

Abstract
Understanding the relationship between protein sequences and structures is essential for accurate protein property prediction. We propose BridgeNet, a pre-trained deep learning framework that integrates sequence and structural information through a novel latent environment matrix, enabling seamless alignment of these two modalities. The model’s modular architecture—comprising sequence encoding, structural encoding, and a bridge module—effectively captures complementary features without requiring explicit structural inputs during inference. Extensive evaluations on tasks such as enzyme classification, Gene Ontology annotation, coenzyme specificity prediction, and peptide toxicity prediction demonstrate its superior performance over state-of-the-art models. BridgeNet provides a scalable and robust solution, advancing protein representation learning and enabling applications in computational biology and structural bioinformatics.
Keywords: protein representation learning, sequence-structure integration, deep learning in bioinformatics, protein property prediction
Introduction
Proteins are fundamental macromolecules that play essential roles in virtually all biological processes, including catalyzing metabolic reactions, transmitting cellular signals, and providing structural stability to cells and tissues [1–5]. Understanding the properties of proteins—such as their structure, function, stability, and interactions—is critical for advancing a wide range of scientific and industrial fields, including biotechnology, synthetic biology, and pharmaceutical development [6–9]. However, the vast diversity and complexity of protein sequences and structures in nature present significant challenges for large-scale characterization [10, 11]. High-throughput and accurate methods for protein property prediction are urgently needed to bridge this gap and accelerate advancements in these fields [12, 13].
Traditional experimental techniques, such as X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and cryo-electron microscopy (Cryo-EM), have been instrumental in elucidating protein structures and functions at atomic resolution [14–17]. These methods provide unparalleled insights into protein conformations and interactions, enabling researchers to study mechanistic details of biological processes. However, these experimental approaches are often labor-intensive, expensive, and highly dependent on sample quality and experimental conditions, which limit their scalability and applicability to the vast number of proteins that remain uncharacterized. Additionally, certain classes of proteins, such as intrinsically disordered proteins (IDPs) or transient complexes, pose inherent challenges to these techniques due to their dynamic nature [18–23].
To overcome the limitations of experimental methods, computational approaches have emerged as powerful tools for protein characterization. These methods leverage the growing availability of protein sequence and structure data to predict various protein properties, including structure, function, and interactions [24, 25]. Early computational techniques were largely reliant on physics-based modeling or sequence homology, such as molecular dynamics simulations and comparative modeling. While these approaches achieved some success, their performance was often constrained by computational cost, limited accuracy, and reliance on homologous templates. The advent of machine learning (ML) and deep learning (DL) has revolutionized the field, enabling more accurate and scalable predictions by learning complex patterns from large-scale biological datasets [26, 27].
Recent breakthroughs in ML and DL have led to the development of advanced sequence-based models, such as ESM (Evolutionary Scale Modeling), TAPE, ProtBERT, and ProteinGPT, which are capable of extracting meaningful representations from protein sequences [28–31]. These models capture evolutionary and biophysical patterns that underlie protein structure and function, significantly improving the accuracy of tasks such as fold classification, functional annotation, and stability prediction [32–34]. On the structure-based side, AlphaFold and its successors have achieved remarkable success in predicting three-dimensional protein structures with near-experimental accuracy, even in cases where no homologous templates are available [35]. These advancements have fundamentally transformed the field, opening new avenues for large-scale protein property prediction. However, despite their success, these models often operate independently on sequence or structure data, which may limit their ability to fully capture the relationship between sequence-intrinsic properties and structure-environmental effects.
However, despite their success, these models often operate independently on sequence or structure data, which may limit their ability to fully capture the relationship between sequence-intrinsic properties and structure-environmental effects [36]. To address this limitation, recent approaches such as SABLE have begun to integrate both sequence and structural information, leveraging the complementary nature of these modalities to obtain more comprehensive protein representations [37]. Nevertheless, such strategies typically treat sequence and structure as separate, complementary sources, and may overlook the fact that they are actually different modalities of the same underlying biological entity. In principle, it is possible to pre-train models using both modalities to learn richer, multi-modal representations, while only relying on the more computationally efficient modality during deployment, thus balancing performance and computational cost [38–40].
In this study, we present BridgeNet, a novel pre-trained model that integrates both protein sequence and structure information during the pre-training process to generate robust and biologically meaningful protein representations. The model is grounded on the biological premise that a protein’s sequence inherently determines its intrinsic properties, while its structure reflects the interplay between these intrinsic properties and environmental factors. By leveraging structural information to guide the encoding process during pre-training, BridgeNet embeds a deep understanding of protein structure within its representations. Importantly, this design enables the model to perform downstream protein property prediction tasks without requiring additional structural information, while still retaining the benefits of structure-informed representations. Extensive experiments across multiple protein property prediction benchmarks demonstrate that BridgeNet significantly outperforms traditional methods and state-of-the-art models. This integrative approach highlights the power of embedding structural insights into sequence-based models, offering a scalable and efficient solution for advancing protein characterization.
Materials and methods
Datasets
Pre-training dataset
Our pre-training dataset is derived from UniRef 50, a comprehensive and widely utilized collection of protein sequences sourced from the UniProt database [41–43]. UniRef 50 clusters protein sequences that share 50% or greater identity into single representative entries, thereby effectively reducing redundancy while preserving the diversity of sequences across millions of proteins. This clustering approach ensures a balance between computational efficiency and biological relevance, making UniRef 50 an ideal resource for pre-training models designed for tasks such as protein function prediction, structural analysis, and a wide range of bioinformatics applications [44–46]. By encompassing a broad spectrum of sequence diversity, this dataset enables the model to generalize effectively across a variety of protein families and functional classes.
For the pre-training process, the pre-training dataset was filtered to exclude sequences longer than 1024 and deduplicated, resulting in a final dataset of 63,919,579 records. Additionally, we randomly selected 10% of the records and downloaded their structural data for structure-enhanced training. The dataset was partitioned into training, validation, and test sets in a 7:1:2 ratio, ensuring a systematic division that supports both model development and rigorous evaluation [47]. This partitioning strategy allows the model to leverage a large proportion of data for training, facilitating the learning of meaningful representations, while reserving sufficient data for validation to monitor overfitting and for testing to provide an unbiased assessment of model performance. The robustness of this approach ensures that the pre-trained model is not only optimized during training but also capable of maintaining high generalization accuracy across unseen protein sequences [48, 49].
