

Abstract
KPC2 (Kip1 ubiquitylation-promoting complex 2) together with KPC1 forms the ubiquitin ligase KPC, which regulates degradation of the cyclin-dependent kinase inhibitor p27 at the G1 phase of the cell cycle. KPC2 contains a ubiquitin-like (UBL) domain, two ubiquitin-associated (UBA) domains, and a heat shock chaperonin-binding (STI1) domain. We now show that KPC2 interacts with KPC1 through its UBL domain, with the 26S proteasome through its UBL and NH2-terminal UBA domains, and with polyubiquitylated proteins through its UBA domains. The association of KPC2 with KPC1 was found to stabilize KPC1 in a manner dependent on the STI1 domain of KPC2. KPC2 mutants that lacked either the NH2-terminal or the COOH-terminal UBA domain supported the polyubiquitylation of p27 in vitro, whereas a KPC2 derivative lacking the STI1 domain was greatly impaired in this regard. Depletion of KPC2 by RNA interference resulted in inhibition of p27 degradation at the G1 phase, and introduction of KPC2 derivatives into the KPC2-depleted cells revealed that the NH2-terminal UBA domain of KPC2 is essential for p27 degradation. These observations suggest that KPC2 cooperatively regulates p27 degradation with KPC1 and that the STI1 domain as well as the UBL and UBA domains of KPC2 are indispensable for its function.
Progression of the cell cycle in eukaryotic cells is controlled by a series of protein complexes composed of cyclins and cyclin-dependent kinases (CDKs) (36). The association of CDK inhibitors (CKIs) with cyclin-CDK complexes is triggered by a variety of antimitogenic signals and results in inhibition of the catalytic activity of these complexes and consequent restraint of cell cycle progression (56). Among the various CKIs identified, p27 plays a pivotal role in the control of cell proliferation (40, 45, 61). We along with others have shown that mice homozygous for deletion of the p27 gene are larger than wild-type animals and exhibit multiple-organ hyperplasia as well as a predisposition to cancer (11, 24, 37). These observations support the notion that p27 is a key determinant of both body and organ size as a result of its role in the control of cell proliferation.
The abundance of p27 is thought to be controlled by multiple mechanisms that operate at the level of the synthesis (transcription and translation), proteolysis, and localization of this protein (1, 7, 16, 18, 32, 33, 44, 51). Degradation of p27 is triggered by its phosphorylation on Thr187 by the cyclin E-CDK2 complex (55, 64) and is executed by the ubiquitin-proteasome system (44, 58). The phosphorylation of Thr187 is required for the binding of p27 to Skp2, the F-box protein component of an SCF ubiquitin ligase (E3) complex, and such binding in turn results in the ubiquitylation and degradation of p27 (3, 59, 62). Consistent with this scenario, we demonstrated that mice deficient in Skp2 exhibit a reduced body size and that Skp2−/− cells show an increased accumulation of p27 (38).
In normal cells, the amount of p27 is high during the G0 phase of the cell cycle but decreases rapidly on reentry of cells into G1 phase. Mitogenic activation of resting lymphocytes or reexposure of serum-deprived embryonic fibroblasts to serum, for example, induces rapid degradation of p27 between 3 and 9 h after stimulation. However, Skp2 is not expressed until the G1-S transition of the cell cycle (12 to 18 h after stimulation), unequivocally later than the degradation of p27 apparent at G0-G1 (15). Moreover, p27 is exported from the nucleus to the cytoplasm at G0-G1 (7, 18, 51, 60), whereas Skp2 is restricted to the nucleus (30, 34). The discrepancies between the temporal and spatial patterns of p27 expression and those of Skp2 expression suggest the existence of a Skp2-independent pathway for the degradation of p27. Indeed, the down-regulation of p27 at the G0-G1 transition occurs normally and is sensitive to proteasome inhibitors in Skp2−/− cells, indicating that p27 is degraded at G0-G1 by a proteasome-dependent, but Skp2-independent, mechanism. In Skp2−/− cells, p27 degradation is impaired only in S and G2 phases, and this defect results in inhibition of Cdc2 kinase by the accumulating p27 (39). Biochemical analysis of crude extracts of Skp2−/− cells revealed the presence in the cytosolic fraction of a Skp2-independent E3 activity that mediates the ubiquitylation of p27 (15). This ubiquitylation was not dependent on the phosphorylation of p27 on Thr187, which is a prerequisite for Skp2-mediated ubiquitylation.
We recently identified a ubiquitin ligase designated KPC (Kip1 ubiquitylation-promoting complex) that consists of KPC1 and KPC2 subunits and which is responsible for the rapid degradation of p27 at the transition from G0 to G1 (19, 26). KPC1 contains a RING finger domain near its COOH terminus and functions as the catalytic subunit. KPC2 contains an NH2-terminal ubiquitin-like (UBL) domain, two ubiquitin-associated (UBA) domains, and a COOH-terminal STI1 (heat shock chaperonin-binding) domain, suggesting that it is a member of the UBL-UBA family of proteins that includes hHR23A and hHR23B and hPLIC1 and hPLIC12 of Homo sapiens as well as Rad23 and Dsk2 of Saccharomyces cerevisiae (6, 13, 17, 25, 48, 50, 53, 65). The UBL domain of these proteins is required for their interaction with the 19S complex of the 26S proteasome (8, 49, 52, 53, 63), whereas the UBA domain interacts with polyubiquitin chains (2, 47, 48, 66). The UBL-UBA proteins are thus thought to deliver polyubiquitylated substrates to the 26S proteasome (5, 48, 49, 63).
To provide insight into the role of KPC2 in KPC-mediated regulation of p27 degradation, we have now characterized the structure-function relation of KPC2. Our data suggest that KPC2 promotes the transfer of p27 molecules that have been ubiquitylated by KPC1 to the 26S proteasome and that it also contributes to stabilization of KPC1. We found that the STI1 domain, whose function has been largely unknown, is required for both the polyubiquitylation of p27 as well as the stabilization of KPC1. KPC2 thus appears to regulate p27 degradation cooperatively with KPC1 at G1 phase of the cell cycle.
MATERIALS AND METHODS
Antibodies.
