

Abstract
Macrophage activation by bacterial lipopolysaccharide (LPS) promotes the secretion of pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), and of secondary mediators, such as leukotrienes and prostaglandins (PGs). Mice lacking the gene encoding the serine/threonine protein kinase Tpl2/Cot produce low levels of TNF-α in response to LPS because of an ERK-dependent post-transcriptional defect, and they are resistant to LPS/d-galactosamine-induced endotoxin shock. In this study we demonstrate that prostaglandin E2 and its regulatory enzyme, COX-2, are also targets of Tpl2-transduced LPS signals in bone marrow-derived mouse macrophages. Thus, LPS-stimulated Tpl2–/– macrophages express low levels of COX-2 and PGE2, compared with wild-type Tpl2+/+ cells. The ability of Tpl2 to regulate COX-2 expression depends on ERK signals that activate p90Rsk and Msk1, which in turn phosphorylate CREB, a key regulator of COX-2 transcription. These data identify physiological targets of Tpl2 signaling downstream of ERK and further implicate Tpl2 in the pathophysiology of inflammation.
Keywords: Cot/COX-2/LPS/signaling/Tpl2
Introduction
Macrophages play a central role in inflammation and the regulation of the immune response. Lipopolysaccharide (LPS), a component of the cell wall of Gram-negative bacteria, activates macrophages to produce pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), and secondary mediators, such as leukotrienes and prostaglandins (PGs). These substances are important regulators of both innate and adaptive immunity. However, their uncontrolled expression can cause acute or chronic inflammatory syndromes. An acute inflammatory syndrome induced by these mediators is the septic shock syndrome, which is characterized by fever, hypotension, disseminated intravascular coagulation and multiple organ failure (Parillo, 1993). Prostaglandin E2 (PGE2), a mediator of this syndrome, is produced by macrophages and contributes to vasodilation, pain and fever (Astiz et al., 1996). The contribution of PGs to this syndrome is underscored by the observation that PGE2 levels are increased in patients with septic shock, and that knock-out (KO) mice defective in PG induction do not develop fever in response to pyrogenic doses of bacterial endotoxin (Li et al., 1999).
The rate-limiting step in PG synthesis is catalyzed by cyclooxygenase (COX), an enzyme that exists in two isoforms encoded by two distinct genes. COX-1 is constitutively expressed in most cell types and plays a role in gastrointestinal and reproductive function (Herschman, 1996). In contrast, COX-2 is expressed at very low levels and is strongly induced by growth factors and pro-inflammatory stimuli, including LPS, as well as by several activated oncogenes (Sheng et al., 1998; Barrios-Rodiles et al., 1999; Caivano and Cohen, 2000; Eliopoulos et al., 2002). The significance of COX-2 in prostaglandin synthesis and inflammation is highlighted by the observation that COX-2 inhibitors block the synthesis of PGE2 and, as a result, they inhibit inflammation and confer analgesia (Meade et al., 1993). Moreover, homozygous deletion of the COX-2 gene in mice leads to alleviation of hepatocellular toxicity caused by LPS administration, suggesting an important physiological role for this enzyme in LPS-induced pathology (Dinchuk et al., 1995). COX-2 expression is regulated at both the transcriptional and the post-transcriptional level. The transcriptional activation of COX-2 is mediated by the binding of the inducible transcription factors NF-κB, NFAT, CREB and c/EBPβ to cis-acting regulatory elements in the COX-2 promoter (Caivano and Cohen, 2000; Wadleigh et al., 2000; Chen et al., 2001; de Gregorio et al., 2001). The specific factors involved in COX-2 activation depend on both the cell type and the stimulus. Thus, NF-κB contributes to COX-2 induction in some, but not all, cell types. Although LPS engages NF-κB, the contribution of NF-κB to COX-2 transactivation in mouse macrophages stimulated with LPS is dispensable (Wadleigh et al., 2000). The factors required for both basal transcription and the induction of COX-2 in this cell type instead appear to be CREB and c/EBPβ (Wadleigh et al., 2000).
The serine/threonine protein kinase Tpl2, also known as Cot, was identified as a target for provirus integration in MoMuLV-induced rat T-cell lymphomas and MMTV-induced mammary carcinomas (Patriotis et al., 1993; Erny et al., 1996). When overexpressed in a variety of cell types, Tpl2 activates ERK, JNK, p38MAPK and the transcription factors NFAT and NF-κB (Ceci et al., 1997; Tsatsanis et al., 1998a,b and references therein). Transgenic mice expressing Tpl2 under the control of a T-cell-specific promoter develop T-cell lymphoblastic lymphomas at an early age (Ceci et al., 1997). Recent studies on cells derived from Tpl2–/– mice showed that the Tpl2 kinase plays a key physiological role in LPS signaling. LPS activates all the signaling pathways that are also activated by Tpl2. However, in LPS-stimulated cells, Tpl2 is required only for the activation of ERK. Because of this signaling defect, LPS-stimulated macrophages from Tpl2–/– mice are defective in the transport of the TNF-α mRNA from the nucleus to the cytoplasm, and they are impaired in the induction of TNF-α by LPS. As a result, Tpl2–/– mice are resistant to LPS/d-galactosamine-induced endotoxin shock (Dumitru et al., 2000).
In this study we have extended our observations on the involvement of Tpl2 in inflammation and endotoxin shock with the demonstration that Tpl2 also affects LPS-induced prostaglandin synthesis. We report that LPS-stimulated Tpl2–/– macrophages exhibit reduced induction of both PGE2 and its regulatory enzyme COX-2, and that the defect in COX-2 induction is, at least in part, transcriptional. The ability of Tpl2 to regulate the LPS-mediated transcriptional induction of COX-2 was found to depend on ERK signals leading to the phosphorylation of CREB via the intermediate kinases p90Rsk and Msk1. These data further implicate Tpl2 in the pathophysiology of inflammation and the endotoxin shock syndrome, and suggest that Tpl2 may provide an excellent pharmacological target against these conditions.
Results
Reduced induction of COX-2 and PGE2 in LPS-treated Tpl2–/– macrophages
PGE2 is induced by LPS in bone marrow-derived macrophages (Reddy and Herschman, 1994). To determine whether PGE2 induction by LPS depends on Tpl2, we measured PGE2 secretion in supernatants of Tpl2+/+ and Tpl2–/– macrophage cultures harvested before and 3, 4.5, 6 and 10 h after stimulation with LPS. The results (Figure 1) showed that Tpl2–/– macrophages are defective in PGE2 induction by LPS. To determine whether this defect resulted from the inability of LPS to induce ERK activation in Tpl2–/– cells, we also measured PGE2 levels in supernatants of LPS-stimulated Tpl2+/+ macrophage cultures pretreated with the MEK inhibitor PD98059. These experiments verified that ERK activation by Tpl2-transduced signals is required for PGE2 induction by LPS.

Fig. 1. LPS-treated Tpl2–/– macrophages secrete reduced levels of PGE2. Culture supernatants were collected at 0, 3, 4.5, 6 or 10 h following LPS stimulation. Unstimulated Tpl2+/+ and Tpl2–/– macrophage cultures secreted similar levels of PGE2. PGE2 levels produced by unstimulated cells were given the arbitrary value of 1. The bar graphs show the fold induction of PGE2 (± SD) compared with control untreated cultures.
