56.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP) Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- D’Souza C, Nakano MM, Zuber P. Identification of comS, a gene of the srfA operon that regulates the establishment of genetic competence in Bacillus subtilis. Proc Natl Acad Sci USA. 1994;91:9397–9401. doi: 10.1073/pnas.91.20.9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagkessamanskaia A, Moscoso M, Henard V, Guiral S, Overweg K, Reuter M, et al. Interconnection of competence, stress and CiaR regulons in Streptococcus pneumoniae: competence triggers stationary phase autolysis of ciaR mutant cells. Mol Microbiol. 2004;51:1071–1086. doi: 10.1111/j.1365-2958.2003.03892.x. [DOI] [PubMed] [Google Scholar]
- Dubnau D. Genetic competence in Bacillus subtilis. Microbiol Rev. 1991a;55:395–424. doi: 10.1128/mr.55.3.395-424.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubnau D. The regulation of genetic competence in Bacillus subtilis. Mol Microbiol. 1991b;5:11–18. doi: 10.1111/j.1365-2958.1991.tb01820.x. [DOI] [PubMed] [Google Scholar]
- Elsner A, Kreikemeyer B, Braun-Kiewnick A, Spellerberg B, Buttaro BA, Podbielski A. Involvement of Lsp, a Member of the LraI-Lipoprotein Family in Streptococcus pyogenes, in Eukaryotic Cell Adhesion and Internalization. Infect Immun. 2002;70:4859–4869. doi: 10.1128/IAI.70.9.4859-4869.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannagan SE, Zitzow LA, Su YA, Clewell DB. Nucleotide sequence of the 18-kb conjugative transposon Tn916 from Enterococcus faecalis. Plasmid. 1994;32:350–354. doi: 10.1006/plas.1994.1077. [DOI] [PubMed] [Google Scholar]
- Hamada S, Ooshima T. Production and properties of bacteriocins (mutacins) from Streptococcus mutans. Arch Oral Biol. 1975;20:641–648. doi: 10.1016/0003-9969(75)90131-4. [DOI] [PubMed] [Google Scholar]
- Hamoen LW, Eshuis H, Jongbloed J, Venema G, van Sinderen D. A small gene, designated comS, located within the coding region of the fourth amino acid-activation domain of srfA, is required for competence development in Bacillus subtilis. Mol Microbiol. 1995;15:55–63. doi: 10.1111/j.1365-2958.1995.tb02220.x. [DOI] [PubMed] [Google Scholar]
- Hamoen LW, Venema G, Kuipers OP. Controlling competence in Bacillus subtilis: shared use of regulators. Microbiology. 2003;149:9–17. doi: 10.1099/mic.0.26003-0. [DOI] [PubMed] [Google Scholar]
- Hansen MC, Palmer RJ, Jr, Udsen C, White DC, Molin S. Assessment of GFP fluorescence in cells of Streptococcus gordonii under conditions of low pH and low oxygen concentration. Microbiology. 2001;147:1383–1391. doi: 10.1099/00221287-147-5-1383. [DOI] [PubMed] [Google Scholar]
- Jenkinson HF, Easingwood RA. Insertional inactivation of the gene encoding a 76-kilodalton cell surface polypeptide in Streptococcus gordonii Challis has a pleiotropic effect on cell surface composition and properties. Infect Immun. 1990;58:3689–3697. doi: 10.1128/iai.58.11.3689-3697.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilian M, Mikkelsen L, Henrichsen J. Taxonomic study of viridans streptococci: description of Streptococcus gordonii sp. nov. and emended descriptions of Streptococcus sanguis (White and Niven 1946), Streptococcus oralis (Bridge and Sneath 1982), and Streptococcus mitis (Andrewes and Horder 1906) Int J Syst Bacteriol. 1989;39:471–484. [Google Scholar]
- Knutsen E, Ween O, Havarstein LS. Two separate quorum-sensing systems upregulate transcription of the same ABC transporter in Streptococcus pneumoniae. J Bacteriol. 2004;186:3078–3085. doi: 10.1128/JB.186.10.3078-3085.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreth J, Merritt J, Bordador C, Shi W, Qi F. Transcriptional analysis of mutacin I (mutA) gene expression in planktonic and biofilm cells of Streptococcus mutans using fluorescent protein and glucuronidase reporters. Oral Microbiol Immunol. 2004;19:252–256. doi: 10.1111/j.1399-302X.2004.00148.x. [DOI] [PubMed] [Google Scholar]
