

Abstract
VE-cadherin is the essential adhesion molecule in endothelial adherens junctions, and the regulation of protein tyrosine phosphorylation is thought to be important for the control of adherens junction integrity. We show here that VE-PTP (vascular endothelial protein tyrosine phosphatase), an endothelial receptor-type phosphatase, co-precipitates with VE-cadherin, but not with β-catenin, from cell lysates of transfected COS-7 cells and of endothelial cells. Co-precipitation of VE-cadherin and VE-PTP required the most membrane-proximal extracellular domains of each protein. Expression of VE-PTP in triple-transfected COS-7 cells and in CHO cells reversed the tyrosine phosphorylation of VE-cadherin elicited by vascular endothelial growth factor receptor 2 (VEGFR-2). Expression of VE-PTP under an inducible promotor in CHO cells transfected with VE-cadherin and VEGFR-2 increased the VE-cadherin-mediated barrier integrity of a cellular monolayer. Surprisingly, a catalytically inactive mutant form of VE-PTP had the same effect on VE-cadherin phosphorylation and cell layer permeability. Thus, VE-PTP is a transmembrane binding partner of VE-cadherin that associates through an extracellular domain and reduces the tyrosine phosphorylation of VE-cadherin and cell layer permeability independently of its enzymatic activity.
Keywords: angiogenesis/cadherin/endothelial permeability/leukocyte extravasation/receptor protein tyrosine phosphatases
Introduction
The regulation of endothelial cell contacts is of central importance for the physiological role of the endothelium as a barrier between blood and tissue and for the formation and maintenance of blood vessels. The control of blood vessel permeability and leukocyte extravasation, as well as the formation and outgrowth of blood vessels, are dependent on the opening and closure, or the dissociation and formation, of inter-endothelial cell junctions (Dejana et al., 2000; Vestweber, 2000). Adherens junctions are essential for the integrity of endothelial cell contacts, and VE-cadherin, the most prominent adhesion molecule of endothelial adherens junctions, is directly involved in the maintenance of these contacts in vitro and in vivo (Lampugnani et al., 1992; Gotsch et al., 1997; Corada et al., 1999). In addition, VE-cadherin has been reported to be essential for vascular endothelial growth factor (VEGF)-mediated endothelial survival during embryonic angiogenesis (Carmeliet et al., 1999).
Like most other cadherins, VE-cadherin is associated with the catenins, intracellular proteins that link the cadherins to the actin cytoskeleton (Kemler, 1993; Huber et al., 1996). β-catenin and plakoglobin can bind directly to the C-terminus of cadherins and link it to α-catenin, which in turn binds either directly or indirectly to microfilaments. This anchoring to the cytoskeleton is generally essential for optimal cadherin-mediated adhesive functions (Aberle et al., 1996). Several different mechanisms have been proposed for the regulation of cadherin adhesive activity, such as changes in the composition of the cadherin–catenin complex, phosphorylation of different components in the complex and alterations in the interaction of the complex with the actin cytoskeleton (Gumbiner, 2000).
Tyrosine phosphorylation of the cadherin–catenin complex has often been proposed as a mechanism that regulates the stability of adherens junctions (Daniel and Reynolds, 1997). Tyrosine phosphorylation of β-catenin by various kinases correlates with the inhibition of cadherin-mediated adhesion (Matsuyoshi et al., 1992; Behrens et al., 1993; Hamaguchi et al., 1993; Shibamoto et al., 1994). The stimulation of the EGF receptor had the same effect, and this receptor was even found to associate with β-catenin (Hoschuetzky et al., 1994). However, it is not yet known whether the phosphorylation of β-catenin is directly responsible for the weakening of cell contacts, and further substrates for these kinases are likely to exist at adherens junctions. Indeed, it has been shown for fibroblasts transfected with an E-cadherin–α-catenin fusion protein that src weakened E-cadherin-mediated cell contacts, even though β-catenin could no longer be involved in this process (Takeda et al., 1995).
In the aforementioned studies, the classic cadherins E-cadherin and N-cadherin were phosphorylated rather weakly or not at all. This is in contrast to HUVECs, in which VE-cadherin, in addition to β-catenin and plakoglobin, was strongly phosphorylated on stimulation with VEGF (Esser et al., 1998). Furthermore, increased confluence of endothelial cells in culture was accompanied by a decrease in VE-cadherin tyrosine phosphorylation (Lampugnani et al., 1997).
The regulation of tyrosine phosphorylation results from the coordinated and competing actions of kinases and phosphatases. The cytoplasmic PTP SHP-2 was reported to bind to β-catenin, a process that could be influenced by thrombin (Ukropec et al., 2000). β-catenin also binds to the receptor PTPs RPTPκ (Fuchs et al., 1996), RPTPβ/ζ (Meng et al., 2000) and LAR (Kypta et al., 1996). The catenin p120ctn was found to bind to, and become dephosphorylated by, SHP-1 and RPTPµ (Zondag et al., 2000). Direct association was also found for cadherins and two PTPs. Cytoplasmic PTP1B (Balsamo et al., 1998) binds to N-cadherin, and RPTPµ was demonstrated to associate with E-cadherin, N-cadherin and R-cadherin (Brady-Kalnay et al., 1998). In each of these examples, the respective phosphatase binds to the cytoplasmic part of the cadherin. Interactions between the extracellular domains of a cadherin and an RPTP have not yet been described.
VE-PTP (vascular endothelial protein tyrosine phosphatase) was recently identified as the mouse homologue of the receptor-type human PTP β (HPTPβ) and was found to be selectively expressed in endothelia based on in situ hybridizaton analysis of embryonic and postnatal tissue sections (Fachinger et al., 1999). In co-precipitation experiments, VE-PTP was found to associate specifically with the tyrosine kinase receptor Tie-2, but not with VEGF receptor 2 (VEGFR-2). The interaction was mediated via the intracellular parts of the proteins and led to the selective dephosphorylation of Tie-2 (Fachinger et al., 1999).
Here, we have tested whether VE-PTP would interact with VE-cadherin. Surprisingly, and in contrast to previously known RPTP–cadherin interactions, we found that VE-PTP and VE-cadherin associate with each other via their membrane-proximal extracellular domains, independently of their cytoplasmic tails. No association was found between VE-PTP and β-catenin. Triple-transfection experiments revealed that VE-PTP reverses VEGFR-2-mediated tyrosine phosphorylation of VE-cadherin. In addition, the expression of VE-PTP via an inducible promotor in VE-cadherin/VEGFR-2-transfected CHO cells reduced VE-cadherin phosphorylation and increased the cell monolayer barrier function. Our results suggest a role for VE-PTP in the regulation of VE-cadherin-mediated cell contacts.
Results
Association of VE-PTP with VE-cadherin, but not with β-catenin
In an attempt to identify an RPTP that would interact with VE-cadherin, we tested whether the endothelial-specific VE-PTP would associate with VE-cadherin in co-immunoprecipitations from double-transfected COS-7 cells. Cells were co-transfected with VE-cadherin and an N-terminally truncated recombinant form of mouse VE-PTP (Flag-VE-PTP) (Fachinger et al., 1999) containing the complete cytoplasmic domain, the transmembrane region, the most membrane-proximal fibronectin type III (FNIII) repeat and a Flag-tag replacing the rest of the extracellular domain (Figure 1A). Immunoprecipitations with an anti-Flag antibody, followed by immunoblotting with anti-VE-cadherin antibodies, demonstrated that VE-cadherin was efficiently co-precipitated with VE-PTP (Figure 1B). No VE-cadherin was precipitated from COS cells that had been transfected with VE-cadherin only (Figure 1B). Another transmembrane protein, junctional adhesion molecule (JAM), could not be co-precipitated with VE-PTP when co-transfected with Flag-VE-PTP (Figure 1B). Interestingly, in contrast to other RPTPs, no interaction was seen with β-catenin, which was tested in similar co-precipitation experiments with COS cells co-transfected with VE-PTP and β-catenin (Figure 1B).