PGE2 synthesis is regulated by COX-2, an enzyme induced by LPS (Caivano and Cohen, 2000; Wadleigh et al., 2000). To determine whether Tpl2 controls the induction of COX-2 by LPS, we measured COX-2 expression in lysates of Tpl2+/+ and Tpl2–/– macrophages harvested at various time points after LPS stimulation. The results showed that COX-2 induction is significantly lower in Tpl2–/– macrophages. Pretreatment of Tpl2+/+ macrophages with the MEK inhibitor PD98059 also blocked the upregulation of COX-2 by LPS (Figure 2A), suggesting that the impairment of COX-2 induction in Tpl2–/– macrophages results from the failure of LPS to activate ERK.

Fig. 2. Tpl2 is required for optimal induction of COX-2 in LPS-stimulated macrophages. (A) Western blot of cell lysates of unstimulated and LPS-stimulated macrophages probed with antibodies against COX-2 (upper panel) or ERK (lower panel). (B) A western blot of lysates of RAW264.7 cells, treated as indicated, was probed with antibodies that recognize phosphorylated ERK (upper panel), total ERK (middle panel) or COX-2 (lower panel).
Overexpressed Tpl2 exhibits constitutive kinase activity and activates ERK in a variety of cell types (Ceci et al., 1997). This observation allowed us to ask whether the Tpl2 activity is sufficient for COX-2 induction. To this end, we probed lysates of RAW264.7 cells engineered to stably overexpress Tpl2 with antibodies to phosphorylated ERK and total ERK and COX-2. The results confirmed that overexpressed Tpl2 constitutively activates ERK and induces COX-2 expression. A 12 h pretreatment with the MEK inhibitor PD98059 confirmed that ERK activation by Tpl2 is MEK dependent (Figure 2B).
Tpl2 regulates COX-2 transcription and promotes stabilization of the COX-2 mRNA
To determine how Tpl2 regulates the expression of COX-2 in LPS-stimulated primary macrophages, we used quantitative RT–PCR to measure the levels of COX-2 RNA in Tpl2+/+, Tpl2–/– and PD98059-pretreated Tpl2+/+ macrophages, harvested before and 1 h after stimulation with LPS. The results (Figure 3A) showed that the induction of COX-2 RNA by LPS was significantly impaired in Tpl2–/– and PD98059-pretreated Tpl2+/+ macrophages. We conclude that Tpl2 regulates the induction of COX-2 by LPS at the RNA level, via an ERK-dependent pathway.

Fig. 3. LPS signals transduced via Tpl2 induce COX-2 transcription and enhance COX-2 mRNA stability. (A) RT–PCR was carried out on RNA derived from the indicated macrophage cultures, using primers specific for COX-2 or for the housekeeping gene HPRT. (B) COX-2 induction by LPS is regulated by Tpl2 at the level of transcription. (a) A nuclear run-on assay carried out using nuclei from Tpl2+/+ and Tpl2–/– macrophages harvested before and 2 h after stimulation with LPS. The experiment was repeated three times with similar results. (b) The mean value and SD of 32P incorporation measured in PhosphorImager units in one experiment carried out in triplicate. The induction of COX-2 transcription by LPS in Tpl2+/+ cells was statistically significant (p < 0.03). (C) Activation of the COX-2 promoter by LPS depends on signals transduced via Tpl2. (a) Schematic diagram of the COX-2 promoter. (b) Activity of a COX-2 promoter (–891 to +7)–luciferase reporter construct in RAW264.7 cells treated as indicated. RLVs were calculated as described in Materials and methods. Similar results were obtained in three independent experiments. (D) The stability of COX-2 mRNA is impaired in LPS-stimulated macrophages. COX-2 mRNA stability was measured as described in Materials and methods.
To determine whether Tpl2 regulates COX-2 expression at the level of transcription, we carried out nuclear run-on assays (Srivastava et al., 1998) using unstimulated and LPS-stimulated Tpl2+/+ and Tpl2–/– macrophages. Cell nuclei harvested before and 2 h after stimulation were incubated with [α-32P]UTP as described in Materials and methods. RNA isolated from these nuclei 20 min later was hybridized to filter-immobilized COX-2 cDNA as well as to control β-actin and TNF-α cDNAs. The results (Figure 3B) showed that COX-2 transcription was induced in Tpl2+/+ macrophages, following stimulation with LPS (p < 0.03). In Tpl2–/– macrophages, we observed that LPS induced a decrease rather than an increase in the amount of 32P incorporation. TNF-α transcription was induced similarly in Tpl2+/+ and Tpl2–/– macrophages. Therefore, the effects of Tpl2-transduced signals on the transcription of COX-2 were gene specific.
The transcriptional induction of COX-2 in LPS-stimulated cells was modest, suggesting that Tpl2-transduced signals may regulate COX-2 both at the level of transcription and post-transcriptionally. To address this question we examined the stability and subcellular localization of the COX-2 mRNA in LPS-stimulated Tpl2+/+ and Tpl2–/– macrophages. The results showed that whereas the subcellular distribution of the message was not affected by Tpl2 (data not shown), its stability was (t1/2wt = 4.4 h, t1/2KO = 2.9 h) (Figure 3D). Given that LPS signals stabilize the COX-2 mRNA in macrophage cell lines (Barrios-Rodiles et al., 1999; Lasa et al., 2000), these data suggest that Tpl2 signals not only increase COX-2 transcription but also promote stabilization of its message.
To calculate the ratio of the steady-state concentration of COX-2 mRNA in LPS-stimulated Tpl2+/+ and Tpl2–/– macrophages, we used the formula

which is based on a zero-order production, first-order degradation model of gene expression (Hargrove 1993). In this formula, CSS is the steady-state concentration of mRNA, λ is the rate of transcription, t1/2 is the half-life of the mRNA and the superscripts refer to the Tpl2 genotype of the cells. Substituting the experimental values for the transcription rate of COX-2 and the half-life of its mRNA in Tpl2+/+ and Tpl2–/– cells revealed that the differential stability of the message in the two cell types amplifies the difference in COX-2 mRNA levels between them. Thus, whereas the ratio
![]()
is 2.1, the ratio
![]()
is 3.2.
In this paper, we focus on the role of Tpl2 in the regulation of COX-2 transcription.
To further explore the role of Tpl2 in the transcriptional induction of COX-2 by LPS, we used reporter assays designed to address the ability of Tpl2 to regulate the activity of the COX-2 promoter. The transfection efficiency of primary macrophages by electroporation, liposome-mediated DNA transfer (Lipofectamine; Gibco) or non-liposomal approaches (Dosper; Roche) was very low. This problem, combined with the observation that Tpl2 induces COX-2 expression in both primary macrophages and RAW264.7 cells (Figure 2B), prompted us to utilize the RAW264.7 macrophage cell line for these experiments. Exponentially growing RAW264.7 cells were electroporated with a reporter construct containing the luciferase gene under the control of the COX-2 promoter (Figure 3Ca) and kinase-inactive or wild-type Tpl2 constructs, as described in Materials and methods. After 24 h, some of the transfected cultures were stimulated with LPS for 12–14 h. The results showed that LPS increased COX-2 promoter activity, whereas the kinase-inactive Tpl2 construct suppressed this activity in a concentration-dependent manner (Figure 3Cb). Ectopic expression of wild-type Tpl-2 activated the COX-2 promoter. Taken together, these data demonstrated the ability of Tpl2 to control LPS-mediated COX-2 expression in mouse macrophages at the level of transcription and confirmed that the effects of Tpl2 on the regulation of COX-2 can be faithfully reproduced in RAW264.7 cells.