Polyclonal antibodies to KPC1 and to KPC2 were described previously (19). Monoclonal antibodies to p27, to glycogen synthase kinase-3β, HSP70, and HSP90 were obtained from BD Biosciences. Monoclonal antibodies (M2 and M5) to the FLAG epitope were from Sigma, and a monoclonal antibody (HA11) to the hemagglutinin (HA) epitope was from Research Diagnostics. A monoclonal antibody to glutathione S-transferase (GST) was from Covance. Monoclonal antibodies to S5a and to the 20S proteasome particle (subunits 1, 2, 3, 5, 6, and 7) as well as polyclonal antibodies to S10 (19S regulator non-ATPase) and to S12 (19S regulator non-ATPase) were obtained from Affiniti (Exeter, United Kingdom). A monoclonal antibody (FK2) to polyubiquitin was obtained from Nippon Bio-Test Laboratories (Tokyo, Japan).
Gel filtration column chromatography.
Lysates of HEK293T or NIH 3T3 cells were applied to a Superose 6 HR 10/30 column (Amersham Biosciences) that had been equilibrated with a solution containing 50 mM Tris-HCl (pH 7.6), 100 mM KCl, 5 mM MgCl2, 2 mM ATP, 0.1% Nonidet P-40 (NP-40), and 20% glycerol. Proteins were eluted with the same solution, and the resulting fractions were subjected to immunoblot analysis with the indicated antibodies.
Production of recombinant proteins in bacteria.
Complementary DNAs encoding wild-type or mutant versions of human KPC2 were subcloned into pGEX6P-1 (Amersham Biosciences), and the GST-KPC2 fusion proteins were expressed in E. coli strain BL21(DE3) pLysS (Novagen) and purified with the use of glutathione beads. Mouse p27, S. cerevisiae Uba1, human UbcH5A, and a GST fusion protein of mouse ubiquitin were prepared as described (19).
Baculovirus expression system.
A cDNA encoding human KPC1 with an NH2-terminal FLAG tag and a COOH-terminal six-histidine (His6) tag as well as cDNAs for wild-type or mutant versions of human KPC2 containing a COOH-terminal HA tag were subcloned into pBacPAK9 (Clontech). Recombinant baculoviruses were generated with the BacPAK baculovirus expression system (Clontech). Recombinant KPC1 and KPC2 were coexpressed in Sf21 cells and copurified as described (19).
Transient expression of recombinant proteins in mammalian cells.
A cDNA encoding human S12 with an NH2-terminal HA tag was cloned into pCGN (23); cDNAs for wild-type or mutant versions of human KPC2 were subcloned into p3xFLAG-CMV-7.1 (Sigma); cDNAs encoding wild-type or mutant versions of human KPC2 tagged at their COOH termini with three copies of the FLAG epitope were subcloned into pcDNA3 (Invitrogen); a cDNA encoding human KPC1 tagged at its NH2-terminus with His6 and FLAG as well as a cDNA for human KPC2 containing COOH-terminal His6 and HA tags were subcloned separately into pCI-neo (Promega). Mammalian cells were transfected with the indicated plasmids by the calcium phosphate method for 24 h.
In vitro assay of ubiquitylation activity.
The ability of purified recombinant KPC complexes to ubiquitylate p27 was assayed as previously described (19).
In vivo binding assay.
HEK293T cells were transfected with the indicated plasmids and then cultured for an additional 4 h in the presence of 10 μM MG132 (Peptide Institute). Cell lysates were subjected to immunoprecipitation and immunoblot analysis as previously described (20).
In vitro binding assay.
The S100 fraction (200 μg of protein) of HeLa cell extracts was incubated in a final volume of 500 μl for 2 h at 4°C with GST fusion proteins of wild-type or mutant versions of KPC2 (1 μg) and glutathione beads (10 μl) in binding buffer (50 mM Tris-HCl [pH 7.9], 150 mM NaCl, 5 mM MgCl2, 0.5% NP-40), after which the beads were washed five times with binding buffer and the precipitated proteins were subjected to immunoblot analysis.
Retroviral expression system.
A cDNA encoding human KPC1 containing His6 and FLAG tags at its NH2-terminus as well as cDNAs encoding wild-type human KPC2 and a series of deletion mutants thereof containing a COOH-terminal HA tag were subcloned into pMX-puro (kindly provided by T. Kitamura, University of Tokyo). Recombinant retroviruses were generated and used to infect NIH 3T3 cells as previously described (19).
RNAi.
RNA interference (RNAi) was performed as described (19). The hairpin sequences specific for mouse KPC2 mRNA and for enhanced green fluorescent protein (EGFP) mRNA (Clontech) corresponded to nucleotides 114 to 134 and 126 to 146 of the respective coding regions.
Pulse-chase analysis.
Pulse-chase analysis combined with immunoprecipitation with antibodies to FLAG was performed as previously described (23).
RESULTS
We recently identified KPC, an E3 composed of KPC1 and KPC2 subunits that is responsible for degradation of p27 in G1 phase. KPC1 contains a RING finger domain near its COOH terminus and functions as the catalytic subunit, whereas KPC2 contains a UBL domain near its NH2 terminus, two UBA domains, and an STI1 domain near its COOH terminus (Fig. 1A). The overall structure of KPC2 is similar to that of the UBL-UBA family members hHR23A and hHR23B (human homolog of S. cerevisiae Rad23) and hPLIC1 and hPLIC2 (human homolog of S. cerevisiae Dsk2), although the position and number of STI1 domains differ among the various molecules. These structural characteristics thus suggested that KPC2 is also a member of this protein family.
FIG. 1.

Structure of KPC2. (A) Domain organization of human KPC1 and KPC2 as well as of hHR23B and hPLIC1. (B) Alignment of the amino acid sequences of the UBL domains of human KPC2 (residues 14 to 96) and hHR23B (residues 1 to 79). Identical (black) and similar (gray) amino acids are shown. Secondary structural elements of the UBL domain of hHR23B are indicated. Asterisks denote the residues in hHR23B that are important for interaction with the COOH-terminal ubiquitin-interacting motif of S5a. (C) Model structures of the UBL domains of human KPC2 and hHR23B (Protein Data Bank code 1UEL). Side chains of the residues of hHR23B important for binding to the COOH-terminal ubiquitin-interacting motif of S5a are shown in stick. The model structure of the UBL domain of KPC2 was built with the program Modeller. The critical amino acids are labeled and numbered.