Fig. 1. Association of VE-cadherin and Flag-VE-PTP in COS-7 cells. (A) Schematic representation of the putative domain structure of VE-PTP and truncated VE-PTP where all extracellular domains, except the most membrane-proximal domain, are replaced by a Flag-tag (Flag-VE-PTP). TM, transmembrane domain. (B) COS-7 cells were transiently transfected either with VE-cadherin alone, with VE-cadherin and Flag-VE-PTP, with JAM alone, with JAM and Flag-VE-PTP, with β-catenin alone or with β-catenin and Flag-VE-PTP. Cell lysates were immunoprecipitated with anti-Flag antibodies, and precipitated proteins were transferred to filters and analysed by immunoblotting with anti-Flag antibodies (top). The same filters were re-probed with anti-VE-cadherin or anti-JAM or anti-β-catenin (middle). Similarly, total cell lysates were immunoblotted for these proteins (bottom). The arrowhead marks the heavy chain of IgG. Molecular mass markers (in kDa) are shown on the left.
Cloning of the full-length mouse VE-PTP cDNA
In order to analyse the interaction of full-length native VE-PTP and VE-cadherin, we cloned the full-length cDNA. The obtained cDNA contained a 5′ untranslated leader of 130 bp, an open reading frame of 5994 bp encoding a polypeptide of 1998 amino acids (Figure 2) and a 3′ untranslated region of 32 bp. Structural prediction programs (SMART, EMBL) allowed us to define 17 extracellular FNIII repeats, one putative transmembrane domain and one intracellular putative phosphatase domain. Sequence comparison revealed 83% identity of the full-length amino acid sequence with the human homologue HPTPβ. The intracellular part of the protein sequence was 94% identical to the corresponding part of HPTPβ and 54% identical to the RPTPs Dep-1 and Byp-1. Thus, the full-length sequence of mouse VE-PTP confirms the close homology to HPTPβ that had already been suggested by the analysis of the partial VE-PTP cDNA clone. VE-PTP belongs to the small subclass III of RPTPs that are characterized by exclusively bearing FNIII-like repeats in the extracellular domain and only a single catalytic domain in the cytoplasmic portion.

Fig. 2. Deduced amino acid sequence of VE-PTP. The predicted signal peptide and the transmembrane region are indicated by bold underlining. The conserved catalytic domain that is homologous to other members of the PTP family is defined by thin underlining. The beginning of each predicted FNIII motif is marked by an arrow.
The cytoplasmic domain of VE-cadherin is not required for association with VE-PTP
Other RPTPs that associate with cadherin–catenin complexes were found to associate with either catenins or, in the case of RPTPµ, the cytoplasmic domain of cadherins. In order to define the domain of VE-cadherin that is required for the interaction with VE-PTP, we generated a truncated form of VE-cadherin (VE-cad extracellular cadherin, VE-cad EC) lacking the cytoplasmic tail. COS cells, co-transfected with full-length VE-PTP and either full-length VE-cadherin or truncated VE-cad EC, were analysed by immunoprecipitation with affinity-purified polyclonal antibodies against VE-PTP followed by immunoblotting with a monoclonal antibody (mAb) against VE-cadherin. We found that full-length VE-cadherin was co-precipitated with full-length VE-PTP (Figure 3B, lane 1), demonstrating that native VE-PTP associates with VE-cadherin. Again, VE- cadherin was not precipitated with anti-VE-PTP antibodies if VE-PTP was not co-transfected (Figure 3B, lane 2). Surprisingly, truncated VE-cadherin lacking the cytoplasmic tail was as efficiently co-precipitated as full-length VE-cadherin (Figure 3B, lane 3), suggesting that the interaction between VE-PTP and VE-cadherin requires either the extracellular part or the transmembrane region of VE-cadherin. In addition, this result rules out the necessity of intracellular VE-cadherin-associated proteins for the association of VE-cadherin and VE-PTP.

Fig. 3. VE-cadherin does not require its cytoplasmic tail for association with full-length VE-PTP. (A) Schematic representation of the domain structure of full-length VE-cadherin and truncated VE-cadherin lacking its complete cytoplasmic domain (VE-cad EC). (B) COS cells were transfected either with full-length VE-PTP and full-length VE-cadherin (VE-PTP/VE-cadherin), with VE-cadherin alone (VE-cadherin), with full-length VE-PTP and truncated VE-cadherin lacking its complete cytoplasmic domain (VE-PTP/VE-cad EC) or with the truncated version of VE-cadherin alone (VE-cad EC). Cell lysates were subjected to immunoprecipitations with anti-VE-PTP antibodies, and precipitated proteins were immunoblotted either with anti-VE-cadherin antibodies (upper) or with anti-VE-PTP antibodies (middle). Note that full-length and truncated VE-cadherin were co-precipitated with VE-PTP. To control transfection efficiency, total cell lysates were immunoblotted for VE-cadherin. The asterisk marks the degradation product of VE-cadherin. Molecular mass markers (in kDa) are shown on the left.
Association of VE-PTP and VE-cadherin is mediated via the membrane-proximal extracellular parts of each protein
To determine which part of VE-PTP interacts with VE-cadherin, three fusion proteins were generated, consisting of parts of VE-PTP and the tight junction protein JAM (Figure 4A). One of them contained the Flag-tagged 17th extracellular FNIII repeat as the only VE-PTP-derived part, fused at its C-terminus to the transmembrane and cytoplasmic domain of JAM (VE–JAM). Two other fusion proteins each contained the Flag-tagged, complete extracellular part of JAM and the complete cytoplasmic part of VE-PTP, in one case connected by the transmembrane region of JAM (JAM–VE1) and in the other case connected by the transmembrane region of VE-PTP (JAM–VE2). In three separate experiments, each of these three Flag-tagged fusion proteins was co-transfected into COS cells together with a truncated form of VE-cadherin that lacked the cytoplasmic domain. Immunoprecipitations were performed with the anti-Flag antibody. As shown in Figure 4B, VE-cadherin was only co-precipitated if the fusion protein containing the extracellular FNIII repeat of VE-PTP was co-transfected. We conclude that the transmembrane domain of VE-PTP is not required and that the 17th, membrane-proximal FNIII repeat is sufficient for the interaction with VE-cadherin.

Fig. 4. The 17th FNIII repeat of VE-PTP is sufficient for binding to VE-cadherin. (A) Schematic representation of the domain structure of full-length VE-PTP (dark grey), full-length JAM (light grey) and three chimeric fusion proteins containing an N-terminal Flag-tag and various parts of VE-PTP and JAM: the cytoplasmic tail of VE-PTP and the extracellular and transmembrane domain of JAM (JAM–VE1), the cytoplasmic and transmembrane domain of VE-PTP fused to the extracellular domain of JAM (JAM–VE2) or the extracellular 17th FNIII repeat of VE-PTP and the cytoplasmic and transmembrane domain of JAM (VE–JAM). (B) COS cells were transfected with truncated VE-cadherin lacking its cytoplasmic tail (VE-cad EC, lanes 1–4) and, in addition, co-transfected with one of the three different VE-PTP– JAM fusion proteins (lanes 1–3). Proteins immunoprecipitated with anti-Flag antibodies were immunoblotted first with anti-VE-cadherin antibodies (top), and then the same filters were re-probed with anti-Flag antibodies (middle). Total cell lysates were immunoblotted with anti-VE-cadherin antibodies (bottom). Note that truncated VE-cadherin was only co-precipitated with the VE-PTP–JAM fusion protein that included the 17th FNIII repeat of VE-PTP (lane 3). The asterisk marks the IgG heavy chain detected by the secondary antibody. Molecular mass markers (in kDa) are shown on the left.
To identify the domain in VE-cadherin that is necessary for the interaction with VE-PTP, we generated a series of truncation mutants lacking one, two, three or four of the N-terminal cadherin domains (Figure 5A). Each of these constructs contained the transmembrane and no cytoplasmic domain, except for the five most membrane-proximal amino acids. Co-transfection with Flag-VE-PTP lacking the 16 N-terminal FNIII repeats, followed by immunoprecipitation with the anti-Flag antibody, resulted in co-precipitation of each of the constructs (Figure 5B), demonstrating that none of the first four N-terminal cadherin domains is necessary for binding. In combination with the finding that the transmembrane domain of VE-PTP is dispensable for binding, our results suggest that the fifth cadherin domain of VE-cadherin associates, either directly or indirectly, with the 17th FNIII repeat of VE-PTP. Due to the membrane-proximal position of both interacting domains, this is likely to be a cis interaction. Thus, VE-PTP is the first candidate for a transmembrane protein that interacts in cis with a cadherin via an extracellular domain.

