

Abstract
We previously identified a small basic leucine zipper (bZIP) protein, Jun dimerization protein 2 (JDP-2), that acts as a coregulator of the N-terminal transcriptional activation domain of progesterone receptor (PR). We show here that JDP-2, through interaction with the DNA binding domain (DBD), induces or stabilizes structure in the N-terminal domain in a manner that correlates with JDP-2 stimulation of transcriptional activity. Circular dichroism spectroscopy experiments showed that JDP-2 interaction caused a significant increase in overall helical content of a two-domain PR polypeptide containing the N-terminal domain and DBD and that the change in structure resides primarily in the N-terminal domain. Thermal melt curves showed that the JDP-2/PR complex is significantly more stable than either protein alone, and partial proteolysis confirmed that JDP-2 interaction alters conformation of the N-terminal domain of PR. Functional analysis of N-terminal domain mutants and receptor chimeras provides evidence that the stimulatory effect of JDP-2 on transcriptional activity of PR is mediated through an interdomain communication between the DBD and the N-terminal domain and that transcriptional activity and functional response to JDP-2 are mediated by multiple elements of the N-terminal domain as opposed to a discrete region.
Steroid hormone nuclear receptors are ligand-dependent transcriptional activators consisting of a carboxy-terminal ligand binding domain (LBD), a centrally located DNA binding domain (DBD), and an amino-terminal domain that is required for full transcription activity. Within these domains, there are at least two transcription activation functions (AF) that provide protein interaction surfaces for coactivators: AF-1 in the N-terminal domain and a conserved ligand-dependent AF-2 in the LBD. AF-2 consists of a hydrophobic cleft, formed by ligand-induced conformational changes in specific α-helices in the LBD, that interacts specifically with LXXLL motifs present in the p160 family of steroid receptor coactivators (SRCs). Secondary coactivators including CBP/p300 and pCAF, which possess histone acetyltransferase activity, and histone methyltransferases such as CARM1 and PRMT1 (33) are recruited by receptors through interaction with SRCs (for reviews, see references 29, 30, 36, 50, and 51). Thus, AF-2-interacting coactivator complexes appear to play a role in receptor-mediated gene activation by promoting chromatin remodeling of target genes.
Compared with AF-2, the transcriptional activation function of the N-terminal domain is less well defined in terms of structure, interacting coactivators, and mechanism of action. The N-terminal domain is the least conserved region of nuclear receptors, both in length and in amino acid sequence (for reviews, see references 25 and 54). Depending on cell and promoter context, AF-1 can activate transcription constitutively independent of the LBD or can synergize with AF-2 to elicit maximal ligand-dependent activity of the receptor (22, 42, 52). With some receptors, such as androgen receptor (AR), AF-1 can be dominant (25, 54). Several diverse proteins have been reported to interact with and enhance the activity of the N-terminal domain of nuclear receptors, including general transcription factors such as TATA binding protein (TBP) and transcription factor IIF (TFIIF), the p68 and p72 DEAD-box RNA helicases, steroid receptor coactivator RNA molecule (SRA), MMS19 (human homolog of yeast DNA repair and TFIIH regulator), and DRIP150 (15, 25, 26, 54, 55, 57, 59). Biophysical and secondary structure analyses have indicated that the N-terminal domain of nuclear receptors including estrogen receptor (ER), AR, retinoic acid receptor (RAR). and glucocorticoid receptor (GR) is not well structured and consists largely of random coil with areas of negative charge density similar to acidic activation domains (12, 23, 24, 47, 55) of other transcription factors such as NF-κB, CREB, p53, Myc, and VP16 (46, 48, 49). Acidic activation domains are intrinsically unstructured and undergo transition from an unfolded to a folded state upon interaction with other stable proteins (46, 48, 49). Similar to acidic activation domains, the N-terminal domains of GR, ER, RAR, and AR have been reported to exhibit an increase in α-helical structure in the presence of α-helical-stabilizing solvents or in response to interaction with certain coregulatory proteins such as CBP, TBP, and TFIIF (4, 12, 23, 24, 47, 55).
Human progesterone receptor (PR) is expressed as two protein isoforms, full-length PR-B and N-terminally truncated PR-A, and the two isoforms have distinct transcription activities (Fig. 1A). In most contexts, PR-B is a strong activator of gene transcription, while PR-A can act as a ligand-dependent transrepressor of PR-B and other steroid receptors including ER. The repressor action of PR-A was mapped to an inhibitory subdomain (ID) of ∼155 amino acids (aa) of the N-terminal domain that does not function in the context of PR-B (Fig. 1A) (for reviews, see references 13 and 30). AF-1 of PR was originally defined as a minimal region of the N-terminal domain between aa 456 and 546 that exhibited constitutive transcriptional activity when linked to the heterologous GAL4 DNA binding domain (40).
FIG. 1.

JDP-2 enhancement of the transcription activity of the N-terminal domain is mediated by the PR DBD. (A) Domain structure of PR and JDP-1/JDP-2, DBD and LBD, AF, ID, and leucine zipper (ZIP). (B) Cos-1 cells were transfected with the 5X-GAL (UAS)-Luc reporter (500 ng) together with GAL4-DBD, GAL4-DBD-JDP-2, or GAL4-DBD-SRC-1 fusion construct (50 ng each). Relative luciferase activity was calculated by setting normalized luciferase activity of reporter alone equal to 1.0 and all other treatment groups as folds relative to 1.0. UAS, upstream activation sequence. (C) Schematic of chimeric receptor constructs including PR DBD-VP16, RARα-N-PR-DBD, ERα-N-PR-DBD, and PR-N-ERα-DBD. (D) Cos-1 cells were transfected with an N-terminal/DBD PR construct (1.5 ng) or the chimeric constructs (2 to 25 ng) together with cognate PRE2-TATA-Luc (200 ng) or ERE3-TATA-Luc (100 ng) reporter genes in the presence or absence of JDP-2 (PCR3.1-JDP-2; 100 ng). The fold coactivation by JDP-2 was calculated by setting relative luciferase activity for each receptor construct equal to 1.0 and corresponding values in the presence of JDP-2 as folds relative to 1.0. Values are averages ± standard errors of the means (SEM) of at least three independent experiments.
Jun dimerization protein 1 (JDP-1) and related JDP-2 are small bZIP proteins containing the leucine zipper and the basic amino acid DNA binding domain common to AP-1 factors but lacking an N-terminal activation domain (1; Fig. 1A). JDP-1 and -2 are reported to function as repressors of AP-1 and can inhibit activity either through binding to AP-1 DNA sites as heterodimers with Jun or by recruitment of histone deacetylases (5, 7, 17-19, 43). We previously reported that JDP-2 also interacts with and enhances the transcriptional activity of human PR (11, 53). JDP-2 is expressed in human female reproductive tissues and PR-positive cell lines, and in cell culture experiments, an inverse correlation was observed between enhancement of PR transactivation by ectopic expression of JDP-2 and endogenous levels of JDP-2. As detected by chromatin immunoprecipitation assay, endogenous JDP-2 is recruited to the promoter of a progesterone-regulated target gene in breast cancer cells in a hormone- and PR-dependent manner (11, 53). JDP-2 acts on PR primarily by enhancing the activity of the amino-terminal domain independent of AF-2 and p160 coactivators; however, it physically interacts with the DBD, not with the transcription activation surfaces in the N-terminal domain (11, 53). In the present paper, we sought to define the mechanism by which JDP-2 modulates transcriptional activity of the amino-terminal domain of PR through binding to the DBD. We show that JDP-2 induces or stabilizes the structure in the N-terminal domain in a manner that correlates with its stimulation of transcriptional activity. The effect of JDP-2 appears to be dependent on a specific interdomain communication between the DBD and N-terminal domains. Additionally, we provide evidence that the originally defined AF-1 sequence region of PR is not sufficient for the transcription activity of the N-terminal domain and response to JDP-2. Instead, multiple elements are required, suggesting that the transcriptional activity of the N-terminal domain involves protein folding that can be integrated allosterically through cofactor binding to the DBD.
MATERIALS AND METHODS
Hormones and antibodies.
R5020 (Promegestone) was obtained from Dupont/New England Nuclear Products (Boston, MA). Rabbit polyclonal antisera recognizing rat JDP-2 have been previously described (1, 53). 1294/H9 is a mouse monoclonal antibody (MAb) that recognizes both A and B isoforms of human PR (11, 53). MAb N559 recognizes an epitope on the N-terminal side of the PR DBD between aa 551 and 564. MAbs to His6 (1162/F6) or glutathione S-transferase (GST) (794/H2) tags have been previously described (6). A rabbit polyclonal antibody was raised against recombinant purified PR DBD.
Mammalian cell expression plasmids and reporter genes.
pCDNA3-his(rat)JDP-1/-2 and pCR3.1-rat JDP-2 that express JDP-1/JDP-2 under the control of the cytomegalovirus promoter and phPR-B that expresses human PR-B under the control of the simian virus 40 promoter have been previously described (53). A GAL4/DBD-SRC-1 fusion protein (pABΔGAL-SRC-1) construct was provided by Sergio Onãte (Roswell Park, Buffalo, N.Y.). A FLAG-PR-DBD-VP16 (PR aa 556 to 642) fusion construct cloned into pCDNA1 was a gift from David Shapiro (University of Illinois). A GAL4/DBD-JDP-2 fusion protein plasmid was constructed by insertion of JDP-2 cDNA into plasmid pM (Clontech, Palo Alto, CA). Luciferase reporter plasmids used were PRE2-TATA-Luc (provided by Zafar Nawaz, Baylor University), MMTV-Luc (provided by Steven Nordeen, University of Colorado Health Sciences Center), 5X-GAL (UAS)-Luc, and ERE3-TATA-Luc (provided by Donald McDonnell, Duke University).
Amino-terminal DBD two-domain expression plasmids of PR, BnDBD (aa 1 to 650), and AnDBD (aa 165 to 650) were cloned into pABΔGAL-AF-1-DBD and ΔGAL-AnDBD, respectively, and are under the control of the Rous sarcoma virus promoter (42). PR N-terminal/DBD two-domain constructs with truncations in the N-terminal domain including 319-DBD (PR aa 319 to 650), 350-DBD (PR aa 350 to 650), 402-DBD (PR aa 402 to 650), and 428-DBD (PR aa 428 to 650) were constructed by PCR amplification and insertion of the amplicon into pABΔGAL. Internal deletions of the N-terminal domain of PR including Δ323-427 and Δ475-534 were constructed in the context of the full-length PR-B expression plasmid and the two-domain pABΔGAL-BnDBD plasmid.