The UBL domain of hHR23B is able to bind to S5a (non-ATPase subunit) of the 19S proteasome complex (12, 17, 65) and folds into an α/β structure that comprises a five-stranded twisted β-sheet with a long α-helical backbone, which closely resembles the structure of ubiquitin (12). Alignment of the amino acid sequences of the UBL domain of human KPC2 (residues 14 to 96) and that of hHR23B (residues 1 to 79) revealed that the residues that form the secondary structural elements in hHR23B are similar in KPC2 (Fig. 1B). Residues responsible for the interaction of hHR23B with S5a (12) are also conserved in the UBL domain of KPC2 (residues Lys62, Ile64, Ala66, Ala67, Val88, and Leu90); Leu8 and Tyr48 of hHR23B, which also contribute to the interaction with S5a, are not conserved in KPC2, however. Comparison of the UBL sequence of KPC2 with sequences of known tertiary structure in the database was performed with the program FUGUE (57). The structure of ubiquitin (Protein Data Base code 1UBQ) was predicted to be the most similar to that of the UBL domain of KPC2, with a Z score of 6.52 and reliability of ≥99%. An additional five proteins containing ubiquitin-like folds yielded Z scores of >4.0. These results thus suggest that residues 14 to 96 of KPC2 adopt a ubiquitin-like fold. We built a structural model of the UBL domain of KPC2 with the program Modeller (29) (Fig. 1C). The model structure indicated that residues in the UBL domain of hHR23B responsible for interaction with the proteasome subunit S5a are similarly positioned in the UBL domain of KPC2 and that the UBL domains of KPC2 and hHR23B share the same fold.
To investigate whether KPC2 interacts with the proteasome, we fractionated NIH 3T3 cell lysates by chromatography with a gel filtration column and examined the elution profiles of KPC2, the non-ATPase subunits S10 and S12 of the 19S proteasome complex, and α subunits of the 20S proteasome by immunoblot analysis. Whereas a small amount of KPC2 coeluted with S10, S12, and 20S proteasome subunits in fractions 9 and 10, most of the protein was detected in fractions 13 to 17 (Fig. 2A). Prior treatment of the cells with the proteasome inhibitor MG132 resulted in a shift in the elution profile of KPC2, with more of the protein coeluting with S10, S12, and α subunits of the proteasome in fractions 8 to 11. These results suggested that treatment of the cells with the proteasome inhibitor promoted the association of KPC2 with the proteasome. Similar observations have been described for the UBL-UBA proteins hPLIC1, hPLIC2, and Rad23 (25, 52).
FIG. 2.

Interaction of KPC2 with the proteasome. (A) Coelution of KPC2 with proteasome subunits on gel filtration chromatography. NIH 3T3 cells were incubated for 6 h in the presence of 10 μM MG132 or vehicle, lysed, and fractionated by gel filtration column chromatography. The resulting fractions (as well as cell lysates [lane L]) were subjected to immunoblot analysis with antibodies to KPC2, to S10 or to S12 subunits of the 19S proteasome complex, and to core α subunits (subunits 1, 2, 3, 5, 6, and 7) of the 20S proteasome. The elution positions of marker proteins (thyroglobulin, 669 kDa; ferritin, 440 kDa; bovine serum albumin, 67 kDa; RNase A, 13.7 kDa) and the void volume (Vo) are indicated. (B) Coimmunoprecipitation of the S5a subunit of the 19S proteasome complex with KPC2. HEK293T cells were incubated in the presence (left and right panels) or absence (right panel) of MG132 for 4 h, lysed, and subjected to immunoprecipitation (IP) with rabbit antibodies to KPC2 or with control rabbit IgG. The resulting precipitates were subjected to immunoblot analysis with antibodies to KPC1, KPC2, S5a, and glycogen synthase kinase-3β (internal control). A portion (1%) of the input lysates was also subjected directly to immunoblot analysis with the same antibodies. (C) Coimmunoprecipitation of S12 and KPC2. HEK293T cells were transfected with expression plasmids encoding KPC2 (tagged with the three-FLAG epitope) or S12 (tagged with the HA epitope), as indicated. They were then incubated with MG132 for 4 h, lysed, and subjected to immunoprecipitation with antibodies to FLAG or to HA. The resulting precipitates were subjected to immunoblot analysis with the same antibodies. A portion (1%) of the input lysates was also subjected directly to immunoblot analysis. (D) Coimmunoprecipitation of S10 with various mutants of KPC2. HEK293T cells expressing the indicated three-FLAG-tagged KPC2 derivatives or transfected with the empty vector (Mock) were incubated with MG132 for 4 h, lysed, and subjected to immunoprecipitation with antibodies to FLAG. The resulting precipitates were subjected to immunoblot analysis with antibodies to S10 or to FLAG. A portion (1%) of the input lysates was also subjected directly to immunoblot analysis with the same antibodies. IB, immunoblot; IP, immunoprecipitation.
We therefore next examined the potential physiological interaction between endogenous KPC2 and the proteasome in HEK293T cells. The cells were incubated with MG132, cell lysates were subjected to immunoprecipitation with antibodies to KPC2 or with control immunoglobulin G (IgG), and the resulting precipitates were subjected to immunoblot analysis. The proteasome subunit S5a and KPC1 were specifically coprecipitated by the antibodies to KPC2 (Fig. 2B). Furthermore, the extent of the association between KPC2 and S5a, but not that of the interaction between KPC2 and KPC1, was markedly reduced in cells not exposed to MG132. To confirm the interaction between KPC2 and the proteasome, we transfected HEK293T cells with expression vectors both for KPC2 tagged at its COOH terminus with the three-FLAG epitope and for S12 tagged at its NH2 terminus with HA. After incubation of the transfected cells with MG132, cell lysates were subjected to immunoprecipitation with antibodies to HA or to FLAG. Immunoblot analysis of the resulting precipitates revealed that KPC2 associated with S12 (Fig. 2C).