Fig. 5. The fifth cadherin domain of VE-cadherin is sufficient for binding to VE-PTP. (A) Schematic representation of the domain structure of truncated VE-cadherin lacking the cytoplasmic part (VE-cad EC) and four truncation mutants with the same cytoplasmic truncation and lacking, in addition, the first (VE2–5), the first two (VE3–5), the first three (VE4–5) or the first four (VE5) extracellular cadherin (EC) domains. (B) COS cells were either transfected with one of the five different truncated VE-cadherin constructs alone or, in addition, co-transfected with the VE–JAM fusion protein containing the Flag-tagged 17th FNIII domain of VE-PTP and the transmembrane and cytoplasmic domains of JAM. Cell lysates were immunoprecipitated with anti-Flag antibodies, precipitated proteins were transfered to a filter and the same filter was first reacted with anti-VE-cadherin antibodies and then with anti-Flag antibodies. Molecular mass markers (in kDa) are shown on the left.
VE-PTP associates with VE-cadherin in endothelial cells
Next, we analysed whether endogenously expressed VE-cadherin and VE-PTP in endothelial cells associate with each other. We first analysed the expression of VE-PTP in bEnd3 cells. As simple immunoblotting of cell lysates with our polyclonal antibodies against VE-PTP was not sensitive enough (data not shown), we performed immunoprecipitations with lysates of 5 × 107 cells followed by immunoblotting with the same antibodies. In this way, we detected two prominent bands of 240 and 260 kDa in a range expected for this large protein, and two additional weak bands at 150 and 160 kDa that are possibly due to proteolytic degradation (Figure 6, lane 1). Blotting of VE-PTP immunoprecipitates with anti-VE-cadherin antibodies revealed that VE-cadherin could be co-precipitated from lysates of endothelial cells (Figure 6, lane 1).
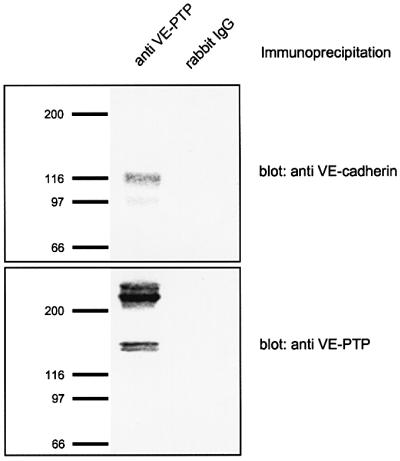
Fig. 6. Endogenous VE-cadherin in mouse endothelioma cells interacts with native VE-PTP. Lysates of bEnd3 mouse endothelioma cells were immunoprecipitated with polyclonal antibodies against VE-PTP (anti-VE-PTP) or control rabbit IgG (rabbit IgG). Precipitated proteins were immunoblotted either with anti-VE-cadherin antibodies (top) or with anti-VE-PTP antibodies (bottom). Molecular mass markers (in kDa) are shown on the left.
In vivo dephosphorylation of VE-cadherin by VE-PTP
To analyse whether VE-cadherin can be dephosphorylated by VE-PTP, we again used COS-7 cells. As transfected VE-cadherin was not tyrosine phosphorylated (Figure 7, lane 4), we co-transfected VEGFR-2. The stimulation of human endothelial cells with VEGF has been reported to induce phosphorylation of VE-cadherin (Esser et al., 1998). In agreement with this, VE-cadherin was tyrosine phosphorylated in VEGFR-2/VE-cadherin co-transfected cells, as revealed by immunoblotting of immunoprecipitated VE-cadherin with a phospho-tyrosine-specific mAb (Figure 7, lane 3). In contrast to the effect of VEGF in HUVECs, we mostly detected no tyrosine phosphorylation or, in some cases, only weak tyrosine phosphorylation of β-catenin in VE-cadherin/VEGFR-2 co-transfected COS-7 cells (Figure 7; data not shown). To test whether VE-PTP would be able to dephosphorylate VEGFR-2-phosphorylated VE-cadherin, we performed triple-transfection experiments with VE-cadherin, VEGFR-2 and either the Flag-VE-PTP construct or a dominant-negative mutant of this construct carrying a point mutation in the phosphatase domain that renders the catalytic domain inactive (Flag-VE-PTP R/A). As shown in Figure 7, only the catalytically active, but not the inactive, form of VE-PTP induced dephosphorylation of VE-cadherin (lanes 1 and 2). Analysis of the same filter with anti-VE-cadherin antibodies demonstrated that the cells expressed VE-cadherin at similar levels (Figure 7). In addition, we controlled for equal transfection efficiency of VEGFR-2 and the VE-PTP constructs by immunoprecipitations followed by immunoblotting (Figure 7). We conclude that VE-PTP is able to cause the dephosphorylation of VE-cadherin following VEGFR-2-induced phosphorylation.
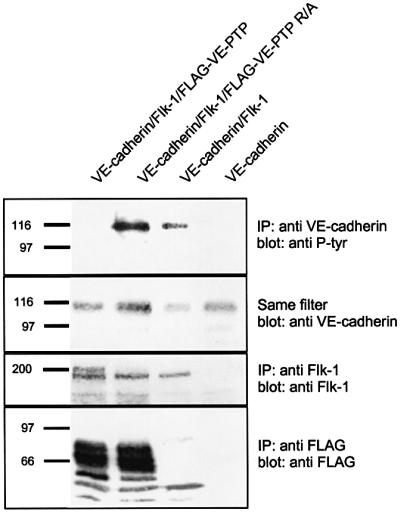
Fig. 7. In vivo dephosphorylation of VE-cadherin by Flag-VE-PTP. COS cells were transfected with VE-cadherin, VEGFR-2 (Flk-1) and Flag-VE-PTP (lane 1); with VE-cadherin, VEGFR-2 (Flk-1) and the dominant negative Flag-VE-PTP R/A mutant (lane 2); with VE-cadherin and VEGFR-2 (Flk-1) (lane 3); or with VE-cadherin alone (lane 4). Cell lysates were immunoprecipitated with either anti-VE-cadherin, anti-VEGFR-2 (Flk-1) or anti-Flag antibodies, and immunoprecipitates were analysed by immunoblotting. Note that VE-cadherin was only phosphorylated in the presence of Flk-1 and that this phosphorylation was reversed by recombinant VE-PTP, but not by its point-mutated version. Molecular mass markers (in kDa) are shown on the left.
Induction of VE-PTP decreases tyrosine phosphorylation of VE-cadherin and increases cell contact integrity
In order to test whether VE-PTP could affect the adhesive function of VE-cadherin, we decided to generate two different types of triple-transfected stable CHO clones. Each expressed VE-cadherin and VEGFR-2 constitutively (under CMV promotors), and one (CHO-F12) expressed Flag-VE-PTP and the other (CHO-RA7) expressed the enzymatically inactive form Flag-VE-PTP R/A from inducible promotors. Induction was based on the activation of a Gal-4-based chimeric transcription factor with the progesterone antagonist mifepristone. In addition, we generated the double-transfected CHO clone CHO-C3, constitutively expressing VE-cadherin and inducibly expressing Flag-VE-PTP. The expression of the transfected proteins was determined by FACS analysis. Figure 8D and E shows the expression of VE-cadherin and VEGFR-2 on non-induced CHO-F12 cells as compared with mock-transfected CHO cells. Analysis of the expression of Flag-VE-PTP with an anti-Flag mAb revealed no significant expression in the absence of mifepristone, moderate expression after 4 h of mifepristone and strong expression after 24 h of mifepristone (Figure 8A–C). The expression level of Flag-VE-PTP in CHO cells after 10 h induction in CHO cells was slightly lower than in bEnd3, as analysed in immunoblots (data not shown). Flag-VE-PTP R/A was similarly inducible in CHO-RA7 cells (Figure 8F).