Protein function prediction
The dataset curated by Ruijie Quan et al. is specifically designed for two critical tasks in bioinformatics: Enzyme Commission (EC) number prediction and Gene Ontology (GO) term prediction, both of which are pivotal in understanding protein functions and their roles in biological processes [50, 51]. The EC number dataset focuses on predicting enzyme classifications based on the chemical reactions they catalyze, providing insights into enzyme functionality and aiding in the systematic annotation of protein roles [52]. In addition, the dataset supports enzyme reaction classification, which aims to assign proteins to specific enzymatic reaction classes according to the full four-level EC hierarchy, enabling detailed characterization of catalytic activities [50]. Detailed information about the datasets can be found in the ‘Protein Function Prediction’ section of the Supplementary Materials.
Coenzyme classification
The dataset for the Coenzyme Classification task was obtained through the work of Ye et al., which carefully curated a collection of non-redundant sequences categorized by their cofactor binding specificities [53]. Cofactors such as NAD+ (nicotinamide adenine dinucleotide) and NADP+ (nicotinamide adenine dinucleotide phosphate) play crucial roles in enzymatic reactions, serving as electron carriers and influencing metabolic pathways. NAD+ is primarily involved in catabolic processes, such as cellular respiration, where it facilitates energy production, whereas NADP+ is key in anabolic processes like biosynthesis and redox reactions. Understanding the binding specificities of enzymes to these cofactors is essential for deciphering metabolic pathways, optimizing enzyme functionality, and advancing applications in fields like bioremediation and sustainable chemical synthesis. Detailed information about the datasets can be found in the ‘Coenzyme Classification’ section of the Supplementary Materials.
Peptide toxicity prediction
Utilizing the methodology proposed by Hossein Ebrahimikondori et al., we performed a comparative analysis on a protein benchmark dataset to advance the field of protein toxicity prediction [54]. This task holds substantial importance in bioinformatics, toxicology, and drug discovery, as the accurate identification of toxic proteins is critical for mitigating adverse effects in pharmaceutical development, ensuring patient safety, and guiding the design of safer therapeutics. By addressing this challenge, researchers can also gain deeper insights into the molecular mechanisms underlying toxicity, which has implications for both fundamental biology and applied sciences. Detailed information about the datasets can be found in the ‘Peptide Toxicity Prediction’ section of the Supplementary Materials.
The framework of the BridgeNet model
Our study is grounded on the foundational premise that the sequence of a protein dictates its intrinsic properties, while its three-dimensional structure determines its functional capabilities. This relationship suggests a transformative potential within certain latent projection spaces, whereby the representation of protein sequences can be converted into the representation of protein structures. This concept underlies the principles behind protein structure prediction tools such as AlphaFold.
In the context of protein representation, when structural and sequence representations can be interconverted within certain latent projection spaces, it becomes unnecessary to input the complete protein structure into the representation network. This approach significantly reduces the time and computational power required during inference. Building upon this understanding, we introduce a novel conceptual framework termed the ‘environment matrix’. We hypothesize that the transformation of protein sequence representations into structural representations is influenced by specific environmental matrices. In an ideal scenario, protein sequences and structures should correspond perfectly; however, environmental factors can lead the same protein to adopt multiple distinct structures. These matrices encompass various factors such as solution conditions (e.g., pH, ionic strength, and temperature), the involvement of molecular chaperones, and post-translational modifications like phosphorylation, methylation, and acetylation.
To address this complexity, we propose ‘BridgeNet’, a deep learning-based framework designed to integrate structural and sequence representations through distinct modules (Fig. 1). Within this framework, the latent environment matrix plays a crucial role in mapping identical sequence representations to diverse conformational states of proteins, thereby acknowledging the multifaceted nature of protein structure determination influenced by environmental conditions. This approach not only enhances our understanding of protein dynamics but also advances the predictive capabilities of computational models in structural biology.
Figure 1.

The overall framework of BridgeNet. (A) the pretraining process of BridgeNet. During pretraining, structural information of proteins is utilized to guide the formation of protein encoding. (B) the complete prediction framework integrating BridgeNet. Protein sequences are encoded via BridgeNet to obtain sequence representations, which are subsequently processed through various downstream task modules to generate corresponding prediction results.
In BridgeNet, the network is divided into three modules: the structure representation module, the sequence representation module, and the Bridge module. These modules are responsible for encoding the protein’s structure, encoding the protein’s sequence, and unifying the sequence and structural representations of proteins through a latent environment matrix, respectively.
Embedding module
In our network, the Sequence Representation Module is primarily responsible for processing protein sequence information, while the Structure Representation Module focuses on the characterization of protein structural information. For sequence encoding, we employ the BLOSUM-62 matrix, which is a widely used amino acid substitution scoring matrix derived from evolutionary information. Specifically, each amino acid residue in the protein sequence is converted into a fixed-length vector by extracting the corresponding row from the BLOSUM-62 matrix. By concatenating the encoding vectors of all residues in order, a sequence representation matrix is constructed, as Fig. 2A shown. This matrix is subsequently flattened and fed into the Sequence Representation Module for further extraction of high-level sequence features.
Figure 2.

Workflow of the embedding module. (A) Illustration of the process by which protein sequences are encoded into BLOSUM-62 encoding vectors using the BLOSUM-62 matrix. (B) Construction of the protein structure graph by establishing edges between residues within a 5 Å distance threshold, resulting in the corresponding adjacency matrix.
For protein structural information, the processing procedure is relatively more complex. Specifically, we first retrieve the corresponding PDB ID based on the UniProt ID of the original data, and then download the PDB structure file of the target protein from the RCSB Protein Data Bank, which is a well-established resource for experimentally determined protein structures. Subsequently, we extract the primary sequence as well as the three-dimensional coordinates of the Cα atom for each residue in the specified chain. Using a distance threshold of 5 Å, we calculate the pairwise spatial distances between all residues, and construct edges between those whose distances are less than or equal to 5 Å, thereby generating the protein structure adjacency matrix (i.e., the edge index of the graph structure). This adjacency matrix, together with the spatial coordinates of the nodes, is used as the input for the graph neural network, as Fig. 2B shown.
The sequence representation module
The Sequence Representation Module is a neural network architecture designed to encode and decode protein sequences through a sophisticated transformer-based framework. This network comprises several key components. The module begins with an input projector, which maps the amino acid dimension to the desired output dimension through a linear layer followed by a ReLU activation function to introduce non-linearity (Eq. 1). At the output stage, the output projector performs the inverse operation by mapping the output dimension back to the amino acid dimension through a linear layer, ensuring compatibility with sequence-level tasks.
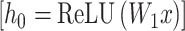 |
(1) |
To incorporate sequence order information, a positional encoding module is employed. This module adds position-specific information to the input sequence, enabling the model to capture the relative and absolute positional relationships between amino acids within the sequence.