Given that the UBL domain of other UBL-UBA proteins is thought to be required for interaction with the proteasome, we examined the role of the UBL domain of KPC2 in its association with the proteasome. Wild-type or mutant forms of KPC2 with a COOH-terminal three-FLAG tag were expressed in HEK293T cells, the cells were incubated with MG132, and cell lysates were subjected to immunoprecipitation and immunoblot analysis (Fig. 2D). Wild-type KPC2 interacted with the endogenous proteasome subunit S10. Unexpectedly, KPC2(ΔUBL), which lacks the UBL domain (see Fig. 7), bound to S10 as well as did wild-type KPC2. KPC2(ΔUBAn), which lacks the NH2-terminal UBA domain, also associated with S10 but to a reduced extent. KPC2(ΔUBL-UBAn), which lacks both the UBL domain and the NH2-terminal UBA domain, did not interact with S10. Deletion of the COOH-terminal UBA domain [KPC2(ΔUBAc) mutant] did not affect the interaction of KPC2 with S10. A mutant consisting of only the NH2-terminal region of KPC2 spanning the UBL and UBAn domains was found to be sufficient for the interaction with S10. Together, these data suggest that, in contrast to other UBL-UBA family proteins, the wider NH2-terminal region of KPC2 is required for interaction with the 26S proteasome. We also confirmed that these alterations of KPC2 structure did not affect the intracellular distribution of this protein (data not shown).
FIG. 7.

Role of KPC2 in the regulation of p27 degradation. Schematic representation of KPC2 deletion mutants generated in the present study. KPC2 contains a UBL domain (residues 14 to 96), two UBA domains (residues 196 to 230 and 289 to 325), and an STI1 domain (residues 352 to 391). The results of experiments examining the abilities of the KPC2 derivatives (i) to bind to the proteasome, to polyubiquitylated proteins, and to KPC1; (ii) to support p27 ubiquitylation; and (iii) to complement the destabilization of KPC1 or impairment of p27 degradation in NIH 3T3 cells expressing a KPC2 shRNA are summarized. ND, not determined.
The UBA domain of Rad23 interacts with polyubiquitin chains (2, 6, 48). To investigate whether KPC2 also binds to polyubiquitylated proteins, we incubated a GST-KPC2 fusion protein with an S100 fraction of HeLa cells. Proteins that associated with GST-KPC2 were precipitated with glutathione beads and subjected to immunoblot analysis with antibodies to polyubiquitin. Like Rad23, KPC2 was found to interact with polyubiquitylated proteins (Fig. 3A). To map the region of KPC2 required for binding to polyubiquitylated proteins, we generated GST fusion proteins containing deletion mutants of KPC2 and subjected them to the same binding assay. KPC2(ΔUBL) and KPC2(ΔSTI1), the latter of which lacks the STI1 domain, both retained the ability to bind to polyubiquitylated proteins (Fig. 3A). In contrast, KPC2(ΔUBAc) bound to polyubiquitylated proteins only weakly, and KPC2(ΔUBAn) had almost completely lost the ability to interact with these proteins.
FIG. 3.
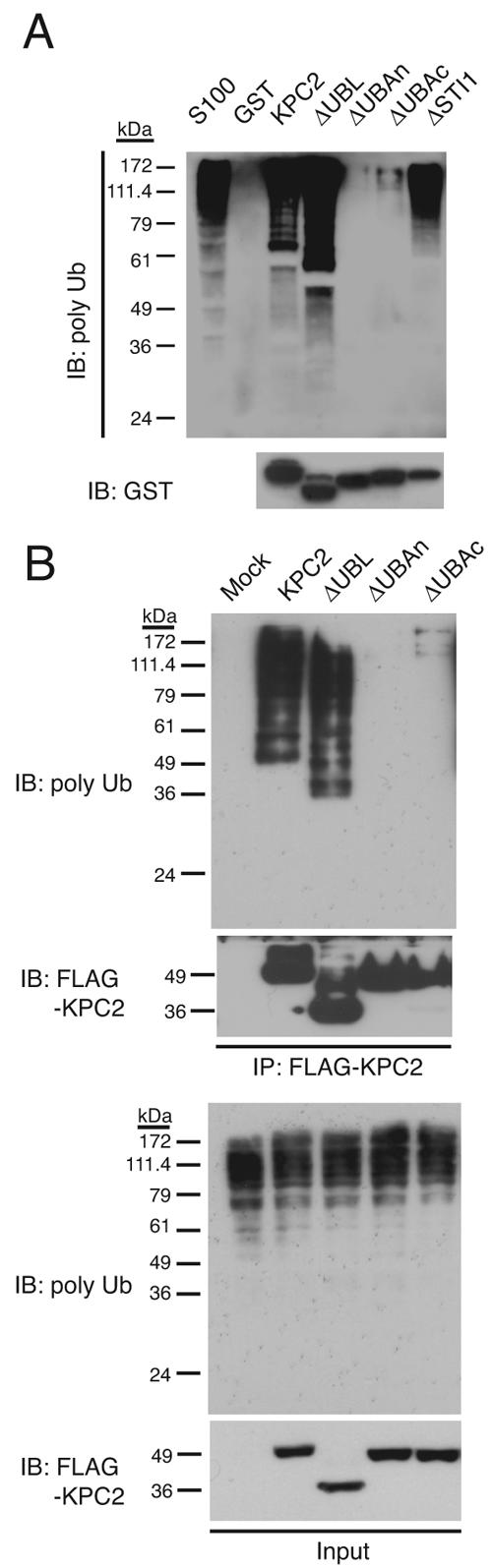
Role of the UBA domains of KPC2 in binding to polyubiquitylated proteins. (A) In vitro binding of KPC2 derivatives to polyubiquitylated proteins. GST and GST fusion proteins of wild-type KPC2 or mutants thereof were incubated with the S100 fraction of HeLa cells for 2 h at 4°C and were then precipitated with glutathione beads. The bead-bound material was subjected to immunoblot analysis with antibodies to polyubiquitin or to GST. The S100 fraction was also subjected directly to immunoblot analysis. (B) In vivo binding of KPC2 derivatives to polyubiquitylated proteins. HEK293T cells transfected with plasmids for wild-type or mutant forms of KPC2 tagged with the three-FLAG epitope were incubated with MG132 for 4 h, lysed, and subjected to immunoprecipitation with antibodies to FLAG. The resulting precipitates were subjected to immunoblot analysis with antibodies to polyubiquitin or to FLAG. A portion (1%) of the input lysates was also subjected directly to immunoblot analysis with the same antibodies. IB, immunoblot; IP, immunoprecipitation; poly Ub, polyubiquitylated.