Fig. 8. FACS analysis of CHO cells stably co-transfected with VE-cadherin, VEGFR-2 and inducible Flag-VE-PTP or Flag-VE-PTP R/A. The triple-transfected CHO-F12 cells were either stimulated with mifepristone for 0 h (A), 4 h (B) or 24 h (C) or left unstimulated (D and E). Cells were analysed by flow cytometry with antibodies against the Flag-tag, recognizing tagged VE-PTP (A–C), VE-cadherin (D) or VEGFR-2 (E). CHO-RA7 cells (F) were analysed with anti-Flag antibodies after 24 h stimulation with mifepristone (bold line) or left unstimulated (grey line). The thin lines indicate signals obtained with mock-transfected CHO cells.
Phosphorylation levels of VE-cadherin were analysed by immunoblotting VE-cadherin immunoprecipitates with anti-phospho-tyrosine antibodies. No tyrosine phosphorylation signal was obtained with VE-cadherin-transfected CHO cells or with CHO-C3 cells, either in the absence or in the presence of mifepristone (data not shown). In contrast, VE-cadherin was tyrosine phosphorylated in CHO-F12 cells in the absence of mifepristone, and the signal vanished if the cells were stimulated for 10 h with the progesterone antagonist (Figure 9E, top). Expression levels of VE-cadherin were unaffected by mifepristone. The induction of the Flag-VE-PTP fusion protein was confirmed by immunoprecipitating and immunoblotting the antigen with anti-Flag antibodies (Figure 9E, middle and bottom). We conclude that the expression of VEGFR-2 in stably transfected CHO cells leads to the phosphorylation of VE-cadherin and that the induction of Flag-VE-PTP reverses this effect. Surprisingly, the induction of Flag-VE-PTP R/A in CHO-RA7 cells reduced tyrosine phosphorylation of VE-cadherin to the same extent as had been found for the wild-type form (Figure 9F), indicating that the enzymatic activity of VE-PTP is dispensable for this effect. Mifepristone did not affect the tyrosine phosphorylation of VE-cadherin in VE-cadherin/VEGFR-2 double transfectants (data not shown).

Fig. 9. VE-PTP and VE-PTP R/A reduce paracellular permeability in VE-cadherin/Flk-1/VE-PTP triple-transfected CHO cells and inhibit tyrosine phosphorylation of VE-cadherin. Paracellular permeability for FITC-dextran was analysed with monolayers of various stably transfected CHO cells. (A) VE-cadherin-transfected CHO cells (dark grey) had a 52 ± 8% reduced permeability compared with mock-transfected CHO (light grey.). (B) No significant difference in permeability was observed if CHO-C3 cells double transfected with VE-cadherin and inducible VE-PTP were either stimulated with mifepristone (dark grey) or mock stimulated (light grey). (C) Analysis of triple-transfected CHO-F12, transfected with VE-cadherin, VEGFR-2 and inducible Flag-VE-PTP. Stimulation with mifepristone for 10 h resulted in a 38 ± 12% reduction of cell permeability (dark grey) as compared with mock-stimulated cells (light grey). All three experiments are representative of nine similar experiments. (D) Analysis of triple-transfected CHO-RA7, transfected with VE-cadherin, VEGFR-2 and inducible Flag-VE-PTP R/A. Stimulation with mifepristone for 10 h resulted in a 38 ± 13% reduction of cell permeability (dark grey) as compared with mock-stimulated cells (light grey). This experiment is representative of five similar experiments. CHO-F12 cells (E) and CHO-RA7 cells (F) were analysed by immunoprecipitations with anti-VE-cadherin antibodies followed by immunoblotting with an antibody against phospho-tyrosine (anti-PY) (top). The same filter was re-probed with an antibody against VE-cadherin, demonstrating equal loading of VE-cadherin (middle). Immunoprecipitations with anti-Flag antibodies, followed by immunoblotting with the same antibody, revealed strong induction of truncated VE-PTP on mifepristone treatment (bottom, only shown for CHO-F12 cells).
To examine the influence of VE-PTP on the function of VE-cadherin, we tested the permeability of monolayers of the transfected CHO cells for FITC-dextran in transwell filter assays. The comparison of mock-transfected and VE-cadherin-transfected CHO cells revealed that VE-cadherin reduced the permeability by 52 ± 8% (Figure 9A), confirming similar results reported previously (Breviario et al., 1995). When the permeability of CHO-C3 monolayers was tested after 10 h treatment with mifepristone and compared with similarly treated CHO-C3 cells in the absence of mifepristone, no effect of the progesteron antagonist was detected (Figure 9B). This demonstrates that neither mifepristone nor the expression of Flag-VE-PTP has any effect on transcellular permeability if the cells lack VEGFR-2. In contrast, analysis of similarly treated CHO-F12 cells revealed that mifepristone stimulation reduced the permeability by 38 ± 12% compared with the same cells mock-treated with medium alone (Figure 9C). A similar reduction of cell layer permeability was achieved by the induction of enzymatically inactive Flag-VE-PTP R/A in CHO-RA7 cells (Figure 9D). Thus, VE-PTP can affect VE-cadherin phosphorylation and cell layer permeability independently of its intrinsic phosphatase activity, arguing for a distinct phosphatase that acts on VE-cadherin and becomes recruited and/or activated by VE-PTP. The fact that Flag-VE-PTP only reduces cell monolayer permeability if the cells co-express the VEGFR-2 kinase argues indirectly for the involvement of this unknown phosphatase activity in the strengthening of intercellular contacts of VE-cadherin-expressing cells.
Mifepristone affected neither the expression of VE-cadherin nor VEGFR-2 in double transfectants lacking VE-PTP (see Supplementary figure 10A available at The EMBO Journal Online) nor the expression of VE-cadherin in the triple transfectants CHO-F12 (Supplementary figure 10B) and CHO-RA7 (data not shown). However, we were surprised to find that VEGFR-2 cell surface expression was significantly reduced in the triple transfectants on the induction of Flag-VE-PTP (Supplementary figure 10C) or Flag-VE-PTP R/A (data not shown). This effect was blocked if cells were cultured in the presence of a proteasome inhibitor while stimulated with mifepristone, arguing for increased VEGFR-2 degradation on induced expression of VE-PTP (Supplementary figure 10D). In theory, these results could provide an alternative mechanism for how VE-PTP could indirectly affect VE-cadherin phosphorylation (and function). However, we could demonstrate that Flag-VE-PTP or Flag-VE-PTP R/A-induced dephosphorylation of VE-cadherin can occur independently of the down-regulation of VEGFR-2, as dephosphorylation of VE-cadherin was still observed in the presence of the proteasome inhibitor (Supplementary figure 11A). Similarly, VE-PTP-induced inhibition of cell layer permeability was still observed in the presence of the proteasome inhibitor, establishing that the functional effect on cell contact integrity can also be observed independently of VEGFR-2 down-regulation (Supplementary figure 11B).
Discussion
Tyrosine phosphorylation of adherens junction components has long been suspected to be involved in the regulation of junction integrity (Warren and Nelson, 1987; Volberg et al., 1991), and a correlation between tyrosine phosphorylation of cadherin–catenin complexes and changes in the stability of inter-endothelial cell contacts has been well documented (Daniel and Reynolds, 1997). Although one PTP and four RPTPs that associate with cadherin–catenin complexes have been described, almost all of them bind to catenins. RPTPµ is the only RPTP known to bind directly to cadherins; however, binding occurs through the cytoplasmic domain, as was shown in the case of E-cadherin (Brady-Kalnay et al., 1998). The interaction between VE-PTP and VE-cadherin that we describe here is the first RPTP–cadherin interaction that occurs via extracellular domains.