- Kroes I, Lepp PW, Relman DA. Bacterial diversity within the human subgingival crevice. Proc Natl Acad Sci USA. 1999;96:14547–14552. doi: 10.1073/pnas.96.25.14547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Morrison DA. Identification of a new regulator in Streptococcus pneumoniae linking quorum sensing to competence for genetic transformation. J Bacteriol. 1999;181:5004–5016. doi: 10.1128/jb.181.16.5004-5016.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YH, Lau PC, Lee JH, Ellen RP, Cvitkovitch DG. Natural genetic transformation of Streptococcus mutans growing in biofilms. J Bacteriol. 2001;183:897–908. doi: 10.1128/JB.183.3.897-908.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YH, Tang N, Aspiras MB, Lau PC, Lee JH, Ellen RP, Cvitkovitch DG. A quorum-sensing signaling system essential for genetic competence in Streptococcus mutans is involved in biofilm formation. J Bacteriol. 2002;184:2699–2708. doi: 10.1128/JB.184.10.2699-2708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loimaranta V, Tenovuo J, Koivisto L, Karp M. Generation of bioluminescent Streptococcus mutans and its usage in rapid analysis of the efficacy of antimicrobial compounds. Antimicrob Agents Chemother. 1998;42:1906–1910. doi: 10.1128/aac.42.8.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo P, Morrison DA. Transient association of an alternative sigma factor, ComX, with RNA polymerase during the period of competence for genetic transformation in Streptococcus pneumoniae. J Bacteriol. 2003;185:349–358. doi: 10.1128/JB.185.1.349-358.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrina FL, Tobian JA, Jones KR, Evans RP, Clewell DB. A cloning vector able to replicate in Escherichia coli and Streptococcus sanguis. Gene. 1982;19:345–353. doi: 10.1016/0378-1119(82)90025-7. [DOI] [PubMed] [Google Scholar]
- Merritt J, Qi F, Goodman SD, Anderson MH, Shi W. Mutation of luxS affects biofilm formation in Streptococcus mutans. Infect Immun. 2003;71:1972–1979. doi: 10.1128/IAI.71.4.1972-1979.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt J, Qi F, Shi W. A unique nine-gene comY operon in Streptococcus mutans. Microbiology. 2005;151:157–166. doi: 10.1099/mic.0.27554-0. [DOI] [PubMed] [Google Scholar]
- Moore WE, Moore LV. The bacteria of periodontal diseases. Periodontol 2000. 1994;5:66–77. doi: 10.1111/j.1600-0757.1994.tb00019.x. [DOI] [PubMed] [Google Scholar]
- Morrison DA. Streptococcal competence for genetic transformation: regulation by peptide pheromones. Microb Drug Resist. 1997;3:27–37. doi: 10.1089/mdr.1997.3.27. [DOI] [PubMed] [Google Scholar]
- Morrison DA, Lee MS. Regulation of competence for genetic transformation in Streptococcus pneumoniae: a link between quorum sensing and DNA processing genes. Res Microbiol. 2000;151:445–451. doi: 10.1016/s0923-2508(00)00171-6. [DOI] [PubMed] [Google Scholar]
- Paster BJ, Boches SK, Galvin JL, Ericson RE, Lau CN, Levanos VA, et al. Bacterial diversity in human subgingival plaque. J Bacteriol. 2001;183:3770–3783. doi: 10.1128/JB.183.12.3770-3783.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestova EV, Havarstein LS, Morrison DA. Regulation of competence for genetic transformation in Streptococcus pneumoniae by an auto-induced peptide pheromone and a two-component regulatory system. Mol Microbiol. 1996;21:853–862. doi: 10.1046/j.1365-2958.1996.501417.x. [DOI] [PubMed] [Google Scholar]
- Peterson SN, Sung CK, Cline R, et al. other authorsIdentification of competence pheromone responsive genes in Streptococcus pneumoniae by use of DNA microarrays. Mol Microbiol. 2004;51:1051–1070. doi: 10.1046/j.1365-2958.2003.03907.x. [DOI] [PubMed] [Google Scholar]