To confirm these in vitro observations, we transfected HEK293T cells with expression vectors for wild-type or mutant forms of KPC2 tagged with the three-FLAG epitope at their NH2 termini. The cells were cultured with MG132 to facilitate the potential interaction of KPC2 with polyubiquitylated proteins, lysed, and subjected to immunoprecipitation with antibodies to FLAG. Consistent with the in vitro binding data, wild-type KPC2 and KPC2(ΔUBL) bound efficiently to polyubiquitylated proteins, whereas KPC2(ΔUBAc) exhibited a greatly reduced level of interaction with such proteins, and KPC2(ΔUBAn) manifested no such association (Fig. 3B). These results indicate that KPC2 binds to polyubiquitylated proteins through its UBA domains, especially the NH2-terminal UBA domain.
We have previously shown that KPC1 and KPC2 are associated and extensively colocalized in the cytoplasm (19). To evaluate the extent of KPC2 association with KPC1 in cells, we fractionated HEK293T cell lysates by chromatography on a gel filtration column and examined the elution profiles of KPC1 and KPC2 by immunoblot analysis (Fig. 4A). KPC2 (50 kDa) coeluted with KPC1 (140 kDa) and peaked in fractions 15 and 16, corresponding to an apparent native molecular mass of between 440 and 100 kDa. We also performed reciprocal coimmunoprecipitation experiments with endogenous proteins and found that substantial proportions of endogenous KPC1 and KPC2 were associated with each other (Fig. 4B). These results suggest that KPC1 and KPC2 exist as a heterodimer or heterotetramer in HEK293T cells.
FIG. 4.

Stabilization of KPC1 by its interaction with KPC2. (A) HEK293T cell lysates were fractionated by gel filtration column chromatography, and the resulting fractions were subjected to immunoblot analysis with antibodies to KPC1 or to KPC2. The asterisk indicates the position of a protein that cross-reacted with the antibodies to KPC2. (B) HEK293T cell lysates were subjected to immunoprecipitation with antibodies to KPC1 or to KPC2 or with control IgG, and the resulting precipitates (as well as 1% of the input lysates) were subjected to immunoblot analysis with antibodies to KPC1 or to KPC2. The asterisk indicates the position of a protein that cross-reacted with the antibodies to KPC1. (C) NIH 3T3 cells stably expressing KPC1 (tagged with the FLAG epitope) with or without KPC2 (tagged with the HA epitope) were treated with cycloheximide (50 μg/ml; CHX) for the indicated times, after which cell lysates were subjected to immunoblot analysis with antibodies to FLAG, to HA, or HSP90 (control). (D) Lysates prepared from NIH 3T3 cells stably expressing shRNAs specific for KPC2 mRNA or for EGFP mRNA (control) were subjected to immunoblot analysis with antibodies to KPC1, KPC2, or HSP70 (control). The asterisk indicates the position of a protein that cross-reacted with the antibodies to KPC1. (E) Lysates of HEK293T cells expressing the indicated three-FLAG-tagged KPC2 derivatives were subjected to immunoprecipitation with antibodies to FLAG, and the resulting precipitates were subjected to immunoblot analysis with antibodies to KPC1 or to FLAG. A portion (1%) of the input lysates was also subjected directly to immunoblot analysis with the same antibodies. IgL, immunoglobulin light chain. (F) NIH 3T3 cells expressing KPC2 or EGFP shRNAs were infected with retroviral vectors encoding the indicated human KPC2 derivatives (tagged with the HA epitope) or with the corresponding empty retrovirus (Mock). Cell lysates were subsequently subjected to immunoblot analysis with antibodies to KPC1, HA, KPC2, or HSP70 (control). The asterisk indicates the position of a protein that cross-reacted with the antibodies to KPC1. (G) HEK293T cells were transfected with a vector for KPC1 (tagged with the FLAG epitope) and either with a vector for wild-type (WT) KPC2, KPC2(ΔSTI1), or KPC2(ΔUBL-STI1), each tagged with the HA epitope, or with the corresponding empty vector (Mock). Cell lysates were subjected to immunoprecipitation (IP) with antibodies to FLAG or to HA, and the resulting precipitates (as well as 1% of the input lysates) were subjected to immunoblot analysis with the same antibodies (upper panel). Alternatively, the transfected cells were labeled with [35S]methionine for 1 h, washed, and incubated for the indicated chase periods, after which cell lysates were subjected to immunoprecipitation with antibodies to FLAG and the resulting precipitates were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and autoradiography (lower left panel). The intensities of the 35S-labeled KPC1 bands were quantified by scanning densitometry (lower right panel). IB, immunoblot.
We observed that the abundance of KPC1 was greater when this protein was coexpressed with KPC2 in transfected cells. To determine whether this observation might be attributable to stabilization of KPC1 by KPC2, we stably transfected NIH 3T3 cells with a vector for FLAG-tagged KPC1 in the absence or presence of a vector for HA-tagged KPC2. The cells were incubated with cycloheximide (50 μg/ml) for various times to block protein synthesis, and the rate of KPC1 degradation was measured by immunoblot analysis (Fig. 4C). KPC1 was more stable in cells expressing both KPC1 and KPC2 than in those expressing KPC1 alone. To investigate whether KPC2 increases the stability of KPC1 under physiological conditions, we examined the effect of depletion of KPC2 by RNAi on the stability of endogenous KPC1 in NIH 3T3 cells (Fig. 4D). Cells infected with a retroviral vector encoding a short hairpin RNA (shRNA) specific for KPC2 mRNA exhibited a >80% decrease in the abundance of KPC2 compared with the amount apparent in cells infected with a control vector. Depletion of KPC2 by RNAi also resulted in a marked decrease in the amount of KPC1 that appeared to be proportional to the reduction in the amount of KPC2. These findings suggest that KPC2 is required for stabilization of KPC1.
To determine the region of KPC2 required for interaction with KPC1, we transfected HEK293T cells with expression vectors for NH2-terminally three-FLAG-tagged KPC2 derivatives and examined the abilities of these proteins to associate with endogenous KPC1 (Fig. 4E). KPC2(ΔUBAn), KPC2(ΔUBAc), and KPC2(ΔSTI1) retained the ability to bind to KPC1, whereas KPC2(ΔUBL) did not, indicating that the interaction between KPC2 and KPC1 is mediated by the UBL domain of KPC2. We also examined various other KPC2 mutants including KPC2(ΔUBA), which lacks the region COOH-terminal to the UBAn domain, and KPC2(ΔUBL-UBAc), which lacks the UBL and UBAc domains (see Fig. 7). All mutants that lacked the UBL domain (ΔUBL, ΔUBL-UBAn, and ΔUBL-UBAc) failed to interact with KPC1 (Fig. 4E), suggesting that this domain is necessary for binding. Given that the UBL domain alone did not associate with KPC1, however, this domain is not sufficient for binding. Consistent with these observations, KPC2(ΔUBA) did not interact with KPC1, whereas UBL-UBAn did, suggesting that the UBAn domain of KPC2 also contributes to the interaction with KPC1.