We found that the induction of VE-PTP in CHO cells expressing VE-cadherin and VEGFR-2 increased the barrier function of VE-cadherin-mediated cell contacts. There are various potential mechanisms by which VE-PTP could mediate this effect. The simplest one would be that it acts as a cell adhesion molecule itself. Although this has not yet been analysed for VE-PTP or its human homologue HPTPβ, other RPTPs such as RPTPµ and RPTPκ have been reported to support homophilic cell adhesion (Brady-Kalnay et al., 1993; Gebbink et al., 1993; Sap et al., 1994). However, even if VE-PTP could mediate homophilic binding, that type of interaction was prevented in our experiments, as the recombinant form of VE-PTP that we induced in CHO cells was devoid of the first 16 of the 17 extracellular FNIII-like repeats.
It is possible that direct binding between the membrane-proximal extracellular domains of VE-PTP and VE-cadherin could directly influence the activity of VE-cadherin. This physical interaction could affect the conformation or the clustering of VE-cadherin, which in turn could influence its adhesive activity. Indeed, in the case of Xenopus C-cadherin, an antibody was described that can increase the adhesive activity of this cadherin, and this antibody was found to bind to the fifth extracellular domain of this cadherin (Zhong et al., 1999). Although this activity-enhancing effect of an mAb is unique in the cadherin field, numerous examples exist for similar effects with various integrins (Hughes and Pfaff, 1998). However, a similar direct effect for the binding of VE-PTP on VE-cadherin conformation or clustering, if it exists, is not sufficient to explain the effects of VE-PTP on cell adhesion, as VE-PTP induction did not affect VE-cadherin-mediated cell contacts in CHO cells that lacked expression of VEGFR-2. Thus, the kinase activity of VEGFR-2 was required for the effect of VE-PTP on cell adhesion, indirectly arguing for the requirement of VE-PTP-related dephosphorylation events in the regulation of cell adhesion.
The fact that an enzymatically inactive mutated form of VE-PTP inhibited VE-cadherin phosphorylation and cell layer permeability, as well as the enzymatically active form, clearly established that the phosphatase activity of VE-PTP is not required for these effects. Our results suggest that VE-PTP recruits and/or activates a phosphatase of yet unknown identity that dephosphorylates VE-cadherin and potentially affects cell layer permeability. Such mechanisms seem to be lacking in COS cells, where enzymatically inactive VE-PTP did not induce dephosphorylation of VE-cadherin (Figure 7). Indeed, additional mechanisms are conceivable. RPTPµ was recently shown to activate E-cadherin function independently of its phosphatase activity, and it was suggested that the intracellular interaction of this RPTP with RACK-1, a receptor for activated protein kinase-C, would be involved in this process (Hellberg et al., 2002).
Another possible mechanism by which VE-PTP might act on VE-cadherin is based on the proteolytic down-regulation of VEGFR-2. Although such a potentially interesting mechanism could be involved in the regulation of VE-cadherin phosphorylation and function, we found that VE-PTP can influence VE-cadherin independently of this effect, as blocking the down-regulation of VEGFR-2 by a proteasome inhibitor did not inhibit the VE-PTP-induced effect on VE-cadherin dephosphorylation and cell layer permeability. Whether and how the expression of VE-PTP is regulated in endothelial cells and whether VE-PTP can influence the expression of VEGFR-2 in such cells is unknown.
In addition to VE-cadherin, β-catenin also became dephosphorylated in CHO cells on the induction of VE-PTP (Figure 9E and F). However, it is unlikely that the dephosphorylation of β-catenin is responsible for the adhesion strengthening effect of VE-PTP, as the induction of VE-PTP in CHO-C3 cells, where β-catenin was constitutively tyrosine phosphorylated, led to the dephosphorylation of β-catenin (data not shown) without affecting cell contact integrity (Figure 9B). If we assume that VEGFR-2 would not phosphorylate tyrosine residues in other positions than are already constitutively phosphorylated on β-catenin in CHO cells, our results suggest that the dephosphorylation of VE-cadherin, but not the dephosphorylation of β-catenin, correlates with the enhancement of cell contact integrity. The identification of the tyrosine residues targeted by VEGFR-2 and the VE-PTP-dependent phosphatase in VE-cadherin will facilitate the clarification of this question in the future. Other components of adherens junctions possibly involved in the adhesion strengthening effect of VE-PTP still need to be investigated.
As cell monolayer permeability was changed by the expression of VE-cadherin, we used permeability as an indirect parameter for influences on VE-cadherin adhesive function. A more direct cell adhesion assay, such as adhesion of VE-cadherin-transfected cells to immobilized VE-cadherin–IgG, could not be established, due to a lack of VE-cadherin-specific binding at appropriate temperature conditions (data not shown). Cell aggregation assays were not considered, as cytoplasmic truncation of VE-cadherin that destroys the interaction with catenins was reported not to affect cell aggregation, although it strongly affects the ability of VE-cadherin to support cell layer integrity and block permeability (Navarro et al., 1995). Therefore, we believe that the influence of VE-PTP on VE-cadherin-dependent cell layer integrity is likely to be mediated by its extracellular interaction with VE-cadherin and influences on its adhesive function. The generation of VE-cadherin and/or VE-PTP mutants that lack the ability to interact with the respective other protein will be a way to elucidate this mechanism.
The interaction of VE-PTP with VE-cadherin occurs via extracellular domains that are directly proximal to the plasma membrane for each of the two proteins. Therefore, it is likely that both proteins interact in cis, either directly or via a third protein that may interconnect the cadherin and the phosphatase via the ectodomains. There are very few non-cadherin proteins known to interact with the extracellular part of a cadherin. The integrin αEβ7 interacts in trans with E-cadherin and mediates adhesion between intra-epithelial lymphocytes and epithelial cells in the gut (Cepek et al., 1994). Furthermore, the soluble protein fibrin has been reported to bind to VE-cadherin (Bach et al., 1998). A transmembrane protein that interacts in cis with a cadherin via extracellular sites has not yet been described. This unique interaction between VE-PTP and VE-cadherin has interesting implications. Antibodies or other reagents that would target the interacting domains might be able to influence the interaction between VE-PTP and VE-cadherin and thereby VE-cadherin-mediated endothelial contacts. In contrast to other RPTPs, it would not be necessary to target the phosphatase activity of VE-PTP in order to influence its function; instead, it might be possible and sufficient to specifically interfere with its association with VE-cadherin to selectively affect VE-cadherin-related functions of this phosphatase.
Our co-precipitation experiments revealed that the 17th FNIII-like domain of VE-PTP was sufficient to support association with VE-cadherin. This domain was a less typical FNIII-like domain than the other 16 of VE-PTP: it is larger and was not designated as an FNIII-like domain in the human homologue HPTPβ (Krueger et al., 1990). Indeed, analysis by various structural prediction programs did not define this domain as an FNIII-like repeat if the complete sequence of VE-PTP was analysed. It designated an FNIII-like repeat only if the sequence between the 16th FNIII-like repeat and the putative transmembrane region was analysed alone. Its structural difference from the rest of the ectodomain of the protein, as well as the fact that it is necessary and sufficient for the association with VE-cadherin, makes this domain an interesting target for reagents that could influence endothelial cell contacts.
The function of the extracellular domains of RPTPs is still not well understood, and only a few examples of ligands of the ectodomains of RPTPs are known (Bixby, 2001). Homophilic binding has been reported for RPTPµ (Brady-Kalnay et al., 1993; Gebbink et al., 1993), RPTPκ (Sap et al., 1994) and RPTPδ (Wang and Bixby, 1999), suggesting direct support of cell adhesion. A complex of laminin and nidogen was found to bind to a splice variant of LAR (O’Grady et al., 1998), an interaction that is possibly linked to changes in cell morphology. The multifunctional growth factor/cytokine pleiotrophin binds to RPTPζ, and binding was shown to deactivate this phosphatase, resulting in increased tyrosine phosphorylation of RPTPζ-associated β-catenin. This is the first example of a ligand–RPTP interaction that leads to a change in physiological substrate phosphorylation. The relevance of these interactions for cell adhesion still needs to be investigated. The only known extracellular cis interaction of an RPTP was described for RPTPα that was shown to associate with contactin, a glycophosphatidylinositol-linked Ig-superfamily member (Zeng et al., 1999). Except for the homophilic interactions, the binding of the ectodomains of RPTPs to extracellular ligands has not yet been shown to affect cell–cell adhesion. Similarly, whether the association of various RPTPs with cadherin–catenin complexes via intracellular protein domains affects cadherin-mediated cell adhesive events has not yet been directly analysed.