Chimeric N-terminal domain DBD constructs, including the N-terminal domain of RARα linked to the DBD of PR (RARα-N-PR-DBD), the N-terminal domain of ERα linked to the DBD of PR (ER-N-PR-DBD), the N-terminal domain of PR-B linked to ER DBD (PR-N-ER-DBD), and VP16 linked to PR-DBD (PR-DBD-VP16), were constructed using PCR splice-overlap extension (SOEing) (16, 56). Receptor N-terminal sequences, or VP16, were amplified by PCR and stitched to the appropriate receptor DBD. A region of PR N-terminal domain linked to PR DBD (aa 350 to 428) was also constructed by PCR SOEing. Chimeric constructs were inserted into pABΔGAL under the control of the Rous sarcoma virus promoter using BglII and HindIII restriction sites designed into 5′ and 3′ primers, respectively. All cloning junctures and truncation and deletion mutations were verified by DNA sequencing, and protein expression was confirmed by Western blot analysis.
Cell culture and transfection.
Cos-1 cells were plated onto 6-well dishes or 100-mm dishes (Falcon, Franklin Lakes, NJ) at a density of 1.6 × 105 or 1.1 × 106 cells, respectively. Cells were transfected using Lipofectamine and Plus reagents (Gibco BRL, Gaithersburg, MD) and grown in Dulbecco's modified Eagle's medium supplemented with 5% charcoal-stripped fetal bovine serum for 24 h followed by treatment for 16 to 20 h with vehicle (ethanol) or 10 nM R5020. Transfected plasmids included constitutively active pCH110 β-galactosidase as an internal control for transfection efficiency, PRE2-TATA-Luc or MMTV-Luc progesterone-responsive luciferase reporter plasmids, and various amounts of PR domain expression vectors with or without pCR3.1-JDP-2. pCR3.1 empty vector was added to maintain a constant number of moles of cytomegalovirus promoter. Cells were washed (40 mM Tris [pH 7.4], 150 mM NaCl, 1 mM EDTA) and lysed directly in the well using 0.3 ml lysis buffer (20 mM K2HPO4 [pH 7.8], 5 mM MgCl2, 0.5% Triton X-100). Luciferase and β-galactosidase activity was assayed as previously described (53). Luciferase was normalized to β-galactosidase activity, and relative activity was calculated by setting the normalized values obtained in cells with reporter alone and no receptor equal to 1.0 and all treatment groups as values relative to 1.0. The fold increases in the presence of ectopically expressed JDP-2 were calculated by setting relative luciferase activity in the absence of JDP-2 equal to 1.0 and corresponding values in the presence of JDP-2 as n-fold increases over 1.0.
Baculovirus expression and purification of PR and PR domains.
Vectors for various domains of PR including AnDBD (PR aa 165 to 688) and the N-terminal domain of PR-A (termed AN) (PR aa 165 to 535), containing N-terminal polyhistidine tags, were previously described (6). Proteins were expressed in Sf9 insect cells, and cell lysis, extraction, and nickel resin affinity purification procedures have been described previously (6). Purified AN and AnDBD used in circular dichroism (CD) analysis were concentrated in an Amicon stirred cell (Millipore, Billerica, MA) under N2 gas pressure.
Expression and purification of JDP-1/JDP-2, PR DBD, and HMGB-1.
Rat JDP-1/JDP-2, PR DBD, and HMGB-1 as GST fusion proteins were expressed from pGEX2T (Amersham Pharmacia, Uppsala, Sweden) vectors in the BL21 strain of Escherichia coli. Protein expression was induced by IPTG (isopropyl-β-d-thiogalactopyranoside) (1.0 mM) for 4 h, and cells were lysed in 4 ml of lysis buffer/1-g cell pellet (50 mM Tris, pH 8.0, 20 mM NaCl, 1 mM MgCl2, 1 mM EDTA, 5 mM dithiothreitol [DTT], 1 μl of Benzonase [Novagen, Stamford, CT]/ml buffer, 1× BugBuster [Novagen], and a protease inhibitor cocktail). JDP-2 was expressed largely as inclusion bodies. Inclusion bodies collected as pellets by centrifugation at 10,000 × g at 4°C for 30 min were washed three times in a wash buffer (50 mM Tris [pH 8.0], 100 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, 1 mM DTT), followed by resuspension in 90 ml of denaturing buffer (20 mM Tris [pH 8.0], 1.0 mM EDTA, 5 mM DTT, 6 M guanidine-HCl) and incubation at 60°C for 10 min. Glycerol was added to 10% total volume, and denatured protein was clarified by centrifugation for 15 min at 25,000 × g at 4°C. Supernatant containing solubilized protein was dialyzed overnight (12 to 16 h) at 4°C against 4 liters of dialysis buffer 1 (50 mM Tris [pH 8.0], 0.5 M NaCl, 10% glycerol, 2 M urea, 1 mM EDTA, 5 mM DTT), followed by dialysis for 6 h at 4°C against 4 liters of dialysis buffer 2 (50 mM Tris [pH 8.0], 1 M NaCl, 10% glycerol, 1 mM MgCl2, 1 mM EDTA, 5 mM DTT). Renatured protein was bound to glutathione-Sepharose (10-ml packed volume) for 2 h at 4°C, and the resins were washed repeatedly (20 mM Tris [pH 8.0], 1.5 M NaCl, 1 mM EDTA, 1 mM DTT, 10% glycerol), followed by exchange into thrombin cleavage buffer (20 mM Tris [pH 8.0], 250 mM NaCl, 2.5 mM CaCl2, 1 mM DTT, 10% glycerol). JDP-2 was cleaved and released by incubation of the resins overnight at 4°C with 400 units of thrombin (Sigma, St. Louis, MO), and EDTA was added to a final concentration of 1.0 mM. Eluted JDP-2 was concentrated in an Amicon stirred cell (Millipore, Billerica, MA) and fractioned by fast protein liquid chromatography on a Superdex-75 size exclusion column (Amersham Pharmacia, Uppsala, Sweden) equilibrated in buffer containing 20 mM Tris (pH 8), 10% glycerol, 1 M NaCl, and 1 mM DTT. Peak JDP-2 fractions were pooled, concentrated, and analyzed by Coomassie blue staining of sodium dodecyl sulfate-10% polyacrylamide gels and by Western blot. The protein concentration of the purified product was determined by UV optical density scans from 220 to 320 nm and by amino acid analysis.
GST-JDP-1 was expressed, cleaved, and purified in a similar manner, except that IPTG induction at an A600 of 0.7 was performed for 3 h. Also, because GST-JDP-1 was expressed largely in soluble form, the denaturation/renaturation steps were eliminated.
PR DBD (aa 562 to 670) was purified as previously described (37). Briefly, expression of pGEX-PR DBD-670 plasmid was induced in E. coli BL21 as described above for GST-JDP-2, followed by similar cell lysis. However, because GST-PR DBD is soluble, the lysates were centrifuged at 100,000 × g to prepare a soluble whole-cell extract in the supernatant fraction. GST PR DBD in whole-cell extracts was bound in batch to glutathione-Sepharose and purified essentially as described above for JDP-2, except that a Superdex-30 column was used as the size exclusion step.
GST-(rat)-HMGB-1 (aa 8 to 165) was expressed from a pGEX2T vector in BL21 cells. This construct expressed both HMG boxes of HMGB-1 minus the N- and C-terminal extensions and is the minimal region required for interaction with PR DBD (38, 39). GST-HMGB-1 expression was induced by IPTG (200 nM) for 2 h at 25°C. After cell lysis and binding to a glutathione-Sepharose resin, the protein was cleaved from the resin with thrombin, purified further by size exclusion chromatography (Superdex-30; Amersham Biosciences), and concentrated. The yield of HMGB-1 was 10 mg/liter of cell culture, and the protein was ∼98% pure as determined by SDS-polyacrylamide gel electrophoresis (PAGE) and Coomassie blue staining.
CD spectroscopy.
CD spectroscopy was performed with a Jasco-810 spectropolarimeter with constant N2 flushing (Jasco Inc., Easton, MD.) (9). A Lauda circulating water bath was used to control the temperature of the optic cell chamber, where rectangular or cylindrical cells of 1-mm or 0.5-mm path length, respectively, were used. The concentration of purified protein stock solutions was determined by absorbance at 280 nm in CD buffer (20 mM Tris [pH 7.4], 1 mM DTT, 150 mM NaCl, 10% glycerol,1 μM ZnCl2, and 1 mM EDTA [for AN, AnDBD, DBD, and HMGB-1] and 20 mM Tris [pH 8.0], 10% glycerol, 1 M NaCl, and 1 mM DTT [for JDP-1 and -2]) and was verified by amino acid analysis with a Beckman (San Ramon, CA) 6300 amino acid analyzer. Molar extinction coefficients used to calculate protein concentration were as follows (values are M−1 cm−1): AnDBD, 24,890; AN, 17,210; PR DBD, 2,460; JDP-2, 1,280; JDP-1, 11,380 and HMGB-1, 20,430. Purified proteins used for CD spectroscopy were not frozen and thawed; they were freshly prepared and maintained at 4°C as concentrated stocks, and small aliquots were diluted directly in CD buffer just prior to analysis. Final concentrations of proteins used in CD scans ranged from 20 μM to 80 μM and are indicated in the figure legends. CD spectra were performed from at least three independent and separate preparations of purified protein. Thus, the spectra are representative of each protein or protein complex. Each protein spectrum was the average of 10 wavelength scans collected at 0.5- or 1-nm intervals from 195 to 250 nm. The uncertainty in molar ellipticity values was ±300° · cm−1 · dmol−1.
Deconvolution of CD spectra of AnDBD in the presence or absence of JDP-2 was achieved using four secondary structural predication algorithm programs (YANG, SELC3, K2D, and VAR) to analyze the spectra of AnDBD alone or of the mixture of AnDBD with JDP-2 (where absorbance of JDP-2 alone was subtracted from the mixture spectrum). Predicted percent values of α-helix, β-sheet, and random coil were averaged between the four programs (the website for the prediction of percent secondary protein structure can be found at http://www.embl-heidelberg.de/∼andrade/k2d/).
Temperature-induced denaturation monitored by circular dichroism.
Protein stability was determined by thermal denaturation by monitoring the decrease in α-helical content at a wavelength of 222 nm with increasing temperature. For thermal melting experiments, data points were taken at 1°C intervals at a scan rate of 60°C per hour. The temperature dependence of the ellipticity θ was fitted to obtain a fraction of the unfolded state, fU(t), using a nonlinear least-squares algorithm assuming a two-state unfolding reaction with pretransition [folded state, θN(t)] and posttransition [unfolded state, θU(t)] baseline corrections (34):
 |
(1) |
where the pre- and posttransition baseline corrections are assumed to be linearly dependent on temperature. The calculated fraction of the unfolded state, fU(t), is used to determine ΔG (41):
 |
(2) |
where ΔGapp is the change in apparent Gibbs free energy of folding described by the Gibbs-Helmholtz equation:
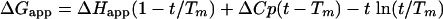 |
(3) |
where t is the observed temperature, Tm is the temperature midpoint of the thermal transition, ΔHapp is the apparent enthalpy of unfolding, and ΔCp is the change in heat capacity change associated with protein unfolding. ΔCp and ΔH are assumed to be constant within the temperature range under this study (20). These thermodynamic parameters were fitted using the program Sigmaplot (SPSS Inc., Chicago, IL), and the derived fraction-folded parameters were used to determine an apparent free-energy stability term, ΔGapp,20, using the following equation:
 |
(4) |
by plotting fraction-folded data and then extrapolating to 20°C.