To delineate the region of KPC2 required for stabilization of KPC1, we expressed HA-tagged wild-type or mutant forms of human KPC2 in NIH 3T3 cells that had been depleted of endogenous KPC2 by transfection with a retroviral vector encoding the shRNA specific for mouse KPC2 mRNA. The KPC2 shRNA specifically down-regulated the expression of endogenous mouse KPC2 without affecting that of ectopic human KPC2. Immunoblot analysis revealed that expression of human wild-type KPC2, KPC2(ΔUBAn), or KPC2(ΔUBAc), but not that of KPC2(ΔUBL), reversed the destabilization of KPC1 induced by RNAi-mediated depletion of endogenous mouse KPC2 (Fig. 4F), suggesting that association of KPC2 with KPC1 is required for the stabilization of KPC1. In contrast, KPC1 seemed to be more unstable in cells expressing KPC2(ΔSTI1) than in those transfected with the corresponding empty vector. We further tested the effect of the STI1 domain of KPC2 on KPC1 stability by pulse-chase analysis (Fig. 4G). HEK293T cells were transiently transfected with a vector for FLAG-tagged KPC1 together either with a vector for HA-tagged wild-type KPC2 or the KPC2(ΔSTI1) mutant or with the empty vector. KPC2(ΔSTI1) promoted KPC1 degradation. KPC2(ΔUBL-STI1), which lacks both the UBL and STI1 domains and did not interact with KPC1, also promoted KPC1 degradation but to a much reduced extent compared with the effect of KPC2(ΔSTI1) (Fig. 4G). These data thus suggest that the STI1 domain of KPC2 contributes to the stabilization of KPC1.
The UBA domain of Rad23 was shown to inhibit assembly of multiubiquitin chains (6, 43). To investigate the role of the domains of KPC2 in p27 ubiquitylation, we coexpressed His6- and FLAG-tagged KPC1 together with HA-tagged wild-type of mutant forms of KPC2 in insect cells and purified the recombinant complexes to near homogeneity for assay of their abilities to catalyze p27 ubiquitylation in vitro in the presence of ATP, Uba1, UbcH5A, and GST-ubiquitin. Monoubiquitylation of p27 was observed in the absence of E3 (presence of only E1 and E2) under these conditions. Polyubiquitylation of p27 was equally pronounced in the presence of wild-type KPC2, KPC2(ΔUBAn), or KPC2(ΔUBAc), but it was greatly reduced in extent in the presence of KPC2(ΔSTI1) (Fig. 5). These data suggest that the STI1 domain of KPC2 is indispensable for the ubiquitylation of p27 mediated by KPC.
FIG. 5.
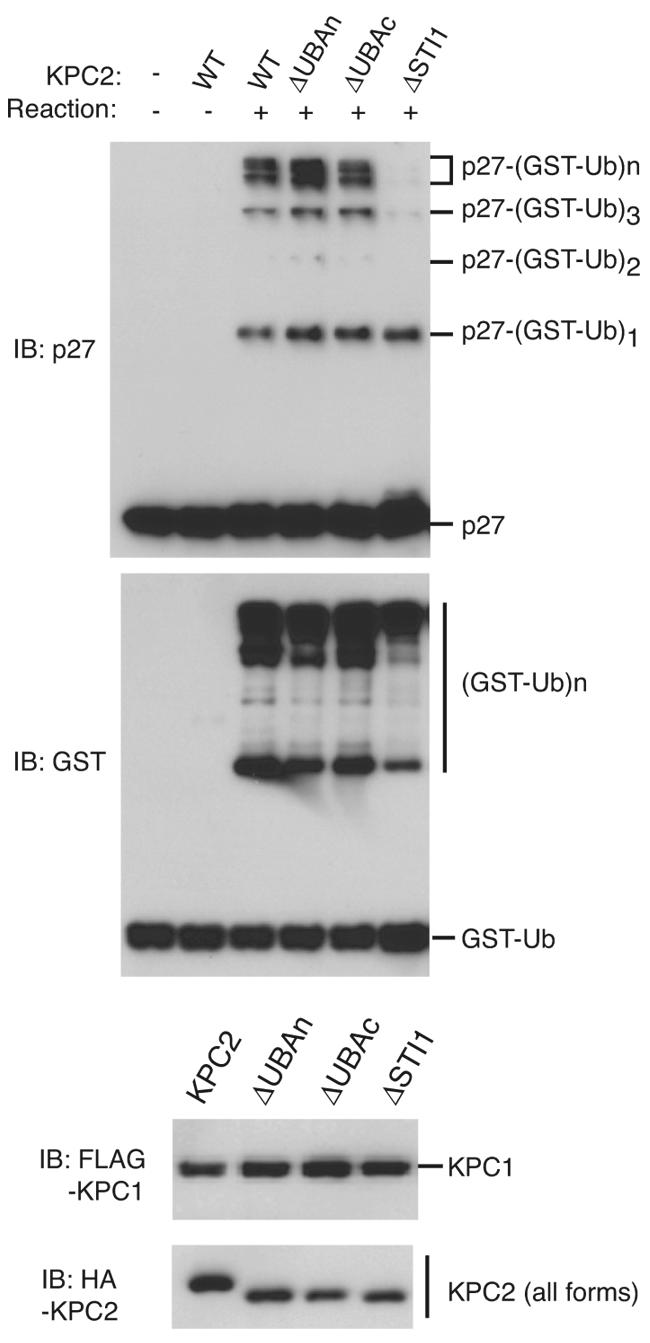
Effect of KPC2 mutants on p27 ubiquitylation in vitro. Recombinant complexes of KPC1 and KPC2 derivatives were assayed for their abilities to mediate polyubiquitylation of p27 in the presence of Uba1, UbcH5A, GST-ubiquitin, and ATP. Reaction products were subjected to immunoblot (IB) analysis with antibodies to p27 or to GST (upper two panels). The bottom panels show immunoblot analysis of the amounts of recombinant KPC1 (tagged with the FLAG epitope) and KPC2 derivatives (tagged with the HA epitope) used in the reactions. WT, wild type; GST-Ub, GST-ubiquitin.