The effect of VE-PTP on cell contacts supported by VE-cadherin could be involved in various physiological processes. One could imagine that leukocytes might bind to VE-PTP, providing a potential mechanism that could allow leukocytes to influence the function of VE-cadherin during leukocyte extravasation (Vestweber, 2000). It is also possible that soluble VE-PTP ligands might exist that could affect the function of VE-cadherin and thereby endothelial permeability or other VE-cadherin-mediated functions involved in angiogenesis (Carmeliet et al., 1999). Even though many details of the mechanism by which VE-PTP influences the strength of VE-cadherin-mediated cell contacts still need to be investigated, our results define a new potential target on the cell surface of endothelial cells by which VE-cadherin-dependent endothelial cell contacts, and possibly other VE-cadherin functions, could be influenced.
Materials and methods
Cell culture and transfection
The mouse endothelioma cell line bEnd3 (Williams et al., 1989) and COS-7 cells were cultured as described previously (Fachinger et al., 1999). CHO cells (CHO DUKX B1) were cultured in α-minimal essential medium (α-MEM; Gibco Life Technologies) supplemented with 10% fetal calf serum, 2 mM l-glutamine and penicillin/streptomycin. Transient transfections were performed using the GeneJammer transfection reagent (Stratagene Europe, Amsterdam, The Netherlands) according to the manufacturer’s instructions. Cells were analysed 36 h after transfection.
Generation of stable cell lines
For stable transfections, 1 × 107 CHO cells were electroporated in 600 µl of phosphate-buffered saline (PBS) containing 20 µg of vector DNA in a 0.4 cm cuvette (950 µF, 0.25 kV). Depending on the resistance gene used, cells were selected with either 800 µg/ml G418 (PAN Biotech, Aidenbach, Germany), 100 µg/ml zeocin (Invitrogen, Groningen, The Netherlands), 200 µg/ml hygromycin (Gibco) or 7 µg/ml blasticidin (Invitrogen) or with a mixture of these antibiotics. CHO-C3 cells were generated by co-transfecting VE-cadherin-transfected CHO cells (Gotsch et al., 1997) with the pSwitch plasmid (Invitrogen; hygromicin resistance) and the expression plasmid pGene/V5-HisA (Invitrogen; zeocin resistance) containing the Flag-VE-PTP construct. Co-transfecting CHO-C3 cells with mouse VEGFR-2 cDNA in pRK5 together with the expression plasmid pEF6 (Invitrogen; blasticidin resistance) gave rise to CHO-F12 cells. VE-cadherin/VEGFR-2 double-transfected CHO cells were generated by transfecting VE-cadherin transfectants (Gotsch et al., 1997) with VEGFR-2. Transfecting these cells with the pSwitch plasmid and the expression plasmid pGene/V5-HisA containing the Flag-VE-PTP R/A construct gave rise to the three different clones CHO-RA7, CHO-RA14 and CHO-RA16. The expression of transfected genes was assessed by FACS analysis.
Antibodies
The rabbit antiserum against VE-PTP was raised against a peptide covering the 13 C-terminal amino acids of mouse VE-PTP and containing an additional N-terminal cysteine. Conjugation to a carrier protein, immunization and antibody affinity purification was performed as described previously (Borges et al., 1997a). Rabbit antiserum C5 and rat mAb 11D4.1 against mouse VE-cadherin have been described previously (Gotsch et al., 1997). The new rat mAb VE-57.2 recognizing the fifth cadherin domain of VE-cadherin was generated against mouse VE-cadherin–Ig as described previously (Gotsch et al., 1997). The following antibodies were commercially obtained: mAbs against β-catenin (Transduction Laboratories, Bluegrass-Lexington, KY), recombinant phospho-tyrosine (RC20H; Transduction Laboratories), VEGFR-2 (BD Pharmingen), Flag-epitope (M2; Sigma-Aldrich, Munich, Germany) and the polyclonal antibodies against pan-cadherin (Sigma-Aldrich). Secondary antibodies were purchased from Dianova (Hamburg, Germany).
DNA constructs
The pFlag-CMV1 vector expressing truncated forms of wild-type VE-PTP (Flag-VE-PTP) and the corresponding trapping mutant (Flag-VE-PTP R/A) have been described previously (Fachinger et al., 1999). For inducible expression, Flag-VE-PTP was excised via the VspI–XbaI sites in pFlag-CMV1 and inserted into the pGene/V5-HisA vector via HindIII–SpeI sites. VE-PTP–JAM fusion constructs were cloned into pFlag-CMV1 by polymerase chain reaction (PCR). Amino acid (aa) positions of JAM-1 are according to accession No. U89912 and in VE-PTP according to the numbering given in Figure 2. JAM–VE1 contained aa 25–261 of JAM-1 (primers JAM7S 5′-CTCTAAGCTT CAAGGCAAGGGTTCGGTG-3′ and JAMR1AS 5′-CTAGATCTATA GGCAAACCAGACGCC-3′) and aa 1645–1997 of VE-PTP (primers VE-PTP3S 5′-GAAGATCTCAGAAAGCTAGCCACAGC-3′ and VE PTP1AS 5′-CCGGTACCATCTTCAGGTGAATTTCTTAA-3′). JAM– VE2 contained aa 25–235 of JAM (primer JAM7S and JAMR2AS 5′-GAAGATCTATTCAGCTCCACAGCATC-3′) and aa 1619–1997of VE-PTP (primer VE-PTP2S 5′-GAAGATCTGGTGTGAGTGCTGG CCTG-3′ and VE-PTP1AS). VE–JAM contained aa 1421–1622 of VE-PTP (primers VE-PTP1S 5′-CGGAATTCAGGTACCTGGTGTCC ATC-3′ and VE-PTP2AS 5′-CTAGATCTTTCAATAACTCCAAAC AA-3′) and aa 235–300 of JAM (primers JAM9S 5′-GAAGATCT GTGGGGGGCATCGTGGCA-3′ and JAM2AS 5′-GCAGTGTCGAC TCACACCAGGAACGACGAGGT-3′).
Full-length VE-PTP was cloned by OriGene Technologies using a Rapid-Screen arrayed cDNA library. This clone was isolated from a mouse liver cDNA library, the library having been constructed from double-purified mRNA using an oligo(dT) primer. A total of 500 000 clones were PCR screened using a primer to the VE-PTP partial cDNA sequence of the published sequence [5′-GTGAGGTGGTTGAAG ATAGC-3′/5′-TTTTTTTTTTTTTTTTTTTTTT(V)-3′]. The longest clone was selected using gene-specific and vector primers (5′-GTGTCTGTACTCCAGGTAG-3′/5′-GCAGAGCTCGTTTAGTGA ACC-3′). This clone contained a DNA fragment with an ∼6 kb fragment encoding the full-length cDNA of VE-PTP. The sequence data have been submitted to the DDBJ/EMBL/GenBank databases under accession No. AY077755.
A series of truncation mutants of VE-cadherin (VE-cad EC) was generated by PCR starting from the VE-cadherin cDNA in pcDNA3 (Breier et al., 1996) and inserted either via BamHI–XbaI sites into pcDNA3 (for VE-cad EC) or via KpnI–ApaI sites into pSecTag2A (Invitrogen). The following primers were used for the PCR: T7-primer (sense) (5′-TAATACGACTCACTATAGGG-3′) and antisense-VECP-30 (5′-CGTCTAGACTACAGGATGATCAGCAAGG-3′) for VE-cad EC. For the other four constructs, the same VE-3′ antisense-primer was used (5′-ATATATGGGCCCCCGGATCCGCCTCCGCAG-3′) and the following sense primers: 5′-GGGGTACCTGTGTTTTCGCACCAGG TA-3′ for VE2–5; 5′-GGGGTACCCGTCTTTACTCAATCCACA-3′ for VE3–5; 5′-GGGGTACCTGTCTTCCAGCGACACTTC-3′ for VE4–5; and 5′-GGGGTACCGGAATTTGCCCAGCCCTAC-3′ for VE5. The resulting constructs contained the following nucleotides with respect to DDBJ/EMBL/GenBank accession No. X83930: VE-cad EC, 1–1899 (aa 1–619); VE2–5, 480–1914 (aa 146–624); VE3–5, 799–1914 (aa 253–624); VE4–5, 1144–1914 (aa 368–624); and VE5, 1465–1914 (aa 475–624). Domain boundaries were chosen according to Overduin et al. (1995) and Shapiro et al. (1995). The full-length cDNA for mouse β-catenin was kindly provided by Dr Jürgen Behrens (University of Erlangen) and cloned into pcDNA3.