Limited proteolytic digestion of PR.
Limited digestion of the two-domain AnDBD PR polypeptide by endoproteinase Arg-C (EndoArgC) protease was carried out essentially as described previously (2, 3). EndoArgC was reconstituted at a concentration of 0.1 mg/ml (50mM Tris [pH 8.0], 10 mM CaCl2, 5 mM EDTA) and was incubated at 37°C over time (0, 5, 10, 15, 30, or 60 min) at a 30:1 ratio (by micrograms) of AnDBD protease in a proteolysis buffer (100 mM Tris [pH 8.0], 10 mM CaCl2, 2% glycerol, 100 mM NaCl, 5 mM DTT). For digestion of the AnDBD in the presence or absence of JDP-2, aliquots containing 1 μM AnDBD were mixed with 7.5 μM JDP-2 (2 μg per reaction) or 2 μg lysozyme as a control protein and incubated as described above for 10 min at 37°C with increasing concentrations of protease (AnDBD:EndoArgC, 12.3:1 to 1.5:1). The reactions were stopped by the addition of EDTA and 80 μl of 2% SDS-β mercaptoethanol sample buffer, followed by heating for 10 min at 80°C. Digested fragments of PR were detected by Western blot with a polyclonal antibody to the PR DBD. A double-stranded 28-bp oligonucleotide containing a single progesterone response element (PRE) was added to some reactions (5′-TTTGAGAACAAACTGTTCTTAAAACGAG3′).
EMSA.
DNA binding activity of purified JDP-2 was analyzed by electrophoretic gel mobility shift (EMSA) assays using a γ-32P-labeled double-stranded oligonucleotide (5′-CGCTTGATGACTCAGCCGGAA-3′) corresponding to an AP-1 element (Santa Cruz Biotechnology, Santa Cruz, CA). Purified JDP-2 (40 nM) or 3 μM purified c-Jun (Promega, Madison, WI) was incubated for 30 min in binding buffer (10 mM Tris [pH 7.8], 50 mM NaCl, 5% glycerol, 1 mM EDTA, 2 mM MgCl2, 5 mM DTT) at room temperature with 0.5 ng of 32P-labeled AP-1 oligonucleotide in a 25-μl reaction mix. Complexes were resolved by native gel electrophoresis using 5% polyacrylamide-0.5× Tris-borate-EDTA gels supplemented with 2.5% glycerol at constant current of 20 mA per gel. Gels were dried, and complexes were detected by autoradiography. DNA binding activity of purified PR DBD, AnDBD polypeptides, and JDP-2 was also assessed by quantitative EMSA with an unlabeled PRE or AP-1 oligonucleotide probe as described above, except that gels were stained with Stains-All (Sigma) to detect both DNA and protein in the gel.
SDS-PAGE and Western blots.
Proteins were separated by 8% or 10% SDS-polyacrylamide gel electrophoresis and analyzed by Western blot as previously described (6, 11). Detection was done by enhanced chemiluminescence (Perkin-Elmer, Boston, MA).
RESULTS
A role for the DNA binding domain in mediating the effect of JDP-2 on the transcription activity of the N-terminal domain of PR.
Because the DBD is the interaction site for JDP-2, it indicates that JDP-2 is mechanistically a different class of coregulatory protein from other coactivators such as SRCs that interact directly with transcription activation domain surfaces. As further evidence of this, JDP-2 does not appear to have an intrinsic transcription activation domain. When JDP-2 was linked to a heterologous DNA binding domain (GAL4 DBD), the JDP-2/GAL4 DBD chimera failed to transactivate a GAL4-controlled luciferase reporter gene in cell cotransfection assays (Fig. 1B). By contrast, SRC-1 linked to GAL4 DBD under the same conditions promoted a strong transactivation (Fig. 1B). The DBD is a highly conserved region of nuclear receptors, and JDP-2 is capable of binding to DBDs of several nuclear receptors including thyroid hormone receptor (TR), GR, AR, and ER. However, ectopically expressed JDP-2 preferentially stimulates transcriptional activity of PR compared to several other nuclear receptors. Of the receptors tested, JDP-2 had no effect on the activity of ER, TR, AR, and RARα and had only a weak stimulatory effect on GR (11, 53; data not shown). To determine whether JDP-2 enhancement of transcription activity may require a specific interdomain communication between the DBD and N terminus of PR, receptor chimeras were constructed by fusing the N-terminal domains of two other nuclear receptors, RARα and ERα, to the PR DBD. Conversely, the N-terminal domain of PR was fused to the ER DBD, and the PR DBD was linked to an unrelated transcription activation domain from VP16, to generate PR-DBD/VP16 (Fig. 1 C). These chimeras are constitutively active when cotransfected into cells with luciferase reporter genes controlled by cognate PREs or estrogen response elements (EREs), and their activity was normalized to 100% (or 1) in the absence of ectopically expressed JDP-2. Transfection conditions that yielded comparable expression levels of these receptor chimeras as determined by immunoblot were determined (data not shown). Transactivation of a PRE-Luc reporter gene mediated by the PR DBD linked to its own N-terminal domain (BnDBD) was enhanced ∼7.5-fold by ectopic expression of JDP-2 (Fig. 1D). Cotransfected JDP-2 also enhanced transactivation of PRE-Luc mediated by chimeric receptors containing the N domains of RARα and ERα linked to PR DBD, although the three- and fivefold stimulatory effects, respectively, were less. In contrast, JDP-2 had a minimal but significant 1.5-fold stimulatory effect on transactivation of ERE-Luc mediated by a receptor chimera containing the N domain of PR linked to ER DBD and on the activity of PR DBD/VP16 (Fig. 1D). These results suggest that the N-terminal domains of other steroid nuclear receptors share interchangeable features with the N domain of PR and are capable of responding to JDP-2 when linked to the PR DBD, whereas activation domains of unrelated transcription factors may not. Additionally, the DBD of PR appears to contain elements lacking in the ER DBD that are required for JDP-2 to transmit a maximal transcription signal to the N-terminal domain.
JDP-2 interaction induces a structural change in PR dependent on the presence of both the DBD and N-terminal domains.
We next used far-UV CD to determine whether JDP-2 interaction with the DBD induces or stabilizes structural changes in the N-terminal domain that might account for the stimulatory effect of JDP-2 on transcription. Because JDP-2 was determined previously to equally stimulate the activity of N-terminal domains of the two PR isoforms, CD spectroscopy was performed only with the N-terminal domain of PR-A (53). The N-terminal domain of PR-A (termed AN [aa 165 to 535]) (Fig. 1A) or the N-terminal domain of PR-A linked to the PR DBD (AnDBD [aa 165 to 688]) (Fig. 1A) was expressed and purified to >95% from the baculovirus system in Sf9 insect cells as previously described (6, 38, 39). The PR DBD (aa 562 to 670), as previously described (37), and JDP-2, as shown in Fig. 2A to C, were expressed as GST fusion proteins in E. coli and purified to near-homogeneity by glutathione-Sepharose affinity chromatography, thrombin cleavage of the GST moiety, and gel filtration to separate free GST and bacterial contaminants. As determined by quantitative EMSAs, where both the purified protein and DNA are detected by staining with Stains-All dye, the majority of purified PR AnDBD polypeptide was functional with respect to DNA binding activity and thus appeared to be properly folded. Assembly of protein-DNA complexes was detected at a ∼2:1 molar ratio of protein to PRE oligonucleotide. This is the expected ratio if the majority of purified AnDBD polypeptide is active, as it should bind as a homodimer to a PRE (Fig. 2E). Similar EMSA results were obtained for the PR DBD polypeptide (data not shown; S. Roemer, L. Sherman, D. P. Edwards, and M. E. Churchill, unpublished data). Because the AN PR polypeptide lacks the DNA binding domain and does not directly bind JDP-2, neither EMSAs nor JDP-2 binding can be used to determine the fraction of purified protein that is functional. However, similar CD spectra of AN and AnDBD polypeptides (Fig. 3) were obtained, suggesting that purified AN and AnDBD polypeptide, which binds DNA quantitatively, exhibit similar secondary structure properties. DNA binding to an AP-1 element was used to determine the fraction of purified JDP-2 that is functional. By standard EMSAs (detection of upshifted 32P DNA), purified JDP-2 bound to the AP-1 element with an efficiency equal to that of commercially prepared purified c-Jun (Fig. 2D). By quantitative EMSA, assembly of the JDP-2/DNA complex was obtained at a ∼2:1 molar ratio of protein to DNA, which is also the expected ratio for binding of a bZIP protein to AP-1 elements (not shown). CD spectra showing a high α-helical content also indicate that purified JDP-2 is properly folded (see below; Fig. 3).
FIG. 2.

Purification and functional analysis of JDP-2 and AnDBD PR polypeptide. (A to C) Purification of bacterial expressed recombinant JDP-2. Expression of JDP-2 as a GST fusion protein was induced in E. coli carrying pGEX2T-JDP-2. Cells were lysed, and insoluble material was pelleted. JDP-2-GST was then denatured/renatured from inclusion bodies and purified by glutathione-Sepharose chromatography followed by thrombin cleavage, release of free JDP-2, and gel filtration by Superdex-75. Panels A to C show Coomassie blue SDS-PAGE analysis of purification fractions. (A) Lane 1, molecular mass (MW) standards; lane 2, lysate (5 μl); lane 3, cleared whole-cell extract (5 μl); lane 4, renatured GST-JDP-2 (50 μl). (B) Eluate from glutathione-Sepharose affinity resin was filtered prior to injection over a Superdex-75 size exclusion column. Other lanes are Superdex-75 column fractions (50 μl) 15 to 26. (C) Fractions corresponding to the JDP-2 elution peak from Superdex-75 were pooled (lane 2), concentrated (lanes 3 to 5), and analyzed compared with purified lysozyme (lanes 6 to 8). (D) EMSA of purified JDP-2. Purified c-Jun (3 μM) or JDP-2 (40 nM) was incubated with 0.5 ng of a γ-32P AP-1 oligonucleotide for 30 min at 25°C. Samples were separated on 5% nondenaturing polyacrylamide gels followed by drying of the gels and detection of protein-DNA complexes by autoradiography. Results are representative of three independent experiments. (E) Quantitative EMSA of purified AnDBD (65 μm to 200 μm) incubated with 65 μm PRE oligonucleotide for 30 min at 4°C. Reactants were electrophoresed on 5% nondenaturing polyacrylamide gels in Tris-acetate-EDTA buffer, pH 8.5, and gels were stained with Stains-All (Sigma) dye. Lane 1, marker dyes; lane 2, free DNA; lane 3, free AnDBD; lanes 4 to 10, various ratios of AnDBD to PRE DNA from 1:1 to 3:1 (lane 7 is a 2:1 ratio [*]).
FIG. 3.