- Podbielski A, Spellerberg B, Woischnik M, Pohl B, Lutticken R. Novel series of plasmid vectors for gene inactivation and expression analysis in group A streptococci (GAS) Gene. 1996;177:137–147. doi: 10.1016/0378-1119(96)84178-3. [DOI] [PubMed] [Google Scholar]
- Qi F, Chen P, Caufield PW. Purification and biochemical characterization of mutacin I from the group I strain of Streptococcus mutans, CH43, and genetic analysis of mutacin I biosynthesis genes. Appl Environ Microbiol. 2000;66:3221–3229. doi: 10.1128/aem.66.8.3221-3229.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi F, Chen P, Caufield PW. The group I strain of Streptococcus mutans, UA140, produces both the lantibiotic mutacin I and a nonlantibiotic bacteriocin, mutacin IV. Appl Environ Microbiol. 2001;67:15–21. doi: 10.1128/AEM.67.1.15-21.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi F, Merritt J, Lux R, Shi W. Inactivation of the ciaH Gene in Streptococcus mutans diminishes mutacin production and competence development, alters sucrose-dependent biofilm formation, and reduces stress tolerance. Infect Immun. 2004;72:4895–4899. doi: 10.1128/IAI.72.8.4895-4899.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichmann P, Hakenbeck R. Allelic variation in a peptide-inducible two-component system of Streptococcus pneumoniae. FEMS Microbiol Lett. 2000;190:231–236. doi: 10.1111/j.1574-6968.2000.tb09291.x. [DOI] [PubMed] [Google Scholar]
- Rimini R, Jansson B, Feger G, et al. other authorsGlobal analysis of transcription kinetics during competence development in Streptococcus pneumoniae using high density DNA arrays. Mol Microbiol. 2000;36:1279–1292. doi: 10.1046/j.1365-2958.2000.01931.x. [DOI] [PubMed] [Google Scholar]
- de Saizieu A, Gardes C, Flint N, Wagner C, Kamber M, Mitchell TJ, et al. Microarray-based identification of a novel Streptococcus pneumoniae regulon controlled by an autoinduced peptide. J Bacteriol. 2000;182:4696–4703. doi: 10.1128/jb.182.17.4696-4703.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah GR, Caufield PW. Enhanced transformation of Streptococcus mutans by modifications in culture conditions. Anal Biochem. 1993;214:343–346. doi: 10.1006/abio.1993.1503. [DOI] [PubMed] [Google Scholar]
- Shi W, Jewett A, Hume WR. Rapid and quantitative detection of Streptococcus mutans with species-specific monoclonal antibodies. Hybridoma. 1998;17:365–371. doi: 10.1089/hyb.1998.17.365. [DOI] [PubMed] [Google Scholar]
- Steinmoen H, Knutsen E, Havarstein LS. Induction of natural competence in Streptococcus pneumoniae triggers lysis and DNA release from a subfraction of the cell population. Proc Natl Acad Sci USA. 2002;99:7681–7686. doi: 10.1073/pnas.112464599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmoen H, Teigen A, Havarstein LS. Competence-induced cells of Streptococcus pneumoniae lyse competence-deficient cells of the same strain during cocultivation. J Bacteriol. 2003;185:7176–7183. doi: 10.1128/JB.185.24.7176-7183.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trieu-Cuot P, Courvalin P. Nucleotide sequence of the Streptococcus faecalis plasmid gene encoding the 3′5′-aminoglycoside phosphotransferase type III. Gene. 1983;23:331–341. doi: 10.1016/0378-1119(83)90022-7. [DOI] [PubMed] [Google Scholar]
- Wen ZT, Burne RA. LuxS-mediated signaling in Streptococcus mutans is involved in regulation of acid and oxidative stress tolerance and biofilm formation. J Bacteriol. 2004;186:2682–2691. doi: 10.1128/JB.186.9.2682-2691.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonezawa H, Kuramitsu HK. Genetic analysis of a unique bacteriocin, Smb, produced by Streptococcus mutans GS5. Antimicrob Agents Chemother. 2005;49:541–548. doi: 10.1128/AAC.49.2.541-548.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida A, Kuramitsu HK. Multiple Streptococcus mutans Genes Are Involved in Biofilm Formation. Appl Environ Microbiol. 2002;68:6283–6291. doi: 10.1128/AEM.68.12.6283-6291.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]