As shown in Eq. 2, for each position index 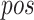 and dimension
and dimension  :
:
 |
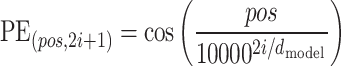 |
(2) |
The core of the architecture consists of transformer encoder and decoder layers. The transformer encoder, composed of multiple stacked layers, processes the input sequence to extract high-level latent representations (Eq. 3). Each encoder layer includes multi-head self-attention mechanisms and feedforward neural networks. The multi-head attention is parameterized by the specified number of attention heads, while the feedforward network is expanded by a predefined factor (expand) to increase the model’s capacity for capturing complex sequence features. The transformer decoder mirrors the encoder structure, enabling the transformation of the encoded representations back into sequence space (Eq. 4).
 |
(3) |
 |
(4) |
This network architecture leverages the capabilities of transformers to model complex dependencies and interactions within protein sequences. By utilizing mechanisms such as multi-head attention and positional encoding, it provides a robust and flexible framework for protein representation learning, enabling the extraction of meaningful sequence features for downstream biological tasks (Eq. 6).
 |
(6) |
The structure representation module
The Structure Representation Module is a novel graph-based neural network architecture designed for encoding and decoding graph-structured data, with a primary focus on protein representations. This model leverages the power of graph convolutional networks (GCNs) to capture intricate structural relationships within graph data, which is particularly relevant in the context of protein–protein interaction networks or residue-level structural representations.
The encoding component of this architecture consists of two graph convolutional layers. The first layer applies a GCN-based transformation to project the input node features from their initial feature dimension to a hidden feature dimension. This transformation is followed by batch normalization, which ensures numerical stability during training, and a ReLU activation function to introduce non-linearity. The second graph convolutional layer takes the hidden representation and maps it to the output feature dimension, which serves as the encoded representation of the graph. Notably, no activation function is applied at this stage, as the encoded representation is directly utilized for downstream tasks that may require a linear feature space.
The decoding component reconstructs node-level features from the graph-level representation. It begins with a fully connected layer that projects the graph-level representation back to the hidden feature dimension. Batch normalization and a ReLU activation function are applied to refine the features and enable effective learning. Subsequently, two additional graph convolutional layers sequentially transform these features back to the original node feature dimension, thereby reconstructing the input graph structure.
To aggregate node-level features into a single graph-level representation, the architecture employs a global mean pooling operation. This mechanism computes the mean of all node features within a graph, producing a compact representation that is invariant to the number of nodes in the graph. Such a representation is particularly advantageous when dealing with graphs of varying sizes, as is common in biological datasets.
The bridge module
At the core of the Bridge module is a linear transformation layer. This layer performs a mapping from the input feature space to an output feature space of identical dimensionality. This absence of a bias component preserves the scale and centrality of the input embeddings, which is particularly advantageous for maintaining the intrinsic properties of the features during their transformation. Such a design choice is critical when the consistent alignment of feature distributions is required across different components of the network (Eq. 7).
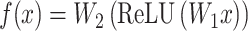 |
(7) |
where 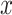 can represent either the Structure Representation or the Sequence Representation. Both
can represent either the Structure Representation or the Sequence Representation. Both  and
and  are transformation matrices of the same shape, which can be regarded as environmental matrices. This transformation does not alter the dimensionality of
are transformation matrices of the same shape, which can be regarded as environmental matrices. This transformation does not alter the dimensionality of  ; rather, the matrices operate on the latent space representations to convert a Structure Representation into a Sequence Representation, or vice versa. It is important to note that the networks employed for converting structure to sequence and sequence to structure are distinct, with independent parameters, although the vector dimensionality remains unchanged during the transformation.
; rather, the matrices operate on the latent space representations to convert a Structure Representation into a Sequence Representation, or vice versa. It is important to note that the networks employed for converting structure to sequence and sequence to structure are distinct, with independent parameters, although the vector dimensionality remains unchanged during the transformation.
Based on this design, the module can be abstracted as an encoder-like structure, where BridgeNet transforms the input from one modality into another. Unlike conventional encoder-decoder frameworks, we do not utilize the hidden encoding representations generated by this structure. Instead, this module functions in conjunction with Eq. 8 as part of the training process, with the objective of enforcing the alignment between structure and sequence representations through this transformation mechanism.
The downstream task module will integrate the sequence encoding module from the pre-trained model with an MLP (Multi-Layer Perceptron) for joint training and generate the corresponding prediction output.
Loss function design and optimization strategies
The training of BridgeNet is divided into two stages. In the first stage, the pretraining phase, the encoding network is simultaneously provided with protein structural and sequential information to facilitate network training. In the second stage, after the completion of pretraining, the encoding network undergoes further task-specific training by incorporating downstream tasks to accomplish various objectives. The overall workflow and loss calculation during the pretraining phase are illustrated in Fig. 3, and the details are provided in the Supplementary Materials.
Figure 3.

Data flow and loss calculation of BridgeNet during the pretraining phase.
The specific number of training epochs was determined based on the convergence of sequence, graph, and bridge losses across epochs, as shown in Fig. S1. Once pretraining was completed, the weights of the sequence representation module were extracted and combined with various downstream task modules for joint training. As Fig. S1 shows, the Graph Rec Loss and Seq Rec Loss show minor changes due to the large dataset, as both submodules quickly stabilize with limited further improvement. In contrast, the significant decrease in Bridge Loss highlights enhanced alignment between structure and sequence. The slight increase in Seq Rec Loss may result from structural guidance introducing challenging-to-recover information, likely due to homologous proteins. This plot corresponds to the loss trends on the validation set during cross-validation. Based on the Bridge Loss trend, pretraining will stop at Epoch 12.
The details of the model’s pretraining and training processes can be found in the ‘Computational Resources and Training Details’ section of the Supplementary Materials.
Results
Protein function prediction
BridgeNet achieves state-of-the-art performance across both enzyme-related and GO-related protein function prediction tasks (Table 1, Fig. 4A). Specifically, it attains an Fmax of 86.6% for EC number prediction, an accuracy of 89.3% for enzyme reaction classification, and Fmax scores of 48.8%, 66.7%, and 49.6% for GO-BP, GO-MF, and GO-CC respectively. Compared to the second-best method, CDConv, BridgeNet improves EC number prediction by 4.6% (Fmax: 82.0% versus 86.6%) and enzyme reaction classification by 0.8% (accuracy: 88.5% versus 89.3%). For GO-MF, the improvement is 1.3% (65.4% versus 66.7%).
Table 1.