We applied RNAi to determine the effect of depletion of endogenous KPC2 on p27 degradation at the transition from G0 to G1 in NIH 3T3 cells. After serum deprivation for 96 h, cells expressing either the mouse KPC2 shRNA or the EGFP shRNA (control) were stimulated to reenter the cell cycle by replating at a density of ∼40% and incubation in complete medium for various times. Cell lysates were then subjected to immunoblot analysis with antibodies to p27. The reduction in the amount of p27 induced by serum restimulation was markedly inhibited in the cells expressing KPC2 shRNA compared with that apparent in the control cells (Fig. 6A). Given that down-regulation of KPC2 expression by KPC2 shRNA also resulted in a reduction in the amount of KPC1, it was unclear whether both KPC1 and KPC2 or just KPC1 is required for p27 degradation at the G0-G1 transition. To address this question, we infected NIH 3T3 cells expressing mouse KPC2 shRNA with retroviruses encoding human KPC2 derivatives and examined the effects of these proteins on p27 degradation (Fig. 6B). Expression of wild-type human KPC2 or KPC2(ΔUBAc) rescued the impairment in p27 degradation as each also rescued the instability of KPC1 (Fig. 4F) induced by depletion of endogenous KPC2, whereas KPC2(ΔSTI1), which destabilized KPC1, did not. In contrast, whereas expression of KPC2(ΔUBAn) stabilized KPC1 (Fig. 4F), it did not reverse the impairment in p27 degradation. Although the ability of KPC2(ΔUBAc) to bind polyubiquitylated proteins is greatly reduced compared with that of wild-type KPC2, the low level of such binding by the mutant might be sufficient to compensate for the loss of endogenous KPC2 in the knocked-down cells when it is overexpressed. It is also possible, however, that deletion of the COOH-terminal UBA domain is less deleterious to the overall structure of KPC2 than is that of the NH2-terminal UBA domain. We were not able to assess the functional significance of the UBL domain of KPC2 in p27 degradation, given that KPC2(ΔUBL) was capable of neither binding to nor stabilizing KPC1. These observations indicate that KPC2 regulates p27 degradation in cooperation with KPC1 and that the NH2-terminal UBA domain of KPC2 is required for p27 degradation, probably as a result of its contribution to interaction with polyubiquitylated p27 and the proteasome.
FIG. 6.

Requirement of KPC2 for p27 degradation. (A) Effect of KPC2 depletion by RNAi on p27 degradation. NIH 3T3 cells stably expressing KPC2 or EGFP shRNAs were synchronized in G0 phase by contact inhibition and serum deprivation and then stimulated to reenter the cell cycle by replating at a density of ∼40% and incubation in complete medium for the indicated times. Cell lysates were then subjected to immunoblot analysis with antibodies to p27 or to HSP70 (control) (left panel). The intensity of the p27 bands was quantitated and plotted (right panel). (B) Complementation of impaired p27 degradation in NIH 3T3 cells expressing mouse KPC2 shRNA by introduction of human KPC2 derivatives. NIH 3T3 cells expressing KPC2 or EGFP shRNAs were infected with retroviral vectors for the indicated human KPC2 derivatives (tagged with the HA epitope) or with the corresponding empty retrovirus (Mock). The cells were then examined for p27 degradation during G1 as in panel A. IB, immunoblot.
DISCUSSION
The amount of the CKI p27 is high in quiescent (G0) cells but rapidly decreases as a result of degradation of the protein by the ubiquitin-proteasome system on entry of cells into the cell cycle. We recently identified a ubiquitin ligase complex, KPC, that is composed of KPC1 and KPC2 subunits and contributes to p27 degradation at the G0-G1 transition (19, 26). Whereas the RING finger protein KPC1 was shown to function as the catalytic subunit, the role of the UBL-UBA protein KPC2 has been unclear. We now demonstrate that KPC2 stabilizes KPC1, recruits polyubiquitylated proteins, and interacts with the 26S proteasome, thereby promoting the degradation of p27 (Fig. 7). On the basis of our present observations, we propose that KPC2 functions to deliver ubiquitylated proteins to the proteasome. The complex of KPC1 (regulator), KPC2 (adapter), and the 26S proteasome (effector) thus appears suited to ensure the rapid and efficient removal of p27 as cells enter the cell cycle from the quiescent state.
Members of the UBL-UBA family of proteins, including hHR23A and hHR23B as well as hPLIC1 and hPLIC2, are thought to deliver polyubiquitylated substrates to the proteasome (5, 8, 47-49, 52, 53, 63). Similar to other UBL-UBA proteins, KPC2 binds to both the proteasome and polyubiquitylated proteins, and these interactions are mediated by the UBL and UBA domains, respectively. KPC2 thus shares the basic biochemical properties of other UBL-UBA proteins. However, our analyses indicate that KPC2 also possesses characteristics not shared by other UBL-UBA proteins. A KPC2 mutant that lacks the UBL domain thus retained the ability to interact with the proteasome, and both UBA domains of KPC2 were found to be required for binding to polyubiquitin chains. In the former instance, it is possible that the KPC2 mutant interacts with endogenous KPC2 or other factors, thereby bypassing the absolute requirement for the UBL domain. In the latter instance, it is possible that the NH2-terminal and COOH-terminal UBA domains of KPC2 are independently required for binding to polyubiquitin or that deletion of one domain affects the function of the other. Consistent with previous observations, we found that a large excess of KPC2 inhibits p27 ubiquitylation (data not shown). However, it remains to be determined whether these results truly reflect the physiological situation.
KPC2 forms a complex with the E3 ligase KPC1 and promotes the degradation of its specific substrate p27 via the ubiquitin-proteasome pathway. hHR23 binds to the E3s E6AP, p300, and anaphase promoting complex/cyclosome, which participate in the degradation of p53 and other cell cycle regulators (14, 27, 54, 69). Rad23 interacts via its UBL domain with the U box-type ubiquitin ligase Ufd2 (22, 49). In addition, PLIC proteins interact with the proteasome and E6AP through their UBL domains (25). The ubiquitin fold has also been recognized in various other proteins, including the Ras binding domain of c-Raf-1 and the PB1 domain of Bem1p, and appears to contribute to protein-protein interactions (9, 68). These observations thus suggest that most UBL-UBA proteins physiologically associate with ubiquitin ligase components as well as the proteasome via their UBL domains and thereby function as a bridge between the ubiquitylation machinery and the proteasome (49, 63).