JDP-2 interaction alters secondary structure of AnDBD PR polypeptide as detected by CD spectroscopy. (A) Experimental CD spectra of purified JDP-2 (80 μM) and AnDBD PR polypeptide (42 μM) determined individually and from a mixture of the two proteins at the same concentrations (AnDBD+JDP-2 exp). The theoretical additive sum of the CD spectra of JDP-2 and AnDBD (theo) is also shown. The arrows indicate the differential between the theoretical and experimental negative absorbance peaks at 208 nm and 222 nm for AnDBD plus JDP-2. (B) Experimental CD spectra of purified JDP-2 (80 μM) and the isolated AN PR (43 μM) polypeptide determined individually and from a mixture of the two polypeptides at the same concentration (AN+JDP-2 exp). The theoretical additive sum of the CD spectra of JDP-2 plus AN polypeptide (theo) is also shown. Arrows indicate little or no difference between theoretical and experimental negative absorbance peaks at 208 nm and 222 nm. (C) Experimental CD spectra of purified JDP-2 (80 μm) and PR-DBD (40 μM) determined individually and from a mixture of the two proteins at the same concentrations (DBD+JDP-2 exp). The theoretical additive sum of a mixture of the two polypeptides is also shown (DBD+JDP-2 theo). Arrows indicate small differences between theoretical and experimental negative absorbance peaks at 208 nm and at 222 nm for the mixture of DBD and JDP-2.
CD detects secondary protein structure in solution, allowing conclusions to be drawn about α-helical, β-sheet, and random coil content of proteins (34, 41). α-Helical conformation is detected by negative absorbance peaks at 208 nm and 222 nm, whereas β-sheet or random coil conformation is detected as negative absorbance peaks at 215 nm or below 210 nm, respectively. We first analyzed spectra of JDP-2, the two-domain PR AnDBD, and single-domain PR AN polypeptides separately, detecting absorbance for all three between approximately 195 nm and 245 nm (Fig. 3A to C). JDP-2 absorbs primarily as two negative peaks at 208 nm and 222 nm, indicating that the purified protein adopts a primarily α-helical conformation in solution, as expected for a bZIP protein (27; Fig. 3A to C). The AnDBD fragment of PR absorbs with a small negative peak at 222 nm but exhibits much more absorbance below 210 nm (Fig. 3A). A similar spectrum was obtained with the single-domain AN polypeptide (Fig. 3B), indicating that both PR polypeptides contain a large amount of random coil with some underlying helical content.
CD analysis of a mixture of two noninteracting proteins, or of two interacting proteins that do not undergo a conformational change, should yield a spectrum that is superimposable (within the accuracy and sensitivity of the instrument) with the theoretical sum of the spectra of the two proteins measured independently. If the observed spectrum of the protein mixture deviates significantly from the theoretical sum, then the overall protein conformation has been changed by interaction between the two proteins. We next compared the theoretical sum (calculated by adding the values of absorbance detected at each wavelength for each protein individually) to the experimental CD spectra collected from a mixture of JDP-2 and PR-AnDBD (Fig. 3A) or JDP-2 and PR-AN (Fig. 3B). AnDBD and AN polypeptides were analyzed at essentially the same molar concentrations (40 μM and 43 μM, respectively), as determined by amino acid analysis, and JDP-2 was added at a twofold molar excess (80 μM). When JDP-2 and AnDBD were mixed, substantial increases in negative absorbance of ∼13 mdeg and ∼14 mdeg, respectively, were observed at 222 nm and 208 nm, compared with the theoretical sum of the individual spectra of JDP-2 and AnDBD (Fig. 3A). In contrast, when JDP-2 and the AN polypeptide were mixed, a minimal difference was detected between the actual and theoretical spectra. The experimental spectra of the two proteins showed a difference of 1 mdeg from the theoretical at 208 nm and a change of 2.5 mdeg at 222 nm (Fig. 3B). Therefore, as expected from previous in vitro binding data that showed that the DBD of PR is the primary binding site for JDP-2 (53), little or no change in protein conformation was detected by CD analysis when JDP-2 and the isolated N-terminal domain (AN) of PR were mixed together.
Although the data in Fig. 3A and B indicate that interaction between JDP-2 and PR-AnDBD results in a change in secondary structure, it cannot distinguish between an effect on the structure of JDP-2, PR, or both proteins (coconformational effects). To attempt to make this distinction, we analyzed structural changes in response to JDP-2 interaction with the isolated PR DBD. We reasoned that if JDP-2 interaction with its primary binding site, the DBD, has no effect on structure, the structural change induced upon JDP-2 interaction with the AnDBD polypeptide should reside in the receptor. The CD spectrum of the PR DBD exhibits two negative peaks of relatively balanced intensity at 208 nm and 222 nm, indicative of the expected predominantly α-helical content of nuclear receptor DBDs (30, 36, 50, 51). When JDP-2 and PR-DBD were mixed, the observed and theoretical sums of the individual spectra exhibited small differences compared with that observed upon JDP-2 interaction with the AnDBD. A change in amplitude of ∼3 mdeg was observed between theoretical and experimental data at the negative 208-nm and 222-nm peaks and a slight shift to the right at wavelengths below 205 nm (compare Fig. 3A and C). These results indicate that structural changes in the DBD or JDP-2 contribute in a minor way to the overall change in structure observed upon JDP-2 interaction with the AnDBD polypeptide. Results with trifluoroethanol (TFE), a solvent that induces maximal formation of α-helices in unstructured polypeptides that have a propensity to form an α-helix, are in further support of this conclusion. TFE (50%, vol/vol) had no effect on the CD spectra of JDP-2, indicating that it is maximally folded in aqueous solution. In contrast, the AnDBD polypeptide exhibited a substantial shift in conformation to more helical content in TFE (data not shown). These data collectively indicate that the substantial change in CD spectrum obtained upon interaction of JDP-2 with the PR AnDBD is dependent on the presence of both the N-terminal domain and the DBD and that changes in secondary structure occur primarily with PR and not with JDP-2.
Quantitative analysis of structural changes in PR induced by JDP-2.
We next analyzed the CD spectra of the AnDBD PR polypeptide when combined with various concentrations of JDP-2. Because changes in JDP-2 structure appear to contribute minimally to the overall change in structure of the AnDBD-JDP-2 complex, the absorbance of JDP-2 itself was mathematically subtracted from all the ellipticity values of the CD spectra (Fig. 4A). A dose-dependent increase in α-helical content that saturated at 2 molecules of JDP-2 per molecule of AnDBD was observed. No additional increase in α-helical conformation was detected at higher ratios of JDP-2 to PR (Fig. 4A). Similar to the experimental data shown in Fig. 3A, JDP-2, at the saturation dose, increased the amplitude of negative absorbance at 208 nm and 222 nm by 12.8 and 13.7 mdeg, respectively. The subtracted CD spectra from Fig. 4A were used for calculations of secondary structure content of the AnDBD in the presence and absence of saturating JDP-2. These subtracted CD spectra were deconvoluted using four different analysis software programs (YANG, SELC3, KJ2D, and VAR), and the percentages of α-helix, β-sheet, and random coil content were calculated as an average value obtained from all four programs. The AnDBD polypeptide in aqueous solution was estimated to contain 30% α-helix. At the saturating dose of JDP-2, the α-helical content of AnDBD increased to 58%, indicating a ∼2-fold increase. JDP-2 also was calculated to induce a decrease in random coil and β-sheet (or β-turn) content of 17% and 11%, respectively.
FIG. 4.

JDP-2 interaction with PR AnDBD polypeptide increases α-helical content in a dose-dependent manner and forms a stable complex. (A) CD spectra of AnDBD alone (42 μM) or a mixture of AnDBD (42 μM) and JDP-2 with various molar ratios of JDP-2 receptor as indicated. The absorbance of JDP-2 alone has been mathematically subtracted from the spectra of the mixed proteins. Arrows indicate differential between theoretical and experimental negative absorbance peaks at 208 nm and 222 nm for AnDBD plus JDP-2. (B) JDP-2 (80 μM) alone or in a mixture with AnDBD (42 μM) was incubated in a temperature-regulated CD cell, and absorbance at 222 nm was monitored as the temperature in the cell was raised from 4°C to 80°C at a controlled rate of 1°C/min. Thermal midpoints (Tm) on each curve are marked with a line. The Tm is the temperature at which half of the sample persists in native conformation and half has unfolded and lost absorbance at 222 nm.
The JDP-2/PR complex exhibits increased structural stability.
Protein-protein interaction that changes conformation, and presumably function, is anticipated to induce formation of a stable complex. Therefore, we asked whether JDP-2 interaction would affect the stability of the PR complex as detected by a change in the thermal melt curve, or the temperature at which protein conformation unfolds. The ellipticities of samples containing the AnDBD polypeptide or JDP-2, alone or in combination, were monitored at a single wavelength (222 nm) by CD spectroscopy to measure the loss of α-helical structure at different elevated temperatures. A melt curve for AnDBD alone was not able to be obtained, as it became unstable at 25°C. The CD spectrum of AnDBD at 25°C indicated a shift from α-helix to β-sheet followed by formation of flocculent precipitate (data not shown), thus demonstrating a nucleating protein aggregation rather than unfolding. In contrast, JDP-2 undergoes a shallow and broad temperature-dependent transition to an unfolded protein without precipitation with a distinct thermal midpoint (Tm) of 45.2°C (Fig. 4B). When JDP-2 and AnDBD were combined, the behavior of the complex was dramatically changed compared to that of individual proteins. The JDP-2/AnDBD complex exhibited a Tm of 57.8°C, and unfolding of the complex resembled that of a highly cooperative two-state transition from a folded to an unfolded protein. This change in the thermal melt curve indicates that the JDP-2/PR complex is more stable than either protein alone.
JDP-2-induced structural alteration in PR correlates with enhanced transcriptional activity of the N-terminal domain.
To determine whether altered receptor structure correlates with the stimulatory effect of JDP-2 on the transcriptional activity of the N-terminal domain, we analyzed the effect of two other DBD-interacting proteins, HMBG-1 and JDP-1, on the structure of the AnDBD PR polypeptide. HMGB-1 interacts with a portion of the DBD termed the C-terminal extension, and this interaction enhances PR binding to specific target DNA sequence (38, 39). HMGB-1 does not affect the transcription activity of the N-terminal domain of PR (6, 38, 39). Consistent with the well-ordered helical structure of HMG boxes, CD spectra of purified HMGB-1 exhibits approximately equivalent negative absorption peaks at 208 and 222 nm (Fig. 5A). The theoretical sum of individual spectra of HMGB-1 and AnDBD and the experimental data collected from a mixture of the two proteins were superimposable at 208 nm and differed by ∼3 mdeg at 222 nm. These data indicate that HMGB-1 interaction has a minimal effect on structure of PR AnDBD compared to JDP-2 (Fig. 5A).
FIG. 5.