The promoter of COX-2 contains binding sites for NFAT, NF-κB, CREB and c/EBPβ (Figure 3Ca). A previously published report used RT–PCR to show that treatment of Jurkat cells with TPA plus ionomycin activates NFAT and induces COX-2 expression via a Tpl-2-dependent process (de Gregorio et al., 2001). Our data addressing the role of NFAT in the induction of COX-2 revealed that treatment of T cells with TPA plus ionomycin or concanavalin A (ConA) induces NFAT binding activity but does not induce detectable levels of COX-2. Furthermore, they showed that LPS induces COX-2 expression in macrophages but does not induce NFAT binding activity (see Supplementary figure 1 available at The EMBO Journal Online). Taken together, these data suggest that NFAT does not contribute significantly to the physiological regulation of COX-2 expression. Previous studies had shown that NF-κB is dispensable for the induction of COX-2 by LPS (Wadleigh et al., 2000). Consistent with this finding, we showed that nuclear extracts from LPS-treated Tpl2+/+ and Tpl2–/– macrophages bind NF-κB-specific oligonucleotides with similar efficiencies (Dumitru et al., 2000; data not shown). The remaining factors, CREB and c/EBPβ, may be required for the induction of COX-2 in response to LPS (Wadleigh et al., 2000). Of these, C/EBPβ was induced in Tpl2+/+ but not in Tpl2–/– cells at 6 h following stimulation. However, the induction of C/EBPβ was not inhibited by the MEK inhibitor PD98059 (see Supplementary figure 2). Given that PGE2 induction is ERK dependent, we conclude that the differential induction of C/EBPβ in Tpl2+/+ and Tpl2–/– cells may not be critical for the induction of COX-2.
Tpl2 regulates the activation of CREB in LPS-stimulated macrophages
CREB is a target of the ERK pathway (Caivano and Cohen, 2000) and ERK phosphorylation is impaired in LPS-stimulated Tpl2–/– macrophages (Dumitru et al., 2000; Figure 4A). We therefore examined whether CREB phosphorylation is differentially regulated in LPS-stimulated macrophages from Tpl2+/+ and Tpl2–/– mice. To this end, lysates of unstimulated and LPS-stimulated wild-type and Tpl2 KO macrophages were probed with an antibody that specifically recognizes CREB phosphorylated at Ser133 or with an antibody that detects both phosphorylated and non-phosphorylated CREB. Parallel western blots were probed with antibodies that recognize phosphorylated and total ERK. The results showed that CREB phosphorylation is more efficient in Tpl2+/+ macrophages (Figure 4B). The transcription factor ATF1, which forms heterodimers and shares a common phosphorylation domain with CREB, was also differentially phosphorylated (Figure 4B).

Fig. 4. Tpl2 regulates the phosphorylation and DNA binding activity of CREB in LPS-stimulated macrophages. (A) Kinetics of ERK phosphorylation in cell lysates from Tpl2+/+ and Tpl2–/– macrophages stimulated with LPS. (B) Kinetics of CREB/ATF1 phosphorylation in cell lysates from Tpl2+/+ and Tpl2–/– macrophages stimulated with LPS. (C) Induction of CREB DNA binding activity by LPS is Tpl2 dependent. Left panel: nuclear extracts of primary Tpl2+/+ and Tpl2–/– macrophages treated with LPS for 0, 30 or 60 min were analyzed by EMSA for binding to an oligonucleotide containing the CREB binding motif of the COX-2 promoter. Right panel: nuclear extracts from Tpl2+/+ cells treated with LPS for 1 h were incubated with excess unlabeled wild-type (wt) or mutant (mt) oligonucleotide probe prior to EMSA.
We next examined whether the differences in CREB phosphorylation in Tpl2+/+ and Tpl2–/– macrophages translate into differences in DNA binding. The results showed that nuclear extracts from Tpl2+/+ macrophages, harvested at 30 min or 1 h after LPS stimulation, exhibit higher CREB-specific DNA binding activity than extracts from Tpl2–/– macrophages (Figure 4C, left). DNA binding was inhibited by excess unlabeled wild-type but not mutant CREB-binding oligonucleotides (Figure 4C, right).
Tpl2 regulates the activation of CREB by LPS via a p90Rsk- and Msk1-dependent pathway
CREB is a substrate for p90Rsk, a kinase that is activated by ERK-mediated phosphorylation at multiple sites, including Ser381, Thr360 and Ser364 (Deak et al., 1998; Xing et al., 1998; Frodin and Gammeltoft, 1999). Here we show that LPS stimulation induces phosphorylation of p90 RSK in Tpl2+/+ but not in Tpl2–/– macrophages, and that phosphorylation of p90RSK at this site correlates with activation of the kinase (Figure 5A and B). Pretreatment with the MEK inhibitor PD98059 blocked the activation of p90Rsk (Figure 5B). However, Ro318220, an inhibitor of Msk1, did not inhibit p90Rsk activation by LPS, suggesting that Msk1 is not a required regulator of p90Rsk.

Fig. 5. Tpl2 transduces LPS signals leading to the phosphorylation and activation of the protein kinases p90Rsk and Msk1. (A) The phosphorylation of p90Rsk in LPS-stimulated Tpl2–/– macrophages is impaired. Lysates of LPS-stimulated macrophages were probed with antibodies to phosphorylated p90Rsk or to total p90Rsk. (B) p90Rsk activation by LPS in Tpl2–/– macrophages is impaired. Kinase activity is expressed in c.p.m. of [32P]ATP incorporated by p90Rsk immunoprecipitates. The data shown are mean values from triplicate determinations in a representative experiment. Four independent experiments gave similar results. PD is PD98059 and Ro is Ro318220. (C) The phosphorylation of Msk1 in LPS-stimulated Tpl2–/– macrophages is impaired. Lysates of LPS-stimulated macrophages were probed with antibodies to phosphorylated Msk1 or to total Msk1. (D) Msk1 activation by LPS in primary macrophages is Tpl2/ERK dependent. In vitro kinase assays were carried out on Msk1 immunoprecipitates using the synthetic peptide EILSRRPSYRK (CREBtide) as substrate. Kinase activity is expressed in c.p.m. of [32P]ATP incorporated in the substrate. The values shown are mean values from triplicate determinations in a representative experiment. Three independent experiments gave similar results.
Msk1 directly phosphorylates CREB, and is activated by a variety of stimuli including LPS (Deak et al., 1998; Caivano and Cohen, 2000). We therefore examined the contribution of Tpl2 in the activation of Msk1 by LPS. The results showed that LPS induces Msk1 phosphorylation at Ser581, and that phosphorylation is more efficient in Tpl2+/+ than in Tpl2–/– macrophages (Figure 5C). Parallel experiments confirmed that phosphorylation correlates with the enzymatic activation of Msk1. Moreover, whereas the Msk1 inhibitor Ro318220 blocks the activation of Msk1 in wild-type macrophages, the MEK inhibitor PD98059 only partially blocks this outcome (Figure 5D).