Performance comparison of methods on protein function prediction (Fmax for EC/GO, accuracy for enzyme reaction). Bold values indicate the best performance for each metric
| Method | Enzyme-related tasks | GO-related tasks | |||
|---|---|---|---|---|---|
| EC number | Enzyme reaction | BP | MF | CC | |
| ResNet [55] | 0.605 | 24.1 | 0.280 | 0.405 | 0.304 |
| LSTM [55] | 0.425 | 11.0 | 0.225 | 0.321 | 0.283 |
| Transformer [55] | 0.238 | 26.6 | 0.264 | 0.211 | 0.405 |
| GCN [56] | 0.320 | 67.3 | 0.252 | 0.195 | 0.329 |
| GAT [57] | 0.368 | 55.6 | 0.284 | 0.317 | 0.385 |
| GVP [58] | 0.489 | 65.5 | 0.326 | 0.426 | 0.420 |
| 3DCNN [59] | 0.077 | 72.2 | 0.240 | 0.147 | 0.305 |
| GraphQA [60] | 0.509 | 60.8 | 0.308 | 0.329 | 0.413 |
| New IEConv [61] | 0.735 | 87.2 | 0.374 | 0.544 | 0.444 |
| GearNet [62] | 0.810 | 85.3 | 0.400 | 0.281 | 0.430 |
| CDConv [63] | 0.820 | 88.5 | 0.453 | 0.654 | 0.479 |
| BridgeNet | 0.866 | 89.3 | 0.488 | 0.667 | 0.496 |
Figure 4.

Performance of BridgeNet on protein function prediction tasks. (A) Comparative analysis of method performance across five protein function prediction tasks. All performance scores are normalized to the range [0, 1] for visual comparison across different tasks. (B) tSNE visualization of sequence and structure representations in the latent space for proteins.
In addition, we performed t-distributed stochastic neighbor embedding (tSNE) to visualize the dimensionality reduction of both structure representations and sequence representations, as shown in Fig. 4B. The results indicate that the distributions of structure and sequence representations exhibit a high degree of overlap in the latent space. Although some subtle differences can still be observed in specific local regions, the overall patterns of the two representations are largely consistent. These findings further confirm that the structure and sequence representations have achieved a high level of unification in the latent space, thus providing a solid foundation for subsequent downstream tasks.
BridgeNet exhibits superior performance compared to the majority of existing methods, including traditional sequence-based approaches such as ResNet, LSTM, and Transformer, as well as advanced graph-based models like GCN, GAT, and GVP [64–67]. This indicates that BridgeNet effectively combines its design components to leverage both sequence and structural information for enhanced predictive performance.
Coenzyme classification
In addition, we conducted a comparative analysis of our proposed method, BridgeNet, against various state-of-the-art approaches in the coenzyme classification task. The results of this analysis are summarized in Table 2, which highlights the performance of BridgeNet across key evaluation metrics.
Table 2.
Performance comparison of methods for coenzyme classification
| Method | ACC | AUC | Precision | Recall |
|---|---|---|---|---|
| LogisticRegression [68] | 0.733 | 0.810 | 0.713 | 0.744 |
| Rossmann-toolbox [69] | 0.793 | 0.842 | 0.801 | 0.813 |
| S2SNet-recommend [70] | 0.78 | 0.821 | 0.782 | 0.722 |
| S2Snet-complete [70] | 0.819 | 0.844 | 0.808 | 0.793 |
| INSIGHT-ESM2 [28] | 0.946 | 0.983 | 0.944 | 0.949 |
| ProteinBERT [71] | 0.937 | 0.943 | 0.866 | 0.922 |
| SABLE [37] | 0.973 | 0.982 | 0.917 | 0.955 |
| BridgeNet | 0.970 | 0.989 | 0.924 | 0.963 |
The performance of BridgeNet was systematically compared with several established methods, including Logistic Regression, Rossmann-toolbox, S2SNet-recommend, S2SNet-complete, INSIGHT-ESM2, ProteinBERT, and SABLE, as summarized in Table 2 and Fig. 5A. Across most evaluation metrics—accuracy (ACC), area under the curve (AUC), and recall—BridgeNet demonstrated superior performance. Notably, BridgeNet achieved the highest AUC (0.989) and recall (0.963), and its accuracy (0.970) was nearly equivalent to that of SABLE (0.973), substantially surpassing both traditional algorithms, such as Logistic Regression (0.733 ACC, 0.810 AUC) and other advanced approaches like S2SNet-complete (0.819 ACC, 0.844 AUC).
Figure 5.

Performance comparison of coenzyme classification methods. (A) Stacked metrics of various methods. (B) Time and memory efficiency of INSIGHT-ESM2 versus BridgeNet.
It is worth noting that while SABLE yielded slightly higher accuracy (0.973), BridgeNet outperformed it in terms of AUC and recall, and also achieved a high precision (0.924). Additionally, although INSIGHT-ESM2 attained the highest precision (0.944), this was accompanied by significantly greater computational requirements, including increased prediction time and memory usage (see Fig. 5B). In contrast, BridgeNet offers a more resource-efficient solution while maintaining leading overall performance, making it particularly advantageous for large-scale applications or resource-constrained environments. Taken together, these results highlight the practical effectiveness and superior comprehensive performance of BridgeNet compared to existing methods.
Peptide toxicity prediction
Based on the methodology established by Hossein Ebrahimikondori et al., we extended their approach to address the peptide toxicity prediction task [54]. The comparative results of this study are detailed in Table 3, which comprehensively evaluates the performance of various methods across key metrics.
Table 3.
Performance comparison of methods for peptide toxicity prediction
| Method | F1-score(%) | MCC(%) | AUC | AUPR |
|---|---|---|---|---|
| BLAST [72] | 80.0 | 80.1 | - | - |
| BLAST-score [72] | 78.9 | 77.5 | 86.8 | 81.8 |
| InterProScan [73] | 34.7 | 40.2 | - | - |
| HmmSearch [74] | 18.5 | 30.7 | - | - |
| ClanTox [75] | 62.0 | 60.4 | 90.3 | 61.2 |
| ToxinPred-RF [76] | 66.7 | 63.8 | 94.8 | 71.6 |
| ToxinPred-SVM [76] | 67.7 | 64.8 | 93.8 | 71.2 |
| Toxify(original) | 71.5 | 69.0 | 93.0 | 74.3 |
| Toxify(re-trained) | 48.6 | 45.0 | 87.2 | 52.4 |
| ToxDL [77] | 80.9 | 79.3 | 98.9 | 91.3 |
| ToxIBTL [78] | 83.0 | 81.6 | 95.3 | 84.7 |
| tAMPer(sequence-only) [54] | 85.4 | 84.2 | 98.8 | 90.8 |
| tAMPer(structure-only) [54] | 63.6 | 60.8 | 94.0 | 67.9 |
| tAMPer [54] | 86.0 | 85.0 | 99.2 | 91.6 |
| BridgeNet | 92.9 | 90.0 | 98.2 | 96.6 |
The results presented in Fig. 6 underscore the superior predictive performance of BridgeNet, which consistently outperforms most existing methods across key evaluation metrics, including F1-score, Matthews Correlation Coefficient (MCC), area under the Receiver Operating Characteristic curve (auROC), and area under the Precision-Recall curve (auPRC). Notably, BridgeNet achieves the highest F1-score of 92.9% and MCC of 90.0%, representing a significant improvement over alternative machine learning-based methods such as Toxify, ToxDL, and ToxIBTL. Furthermore, BridgeNet demonstrates highly competitive performance in auPRC (96.6%), underscoring its robustness in capturing subtle patterns of peptide toxicity and effectively balancing sensitivity and specificity.