JDP-1 and HMGB-1 interactions have minimal influence on conformation of the AnDBD PR polypeptide by CD analysis. (A) CD spectra of HMGB-1 (40 μM) and AnDBD (20 μM) alone or the theoretical (theo) and experimentally determined (exp) spectra of a mixture of AnDBD (20 μM) and HMG-1 (40 μM). (B) CD spectra of JDP-1 (40 μM) or AnDBD (20 μM) alone or the theoretical and experimentally determined (exp) spectra of a mixture of AnDBD (20 μM) and JDP-1 (40 μM). (C) Cos-1 cells were cotransfected with human PR-B plasmid (1.5 ng) and the progesterone-responsive reporter PRE2-TATA-Luc (200 ng) in the presence or absence of JDP-1 or JDP-2 plasmids (200 ng). Coactivation was calculated by setting luciferase induction of each receptor construct alone in each treatment group equal to 1.0 and corresponding values in the presence of JDP-2 or JDP-2 as values relative to 1.0. Values are averages (±SEM) of three independent experiments.
JDP-1 and -2 are related proteins with a similar domain structure (Fig. 1A), and they both interact directly with the PR DBD (53). Purified JDP-1 generated a CD spectrum similar to that of JDP-2 showing predominant negative deflections at 208 nm and 222 nm, indicative of α-helical content. When mixed with the AnDBD polypeptide, the theoretical sum of the spectra of AnDBD and JDP-1 and the observed spectrum also showed minimal differences (Fig. 5B) compared to the effect of JDP-2 interaction (Fig. 3A). Unlike the stimulatory effect of JDP-2 on transcription activity of PR, ectopically expressed JDP-1 had no effect on transcription activity of the N-terminal domain of PR in a cotransfection assay using a PRE-Luc reporter gene as the end-point response (Fig. 5C). Thus, with these three DBD-interacting proteins, a correlation was observed between structural alteration and stimulation of the transcription activity of the N-terminal domain of PR.
JDP-2 interaction alters protease cleavage of the N domain of PR.
CD analysis cannot determine where a conformational change takes place in a protein; it is only capable of detecting changes in the overall secondary structure. To attempt to localize the conformational change within PR, and to provide an independent approach to confirm effects of JDP-2 on PR structure, partial proteolytic digestion assays were performed with the purified PR AnDBD polypeptide in the presence and absence of JDP-2. Based on amino acid sequence, the AnDBD polypeptide contains 29 potential EndoArgC cleavage sites. However, previous studies by Bain et al. (2, 3) demonstrated that only seven of these sites are accessible in the native two-domain PR polypeptide in aqueous solution, suggesting that there is some degree of ordered folding of the N-terminal domain. The schematic in Fig. 6A shows the reported (2, 3) EndoArgC cleavage sites (numbered 1 to 7) in the N terminus and DBD of native PR (Fig. 6A). Digestion with EndoArgC proceeds from the N terminus generating six fragments of decreasing size that retain the DBD (indicated in Fig. 6A by the boundaries of the numbered cleavage sites). EndoArgC digestion assays were performed at different times between 0 and 60 min with a single concentration of protease (Fig. 6B) or with increasing concentrations of protease for a single time point of 10 min (Fig. 6C). The limited digestion fragments generated were analyzed by Western blot using an antibody to the PR DBD. We first tested the effect of an oligonucleotide that contains a single PRE on the protease digestion pattern of AnDBD. In confirmation of a previous report, arginine 634 in the hinge region between DBD and LBD (3) was protected against proteolysis by the presence of PRE, generating a novel EndoArgC fragment containing sequences between aa 402 and 688 (Fig. 6A and B). To analyze the influence of JDP-2, increasing concentrations of EndoArgC were used at a single time point, and noninteracting lysozyme of a comparable molecular weight was used as a negative control for general protein effects. The addition of JDP-2 resulted in increased protection of fragments 4/7 and 5/7 containing sequences between aa 402 and 634 and 428 and 634, respectively (Fig. 6C). These data indicate that JDP-2 altered conformation over a fairly broad region containing the DBD and the N-terminal domain out to aa 402 and that DNA and JDP-2 induce distinct conformational changes in PR (Fig. 6A to C).
FIG. 6.

JDP-2 interaction alters protease digestion of the PR AnDBD polypeptide. (A) Schematic of EndoArgC cleavage sites (sites 1 to 7 at amino acids positions indicated) in native AnDBD PR polypeptide that generates six unique fragments with the indicated N- and C-terminal boundaries. PR fragments stabilized by PRE DNA spans aa 402 to 688 (fragment 4>7), while JDP-2 stabilizes two fragments spanning aa 402 to 634 (fragment 4/7) and aa 428 to 643 (fragment 5/7). (B) Purified AnDBD (1 μM) was incubated for 30 min on ice in the presence or absence of synthetic PRE oligonucleotide (1 μM) in a cleavage buffer containing DTT (5 mM) and Ca2+ (10 mM). EndoArgC protease was added at a 30:1 AnDBD-protease ratio by microgram of protein and incubated at 37°C for 0, 5, 10, 15, 30, or 60 min. Fragments of AnDBD generated were detected by Western blot with an antibody to PR DBD. (C) Purified AnDBD was incubated as described above in the presence of purified JDP-2 (7.5 μM, 2 μg) or lysozyme control protein (2 μg). Increasing amounts of EndoArgC protease were added (AnDBD-protease, 12.3:1 to 1.5:1, by microgram of protein) and incubated for 10 min at 37°C. Fragments were detected as described above (B). #, fragments stabilized by PRE; *, fragments stabilized by JDP-2. Results are representative of three independent experiments.
Functional mapping of sequences in the N-terminal domain of PR required for transcriptional activity and stimulation by JDP-2.
To map the region of the N-terminal domain that functionally responds to JDP-2, a series of truncation and internal deletion mutations were introduced into the N-terminal domain of the AnDBD polypeptide (Fig. 7A and 8A). These constructs were analyzed for constitutive transcriptional activity and response to ectopic expression of JDP-2 in cells cotransfected with a PRE-controlled luciferase reporter gene. Activity curves were generated over a wide range of transfected PR plasmid concentrations. Because the activity of each PR polypeptide varied, activation curves were plotted on two scales, each with the intermediate activity polypeptide at aa 402 to 650 for comparison (Fig. 7B). Based on Western blot with a MAb (N559) that detects an epitope (aa 551 to 564) on the N-terminal side of DBD present in all the constructs, each of these PR fragments were produced as stable proteins at comparable levels of expression (Fig. 7D). As expected, BnDBD had substantially higher (∼9- to 10-fold) activity than AnDBD, while truncation to aa 319, which removes the ID (13), exhibited ∼3-fold increased activity over that of AnDBD (Fig. 7B). Truncation to aa 350 further increased activity by another twofold, such that the construct at aa 350 to 650 has ∼6-fold higher activity than AnDBD and only twofold less activity than BnDBD (Fig. 7B). These data are consistent with previous reports of an ID within the first ∼155 amino acids of PR-A but suggest that the boundary of ID may extend farther than previously reported, from aa 319 to 350. Further truncation of the N-terminal domain to aa 402 (402 to 650), 428 (428 to 650), and 450 (450 to 650) resulted in substantial reductions in activity to levels comparable to or below that of the weak activity of AnDBD (Fig. 7B). The shortest truncation construct (aa 450 to 650) is of interest because it contains the previously defined AF-1. AF-1 was originally defined as a sequence region (aa 456 to 546) of the N-terminal domain capable of conferring transcriptional activity when linked to the heterologous GAL4 DBD (40). However, the AF-1 sequence region was weakly active in the context of PR's own DBD and was not sufficient to account in total for the activity of the N-terminal domain (Fig. 7B). These results indicate that substantial transcription activity resides outside the originally defined AF-1 in an uncharacterized region of the N domain between aa 350 and 428 (Fig. 7A).
FIG. 7.

Sequences required for transcriptional activity of the N-terminal domain of PR and stimulation by JDP-2. (A) Schematic of the truncation mutants of the N-terminal domain (N-term) in the context of N domain/DBD polypeptide. (B) Cos-1 cells were cotransfected with PRE2-TATA-Luc (200 ng) and various amounts of the two-domain PR constructs (2 to 50 ng), and relative luciferase activity was calculated to generate activation curves for each PR construct. The activation curve of aa 402 to 650 (open triangles) is graphed on both the left and right panels as a reference for the ∼10-fold difference in activity of PR constructs grouped in the two panels. (C) Cos-1 cells were transfected with PRE2-TATA-Luc (200 ng) together with PR constructs in the presence or absence of pCR3.1-JDP-2 (100 ng) using a single dose of PR that gives equivalent protein expression by Western blot as follows: AnDBD, 1.5 ng; aa 319 to 650, 2 ng; aa 350 to 650, 1 ng; aa 402 to 650, 1 ng; aa 428 to 650, 2 ng; or aa 450 to 650, 15 ng. Coactivation was calculated by setting relative luciferase activity of each PR construct alone equal to 1.0 and corresponding values in the presence of JDP-2 as values relative to 1.0. Values are averages ±SEM of at least three experiments. (D) Relative protein expression level of two-domain PR polypeptides as detected by Western blot using the N559 MAb to a region in the N-terminal domain of PR between aa 551 and 564 that is present in all constructs.
FIG. 8.

Functional analysis of PR N-terminal domain deletion mutations. (A) Schematic of PR constructs containing internal deletions. (B) Cos-1 cells were transfected with PRE2-TATA-Luc (200 ng) together with various doses of wild-type (WT) or internal deletion PR constructs (2 to 50 ng). (C) Cos-1 cells were transfected with PRE2-TATA-Luc (200 ng) together with wild-type (0.5 ng), Δ323-427 (0.5 ng) or Δ475-534 (0.5 ng), or 350-428-DBD (10 ng) PR constructs in the presence or absence of pCR3.1-JDP-2 (100 ng). Relative luciferase activity and coactivation were calculated as described in the legend to Fig. 7. Values are averages (±SEM) of at least three independent experiments. (D) Relative protein expression levels of PR constructs as detected by Western blot using N559 MAb.
The ability of JDP-2 to enhance the activity of the N-terminal domain truncation constructs was tested in cotransfection assays using a single concentration of receptor expression plasmid. As shown in Fig. 7C, the activity of the AnDBD polypeptide containing the full-length N-terminal domain of PR-A and truncations down to aa 350 to 650 (Fig. 7C) were stimulated 15- to 19-fold by JDP-2. With further truncations to aa 450 to 650, the effect of JDP-2 was reduced only slightly to 8- to 12-fold stimulations (Fig. 7C). Because the basal activity of each of these PR polypeptides varies widely (Fig. 7B and C), the maximal activity in the presence of ectopically expressed JDP-2 reflects a similar range of activities (data not shown).