Immunoprecipitation and immunoblots
Confluent monolayers of bEnd3 cells were rinsed twice with PBS and collected by scraping in PBS supplemented with 2 mM DTT and protease inhibitors (2 mM PMSF, 2 µg/ml pepstatin A, 20 µg/ml aprotinin, 20 µg/ml leupeptin) followed by centrifugation and extraction for 1 h on ice in lysis buffer containing 50 mM Tris–HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, 1 mM CaCl2, 0.5 mM DTT, 1 mM PMSF, 1 µg/ml pepstatin A, 10 µg/ml leupeptin and 10 µg/ml aprotinin. Insoluble material was pelleted by two consecutive centrifugation steps (30 000 g for 30 min, 90 000 g for 1 h). For immunoprecipitations with COS-7 or CHO cells, cells were lysed directly on the culture dish and insoluble material was removed by centrifugation at 20 800 g for 15 min. When tyrosine phosphorylation was analysed, the lysis buffer was supplemented with 1% pervanadate stock solution that had been generated 15 min before by mixing 50 mM orthovanadate with 20 mM hydrogen peroxide. Cell lysates were incubated with affinity-purified antibodies coupled to protein A– or G–Sepharose (Amersham Pharmacia Biotech, Freiburg, Germany) for 3 h or overnight. Immunocomplexes were washed five times with lysis buffer and analysed by SDS–PAGE and fluorography. Immunoblots were performed essentially as described previously (Borges et al., 1997b), except for the new blocking buffer: 1% bovine serum albumin and 0.1% Tween-20 in Tris-buffered saline.
Permeability assay
CHO cells were seeded at a density of 1.5 × 104 cells in 200 µl/well onto 6.5 mm diameter transwell filters with a pore size of 0.4 µm (Costar 3413, Corning), according to Breviario et al. (1995). After 3 days, the medium was replaced by CHO medium supplemented with or without 1 × 10–8 M mifepristone for 10 h. For the assay, the medium was replaced by 150 µl of α-MEM with FITC-dextran (42 kDa; Sigma) at a final concentration of 1 mg/ml. Lower chambers contained 700 µl of α-MEM; incubations were performed for 1 h at 37°C and 10% CO2. Optical density of the lower chamber solution was measured at 492 nm excitation and 520 nm emission in a Fluoromax-2 fluorimeter (Jobin-Yvon, Grasbrunn, Germany). Treatment of cells with proteasome inhibitor MG132 (Alexis, Grünberg, Germany) was performed at 2 µM diluted into the medium from a 4 mM stock solution in DMSO.
Flow cytometry
Flow cytometry analysis was performed essentially as described previously (Borges et al., 1997a). First antibodies were incubated at a concentration of 10 µg/ml and were detected with DTAF conjugated secondary antibodies at a 1:50 dilution.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank Silvia Hennig for excellent technical assistance. This work was supported by a grant from the Deutsche Forschungsgemeinschaft (VE-186/2-2) (to D.V.) and by the Max-Planck-Society.
References
- Aberle H., Schwartz,H. and Kemler,R. (1996) Cadherin–catenin complex: protein interactions and their implications for cadherin function. J. Cell. Biochem., 61, 514–523. [DOI] [PubMed] [Google Scholar]
- Bach T.L., Barsigian,C., Yaen,C.H. and Martinez,J. (1998) Endothelial cell VE-cadherin functions as a receptor for the β15–42 sequence of fibrin. J. Biol. Chem., 273, 30719–30728. [DOI] [PubMed] [Google Scholar]
- Balsamo J., Arregui,C., Leung,T. and Lilien,J. (1998) The nonreceptor protein tyrosine phosphatase PTP1B binds to the cytoplasmic domain of N-cadherin and regulates the cadherin–actin linkage. J. Cell Biol., 143, 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens J., Vakaet,L., Friis,R., Winterhager,E., Van Roy,F., Mareel,M.M. and Birchmeier,W. (1993) Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/β-catenin complex in cells transformed with a temperature-sensitive v-src gene. J. Cell Biol., 120, 757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bixby J.L. (2001) Ligands and signaling through receptor-type tyrosine phosphatases. IUBMB Life, 51, 157–163. [DOI] [PubMed] [Google Scholar]
- Borges E., Tietz,W., Steegmaier,M., Moll,T., Hallmann,R., Hamann,A. and Vestweber,D. (1997a) P-selectin glycoprotein ligand-1 (PSGL-1) on T helper 1 but not on T helper 2 cells binds to P-selectin and supports migration into inflamed skin. J. Exp. Med., 185, 573–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges E., Pendl,G., Eytner,R., Steegmaier,M., Zöllner,O. and Vestweber,D. (1997b) The binding of T cell-expressed P-selectin glycoprotein ligand-1 to E- and P-selectin is differentially regulated. J. Biol. Chem., 272, 28786–28792. [DOI] [PubMed] [Google Scholar]
- Brady-Kalnay S.M., Flint,A.J. and Tonks,N.K. (1993) Homophilic binding of PTPµ, a receptor-type protein tyrosine phosphatase, can mediate cell–cell aggregation. J. Cell Biol., 122, 961–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady-Kalnay S.M. et al. (1998) Dynamic interaction of PTPµ with multiple cadherins in vivo. J. Cell Biol., 141, 287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breier G., Breviario,F., Caveda,L., Berthier,R., Schnurch,H., Gotsch,U., Vestweber,D., Risau,W. and Dejana,E. (1996) Molecular cloning and expression of murine vascular endothelial-cadherin in early stage development of cardiovascular system. Blood, 87, 630–641. [PubMed] [Google Scholar]
- Breviario F. et al. (1995) Functional properties of human vascular endothelial cadherin (7B4/cadherin-5), an endothelium-specific cadherin. Arterioscler. Thromb. Vasc. Biol., 15, 1229–1239. [DOI] [PubMed] [Google Scholar]
- Carmeliet P. et al. (1999) Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell, 98, 147–157. [DOI] [PubMed] [Google Scholar]
- Cepek K.L., Shaw,S.K., Parker,C.M., Russell,G.J., Morrow,J.S., Rimm,D.L. and Brenner,M.B. (1994) Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the αEβ7 integrin. Nature, 372, 190–193. [DOI] [PubMed] [Google Scholar]
- Corada M. et al. (1999) Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc. Natl Acad. Sci. USA, 96, 9815–9820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel J.M. and Reynolds,A.B. (1997) Tyrosine phosphorylation and cadherin/catenin function. BioEssays, 19, 883–891. [DOI] [PubMed] [Google Scholar]
- Dejana E., Lampugnani,M.G., Martinez-Estrada,O. and Bazzoni,G. (2000) The molecular organization of endothelial junctions and their functional role in vascular morphogenesis and permeability. Int. J. Dev. Biol., 44, 743–748. [PubMed] [Google Scholar]
- Esser S., Lampugnani,M.G., Corada,M., Dejana,E. and Risau,W. (1998) Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J. Cell Sci., 111, 1853–1865. [DOI] [PubMed] [Google Scholar]
- Fachinger G., Deutsch,U. and Risau,W. (1999) Functional interaction of vascular endothelial-protein-tyrosine phosphatase with the angiopoietin receptor Tie-2. Oncogene, 18, 5948–5953. [DOI] [PubMed] [Google Scholar]
- Fuchs M., Muller,T., Lerch,M.M. and Ullrich,A. (1996) Association of human protein-tyrosine phosphatase κ with members of the armadillo family. J. Biol. Chem., 271, 16712–11679. [DOI] [PubMed] [Google Scholar]