Figure 6.

Performance comparison between BridgeNet and other methods across four key metrics for peptide toxicity prediction.
Although BridgeNet achieves slightly lower auROC (98.2%) compared to the second-best performing method, tAMPer, which attains an auROC of 99.2%, it surpasses tAMPer in F1-score (92.9% versus 86.0%), MCC (90.0% versus 85.0%), and auPRC (96.6% versus 91.6%). This indicates that while tAMPer marginally excels in distinguishing toxic from non-toxic peptides based on auROC, BridgeNet demonstrates superior overall classification accuracy and precision-recall trade-offs, which are particularly critical in high-stakes predictive tasks such as toxicity prediction.
In contrast, traditional sequence-based approaches, such as BLAST and InterProScan, exhibit substantially inferior performance, with significantly lower F1-scores and MCC values. These results highlight the inherent limitations of non-machine learning (non-ML) methods in addressing the complexity of peptide toxicity prediction and emphasize the advantages of advanced ML-based algorithms.
Overall, the results establish BridgeNet as a new benchmark in peptide toxicity prediction, setting a higher performance standard among both ML- and non-ML-based methods. Its ability to achieve superior outcomes across multiple metrics demonstrates its potential as a powerful tool for addressing challenges in peptide analysis and related computational biology tasks. Future research may focus on further optimizing the model to improve its generalizability and applicability across diverse datasets and peptide families, as well as exploring strategies to enhance auROC performance without compromising other metrics.
Ablation study
To validate the effectiveness of the sequence-structure alignment mechanism, a series of ablation studies were conducted. Specifically, we compared the performance of the original BridgeNet framework with two simplified variants: one utilizing only structural encoding and the other using only sequence encoding. The ablation study demonstrates that the BridgeNet (Full Model) consistently outperforms its simplified variants, BridgeNet (Sequence) and BridgeNet (Structure), across all three subtasks: Protein Function Prediction, Coenzyme Classification, and Peptide Toxicity Prediction (Fig. S2). These results highlight the effectiveness of the sequence-structure alignment mechanism, which enables the Full Model to integrate complementary information from both sequence and structure data, yielding significant performance gains across diverse predictive tasks. More details for the impact of each module of BridgeNet on the performance of the aforementioned downstream tasks can be found in the ‘Ablation Study’ section of the Supplementary Materials.
Discussion
BridgeNet represents a significant advancement in protein representation learning by integrating sequence and structural information into a unified framework. This approach addresses a critical gap in existing methodologies, which often rely on either sequence- or structure-based data in isolation, thereby limiting their capacity to capture the intricate relationships between intrinsic protein properties and their functional manifestations. By bridging these two dimensions, BridgeNet provides a comprehensive and biologically meaningful representation of proteins, enabling robust performance across diverse bioinformatics tasks.
Our results demonstrate that BridgeNet achieves state-of-the-art or near state-of-the-art performance in multiple tasks, including protein function prediction, coenzyme classification, and peptide toxicity prediction. Notably, in the coenzyme classification task, while BridgeNet’s performance was slightly behind the highest-performing method, it offered significant computational advantages, such as reduced memory and time requirements. This balance between predictive accuracy and computational efficiency highlights BridgeNet’s suitability for large-scale applications and resource-constrained environments, making it a practical tool for a wide range of scientific and industrial use cases.
A key innovation of BridgeNet lies in its ability to incorporate environmental factors into its representations through the introduction of a latent environment matrix. This design enables the model to account for the dynamic nature of protein structures influenced by conditions such as pH, temperature, and post-translational modifications. This adaptability makes BridgeNet particularly valuable for studying complex systems, including intrinsically disordered proteins and conformationally flexible biomolecules, which are often challenging for traditional methods to characterize.
Looking ahead, there are several avenues for future development. First, extending BridgeNet to incorporate additional data modalities, such as protein–protein interactions, evolutionary information, or small-molecule binding data, could further enhance its predictive power and applicability. Second, improving the interpretability of the model’s learned representations could provide deeper insights into the underlying biological mechanisms, facilitating experimental validation and hypothesis generation. Techniques such as attention visualization or feature attribution analyses may help elucidate how sequence and structural features contribute to specific predictions.
Furthermore, the scalability of BridgeNet could be enhanced by leveraging recent advancements in protein structure prediction, such as AlphaFold, to expand its applicability to proteins with limited structural data. Fine-tuning the model on domain-specific datasets, such as those focused on antibody design or enzyme engineering, may also unlock new opportunities for specialized applications.
In summary, BridgeNet is a versatile and efficient tool that advances the integration of sequence and structural data for protein characterization. Its robust architecture and broad applicability position it as a valuable resource for the bioinformatics and structural biology communities. By addressing current limitations and exploring new directions, BridgeNet has the potential to drive further innovation in the computational study of proteins.
Conclusion
In this study, we introduced BridgeNet, a novel pre-trained model that integrates protein sequence and structure information to generate unified and biologically meaningful protein representations. By leveraging the intrinsic relationship between sequence and structure and minimizing their representational discrepancy, BridgeNet effectively captures both the inherent properties of proteins and the environmental influences on their structure. Comprehensive evaluations across diverse protein property prediction tasks, including peptide toxicity and enzyme function classification, demonstrate that BridgeNet outperforms existing state-of-the-art methods. These results highlight the effectiveness of integrating sequence and structure information for protein characterization. BridgeNet not only advances the field of protein representation learning but also provides a scalable and robust framework with broad applications in computational biology, structural bioinformatics, and beyond.