To determine whether p90Rsk and Msk1 signals are responsible for CREB phosphorylation in response to LPS, Tpl2+/+ macrophages were pretreated with either PD98059 or Ro318220 and then stimulated with LPS. Western blotting of cell lysates harvested 30 min later, using antibodies specific for the phosphorylated and non-phosphorylated forms of ERK or CREB/ATF1, revealed that whereas PD98059 suppressed ERK phosphorylation, Ro318220 did not (Figure 6A). However, both compounds were found to impair CREB/ATF1 phosphorylation (Figure 6B).
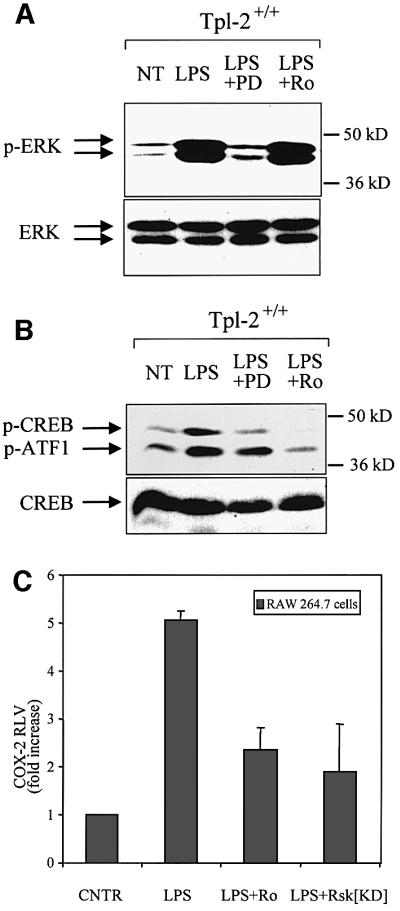
Fig. 6. The phosphorylation of CREB and the activation of the COX-2 promoter by LPS depend on signals transduced by ERK, p90Rsk and Msk1. (A) Lysates of wild-type macrophages treated as indicated were probed with antibodies to phosphorylated ERK or total ERK. PD is PD58059 and Ro is Ro318220. (B) Both PD98059 and Ro318220 inhibit LPS-induced phosphorylation of CREB/ATF1 in Tpl2+/+ macrophages. Cell lysates from cultures identical to those described in (A) were probed with antibodies to phosphorylated CREB and ATF1 or to total CREB. (C) Both Msk1 and p90Rsk contribute to LPS-induced transactivation of the COX-2 promoter. The activity of a COX-2 promoter–luciferase reporter construct in RAW264.7 cells treated as indicated. Data are presented as the mean ± SD of the fold increase of the RLV in stimulated versus unstimulated control cultures.
To determine whether p90Rsk and Msk1 regulate the transcriptional activity of the COX-2 promoter, we transiently transfected RAW264.7 cells with the COX-2 promoter–luciferase reporter construct. Some of the cultures were co-transfected with a kinase-inactive, dominant-negative mutant of p90Rsk, while others were pretreated with the Msk1 inhibitor Ro318220. Luciferase reporter assays were carried out using cell lysates harvested before and after stimulation with LPS. The results showed that inhibition of either kinase interferes with the LPS-mediated induction of the COX-2 promoter (Figure 6C). Therefore, both p90Rsk and Msk1 contribute to the COX-2 transcriptional induction by LPS.
The antibody we used to determine CREB phosphorylation detects only the phosphorylated Ser133 site. Our findings therefore suggest that both Msk1 and p90Rsk phosphorylate CREB at Ser133. The finding that both kinases are required for the phosphorylation of this site suggests that either the two kinases regulate each other or that, in addition to Ser133, they may phosphorylate other non-overlapping sites that affect the efficiency of phosphorylation at Ser133. The regulation of p90Rsk by Msk1 was excluded by the finding that the Msk1 inhibitor Ro318220 does not affect the activation of p90Rsk by LPS (Figure 5B). To address the possibility that p90Rsk is a regulator of Msk1, we co-transfected 293 cells with Msk1 and wild-type, myristoylated or catalytically inactive p90Rsk constructs and examined whether the co-transfected constructs affected the phosphorylation and kinase activity of Msk1. The results showed that p90Rsk constructs neither activate nor inhibit the activity of Msk1 (Figure 7). Epidermal growth factor (EGF), used as a positive control, strongly activated Msk1, but had no effect on a catalytically inactive Msk1 mutant.

Fig. 7. P90Rsk does not regulate Msk1. HEK293 cells were transiently transfected with the indicated FLAG-tagged Msk1 constructs and with the indicated p90Rsk constructs. Control cells were stimulated with EGF. Anti-FLAG immunoprecipitates of lysates derived from these cultures were examined for Msk1 kinase activity. Mean values (± SD) from three independent experiments are shown.
Discussion
LPS, a component of the cell wall of Gram-negative bacteria, is the triggering factor for multiple organ failure during septic shock (Parillo, 1993). The effects of LPS are caused by host-produced cytokines, such as TNF-α and IL-1β, and lipid mediators such as PGE2, which act in an autocrine or paracrine manner to induce and amplify the host response. A major source of TNF-α, IL-1β and PGE2 during sepsis is the macrophages. Our recent work demonstrated that macrophages of Tpl2 KO mice exhibit a post-transcriptional defect in LPS-induced TNF-α synthesis. As a result, these mice are resistant to LPS/d-galactosamine-induced endotoxin shock (Dumitru et al., 2000). In this study we showed that PGE2 synthesis and release from stimulated macrophages is also regulated by Tpl2. Specifically, we showed that LPS-treated Tpl2–/– macrophages release lower levels of PGE2 than Tpl2+/+ macrophages. The synthesis of PGE2 depends on COX-2, which also failed to be induced by LPS in the KO macrophages. In agreement with these data, COX-2 was induced in RAW264.7 macrophages overexpressing Tpl2. These observations combined suggest that Tpl2 is both necessary and sufficient for the induction of COX-2 by LPS. The induction of COX-2 by LPS in wild-type macrophages was inhibited by the MEK inhibitor PD98059, suggesting that COX-2 induction is regulated via a Tpl2/ERK-dependent pathway. PD98059 had been shown earlier to also inhibit TNF-α-induced COX-2 transactivation in epithelial cell lines (Chen et al., 2001). The preceding data combined highlight the importance of the Tpl2/ERK pathway in the induction of COX-2 by LPS and cytokine signals, and suggest that this pathway contributes to the induction of COX-2 in more than one cell type. Tpl2-transduced signals induced COX-2 by regulating both its transcription and the stability of its message. Another enzyme that contributes to prostaglandin synthesis is cytoplasmic phospholipase A2 (cPLA2), which cleaves membrane lipids to release arachidonic acid, the COX-2 target (Syrbu et al., 1999). Although we have not yet fully addressed the regulation of cPLA2, our preliminary data indicate that its phosphorylation in response to LPS in Tpl2–/– macrophages is impaired (our unpublished data).
The data presented in this paper focus on the role of Tpl2-transduced signals on COX-2 transcription. Signals transduced via the Tpl2/ERK pathway regulate COX-2 transcription by regulating CREB phosphorylation and DNA binding following stimulation with LPS. Two kinases targeted by ERK, namely p90Rsk and Msk1, directly phosphorylate CREB and ATF1, a CREB-related transcription factor that forms heterodimers with CREB (Deak et al., 1998; Xing et al., 1998; Frodin and Gammeltoft, 1999). Interestingly, LPS failed to induce phosphorylation and activation of p90Rsk in Tpl2–/– macrophages as well as in Tpl2+/+ macrophages pretreated with the MEK inhibitor PD98059, suggesting that p90Rsk is a bona fide target for the Tpl2/ERK MAPK pathway. Msk1 phosphorylation and activation, on the other hand, was significantly inhibited but not abolished in LPS-stimulated Tpl2–/– macrophages. The residual Msk1 phosphorylation in these cultures may be due to p38MAPK, which also phosphorylates Msk1 (Deak et al., 1998) and is activated normally in LPS-stimulated Tpl2–/– macrophages (Dumitru et al., 2000).