- Gebbink M.F., Zondag,G.C., Wubbolts,R.W., Beijersbergen,R.L., van Etten,I. and Moolenaar,W.H. (1993) Cell–cell adhesion mediated by a receptor-like protein tyrosine phosphatase. J. Biol. Chem., 268, 16101–16104. [PubMed] [Google Scholar]
- Gotsch U., Borges,E., Bosse,R., Böggemeyer,E., Simon,M., Mossmann,H. and Vestweber,D. (1997) VE-cadherin antibody accelerates neutrophil recruiment in vivo. J. Cell Sci., 110, 583–588. [DOI] [PubMed] [Google Scholar]
- Gumbiner B.M. (2000) Regulation of cadherin adhesive activity. J. Cell Biol., 148, 399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaguchi M., Matsuyoshi,N., Ohnishi,Y., Gotoh,B., Takeichi,M. and Nagai,Y. (1993) p60v-src causes tyrosine phosphorylation and inactivation of the N-cadherin–catenin cell adhesion system. EMBO J., 12, 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellberg C.B., Burden-Gulley,S.M., Pietz,G.E. and Brady-Kalnay,S.M. (2002) Expression of the receptor protein-tyrosine phosphatase, PTPµ, restores E-cadherin-dependent adhesion in human prostate carcinoma cells. J. Biol. Chem., 277, 11165–11173. [DOI] [PubMed] [Google Scholar]
- Hoschuetzky H., Aberle,H. and Kemler,R. (1994) β-catenin mediates the interaction of the cadherin–catenin complex with epidermal growth factor receptor. J. Cell Biol., 127, 1375–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber O., Bierkamp,C. and Kemler,R. (1996) Cadherins and catenins in development. Curr. Opin. Cell Biol., 8, 685–691. [DOI] [PubMed] [Google Scholar]
- Hughes P.E. and Pfaff,M. (1998) Integrin affinity modulation. Trends Cell Biol., 8, 359–364. [DOI] [PubMed] [Google Scholar]
- Kemler R. (1993) From cadherins to catenins: cytoplasmic protein interactions and regulation of cell adhesion. Trends Genet., 9, 317–321. [DOI] [PubMed] [Google Scholar]
- Krueger N.X., Streuli,M. and Saito,H. (1990) Structural diversity and evolution of human receptor-like protein tyrosine phosphatases. EMBO J., 9, 3241–3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kypta R.M., Su,H. and Reichardt,L.F. (1996) Association between a transmembrane protein tyrosine phosphatase and the cadherin–catenin complex. J. Cell Biol., 134, 1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampugnani M.G., Resnati,M., Raiteri,M., Pigott,R., Pisacane,A., Houen,G., Ruco,L.P. and Dejana,E. (1992) A novel-endothelial specific membrane protein is a marker of cell–cell contacts. J. Cell Biol., 118, 1511–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampugnani M.G., Corada,M., Andriopoulou,P., Esser,S., Risau,W. and Dejana,E. (1997) Cell confluence regulates tyrosine phosphorylation of adherens junction components in endothelial cells. J. Cell Sci., 110, 2065–2077. [DOI] [PubMed] [Google Scholar]
- Matsuyoshi N., Hamaguchi,M., Taniguchi,S., Nagafuchi,A., Tsukita,S. and Takeichi,M. (1992) Cadherin-mediated cell–cell adhesion is perturbed by v-src tyrosine phosphorylation in metastatic fibroblasts. J. Cell Biol., 118, 703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng K., Rodriguez-Pena,A., Dimitrov,T., Chen,W., Yamin,M., Noda,M. and Deuel,T.F. (2000) Pleiotrophin signals increased tyrosine phosphorylation of β-catenin through inactivation of the intrinsic catalytic activity of the receptor-type protein tyrosine phosphatase β/ζ. Proc. Natl Acad. Sci. USA, 97, 2603–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro P., Caveda,L., Breciario,F., Mandoteanu,I., Lampugnani,M.G. and Dejana,E. (1995) Catenin-dependent and -independent functions of vascular endothelial cadherin. J. Biol. Chem., 270, 30965–30970. [DOI] [PubMed] [Google Scholar]
- O’Grady P., Thai,T.C. and Saito,H. (1998) The laminin–nidogen complex is a ligand for a specific splice isoform of the transmembrane protein tyrosine phosphatase LAR. J. Cell Biol., 141, 1675–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overduin M., Harvey,T.S., Bagby,S., Tong,K.I., Yau,P., Takeichi,M. and Ikura,M. (1995) Solution structure of the epithelial cadherin domain responsible for selective cell adhesion. Science, 267, 386–389. [DOI] [PubMed] [Google Scholar]
- Sap J., Jiang,Y.P., Friedlander,D., Grumet,M. and Schlessinger,J. (1994) Receptor tyrosine phosphatase RPTP-κ mediates homophilic binding. Mol. Cell. Biol., 14, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro L. et al. (1995) Structural basis of cell–cell adhesion by cadherins. Nature, 374, 327–337. [DOI] [PubMed] [Google Scholar]
- Shibamoto S., Hayakawa,M., Takeuchi,K., Hori,T., Oku,N., Miyazawa,K., Kitamura,N., Takeichi,M. and Ito,F. (1994) Tyrosine phosphorylation of β-catenin and plakoglobin enhanced by hepatocyte growth factor and epidermal growth factor in human carcinoma cells. Cell Adhes. Commun., 1, 295–305. [DOI] [PubMed] [Google Scholar]
- Takeda H., Nagafuchi,A., Yonemura,S., Tsukita,S., Behrens,J., Birchmeier,W. and Tsukita,S. (1995) V-src kinase shifts the cadherin-based cell adhesion from the strong to the weak state and β catenin is not required for the shift. J. Cell Biol., 131, 1839–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukropec J.A., Hollinger,M.K., Salva,S.M. and Woolkalis,M.J. (2000) SHP2 association with VE-cadherin complexes in human endothelial cells is regulated by thrombin. J. Biol. Chem., 275, 5983–5986. [DOI] [PubMed] [Google Scholar]
- Vestweber D. (2000) Molecular mechanisms that control endothelial cell contacts. J. Pathol., 190, 281–291. [DOI] [PubMed] [Google Scholar]
- Volberg T., Geiger,B., Dror,R. and Zick,Y. (1991) Modulation of intercellular adherens-type junctions and tyrosine phosphorylation of their components in RSV-transformed cultured chick lens cells. Cell Regul., 2, 105–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. and Bixby,J.L. (1999) Receptor tyrosine phosphatase-δ is a homophilic, neurite-promoting cell adhesion molecule for CNS neurons. Mol. Cell. Neurosci., 14, 370–384. [DOI] [PubMed] [Google Scholar]
- Warren S.L. and Nelson,W.J. (1987) Nonmitogenic morphoregulatory action of pp60v-src on multicellular epithelial structures. Mol. Cell Biol., 7, 1326–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R.L., Risau,W., Zerwes,H.G., Drexler,H., Aguzzi,A. and Wagner,E.F. (1989) Endothelioma cells expressing the polyoma middle T oncogene induce hemangiomas by host cell recruitment. Cell, 57, 1053–1063. [DOI] [PubMed] [Google Scholar]
- Zeng L., D’Alessandri,L., Kalouse,M.B., Vaughan,L. and Pallen,C.J. (1999) Protein tyrosine phosphatase α (PTPα) and contactin form a novel neuronal receptor complex linked to the intracellular tyrosine kinase fyn. J. Cell Biol., 147, 707–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y., Brieher,W.M. and Gumbiner,B.M. (1999) Analysis of C-cadherin regulation during tissue morphogenesis with an activating antibody. J. Cell Biol., 144, 351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zondag G.C., Reynolds,A.B. and Moolenaar,W.H. (2000) Receptor protein-tyrosine phosphatase RPTPµ binds to and dephosphorylates the catenin p120ctn. J. Biol. Chem., 275, 11264–11269. [DOI] [PubMed] [Google Scholar]