Key Points
State-of-the-Art Performance: Extensive experiments across diverse downstream tasks, including enzyme classification, Gene Ontology annotation, coenzyme specificity prediction, and peptide toxicity prediction, demonstrate that BridgeNet consistently outperforms existing state-of-the-art models, achieving superior accuracy and generalization capabilities.
Scalable and Robust Design: The modular architecture of BridgeNet—comprising sequence encoding, structural encoding, and a bridge module—facilitates efficient training and inference, eliminating the need for explicit structural inputs during downstream predictions while retaining structure-informed representations.
Broad Applicability: BridgeNet provides a scalable solution for protein characterization, with potential applications in computational biology, structural bioinformatics, drug discovery, and enzyme engineering, paving the way for advancements in both basic and applied sciences.
Supplementary Material
Acknowledgements
This research is supported by the Science and Technology Development Fund of Macau (0004/2025/RIA1) and Macao Polytechnic University (RP/CAI-01/2023) with Submission Code fca.380c.3a45.b.
Contributor Information
Yilin Ye, Faculty of Applied Sciences, Macao Polytechnic University, R. de Luís Gonzaga Gomes, Macao, 999078, China.
Hongliang Duan, Faculty of Applied Sciences, Macao Polytechnic University, R. de Luís Gonzaga Gomes, Macao, 999078, China.
Yuguang Mu, School of Biological Sciences, Nanyang Technological University, 50 Nanyang Avenue, 639798, Singapore.
Lei Wu, College of Mechanical and Electronic Engineering, China University of Petroleum (East China), 66 Changjiang West Road, Huangdao District, Qingdao, 266580, China.
Jingjing Guo, Faculty of Applied Sciences, Macao Polytechnic University, R. de Luís Gonzaga Gomes, Macao, 999078, China.
Author contribution
J.G. conceived and designed the study. Y.Y. collected the data and developed the model. Y.Y., Y.M., H.D., L.W., and J.G. wrote and revised the manuscript.
Data availability
The implementation of the proposed method will be made publicly available on GitHub at the following repository: https://github.com/iRinYe/BridgeNet.
References
- 1. Wang X, Gan M, Wang Y. et al. Comprehensive review on lipid metabolism and RNA methylation: Biological mechanisms, perspectives and challenges. Int J Biol Macromol 2024;270:132057. 10.1016/j.ijbiomac.2024.132057. [DOI] [PubMed] [Google Scholar]
- 2. Sies H, Mailloux RJ, Jakob U. Fundamentals of redox regulation in biology. Nat Rev Mol Cell Biol 2024;25:701–19. 10.1038/s41580-024-00730-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cai R, Shan Y, Du F. et al. Injectable hydrogels as promising in situ therapeutic platform for cartilage tissue engineering. Int J Biol Macromol 2024;261:129537. 10.1016/j.ijbiomac.2024.129537. [DOI] [PubMed] [Google Scholar]
- 4. Zacco E, Broglia L, Kurihara M. et al. RNA: The unsuspected conductor in the Orchestra of Macromolecular Crowding. Chem Rev 2024;124:4734–77. 10.1021/acs.chemrev.3c00575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chang W, Chen L, Chen K. The bioengineering application of hyaluronic acid in tissue regeneration and repair. Int J Biol Macromol 2024;270:132454. 10.1016/j.ijbiomac.2024.132454. [DOI] [PubMed] [Google Scholar]
- 6. Akinsemolu AA, Onyeaka H, Odion S. et al. Exploring bacillus subtilis: Ecology, biotechnological applications, and future prospects. J Basic Microbiol 2024;64:2300614. 10.1002/jobm.202300614. [DOI] [PubMed] [Google Scholar]
- 7. Ali SS, Alsharbaty MHM, Al-Tohamy R. et al. A review of the fungal polysaccharides as natural biopolymers: Current applications and future perspective. Int J Biol Macromol 2024;273:132986. 10.1016/j.ijbiomac.2024.132986. [DOI] [PubMed] [Google Scholar]
- 8. Huo D, Wang X. A new era in healthcare: The integration of artificial intelligence and microbial. Med Nov Technol Devices 2024;23:100319. 10.1016/j.medntd.2024.100319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lu X, Qian S, Wu X. et al. Research Progress of protein complex systems and their application in food: A review. Int J Biol Macromol 2024;265:130987. 10.1016/j.ijbiomac.2024.130987. [DOI] [PubMed] [Google Scholar]
- 10. Townsend DR, Towers DM, Lavinder JJ. et al. Innovations and trends in antibody repertoire analysis. Curr Opin Biotechnol 2024;86:103082. 10.1016/j.copbio.2024.103082. [DOI] [PubMed] [Google Scholar]
- 11. Eren AM, Banfield JF. Modern microbiology: Embracing complexity through integration across scales. Cell 2024;187:5151–70. 10.1016/j.cell.2024.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Abdelkader GA, Kim J-D. Advances in protein-ligand binding affinity prediction via deep learning: A comprehensive study of datasets, data preprocessing techniques, and model architectures. Curr Drug Targets 2024;25:1041–65. 10.2174/0113894501330963240905083020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhao N, Wu T, Wang W. et al. Review and comparative analysis of methods and advancements in predicting protein complex structure. Interdiscip Sci Comput Life Sci 2024;16:261–88. 10.1007/s12539-024-00626-x. [DOI] [PubMed] [Google Scholar]
- 14. Kousar F, Khan MI, Khokhar AM. et al. Identification of the structure of a protein by x-ray crystallography and NMR spectroscopy. Res Med Sci Rev 2024;2:1621–33. [Google Scholar]
- 15. Desani GV. Structural biology techniques in drug discovery: From crystal structures to therapeutic applications. Multidisc J Mol Biol Biochem 2024;1:16–23. [Google Scholar]
- 16. Zhang X, Li S, Zhang K. Cryo-EM: A window into the dynamic world of RNA molecules. Curr Opin Struct Biol 2024;88:102916. 10.1016/j.sbi.2024.102916. [DOI] [PubMed] [Google Scholar]
- 17. Chen L, Ruan X, Li X. et al. Molecular interactions in biological systems: Technological applications and innovations. Comput Mol Biol 2024;14:21. 10.5376/cmb.2024.14.0021. [DOI] [Google Scholar]
- 18. Orand T, Jensen MR. Binding mechanisms of intrinsically disordered proteins: Insights from experimental studies and structural predictions. Curr Opin Struct Biol 2025;90:102958. 10.1016/j.sbi.2024.102958. [DOI] [PubMed] [Google Scholar]
- 19. Giraldo-Castaño MC, Littlejohn KA, Avecilla ARC. et al. Programmability and biomedical utility of intrinsically-disordered protein polymers. Adv Drug Deliv Rev 2024;212:115418. 10.1016/j.addr.2024.115418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gupta MN, Uversky VN. Protein structure–function continuum model: Emerging nexuses between specificity, evolution, and structure. Protein Sci 2024;33:e4968. 10.1002/pro.4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garg A, González-Foutel NS, Gielnik MB. et al. Design of Functional Intrinsically Disordered Proteins. Protein Eng Des Sel 2024;37:gzae004. 10.1093/protein/gzae004. [DOI] [PubMed] [Google Scholar]