The preceding findings led us to address the role of p90Rsk and Msk1 in the phosphorylation of CREB. The results showed that both kinases are required for the full phosphorylation of CREB at Ser133. Several possible explanations were considered for this puzzling finding. (i) The two kinases may be transregulated. This explanation was excluded, however, because the Msk1 inhibitor Ro318220 did not interfere with the activation of p90Rsk in response to LPS and a catalytically active mutant of p90Rsk did not activate Msk1. (ii) Neither of the two kinases is sufficient to fully phosphorylate CREB at Ser133, suggesting that their activities toward the phosphorylation of this site are additive. This appears unlikely, however, because the inhibition of Msk1 almost completely blocked Ser133 phosphorylation. In agreement with these data, TPA and EGF failed to induce CREB phosphorylation in Msk1 KO embryonic stem cells (Arthur and Cohen, 2000). (iii) In addition to Ser133, the two kinases may phosphorylate other non-overlapping sites that regulate the efficiency of Ser133 phosphorylation. A second phosphorylation site at Ser142, targeted by casein kinase II and Ca2+-calmodulin kinase II, has indeed been identified. However, there is no evidence to date that phosphorylation of Ser133 and Ser142 are interdependent. (iv) One of the two kinases may inhibit the phosphatase that dephosphorylates Ser133. Two phosphatases, PP1 and PP2A, have the potential to dephosphorylate this site. (v) The two kinases may regulate other molecules that bind CREB. Such molecules may function similar to the HTLV-1 Tax protein, which interacts with CREB and promotes its binding to DNA (for a review, see Mayr and Montminy, 2001). Further studies addressing this issue are in progress.
Similar to the phosphorylation of CREB, the activity of the COX-2 promoter following LPS stimulation also depends on the combined action of p90Rsk and Msk1. Inhibition of either kinase inhibited the activation of the promoter by LPS. This correlation suggests that the differential activation of the COX-2 promoter in wild-type and Tpl2 KO macrophages may depend on the differential phosphorylation of CREB via the Tpl2/ERK/p90Rsk-Msk1 pathway. However, p90Rsk and Msk1 may exert additional effects on transcription. Specifically, Msk1, and to a lesser degree the p90Rsk homolog Rsk2, phosphorylate histone H3 at Ser10 and Ser28, and HMG14 at Ser6, and contribute to the nucleosomal response associated with the induction of early response genes (Sassone-Corsi et al., 1999; Thomson et al., 1999). Moreover, p90Rsk phosphorylates the CREB binding histone acetyltransferase CBP (Merienne et al., 2001). The differential ability of Msk1 and p90Rsk to phosphorylate these targets in wild-type and Tpl2 KO cells may also explain why both kinases are required for the activation of the COX-2 promoter by LPS.
C/EBPβ is activated by ERK-mediated phosphorylation in cultured hepatocytes but not in LPS-stimulated macrophages (Nakajima et al., 1993; Studley and Smale, 1999). LPS stimulation of macrophages, instead, induces C/EBPβ expression (Caivano and Cohen, 2000). Our studies confirmed that LPS does not induce phosphorylation and does not increase the DNA binding activity of C/EBPβ in primary macrophages at 1 h following stimulation. In addition, they showed that Tpl2 transduces ERK- independent signals that promote the delayed induction of COX-2 in response to LPS (see Supplementary figure 2). However, given that COX-2 induction and PGE2 synthesis in LPS-simulated macrophages are MEK dependent, C/EBPβ may not play a critical role in COX-2 induction and prostaglandin synthesis in LPS-stimulated primary macrophages.
The preceding data define the role of Tpl2 in an LPS-triggered pathway that stimulates the expression of COX-2 and PGE2 in macrophages. According to the proposed model (Figure 8), LPS triggers a Tpl2-dependent ERK pathway that leads to the phosphorylation and activation of Msk1 and p90Rsk. These kinases phosphorylate CREB, which is critical for COX-2 transactivation. This model addresses the role of Tpl2 in the transcriptional induction of COX-2. However, Tpl2 also promotes the stabilization of the COX-2 mRNA. mRNA stability is often regulated by AU-rich elements in the 3′ untranslated region of the RNA (3′AREs), and COX-2 transcripts possess several 3′AREs (Lasa et al., 2000). Our earlier studies had shown that Tpl2-mediated ERK activation signals play a critical role in the post-transcriptional regulation of TNF-α expression by targeting the TNF-α mRNA 3′AREs. However, in the case of TNF-α, these signals regulate the nucleocytoplasmic RNA transport rather than the stability of the RNA (Dumitru et al., 2000).

Fig. 8. Model of Tpl2-mediated COX-2 transactivation and PGE2 production in response to LPS. LPS engages a Tpl2-dependent pathway, which leads to the activation of ERK and p90RSK downstream of MEK. The same signals, in combination with p38 MAPK-transduced signals, activate Msk1. p90Rsk and Msk1, in turn, phosphorylate and activate CREB, which is critical for the transactivation of COX-2, a key enzyme for the biosynthesis of PGE2. The interrupted line connecting Tpl2 with p38MAPK indicates that non-obligatory signals transduced via Tpl2 may contribute to the activation of p38MAPK by LPS.
The data presented in this report addressed the role of Tpl2 in the transduction of LPS signals that regulate the expression of COX-2 and the synthesis of PGE2 in macrophages. Given the well established role of these molecules in inflammation, the findings reported here clearly demonstrate that Tpl2 plays an important role in the transduction of inflammatory signals. However, these molecules also contribute to oncogenesis. Thus, COX-2 inhibitors decrease the incidence of colon and mammary adenocarcinomas in humans and in genetically susceptible mice (Elder and Paraskeva, 1998; Jones et al., 1999). On the opposite side, intestinal bacterial infections increase the expression of COX-2 in macrophages and other interstitial cells in the intestines, and enhance colon tumorigenesis in Apcmin/+ mice (Newman et al., 2001). In agreement with these findings, Apc–/+/COX-2–/– double mutant mice showed an 85% decrease in polyp induction by comparison with single APC–/+ mutants (Oshima et al., 1996). Finally, Wnt-1, a known oncogene, induces COX-2 expression in mammary epithelial cell lines (Howe et al., 1999). This may contribute to the oncogenic potential of Wnt-1 because expression of COX-2 in mammary and lung carcinoma cell lines correlates with their invasive and metastatic potential (Kundu et al., 2001). The oncogenic effects of COX-2 are prostaglandin mediated in that the COX-2-dependent oncogenic effects of the APCΔ716 mutation are blocked in mice carrying a mutant prostaglandin receptor (EP2) gene (Sonoshita et al., 2001).