To further delineate sequences in the N-terminal domain important for transcription activity and response to JDP-2, we analyzed the internal deletion mutants shown in Fig. 8A. By immunoblot, these constructs also expressed stable protein at comparable levels (Fig. 8D). Deletion of sequences between aa 323 and 427 (Δ323-427) resulted in a substantial reduction (∼5-fold) of activity compared to that of the full-length N-terminal domain of PR-A (Fig. 8B). In contrast, deletion of amino acids 475 to 534 (Δ475-534), the center 60 residues of the previously defined AF-1 (40), had no effect on activity. Thus, in the context of PR's own DBD, AF-1 is dispensable and the region between aa 323 and 427 is more important for transcription activity of the N-terminal domain. However, when the region between aa 350 and 428 was linked directly to PR DBD (350-428-DBD), it had minimal activity itself, indicating that this sequence region alone is also not sufficient to function as an activation domain (Fig. 8B). Similar to results with truncation mutants, the activities of the internal deletion constructs were all stimulated substantially and to approximately the same extent by ectopic expression of JDP-2 (Fig. 8C).
When the same mutations were introduced in the context of full-length PR-B and analyzed for effects on hormone-dependent gene activation, similar results were obtained (Fig. 9A). Each of the PR constructs expressed stable polypeptides at comparable levels of protein (Fig. 9D). An internal deletion of Δ475-534 (removes AF-1) did not reduce hormone-dependent transcription activity of PR, whereas the deletion of Δ323-427 resulted in a significant loss of hormone-dependent activity (Fig. 9B). Truncation to aa 350 (350-DhLBD) also resulted in substantial reduction of hormone-dependent PR transactivation, and a construct consisting of aa 350 to 428 linked to DBD-hLBD exhibited weak hormone-dependent activity. Ectopic expression of JDP-2 equally enhanced hormone-dependent transactivation mediated by PR-B and each of the mutant PRs (Fig. 9C). Taken together, results of these functional analyses in Fig. 7 to 9 indicate that cooperation between multiple elements, as opposed to a discrete sequence region, is required for full transcriptional activity of the N-terminal domain and that multiple elements can mediate the stimulatory effect of JDP-2.
FIG. 9.

Functional analysis of N-terminal domain mutations in the context of full-length PR. (A) Schematic of mutations in the N-terminal domain of PR-B. (B) Cos-1 cells were transfected with PRE2-TATA-Luc (200 ng) together with various doses of wild-type (WT) or mutant PR-B constructs (1.5 to 25 ng). Cells were treated with vehicle or 10 nM R5020 for the final 24 h of transfection. (C) Cos-1 cells were transfected with PRE2-TATA-Luc (200 ng) together with wild-type PR-B, Δ323-427, Δ475-534, 350-DhLBD, or 350-428-DhLBD (1.5 ng each) in the presence or absence of pCR3.1-JDP-2 (100 ng). Cells were treated for the final 24 h of transfection with vehicle (white bars) or 10 nM R5020 (dark bars). Luciferase induction and coactivation were calculated as described in the legends to Fig. 1 and 5. Values are averages (±SEM) of at least three independent experiments. (D) Relative protein expression of PR constructs as detected by Western blot using MAb N559.
DISCUSSION
The present study provides insights into the mechanism by which JDP-2 stimulates the transcriptional activity of the N-terminal domain of progesterone receptor through binding to the receptor DNA binding domain. A combination of data from biophysical, partial proteolysis, and functional mutagenesis experiments supports the conclusion that JDP-2 induces a more ordered structure of the N-terminal domain required for transcriptional activity mediated by this region of receptor. Furthermore, the effect of JDP-2 binding to the DBD on structure and function of the N-terminal domain appears to be propagated through an interdomain communication (Fig. 10). The mechanism by which increased folding of the N-terminal domain modulates transcriptional activity of PR is not known. Because it does not contain intrinsic transcriptional activity (Fig. 1B), it is likely that JDP-2 bound to the DBD is not a platform for recruiting other coactivators but instead allosterically produces a protein interaction surface for other coactivators to directly mediate the transcriptional activity of the N-terminal domain (Fig. 10). The nature of such cofactors is not known, and their identification is a future goal.
FIG. 10.

Proposed mechanism of JDP-2 stimulation of transcriptional activity of the N-terminal domain of PR. Compared to the highly structured DBD and LBD, the N-terminal domain of PR is unstructured, consisting largely of random coil in solution. JDP-2 interaction with the DBD transmits an interdomain signal to the N-terminal region, resulting in the induction or stabilization of secondary structure elements as evidenced by a twofold increase in α-helical content and a decrease in random coil. Altered structure of the N-terminal domain is proposed to create interaction surfaces for other coactivators that mediate transcriptional activity of the N-terminal domain.
CD spectroscopy is a powerful tool to detect changes in secondary protein in response to the interaction of two proteins. However, it can be difficult to determine whether one or both proteins undergo structural change. In the present study, several findings provide evidence that the observed change in structural conformation upon JDP-2 interaction with the AnDBD polypeptide occurs primarily in PR and not in JDP-2. JDP-2 and its minimal binding site, the isolated PR DBD, are well-structured polypeptides that adopt a primarily α-helical conformation, and neither JDP-2 nor the isolated DBD undergoes a significant structural change when they bind to each other, compared with the magnitude of change observed by JDP-2 interaction with the AnDBD polypeptide (Fig. 3). Thus, the structural stability of JDP-2 in solution and when bound to isolated DBD indicates that protein conformation change occurs primarily in the less-well-structured AnDBD polypeptide. Quantitative analysis of deconvoluted CD spectra estimated that JDP-2 produced an overall increase in α-helical content of the AnDBD from 30% to 58% and a decrease in random coil and β-sheet of 17% and 11%, respectively.
Whether the conformational change in the AnDBD polypeptide is localized to the N-terminal domain, the DBD, or both is more difficult to interpret from these analyses. Several findings support the conclusion that JDP-2 interaction alters the structure of the N domain. As determined by CD analysis, the N-terminal domain is intrinsically less structured than the DBD and potentially more susceptible to changes in protein folding. JDP-2 interaction had a small effect on the structure of the isolated DBD and little or no effect on the isolated amino-terminal domain (AN). Substantial structural change induced upon JDP-2 interaction was dependent on the presence of both the N-terminal domain and DBD of PR (Fig. 3A to C). An independent approach of analyzing structural conformation by partial proteolytic mapping confirmed that JDP-2 can alter structural conformation of the N domain. The presence of JDP-2 altered susceptibility of regions of the N domain of PR to proteolysis. However, these data cannot rule out the possibility that JDP-2 interaction also alters the structure of the DBD in the context of the two-domain PR polypeptide, even though JDP-2 had a minimal effect upon binding to the isolated PR DBD. Suggestive of this possibility, the partial proteolysis data showed that altered susceptibility of PR to protease digestion in the presence of JDP-2 mapped to a broad region of the AnDBD polypeptide that covered both the DBD and a portion of the N-terminal domain (Fig. 6).
That the effect of JDP-2 on the structure of the PR AnDND polypeptide is functionally meaningful was suggested by several results. The effect of JDP-2 on increasing α-helical content of the AnDBD PR polypeptide was dose responsive and saturated at a 2:1 molar ratio with PR. JDP-2 is capable of homodimerization, so one might expect a functional protein complex to consist of two molecules of JDP-2 per molecule of PR. Second, the JDP-2/PR complex is significantly more thermostable than either protein alone, suggesting that the protein conformation induced or stabilized by JDP-2 interaction is functional. The PR N-terminal domain has previously been suggested to exist in multiple conformations (2). Thus, JDP-2 in the complex may limit the fluctuation of conformations and promote organization of a stable conformation required for activity. Third, a correlation was observed between protein-induced conformational change in the AnDBD polypeptide and stimulation of transcription activity of the N-terminal domain. Of the three different DBD-interacting proteins tested, only JDP-2 altered the structure and stimulated transcriptional activity of the N-terminal domain. Two other proteins, JDP-1 and HMGB-1, failed to alter the structure of the AnDBD polypeptide, and neither protein influences the transcription activity of the N-terminal domain.
In addition to its role in recognition of specific DNA elements, there is increasing evidence that the DNA binding domain of nuclear receptors contains interaction motifs for certain classes of coregulatory proteins. In addition to JDP-2, several other coregulatory proteins have been reported to interact with the DBD, including pCAF (CBP-associated factor), PGC-1 (PPARγ coactivator), GT198 (tissue-specific bZIP protein), SNURF (small nuclear ring finger protein) (21, 44, 45), and a novel corepressor, PSF (35). HMGB-1 and -2, members of the nonhistone family of high-mobility group chromatin proteins, interact with the carboxy-terminal extension region of the DBD of the steroid class of receptors. Rather than influencing the activity of AFs, HMGB-1 and -2 facilitate receptor interaction with sequence-specific target DNA (38, 39). The relationship between the bZIP proteins GT198 and JDP-2 is not known, and whether GT198 acts on the N-terminal domain or AF-2 was not reported (21). GT198 exhibits a highly tissue-specific expression pattern and was shown to interact with and enhance the transcriptional activity of a much broader range of nuclear receptors than observed for JDP-2. Similar to JDP-2, PGC-1 as a GAL4-DBD fusion protein does not possess intrinsic transcriptional activity, and it appears to work by inducing a protein conformational change that in turn facilitates recruitment of SRC-1 and p300 as secondary coactivators. However, PGC-1 does not alter the structure of the receptor; it was reported to alter structural conformation of secondary coactivators in a manner associated with their activation (45).
DNA has been reported to act as a ligand and to be able to induce conformational changes in the DBD itself and/or transcription activation surfaces of nuclear receptors (14, 28, 31, 58). Interestingly, different DNA response elements can induce subtle but distinct conformations in receptors with the consequence of influencing coregulatory protein interactions and receptor function (14, 28, 58). Thus, in addition to providing interaction surfaces for coregulatory proteins, the DBD, in response to binding specific DNA, can transmit conformational changes to other regions of the receptor. Interestingly, JDP-2 and DNA interactions with the DBD have different effects on partial proteolytic cleavage patterns of the AnDBD polypeptide (Fig. 6). DNA binding altered the sensitivity of the hinge region to partial proteolysis, whereas JDP-2 interaction altered proteolysis of the DBD and a portion of the N-terminal domain, suggesting that DNA and JDP-2 affect structural conformations in different regions of the receptor.
Other transcriptional coregulatory proteins have been reported to directly bind and alter the structural conformation of the N-terminal domain of steroid receptors. The novelty of JDP-2 is that it affects the structure and function of the N-terminal domain through binding to the DBD. Examples of coregulatory proteins that directly interact with the N-terminal domain of steroid receptors include TFIIF, TBP, CBP, and SRC-1 (24, 47, 55). Interaction with TFIIF resulted in decreased protease sensitivity of the N-terminal domain of AR, indicating that the conformation of AR was changed as a result of TFIIF interaction (47). CD analysis detected an overall decrease in randomness and an increase in both α-helix and β-sheet within the N-terminal domain of ERα upon interaction with TBP. The influence of TBP was specific to ERα, having no effect on the N domain of ERβ (55). In the presence of trimethylamine N-oxide (TMAO; α-helix-inducing solvent), the GR N-terminal domain adopts a more α-helical conformation, and TMAO correspondingly increased the association of CBP, TBP, and SRC-1 with the N-terminal domain of GR (24).