- 22. Vemulapalli S. Targeting intrinsically disordered proteins (IDPs) in drug discovery. In: Rudrapal M, (ed.), Computational Methods for Rational Drug Design. John Wiley & Sons, Ltd, 2025, 493–517. [Google Scholar]
- 23. Maiti S, Singh A, Maji T. et al. Experimental methods to study the structure and dynamics of intrinsically disordered regions in proteins. Curr Res Struct Biol 2024;7:100138. 10.1016/j.crstbi.2024.100138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang Y, Li S, Meng K. et al. Machine learning for sequence and structure-based protein–ligand interaction prediction. J Chem Inf Model 2024;64:1456–72. 10.1021/acs.jcim.3c01841. [DOI] [PubMed] [Google Scholar]
- 25. Srivastava G, Liu M, Ni X. et al. Machine learning techniques to infer protein structure and function from sequences: A comprehensive review. In: Kloczkowski A, Kurgan L, Faraggi E, (eds.), Prediction of Protein Secondary Structure. New York, NY: Springer US, 2025, 79–104. [DOI] [PubMed] [Google Scholar]
- 26. Asif S, Wenhui Y, ur-Rehman S. et al. Advancements and prospects of machine learning in medical diagnostics: Unveiling the future of diagnostic precision. Arch Computat Methods Eng 2024;32:853–83. 10.1007/s11831-024-10148-w. [DOI] [Google Scholar]
- 27. Rane NL, Paramesha M, Choudhary SP. et al. Machine learning and deep learning for big data analytics: A review of methods and applications. Partn Univ Int Innov J 2024;2:172–97. 10.5281/zenodo.12271006. [DOI] [Google Scholar]
- 28. Xu L. Deep learning for protein-protein contact prediction using evolutionary scale modeling (ESM) feature. In: Jin H, Pan Y, Lu J, (eds.), Artificial Intelligence and Machine Learning. Singapore: Springer Nature, 2024, 98–111. [Google Scholar]
- 29. Rao R, Bhattacharya N, Thomas N. et al. Evaluating protein transfer learning with TAPE. Adv Neural Inf Process Syst 2019;32:9689–701. [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang Y, Zhu G, Li K. et al. HLAB: Learning the BiLSTM features from the ProtBert-encoded proteins for the class I HLA-peptide binding prediction. Brief Bioinform 2022;23:bbac173. 10.1093/bib/bbac173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xiao Y, Sun E, Jin Y. et al. ProteinGPT: Multimodal LLM for protein property prediction and structure understanding arXiv. 2024. 10.48550/arXiv.2408.11363. [DOI]
- 32. Gladstone Sigamani G, Vincent PMDR. Multimodal neural network for enhanced protein stability prediction by integration of contact scores and spatial maps. Results Eng 2024;24:103440. 10.1016/j.rineng.2024.103440. [DOI] [Google Scholar]
- 33. Listov D, Goverde CA, Correia BE. et al. Opportunities and challenges in design and optimization of protein function. Nat Rev Mol Cell Biol 2024;25:639–53. 10.1038/s41580-024-00718-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rahimzadeh F, Mohammad Khanli L, Salehpoor P. et al. Unveiling the evolution of policies for enhancing protein structure predictions: A comprehensive analysis. Comput Biol Med 2024;179:108815. 10.1016/j.compbiomed.2024.108815. [DOI] [PubMed] [Google Scholar]
- 35. OpenProteinSet: Training data for structural biology at scale. https://proceedings.neurips.cc/paper_files/paper/2023/hash/0eb82171240776fe19da498bef3b1abe-Abstract-Datasets_and_Benchmarks.html (accessed 2025-01-19).
- 36. Zhou L, Tao C, Shen X. et al. Unlocking the potential of enzyme engineering via rational computational design strategies. Biotechnol Adv 2024;73:108376. 10.1016/j.biotechadv.2024.108376. [DOI] [PubMed] [Google Scholar]
- 37. Li J, Chen X, Huang H. et al. Sable: Bridging the gap in protein structure understanding with an empowering and versatile pre-training paradigm. Brief Bioinform 2025;26:bbaf120. 10.1093/bib/bbaf120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang C, Wang Q, Li Y. et al. The historical evolution and significance of multiple sequence alignment in molecular structure and function prediction. Biomolecules 2024;14:1531. 10.3390/biom14121531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kumar N, Srivastava R. Deep learning in structural bioinformatics: Current applications and future perspectives. Brief Bioinform 2024;25:bbae042. 10.1093/bib/bbae042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Israr J, Alam S, Siddiqui S. et al. Advances in structural bioinformatics. In: Singh V, Kumar A, (eds.), Advances in Bioinformatics. Singapore: Springer Nature, 2024, 35–70. [Google Scholar]
- 41. Suzek BE, Huang H, McGarvey P. et al. UniRef: Comprehensive and non-redundant UniProt reference clusters. Bioinformatics 2007;23:1282–8. 10.1093/bioinformatics/btm098. [DOI] [PubMed] [Google Scholar]
- 42. Strodthoff N, Wagner P, Wenzel M. et al. UDSMProt: Universal deep sequence models for protein classification. Bioinformatics 2020;36:2401–9. 10.1093/bioinformatics/btaa003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dohan, D.; Gane, A.; Bileschi, M. L.; Belanger, D.; Colwell, L. Improving protein function annotation via unsupervised pre-training: Robustness, efficiency, and insights. In Proceedings of the 27th ACM SIGKDD Conference on Knowledge Discovery & Data Mining; KDD’21; Association for Computing Machinery: New York, NY, USA, 2021; pp 2782–2791. [Google Scholar]
- 44. The UniProt Consortium . UniProt: The universal protein knowledgebase in 2025. Nucleic Acids Res 2025;53:D609–17. 10.1093/nar/gkae1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lau AM, Bordin N, Kandathil SM. et al. Exploring structural diversity across the protein universe with the encyclopedia of domains. Science 2024;386:eadq4946. 10.1126/science.adq4946. [DOI] [PubMed] [Google Scholar]
- 46. Barone F, Russo ET, Villegas Garcia EN. et al. Protein family annotation for the unified human gastrointestinal proteome by DPCfam clustering. Sci Data 2024;11:568. 10.1038/s41597-024-03131-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen Y, Xu Y, Liu D. et al. An end-to-end framework for the prediction of protein structure and fitness from single sequence. Nat Commun 2024;15:7400. 10.1038/s41467-024-51776-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gao X, Cao C, He C. et al. Pre-training with a rational approach for antibody sequence representation. Front Immunol 2024;15:15. 10.3389/fimmu.2024.1468599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu Y, Liu D, Gong H. Improving the prediction of protein stability changes upon mutations by geometric learning and a pre-training strategy. Nat Comput Sci 2024;4:840–50. 10.1038/s43588-024-00716-2. [DOI] [PubMed] [Google Scholar]
- 50. Quan R, Wang W, Ma F. et al. Clustering for protein representation learning.