Tpl2 contributes to the transduction of signals that originate in the T-cell receptor and activate the transcription factor NFAT (Tsatsanis et al., 1998a). Thus, ConA or anti-CD3 stimulation engages a Tpl2-dependent pathway that activates NFAT and promotes IL-2 secretion and cell cycle entry in T-cell lines. Tpl2-induced NFAT activation was also implicated recently in COX-2 induction in PMA- plus ionomycin-activated Jurkat cells (de Gregorio et al., 2001). This led us to examine the role of NFAT in COX-2 induction in both T cells and macrophages. Our data showed that TPA and ionomycin induced a robust activation of NFAT in Jurkat cells, but failed to induce detectable levels of COX-2. Similarly, ConA, which is known to activate NFAT, failed to induce COX-2 expression in normal T cells. In the same experiment, LPS induced a dramatic increase in COX-2 expression in Tpl2+/+ macrophages in the absence of NFAT activation. We therefore conclude that NFAT does not play an important role in the physiological regulation of COX-2 by Tpl2.
In summary, the data presented here define a pathway that is triggered by LPS and links Tpl2, and its downstream target MEK, with transcription. Moreover, they show that Tpl2 regulates the induction of COX-2 and its product PGE2, in response to LPS. PGE2 is known to contribute to the pathophysiology of sepsis (Astiz et al., 1996), chronic inflammation (Tilley et al., 2001) and neoplasia. The findings presented here, therefore, further implicate Tpl2 in these processes and suggest that it is an excellent target for the development of drugs against septic shock, other inflammatory syndromes and cancer.
Materials and methods
Cell culture, stimulation and PGE2 ELISA
Bone marrow-derived macrophages (BMDM) were cultured as described previously (Dumitru et al., 2000). At day 6, adherent macrophages were replated in six-well plates and used for experiments 18 h later (Dumitru et al., 2000). PD98059 (20 µM; Calbiochem) and Ro318220 (3 µM; Calbiochem) were applied 45 min prior to LPS stimulation. PGE2 was measured in macrophages plated at 2 × 106 cells per dish, using a commercially available PGE2 enzyme immunoassay kit (Cayman Chemicals, An Arbor, MI).
RAW264.7 macrophages were cultured in DMEM supplemented with 10% FBS. To inhibit Msk1 in this cell line we used 5 µM Ro318220. Jurkat cells were cultured in RPMI 1640 and were stimulated with TPA (100 ng/ml) and ionomycin (100 nM). Mouse splenocytes were cultured as described previously (Dumitru et al., 2000).
Nuclear run-on assay
Nuclear run-on assays were carried out as described previously (Srivastava et al., 1998). Briefly, 108 BMDM were either left untreated or treated with LPS from Salmonella typhimurium (1 µg/ml). After 2 h, cells were washed with ice-cold PBS and lysed in an NP-40 lysis buffer. Nuclei isolated from the lysed cells were washed twice in lysis buffer and collected by centrifugation at 1000 g. Following this, 107 nuclei were resuspended in 200 µl of reaction buffer. The in vitro elongation reaction was initiated with the addition of ribonucleotides (ATP, GTP and CTP) to a final concentration of 0.33 mM each, plus 100 µCi [α-32P]UTP. The reaction was carried out for 10 min at 25°C. The labeled RNA was hybridized to 5 µg of filter-immobilized denatured COX-2 cDNA (Oxford Biomedical Research, Oxford, MI), 5 µg of mouse cytoplasmic β-actin cDNA and 5 µg of TNF-α cDNA blotted onto nylon membranes (ZetaProbe GTG; Bio-Rad, Hercules, CA). Filters were washed stringently in 0.1× SSC/0.1% SDS at 65°C. The intensity of hybridization was measured by a PhosphorImager (Molecular Dynamics).
Transfections and reporter assays
COX-2 promoter (–891 to +7 bp)–luciferase reporter constructs were provided by Drs L.-H.Wang and Harvey R.Herschman. RAW264.7 cells (4 × 106) were electroporated (270 V, 950 µF) with 10 µg of the COX-2 luciferase reporter and 2 µg of a Renilla luciferase expression construct (Promega) alone or in combination with wild-type Tpl2, kinase-inactive Tpl2 (K167M) or kinase-inactive p90Rsk (K112R/K464R) (Shimamura et al., 2000) constructs. Transfection efficiency was monitored by electroporation of a GFP expression construct (10 µg) and was ∼5–6%. The relative luciferase value (RLV) was defined as the ratio of the luciferase activity divided by the activity of Renilla luciferase in transfected cell lysates. The RLV of unstimulated cultures was given the arbitrary value of 1. The fold induction was calculated by dividing the RLV of a given culture by the RLV of the unstimulated control. Each experiment was repeated a minimum of three times.
RNA stability assay
Tpl2+/+ and Tpl2–/– macrophages were stimulated with LPS (1 µg/ml). After 1 h, the cells were treated with actinomycin D (10 µg/ml). mRNA was isolated from cell lysates harvested 1, 3, 6 and 9 h later. COX-2 mRNA levels were measured by RT–PCR.
Electrophoretic mobility shift assay (EMSA)
EMSAs were performed as described previously (Eliopoulos et al., 1999). Briefly, cells were resuspended in buffer A [10 mM HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 1 mM DTT, 0.1% (v/v) NP-40, 0.1 mM sodium orthovanadate, 10 mM sodium glycerophosphate and complete proteinase inhibitor mixture] on ice and then spun at 13 500 g for 1 min at 4°C. The nuclear pellets were resuspended in buffer B (20 mM HEPES pH 7.9, 1.5 mM MgCl2, 420 mM NaCl, 1 mM DTT, 0.1 mM sodium o-vanadate, 10 mM sodium glycerophosphate and complete proteinase inhibitor mixture) and incubated at 4°C for 15 min. Following this, they were centrifuged at 13 500 g for 15 min at 4°C. Protein concentration in the supernatants was determined using the Bio-Rad protein assay kit.. The oligonucleotides used as probes were: 5′-GAAGCAGAGAGGGGGA AAAGTTGGTGGG-3′ (NFAT/COX-2), 5′-GAGTCACCACTACGTC ACGTGGAGTCCGCT-3′ (CREB/COX-2) and 5′-GAGTCACCACT GATTCACGTGGAGTCCGCT-3′ (mtCREB/COX-2; underlined bases indicate mutations).
Immunoblotting
SDS–PAGE and immunoblotting were carried out as described previously (Dumitru et al., 2000). The antibodies used were the c/EBPβ-specific antibody C-19 (Santa Cruz Biotechnology), the cyclin D2-specific antibody M20 (Santa Cruz Biotechnology), polyclonal antibodies against Msk1 phosphorylated at Ser581 (Cell Signaling, Beverly, MA) or against total Msk1 (Upstate Biotechnology), the COX-2-specific antibody C-20 (Santa Cruz Biotechnology), antibodies against CREB phosphorylated at Ser133 and ATF1 phosphorylated at Ser63 or against total CREB (Cell Signaling), antibodies against p90Rsk phosphorylated at Ser381 or against total p90Rsk (Cell Signaling), and antibodies against phosphorylated or total ERK (Cell Signaling).