Because the effect of JDP-2 on the structure and function of the N-terminal domain occurs through binding of JDP-2 to the DBD, it suggests that there is a functional integration between the N-terminal domain of PR and its own DBD that may not be mediated by heterologous DBDs or transcriptional activation domains. To test this concept, we analyzed various chimeric receptors where either the DBD or transcriptional activation domain were swapped between different nuclear receptors, or the DBD and transcription activation domains were substituted with similar functional domains of an unrelated transcription factor. Despite the fact that JDP-2 can interact with the ER DBD, it had a very weak effect at best on transcription activity of an ER DBD-PR N-terminal domain chimeric receptor, and the PR DBD fused to the heterologous VP16 activation domain (Fig. 1D). A chimeric receptor containing the GAL4 DBD substituted for PR DBD also was not responsive to JDP-2 (53). These results suggest that the PR DBD plays an active and specific role in modulating the transcription activity of the N-terminal domain in response to binding JDP-2. There are examples with other transcription factors where the transcriptional activation domain is only regulated appropriately in the context of the homologous DBD and not when fused to a heterologous DBD (8, 10, 28, 32). Oct-4, a member of the POU transcription factor family, possesses an activation domain in the C terminus that, in the context of its own POU DBD, transactivates in a cell-specific manner. However, fusing this activation domain to either GAL4-DBD or to the POU domain of Pit-1 resulted in the transactivation in all cell types examined, indicating that tissue-specific regulation requires the DBD of Oct-1 specifically (8). Similarly, USF2 of the USF family of basic helix-loop-helix transcription factors contains an activation domain referred to as USR (USF-specific region), which activates transcription only when placed in its natural context upstream of the USF2 basic region and not when fused to the GAL4 DBD (32). Within the nuclear receptor family, GR-mediated repression of specific GRE elements, termed negative glucocorticoid response elements, requires specific communication between the receptor DBD and AFs to elicit the appropriate repressive response (10).
Although heterologous DBDs and activation domains of other transcription factors were unable to mediate the coactivation effect of JDP-2, the N-terminal domains of at least two other nuclear receptors (ERα and RARα) were found to be capable of responding to the activating signal induced by JDP-2, albeit not as efficiently as the PR N domain (Fig. 1D). These results suggest that despite a lack of sequence homology, the N-terminal domains of nuclear receptors have a shared propensity to undergo a shift in conformation from an unfolded inactive state to a more ordered active state in response to binding a coregulatory protein.
How JDP-2 binding to the DBD transmits an effect on structural conformation of the N-terminal domain is not known. One possibility is that JDP-2 propagates a conformational change along the peptide backbone between the two domains. The lack of direct binding of JDP-2 to the N-terminal domain would favor this mechanism. However, our data cannot rule out the presence of secondary protein contacts between JDP-2 and the N-terminal domain in the context of the AnDBD polypeptide or full-length PR. Binding of JDP-2 to the DBD as a primary interaction site may alter conformation of the two-domain AnDBD (or full-length PR) in a manner that promotes a secondary interaction between JDP-2 and the N-terminal domain. Cocrystal structure of the AnDBD/JDP-2 complex may be needed to decipher between these mechanisms.
The AF-1 of progesterone receptor was originally defined as a sequence of the N-terminal domain linked to a heterologous DBD that was required for mediating transcription activity (40). However, when we analyzed a series of truncation and internal deletion mutations of the N-terminal domain, the originally defined AF-1 sequence region was dispensable for both constitutive activity of the AnDBD polypeptide as well as hormone-dependent activity of full-length PR. The AF-1 sequence region was also not required for the stimulatory effect of JDP-2 on either the constitutive transcription activity of the two-domain AnDBD PR polypeptide (Fig. 8) or hormone-dependent activity of full-length PR (Fig. 9). The AF-1 sequence alone linked to PR DBD does mediate weak transcription activity; however, it is only a fraction of the total activity of the N-terminal domain. Maximal transcription activity of the N-terminal domain of PR-A in the context of the receptor's own DBD required sequences between aa 350 and 428 (Fig. 7). However, this region alone (aa 350 to 428), when linked to PR DBD, had minimal activity, suggesting that it also does not function autonomously without interactions from other regions of N-terminal domain (Fig. 8). Thus, we conclude that the originally defined AF-1 sequence region is not sufficient for mediating transcription activity of the N-terminal domain of PR in the context of its own DBD and that significant transcription activity resides in other regions of the N domain between aa 350 and 428 (Fig. 7A). The previously reported failure to detect transcription activity in this region (40) by the classical approach of linking sequence regions to the GAL4 DBD is likely due to the active participation of the PR DBD in controlling structure and activity of the N-terminal domain that is not fully mimicked with the heterologous GAL4 DBD. Alternatively, our experiments were done in a single cell line, and autonomous activity of AF-1 sequences may be cell type specific.
We were unable to define a discrete region of the N-terminal domain of PR responsible for enhancement of transcription by JDP-2. Only partial reduction in response to JDP-2 was observed by the truncation or deletion of the region between aa 350 and 428, whereas deletion or truncation of other regions, including the originally defined AF-1, had either no effect or no further reduction below that obtained by removal of aa 350 to 428. These results are consistent with the conclusion that multiple elements of the N-terminal domain mediate the stimulatory effect of JDP-2 on transcription. Further insights into the mechanism of action of JDP-2 and the role of induced protein folding in mediating the activity of the N-terminal domain may require crystal structure of the N-terminal domain/DBD polypeptide complexed to JDP-2.
Acknowledgments
We acknowledge the UC Cancer Center Tissue Culture Core Laboratory and Kurt Christensen for technical support with expression of recombinant baculovirus proteins in insect cells and Mair Churchill, Sarah Roemer, and Purnima Mungalachetty for providing purified HMGB-1 protein.
This work was supported by NIH Public Health Service grants CA46938 and DK49030 (D.P.E.) and NIH grant RO1 GM061855 (R.S.H.).
REFERENCES
- 1.Aronheim, A., E. Zandi, H. Hennemann, S. Elledge, and M. Karin. 1997. Isolation of an AP-1 repressor by a novel method for detecting protein-protein interactions. Mol. Cell. Biol. 17:3094-3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bain, D., M. Franden, J. McManaman, G. Takimoto, and K. Horwitz. 2001. The N-terminal region of human progesterone B-receptors. J. Biol. Chem. 276:23825-23831. [DOI] [PubMed] [Google Scholar]
- 3.Bain, D., M. Franden, J. McManaman, G. Takimoto, and K. Horwitz. 2000. The N-terminal region of the human progesterone A-receptor. J. Biol. Chem. 275:7313-7320. [DOI] [PubMed] [Google Scholar]
- 4.Baskakov, I., R. Kumar, G. Srinivasan, Y.-S. Ji, D. Bolen, and E. Thompson. 1999. Trimethylamine N-oxide-induced cooperative folding of an intrinsically unfolded transcription-activating fragment of human glucocorticoid receptor. J. Biol. Chem. 274:10693-10696. [DOI] [PubMed] [Google Scholar]
- 5.Blazek, E., S. Wasmer, U. Kruse, A. Aronheim, M. Aoki, and P. Vogt. 2003. Partial oncogenic transformation of chicken embryo fibroblasts by Jun dimerization protein 2, a negative regulator of TRE- and CRE-dependent transcription. Oncogene 22:2151-2159. [DOI] [PubMed] [Google Scholar]
- 6.Boonyaratanakornkit, V., V. Melvin, P. Prenderast, M. Altmann, L. Ronfani, M. Bianchi, L. Taraseviciene, S. Nordeen, E. Allegretto, and D. Edwards. 1998. High-mobility group chromatin proteins 1 and 2 functionally interact with steroid hormone receptors to enhance their DNA binding in vitro and transcriptional activity in mammalian cells. Mol. Cell. Biol. 18:4471-4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bower, K., R. Zeller, W. Wachsman, T. Martinez, and K. McGuire. 2002. Correlation of transcriptional repression by p21SNFT with changes in DNA-NF-AT complex interactions. J. Biol. Chem. 277:34967-34977. [DOI] [PubMed] [Google Scholar]
- 8.Brehm, A., K. Ohbo, and H. Scholer. 1997. The carboxy-terminal transactivation domain of Oct-4 acquires cell specificity through the POU domain. Mol. Cell. Biol. 17:154-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen, Y.-H., J. Yang, and K. Chau. 1974. Determination of the helix and beta form of proteins in aqueous solution by circular dichroism. Biochemistry 13:3350-3359. [DOI] [PubMed] [Google Scholar]
- 10.Drouin, J., Y. Sun, M. Chamberland, Y. Gauthier, A. De Lean, M. Nemer, and T. Schmidt. 1993. Novel glucocorticoid receptor complex with DNA element of the hormone-repressed POMC gene. EMBO J. 12:145-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edwards, D., S. Wardell, and V. Boonyaratanakornkit. 2003. Progesterone receptor interacting coregulatory proteins and cross talk with cell signaling pathways. J. Steroid Biochem. Mol. Biol. 83:173-186. [DOI] [PubMed] [Google Scholar]
- 12.Folkers, G. E., E. C. van Heerde, and P. T. van der Saag. 1995. Activation function 1 of retinoic acid receptor β2 is an acidic activator resembling VP16. J. Biol. Chem. 270:23552-23559. [DOI] [PubMed] [Google Scholar]
- 13.Giangrande, P. H., and D. P. McDonnell. 1999. The A and B isoforms of the human progesterone receptor: two functionally different transcription factors encoded by a single gene. Recent Prog. Hormone Res. 45:291-313. [PubMed] [Google Scholar]
- 14.Hall, J., D. McDonnell, and K. Korach. 2002. Allosteric regulation of estrogen receptor structure, function, and coactivator recruitment by different estrogen response elements. Mol. Endocrinol. 16:469-486. [DOI] [PubMed] [Google Scholar]