- 51. Gligorijević V, Renfrew PD, Kosciolek T. et al. Structure-based protein function prediction using graph convolutional networks. Nat Commun 2021;12:168. 10.1038/s41467-021-23303-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ghosh S, Baltussen MG, Ivanov NM. et al. Exploring emergent properties in enzymatic reaction networks: Design and control of dynamic functional systems. Chem Rev 2024;124:2553–82. 10.1021/acs.chemrev.3c00681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ye Y, Jiang H, Xu R. et al. The INSIGHT platform: Enhancing NAD(P)-dependent specificity prediction for Co-factor specificity engineering. Int J Biol Macromol 2024;278:135064. 10.1016/j.ijbiomac.2024.135064. [DOI] [PubMed] [Google Scholar]
- 54. Ebrahimikondori H, Sutherland D, Yanai A. et al. Structure-aware deep learning model for peptide toxicity prediction. Protein Sci 2024;33:e5076. 10.1002/pro.5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rao R, Bhattacharya N, Thomas N. et al. Evaluating protein transfer learning with TAPE. Adv Neural Inf Process Syst 2019;32:9689–701. [PMC free article] [PubMed] [Google Scholar]
- 56. [1609.02907] semi-supervised classification with graph convolutional networks. https://arxiv.org/abs/1609.02907 (accessed 2025-02-27).
- 57. [1710.10903] graph attention networks. https://arxiv.org/abs/1710.10903 (accessed 2025-02-27).
- 58. [2009.01411] learning from protein structure with geometric vector Perceptrons. https://arxiv.org/abs/2009.01411 (accessed 2025-02-27).
- 59. Derevyanko G, Grudinin S, Bengio Y. et al. Deep convolutional networks for quality assessment of protein folds. Bioinformatics. 2018;34:4046–53. 10.1093/bioinformatics/bty494. [DOI] [PubMed] [Google Scholar]
- 60. Baldassarre F, Hurtado DM, Elofsson A. et al. GraphQA: Protein model quality assessment using graph convolutional networks. Bioinformatics. 2021;37:360–6. 10.1093/bioinformatics/btaa714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. [2205.15675] contrastive representation learning for 3D protein structures. https://arxiv.org/abs/2205.15675 (accessed 2025-02-27).
- 62. [2203.06125] protein representation learning by geometric structure Pretraining. https://arxiv.org/abs/2203.06125 (accessed 2025-02-27).
-
63.
9_continuous_discrete_convolutio.pdf - Google
 . https://drive.google.com/file/d/1937AHyN3tt1sodrTZiU-D3KIdFkE9dxK/view (accessed 2025-02-27).
. https://drive.google.com/file/d/1937AHyN3tt1sodrTZiU-D3KIdFkE9dxK/view (accessed 2025-02-27).
- 64. Song Z, Zang Z, Wang Y. et al. Set-CLIP: Exploring aligned semantic from low-alignment multimodal data through a distribution view arXiv. 2024. 10.48550/arXiv.2406.05766. [DOI]
- 65. Hu B, Tan C, Xu Y. et al. ProtGO: Function-guided protein modeling for unified representation learning. 2024.
- 66. Ye Q, Li J, Chen X. et al. Sable: Bridging the gap in protein structure understanding with an empowering and versatile pre-training paradigm. Res Square 2024. 10.21203/rs.3.rs-4647798/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hu B, Tan C, Wu L. et al. Advances of deep learning in protein science: A comprehensive survey arXiv. 2024. 10.48550/arXiv.2403.05314. [DOI]
- 68. Logistic regression-guided identification of cofactor specificity-contributing residues in enzyme with sequence datasets partitioned by catalytic properties. ACS Synthetic Biology 2022;11:3973–85. 10.1021/acssynbio.2c00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rossmann-toolbox: A deep learning-based protocol for the prediction and design of cofactor specificity in Rossmann fold proteins . Brief Bioinform 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Liu Y, Munteanu CR, Kong Z. et al. Identification of coenzyme-binding proteins with machine learning algorithms. Comput Biol Chem 2019;79:185–92. 10.1016/j.compbiolchem.2019.01.014. [DOI] [PubMed] [Google Scholar]
- 71. Brandes N, Ofer D, Peleg Y. et al. ProteinBERT: A universal deep-learning model of protein sequence and function. Bioinformatics 2022;38:2102–10. 10.1093/bioinformatics/btac020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Altschul SF, Madden TL, Schäffer AA. et al. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 1997;25:3389–402. 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Quevillon E, Silventoinen V, Pillai S. et al. InterProScan: Protein domains identifier. Nucleic Acids Res 2005;33:W116–20. 10.1093/nar/gki442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Potter SC, Luciani A, Eddy SR. et al. HMMER web server: 2018 update. Nucleic Acids Res 2018;46:W200–4. 10.1093/nar/gky448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. ClanTox: A classifier of short animal toxins . Nucleic Acids Res 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gupta S, Kapoor P, Chaudhary K. et al. In Silico approach for predicting toxicity of peptides and proteins. PloS One 2013;8:e73957. 10.1371/journal.pone.0073957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Pan X, Zuallaert J, Wang X. et al. ToxDL: Deep learning using primary structure and domain embeddings for assessing protein toxicity. Bioinformatics. 2020;36:5159–68. 10.1093/bioinformatics/btaa656. [DOI] [PubMed] [Google Scholar]
- 78. Wei L, Ye X, Sakurai T. et al. ToxIBTL: Prediction of peptide toxicity based on information bottleneck and transfer learning. Bioinformatics. 2022;38:1514–24. 10.1093/bioinformatics/btac006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The implementation of the proposed method will be made publicly available on GitHub at the following repository: https://github.com/iRinYe/BridgeNet.