RT–PCR
Total cell RNA (2 µg) was reverse transcribed using the Ambion RetroScript kit. PCR was performed using the oligonucleotides 5′-GTATCAGAACCGCATTGCCTCTGA-3′ and 5′-CGGCTTCCAGTAT TGAGGAGAACAGAT-3′ derived from the murine COX-2 gene and 5′-GTTGGATACAGGCCAGACTTTGTTG-3′ and 5′-GAGGGTAG GCTGGCCTATAGGCT-3′ derived from the hypoxanthine-guanine phosphoribosyltransferase (HPRT) gene. Conditions for PCR amplification were: denaturing at 94°C for 30 s, annealing at 60°C for 1 min and extension at 68°C for 1 min. PCR was carried out for 24 cycles using DNA polymerase (5 U/ml; Roche). RT–PCR gave rise to a single 746 bp COX-2 band and a single 352 bp HPRT band.
Kinase assays
p90Rsk kinase assays were performed using 250 µg of cell lysates and the S6 kinase assay kit from Upstate Biotechnology. For Msk1 kinase assays, cells were lysed by incubating them for 15 min in ice-cold Triton X-100 lysis buffer containing 1 µM microcystin (Sigma). Cell lysates (1 mg) were precleared by incubation with Sepharose G beads for 30 min. Msk1 was immunoprecipitated from these lysates with an antibody pre-absorbed with Sepharose G beads. Immunoprecipitates were washed twice with Triton X-100 lysis buffer and twice with a Tris/EGTA/2-mercaptoethanol-containing buffer. The washed pellets were resuspended in 10 µl of lysis buffer. The kinase reaction was performed in a volume of 50 µl containing 17 µM PKI (PKA inhibitor peptide TTYADFIASGRTGRRNAIHD), 100 µM Msk1 substrate peptide [EILSRRPSYRK (CREBtide) (Deak et al., 1998)], 167 µM [γ-32P]ATP (specific activity ∼500 000 c.p.m./nmol, 10 µM cold ATP and 16.7 mM magnesium acetate. After 10 min at 30°C, the reaction was terminated by pipetting 40 µl of the assay mixture onto a 2 × 2 cm square phosphocellulose paper (Upstate Biotechnology) that binds CREBtide but not ATP. The phosphocellulose paper was washed three times in 0.5% phosphoric acid and once in acetone, and it was counted in a scintillation counter. Kinase assays were repeated a minimum of three times.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We are grateful to Drs H.Herschman, J.Blenis, L.-H.Wang and D.Alessi for providing us with constructs. Moreover, we are grateful to Dr D.Alessi for helpful suggestions on the Msk1 kinase assay. The work was supported by Public Health Service grant R01 CA38047. A.G.E. is supported by a Medical Research Council (MRC, UK) Career Development Award. C.D.D. is a Special Fellow of the Leukemia and Lymphoma Society.
References
- Arthur J.S. and Cohen,P. (2000) MSK1 is required for CREB phosphorylation in response to mitogens in mouse embryonic stem cells. FEBS Lett., 482, 44–48. [DOI] [PubMed] [Google Scholar]
- Astiz M., Saha,D., Lustbader,D., Lin,R. and Rackow,E. (1996) Monocyte response to bacterial toxins, expression of cell surface receptors, and release of anti-inflammatory cytokines during sepsis. J. Lab. Clin. Med., 128, 594–600. [DOI] [PubMed] [Google Scholar]
- Barrios-Rodiles M., Tiraloche,G. and Chadee,K. (1999) Lipopoly saccharide modulates cyclooxygenase-2 transcriptionally and posttranscriptionally in human macrophages independently from endogenous IL-1β and TNF-α. J. Immunol., 163, 963–969. [PubMed] [Google Scholar]
- Caivano M. and Cohen,P. (2000) Role of mitogen-activated protein kinase cascades in mediating lipopolysaccharide-stimulated induction of cyclooxygenase-2 and IL-1β in RAW264 macrophages. J. Immunol., 164, 3018–3025. [DOI] [PubMed] [Google Scholar]
- Ceci J.D., Patriotis,C.P., Tsatsanis,C., Makris,A.M., Kovatch,R., Swing,D.A., Jenkins,N.A., Tsichlis,P.N. and Copeland,N.G. (1997) Tpl-2 is an oncogenic kinase that is activated by carboxy-terminal truncation. Genes Dev., 11, 688–700. [DOI] [PubMed] [Google Scholar]
- Chen C.C., Sun,Y.T., Chen,J.J. and Chang,Y.J. (2001) Tumor necrosis factor-α-induced cyclooxygenase-2 expression via sequential activation of ceramide-dependent mitogen-activated protein kinases, and IκB kinase 1/2 in human alveolar epithelial cells. Mol. Pharmacol., 59, 493–500. [DOI] [PubMed] [Google Scholar]
- de Gregorio R., Iniguez,M.A., Fresno,M. and Alemany,S. (2001) Cot kinase induces cyclooxygenase-2 expression in T cells through activation of the nuclear factor of activated T cells. J. Biol. Chem., 276, 27003–27009. [DOI] [PubMed] [Google Scholar]
- Deak M., Clifton,A.D., Lucocq,L.M. and Alessi,D.R. (1998) Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J., 17, 4426–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinchuk J.E. (1995) Renal abnormalities and an altered inflammatory response in mice lacking cyclooxygenase II. Nature, 378, 406–409. [DOI] [PubMed] [Google Scholar]
- Dumitru C.D. et al. (2000) TNF-α induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway. Cell, 103, 1071–1083. [DOI] [PubMed] [Google Scholar]
- Elder D.J. and Paraskeva,C. (1998) COX-2 inhibitors for colorectal cancer. Nat. Med., 4, 392–393. [DOI] [PubMed] [Google Scholar]
- Eliopoulos A.G., Gallagher,N.J., Blake,S.M.S., Dawson,C.W. and Young,L.S. (1999) Activation of the p38 MAPK pathway by Epstein–Barr virus encoded latent membrane protein 1 (LMP1) co-regulates interleukin-6 and interleukin-8 production. J. Biol. Chem., 274, 16085–16096. [DOI] [PubMed] [Google Scholar]
- Eliopoulos A.G., Davies,C., Blake,S.M.S., Murray,P., Najafipour,S., Tsichlis,P.N. and Young,L.S. (2002) The oncogenic protein kinase Tpl-2/cot contributes to Epstein–Barr virus-encoded latent infection membrane protein 1-induced NF-κB signaling downstream of TRAF2. J. Virol., 76, 4567–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erny K.M., Peli,J., Lambert,J.F., Muller,V. and Diggelmann,H. (1996) Involvement of the Tpl-2/cot oncogene in MMTV tumorigenesis. Oncogene, 13, 2015–2020. [PubMed] [Google Scholar]
- Frodin M. and Gammeltoft,S. (1999) Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol. Cell. Endocrinol., 151, 65–77. [DOI] [PubMed] [Google Scholar]
- Hargrove J.L. (1993) Microcomputer-assisted kinetic modeling of mammalian gene expression. FASEB J., 7, 1163–1170. [DOI] [PubMed] [Google Scholar]
- Herschman H.R. (1996) Prostaglandin synthase 2. Biochim. Biophys. Acta, 1299, 125–140. [DOI] [PubMed] [Google Scholar]
- Howe L.R., Subbaramaiah,K., Chung,W.J., Dannenberg,A.J. and Brown,A.M. (1999) Transcriptional activation of cyclooxygenase-2 in Wnt-1-transformed mouse mammary epithelial cells. Cancer Res., 59, 1572–1577. [PubMed] [Google Scholar]