- 15.Hittelman, A., D. Burakov, J. Ineguez-Lluhi, L. Freedman, and M. Garabedian. 1999. Differential regulation of glucocorticoid receptor transcriptional activation via AF-1-associated proteins. EMBO J. 18:5380-5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horton, R., H. Hunt, S. Ho, J. Pullen, and L. Pease. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61-68. [DOI] [PubMed] [Google Scholar]
- 17.Iacobelli, M., W. Wachsman, and K. McGuire. 2000. Repression of IL-2 promoter activity by the novel basic leucine zipper p21SNFT protein. J. Immunol. 165:860-868. [DOI] [PubMed] [Google Scholar]
- 18.Jin, C., H. Li, T. Murata, K. Sun, M. Horikoshi, R. Chiu, and K. Yokoyama. 2002. JDP2, a repressor of AP-1, recruits a histone deacetylase 3 complex to inhibit the retinoic acid-induced differentiation of F9 cells. Mol. Cell. Biol. 22:4815-4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin, C., H. Ugai, J. Song, T. Murata, F. Nili, K. Sun, M. Horikoshi, and K. Yokoyama. 2001. Identification of mouse Jun dimerization protein 2 as a novel repressor of ATF-2. FEBS Lett. 489:31-41. [DOI] [PubMed] [Google Scholar]
- 20.Johnson, W. J. 1992. Analysis of circular dichroism spectra. Methods Enzymol. 210:426-447. [DOI] [PubMed] [Google Scholar]
- 21.Ko, L., G. Cardona, A. Henrion-Caude, and W. Chin. 2002. Identification and characterization of a tissue-specific coactivator, GT198, that interacts with the DNA-binding domains of nuclear receptors. Mol. Cell. Biol. 22:357-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kraus, W., E. McInerney, and B. Katzenellenbogen. 1995. Ligand-dependent, transcriptionally productive association of the amino- and carboxyl-terminal regions of a steroid hormone nuclear receptor. Proc. Natl. Acad. Sci. USA 92:12314-12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar, R., I. Baskakov, G. Srinivasan, D. Bolen, J.-C. Lee, and E. Thompson. 1999. Interdomain signaling in a two-domain fragment of the human glucocorticoid receptor. J. Biol. Chem. 274:24737-24741. [DOI] [PubMed] [Google Scholar]
- 24.Kumar, R., J.-C. Lee, D. Bolen, and E. Thompson. 2001. The conformation of the glucocorticoid receptor AF1/tau1 domain induced by osmolyte binds co-regulatory proteins. J. Biol. Chem. 276:18146-18152. [DOI] [PubMed] [Google Scholar]
- 25.Kumar, R., and E. Thompson. 2003. Transactivation functions of the N-terminal domains of nuclear hormone receptors: protein folding and coactivator interactions. Mol. Endocrinol. 17:1-10. [DOI] [PubMed] [Google Scholar]
- 26.Lanz, R., N. McKenna, S. Onãte, U. Albrecht, J. Wong, S. Tsai, M.-J. Tsai, and B. O'Malley. 1999. A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell 97:17-27. [DOI] [PubMed] [Google Scholar]
- 27.Lavigne, P., L. Kondejewski, M. J. Houston, F. Sonnichsen, B. Lix, B. Skyes, R. Hodges, and C. Kay. 1995. Preferential heterodimeric parallel coiled-coil formation by synthetic Max and c-Myc leucine zippers: a description of putative electrostatic interactions responsible for the specificity of heterodimerization. J. Mol. Biol. 254:505-520. [DOI] [PubMed] [Google Scholar]
- 28.Lefstin, J., and K. Yamamoto. 1998. Allosteric effects of DNA on transcriptional regulators. Nature 392:885-888. [DOI] [PubMed] [Google Scholar]
- 29.Leo, C., and J. Chen. 2000. The SRC family of nuclear receptor coactivators. Gene 245:1-11. [DOI] [PubMed] [Google Scholar]
- 30.Li, X., and B. W. O'Malley. 2003. Unfolding the action of progesterone receptors. J. Biol. Chem. 278:39261-39264. [DOI] [PubMed] [Google Scholar]
- 31.Loven, M., J. Wood, and A. Nardulli. 2001. Interaction of estrogen receptors α and β with estrogen response elements. Mol. Cell. Endocrinol. 181:151-163. [DOI] [PubMed] [Google Scholar]
- 32.Luo, X., and M. Sawadogo. 1996. Functional domains of the transcription factor USF2: atypical nuclear localization signals and context-dependent transcriptional activation domains. Mol. Cell. Biol. 16:1367-1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma, H., H. Hong, S.-M. Huang, R. Irvine, P. Webb, P. Kushner, G. Coetzee, and M. Stallcup. 1999. Multiple signal input and output domains of the 160-kilodalton nuclear receptor coactivator proteins. Mol. Cell. Biol. 19:6164-6173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Makhatadze, G., and P. Privalov. 1995. Energetics of protein structure. Adv. Protein Chem. 47:307-425. [DOI] [PubMed] [Google Scholar]
- 35.Mathur, M., P. Tucker, and H. Samuels. 2001. PSF is a novel corepressor that mediates its effect through Sin3A and the DNA binding domain of nuclear receptors. Mol. Cell. Biol. 21:2298-2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McKenna, N., and B. O'Malley. 2002. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 108:465-474. [DOI] [PubMed] [Google Scholar]
- 37.Melvin, V., and D. Edwards. 2001. Expression and purification of recombinant human progesterone receptor in baculovirus and bacterial systems. Methods Mol. Biol. 176:39-54. [DOI] [PubMed] [Google Scholar]
- 38.Melvin, V., C. Harrell, J. Adelman, W. Kraus, M. Churchill, and D. Edwards. 2004. The role of the C-terminal extension (CTE) of the estrogen receptor alpha and beta DNA binding domain in DNA binding and interaction with HMGB. J. Biol. Chem. 279:10882-10887. [DOI] [PubMed] [Google Scholar]
- 39.Melvin, V., S. Roemer, M. Churchill, and D. Edwards. 2002. The C-terminal extension (CTE) of the nuclear hormone receptor DNA binding domain determines interactions and functional response to HMG-1/-2 co-regulatory proteins. J. Biol. Chem. 277:25115-25124. [DOI] [PubMed] [Google Scholar]
- 40.Meyer, M.-E., C. Quirin-Stricker, T. Lerouge, M.-T. Bocquel, and H. Gronemeyer. 1992. A limiting factor mediates the differential activation of promoters by the human progesterone receptor isoforms. J. Biol. Chem. 267:10882-10887. [PubMed] [Google Scholar]
- 41.Miura, T., and G. J. Thomas. 1995. Optical and vibrational spectroscopic methods, p. 261-315. In J. Glasel and M. Deutscher, Introduction to biophysical methods for protein and nucleic acid research. Academic Press, Inc., San Diego, Calif.
- 42.Onãte, S., V. Boonyaratanakornkit, T. Spencer, S. Tsai, M.-J. Tsai, D. Edwards, and B. O'Malley. 1998. The steroid receptor coactivator-1 contains multiple receptor interacting and activation domains that cooperatively enhance the activation function 1 (AF1) and AF2 domains of steroid receptors. J. Biol. Chem. 273:12101-12108. [DOI] [PubMed] [Google Scholar]
- 43.Pan, J., C. Jin, T. Murata, and K. Yokoyama. 2003. Sequence specific transcription factor, JDP2 interactions with histone and inhibits p300-mediated acetylation. Nucleic Acids Res. 3:305-306. [DOI] [PubMed] [Google Scholar]
- 44.Poukka, H., U. Karvonen, N. Yoshikawa, H. Tanaka, J. Palvimo, and O. Janne. 2000. The RING finger protein SNURF modulates nuclear trafficking of the androgen receptor. J. Cell Sci. 113:2991-3001. [DOI] [PubMed] [Google Scholar]
- 45.Puigserver, P., G. Adelmant, Z. Wu, M. Fan, J. Xu, B. O'Malley, and B. Spiegelman. 1999. Activation of PPARγ coactivator-1 through transcription factor docking. Science 286:1368-1372. [DOI] [PubMed] [Google Scholar]
- 46.Radhakrishnan, I., G. Perez-Alvarado, D. Parker, H. Dyson, M. Montminy, and P. Wright. 1997. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: a model for activator:coactivator interactions. Cell 91:741-752. [DOI] [PubMed] [Google Scholar]
- 47.Reid, R., S. Kelly, K. Watt, N. Price, and I. McEwan. 2002. Conformational analysis of the androgen receptor amino-terminal domain involved in transactivation. J. Biol. Chem. 277:20079-20086. [DOI] [PubMed] [Google Scholar]
- 48.Schmitz, M., M. dos Santos Silva, H. Altmann, M. Czisch, T. Holak, and P. Baeuerle. 1994. Structural and functional analysis of the NF-κB p65 C terminus. J. Biol. Chem. 269:25613-25620. [PubMed] [Google Scholar]
- 49.Shen, F., S. Triezenberg, P. Hensley, D. Porter, and J. Knutson. 1996. Transcriptional activation domain of the herpesvirus protein VP16 becomes conformationally constrained upon interaction with basal transcription factors. J. Biol. Chem. 271:4827-4837. [DOI] [PubMed] [Google Scholar]
- 50.Smith, C., and B. O'Malley. 2004. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocrine Rev. 25:45-71. [DOI] [PubMed] [Google Scholar]
- 51.Steinmetz, A., J.-P. Renaud, and D. Moras. 2001. Binding of ligands and activation of transcription by nuclear receptors. Annu. Rev. Biophys. Biomol. Structure 30:329-359. [DOI] [PubMed] [Google Scholar]
- 52.Tora, L., J. White, C. Brou, D. Tasset, N. Webster, E. Scheer, and P. Chambon. 1989. The human estrogen receptor has two independent nonacidic transcriptional activation functions. Cell 59:477-487. [DOI] [PubMed] [Google Scholar]
- 53.Wardell, S., V. Boonyaratanakornkit, J. Adelman, A. Aronheim, and D. Edwards. 2002. Jun dimerization protein 2 functions as a progesterone receptor N-terminal domain coactivator. Mol. Cell. Biol. 22:5451-5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Warnmark, A., E. Treuter, A. Wright, and J.-A. Gustafsson. 2003. Activation functions 1 and 2 of nuclear receptors: molecular strategies for transcriptional activation. Mol. Endocrinol. 17:1901-1909. [DOI] [PubMed] [Google Scholar]
- 55.Warnmark, A., A. Wikstrom, A. Wright, J.-A. Gustafsson, and T. Hard. 2001. The N-terminal regions of estrogen receptor alpha and beta are unstructured and show different TBP binding properties. J. Biol. Chem. 276:45939-45944. [DOI] [PubMed] [Google Scholar]
- 56.Warrens, A., M. Jones, and R. Lechler. 1997. Splicing by overlap extension by PCR using asymmetric amplification: an improved technique for the generation of hybrid proteins of immunological interest. Gene 186:29-35. [DOI] [PubMed] [Google Scholar]
- 57.Watanabe, M., J. Yanagisawa, H. Kitagawa, K. Takeyama, S. Ogawa, Y. Aroa, M. Suzawa, Y. Kobayashi, T. Yano, H. Yoshikawa, Y. Masuhiro, and S. Kato. 2001. A subfamily of RNA-binding DEAD-box proteins acts as an estrogen receptor alpha coactivator through the N-terminal activation domain (AF-1) with an RNA coactivator, SRA. EMBO J. 20:1341-1352. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 58.Wood, J., V. Likhite, M. Loven, and A. Nardulli. 2001. Allosteric modulation of estrogen receptor conformation by different estrogen response elements. Mol. Endocrinol. 15:1114-1126. [DOI] [PubMed] [Google Scholar]
- 59.Wu, X., H. Li, and J. Chen. 2001. The human homologue of the yeast DNA repair and TFIIH regulator MMS19 is an AF-1-specific coactivator of estrogen receptor. J. Biol. Chem. 276:23962-23968. [DOI] [PubMed] [Google Scholar]