- Jones M.K., Wang,H., Peskar,B.M., Levin,E., Itani,R.M., Sarfeh,I.J. and Tarnawski,A.S. (1999) Inhibition of angiogenesis by nonsteroidal anti-inflammatory drugs: insight into mechanisms and implications for cancer growth and ulcer healing. Nat. Med., 5, 1418–1423. [DOI] [PubMed] [Google Scholar]
- Kundu N., Yang,Q., Dorsey,R. and Fulton,A.M. (2001) Increased cyclooxygenase-2 (cox-2) expression and activity in a murine model of metastatic breast cancer. Int. J. Cancer, 93, 681–686. [DOI] [PubMed] [Google Scholar]
- Lasa M., Mahtani,K.R., Finch,A., Brewer,G., Saklatvala,J. and Clark,A.R. (2000) Regulation of cyclooxygenase 2 mRNA stability by the mitogen-activated protein kinase p38 signaling cascade. Mol. Cell. Biol., 20, 4265–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Wang,Y., Matsumura,K., Ballou,L.R., Morham,S.G. and Blatteis,C.M. (1999) The febrile response to lipopolysaccharide is blocked in cyclooxygenase-2–/–, but not in cyclooxygenase-1–/– mice. Brain Res., 825, 86–94. [DOI] [PubMed] [Google Scholar]
- Mayr B. and Montminy,M. (2001) Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol., 2, 599–609. [DOI] [PubMed] [Google Scholar]
- Meade E.A., Smith,W.L. and DeWitt,D.L. (1993) Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other non-steroidal anti-inflammatory drugs. J. Biol. Chem., 268, 6610–6614. [PubMed] [Google Scholar]
- Merienne K., Pannetier,S., Harel-Bellan,A. and Sassone-Corsi,P. (2001) Mitogen-regulated rsk2–cbp interaction controls their kinase and acetylase activities. Mol. Cell. Biol., 21, 7089–7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima T., Kinoshita,S., Sasagawa,T., Sasaki,K., Naruto,M., Kishimoto,T. and Akira,S. (1993) Phosphorylation at threonine-235 by a ras-dependent mitogen-activated protein kinase cascade is essential for transcription factor NF-IL6. Proc. Natl Acad. Sci. USA, 90, 2207–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman J.V., Kosaka,T., Sheppard,B.J., Fox,J.G. and Schauer,D.B. (2001) Bacterial infection promotes colon tumorigenesis in ApcMin/+ mice. J. Infect. Dis., 184, 227–230. [DOI] [PubMed] [Google Scholar]
- Oshima M., Dinchuk,J.E., Kargman,S.L., Oshima,H., Hancock,B., Kwong,E., Trzaskos,J.M., Evans,J.F. and Taketo,M.M. (1996) Suppression of intestinal polyposis in ApcΔ716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell, 87, 803–809. [DOI] [PubMed] [Google Scholar]
- Parillo J.E. (1993) Pathogenic mechanisms of septic shock. N. Engl. J. Med., 328, 1471–1477. [DOI] [PubMed] [Google Scholar]
- Patriotis C., Makris,A., Bear,S.E. and Tsichlis,P.N. (1993) Tumor progression locus 2 (Tpl-2) encodes a protein kinase involved in the progression of rodent T-cell lymphomas and in T-cell activation. Proc. Natl Acad. Sci. USA, 90, 2251–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy S.T. and Herschman,H.R. (1994) Ligand-induced prostaglandin synthesis requires expression of the TIS10/PGS-2 prostaglandin synthase gene in murine fibroblasts and macrophages. J. Biol. Chem., 269, 15473–15480. [PubMed] [Google Scholar]
- Sassone-Corsi P., Mizzen,C.A., Cheung,P., Crosio,C., Monaco,L., Jacquot,S., Hanauer,A. and Allis,C.D. (1999) Requirement of Rsk-2 for epidermal growth factor-activated phosphorylation of histone H3. Science, 285, 886–891. [DOI] [PubMed] [Google Scholar]
- Sheng H., Williams,C.S., Shao,J., Liang,P., DuBois,R.N. and Beauchamp,R.D. (1998) Induction of cyclooxygenase-2 by activated Ha-ras oncogene in Rat-1 fibroblasts and the role of mitogen-activated protein kinase pathway. J. Biol. Chem., 273, 22120–22127. [DOI] [PubMed] [Google Scholar]
- Shimamura A., Ballif,B.A., Richards,S.A. and Blenis,J. (2000) Rsk1 mediates a MEK–MAP kinase cell survival signal. Curr. Biol., 10, 127–135. [DOI] [PubMed] [Google Scholar]
- Sonoshita M., Takaku,K., Sasaki,N., Sugimoto,Y., Ushikubi,F., Narumiya,S., Oshima,M. and Taketo,M.M. (2001) Acceleration of intestinal polyposis through prostaglandin receptor EP2 in Apc(Δ716) knockout mice. Nat. Med., 7, 1048–1051. [DOI] [PubMed] [Google Scholar]
- Srivastava R.A.K. and Schonfeld,G. (1998) Measurement of rate of transcription in isolated nuclei by nuclear ‘run-off’ assay. In Rapley,R. and Manning,D.L. (eds), Methods in Molecular Biology: RNA Isolation and Characterization Protocols, Vol. 86. Humana Press, Totowa, NJ. [DOI] [PubMed]
- Studley M. and Smale,S.T. (1999) C/EBPβ regulation and its role in IL-12 induction in activated macrophages. Cold Spring Harb. Symp. Quant. Biol., 64, 206. [Google Scholar]
- Syrbu S.I., WatermanW.H., Molski,T.F., Nagarkatti,D., Hajjar,J.J. and Sha’afi,R.I. (1999) Phosphorylation of cytosolic phospholipase A2 and the release of arachidonic acid in human neutrophils. J. Immunol., 162, 2334–2340. [PubMed] [Google Scholar]
- Thomson S., Clayton,A.L., Hazzalin,C.A., Rose,S., Barratt,M.J. and Mahadevan,L.C. (1999) The nucleosomal response associated with immediate-early gene induction is mediated via alternative MAP kinase cascades: MSK1 as a potential histone H3/HMG-14 kinase. EMBO J., 18, 4779–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilley S.L., Coffman,T.M. and Koller,B.H. (2001) Mixed messages: modulation of inflammation and immune responses by prostaglandins and thromboxanes. J. Clin. Invest., 108, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsatsanis C., Patriotis,C., Bear,S.E. and Tsichlis,P.N. (1998a) The Tpl-2 protooncoprotein activates the nuclear factor of activated T cells and induces interleukin 2 expression in T cells. Proc. Natl Acad. Sci. USA, 95, 3827–3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsatsanis C., Patriotis,C. and Tsichlis,P.N. (1998b) Tpl-2 induces IL-2 expression in T cell lines by triggering multiple signaling pathways that activate NFAT and NF-κB. Oncogene, 17, 2609–2618. [DOI] [PubMed] [Google Scholar]
- Wadleigh D.J., Reddy,S.T., Kopp,E., Ghosh,S. and Herschman,H.R. (2000) Transcriptional activation of the cyclooxygenase-2 gene in endotoxin-treated RAW 264.7 macrophages. J. Biol. Chem., 275, 6259–6266. [DOI] [PubMed] [Google Scholar]
- Xing J., Kornhauser,J.M., Xia,Z., Thiele,E.A. and Greenberg,M.E. (1998) Nerve growth factor activates extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways to stimulate CREB serine 133 phosphorylation. Mol. Cell. Biol., 18, 1946–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]