The STI1 domain has been identified in several UBL-UBA proteins other than KPC2. The STI1 domain of Dsk2 is required for its interaction with the ATPase domain of the truncated HSP70-like protein Stch (21). hHR23B stabilizes the protein XPC (xeroderma pigmentosum group C), which contributes to the recognition of DNA damage for nucleotide excision repair, in a manner dependent on its STI1 domain (31, 42); indeed, a small polypeptide corresponding to the STI1 domain of hHR23B is sufficient to stimulate the activity of XPC in nucleotide excision repair. Together with our finding that the STI1 domain of KPC2 is indispensable for stabilization of KPC1, these observations suggest that the STI1 domains of UBL-UBA proteins may perform a chaperone-like function for their binding partners. It is possible that the STI1 domain of KPC2 plays a role in maintaining the normal structure of KPC1. Deletion of this domain may thus result in a conformational change of KPC1 that leads to impairment of p27 binding, p27 ubiquitylation, or both.
KPC is responsible for degradation of p27 at the G0-G1 transition, and we have now shown that the UBL-UBA protein KPC2 plays an important role in this process. Mutations in the p27 gene appear to be rare in human cancers. However, a reduced abundance of p27 in a subset of various cancers correlates well with poor prognosis (4, 28, 35, 46). Furthermore, the loss of p27 alleles in mice increases the sensitivity of these animals to cancer-inducing agents (10). Elucidation of the molecular mechanism by which KPC regulates p27 degradation may thus provide insight into the altered expression of this CKI in tumor cells as well as into whether such altered expression is a cause or a consequence of cell transformation. Indeed, Skp2 is overexpressed in many human cancer cells, suggesting that p27 degradation mediated by Skp2 may be related to carcinogenesis (41, 67). It is thus possible that the KPC pathway of p27 degradation is also deregulated in cancer cells.
Acknowledgments
We thank T. Kitamura for pMX-puro, R. Yada and N. Nishimura for technical assistance; and M. Kimura and A. Ohta for help in preparation of the manuscript.
This work was supported in part by a grant from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
REFERENCES
- 1.Agrawal, D., P. Hauser, F. McPherson, F. Dong, A. Garcia, and W. J. Pledger. 1996. Repression of p27kip1 synthesis by platelet-derived growth factor in BALB/c 3T3 cells. Mol. Cell. Biol. 16:4327-4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bertolaet, B. L., D. J. Clarke, M. Wolff, M. H. Watson, M. Henze, G. Divita, and S. I. Reed. 2001. UBA domains of DNA damage-inducible proteins interact with ubiquitin. Nat. Struct. Biol. 8:417-422. [DOI] [PubMed] [Google Scholar]
- 3.Carrano, A. C., E. Eytan, A. Hershko, and M. Pagano. 1999. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol. 1:193-199. [DOI] [PubMed] [Google Scholar]
- 4.Catzavelos, C., N. Bhattacharya, Y. C. Ung, J. A. Wilson, L. Roncari, C. Sandhu, P. Shaw, H. Yeger, I. Morava-Protzner, L. Kapusta, E. Franssen, K. I. Pritchard, and J. M. Slingerland. 1997. Decreased levels of the cell-cycle inhibitor p27Kip1 protein: prognostic implications in primary breast cancer. Nat. Med. 3:227-230. [DOI] [PubMed] [Google Scholar]
- 5.Chen, L., and K. Madura. 2002. Rad23 promotes the targeting of proteolytic substrates to the proteasome. Mol. Cell. Biol. 22:4902-4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen, L., U. Shinde, T. G. Ortolan, and K. Madura. 2001. Ubiquitin-associated (UBA) domains in Rad23 bind ubiquitin and promote inhibition of multi-ubiquitin chain assembly. EMBO Rep. 2:933-938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Connor, M. K., R. Kotchetkov, S. Cariou, A. Resch, R. Lupetti, R. G. Beniston, F. Melchior, L. Hengst, and J. M. Slingerland. 2003. CRM1/Ran-mediated nuclear export of p27Kip1 involves a nuclear export signal and links p27 export and proteolysis. Mol. Biol. Cell 14:201-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elsasser, S., R. R. Gali, M. Schwickart, C. N. Larsen, D. S. Leggett, B. Muller, M. T. Feng, F. Tubing, G. A. Dittmar, and D. Finley. 2002. Proteasome subunit Rpn1 binds ubiquitin-like protein domains. Nat. Cell Biol. 4:725-730. [DOI] [PubMed] [Google Scholar]
- 9.Emerson, S. D., V. S. Madison, R. E. Palermo, D. S. Waugh, J. E. Scheffler, K. L. Tsao, S. E. Kiefer, S. P. Liu, and D. C. Fry. 1996. Structure of the Ras-binding domain of c-Raf-1 as determined by NMR spectroscopy and identification of the region that interacts with Ras. Drug Des. Discov. 13:83-93. [PubMed] [Google Scholar]
- 10.Fero, M. L., E. Randel, K. E. Gurley, J. M. Roberts, and C. J. Kemp. 1998. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature 396:177-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fero, M. L., M. Rivkin, M. Tasch, P. Porter, C. E. Carow, E. Firpo, K. Polyak, L. H. Tsai, V. Broudy, R. M. Perlmutter, K. Kaushansky, and J. M. Roberts. 1996. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27Kip1-deficient mice. Cell 85:733-744. [DOI] [PubMed] [Google Scholar]
- 12.Fujiwara, K., T. Tenno, K. Sugasawa, J. G. Jee, I. Ohki, C. Kojima, H. Tochio, H. Hiroaki, F. Hanaoka, and M. Shirakawa. 2004. Structure of the ubiquitin-interacting motif of S5a bound to the ubiquitin-like domain of HR23B. J. Biol. Chem. 279:4760-4767. [DOI] [PubMed] [Google Scholar]
- 13.Funakoshi, M., T. Sasaki, T. Nishimoto, and H. Kobayashi. 2002. Budding yeast Dsk2p is a polyubiquitin-binding protein that can interact with the proteasome. Proc. Natl. Acad. Sci. USA 99:745-750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grossman, S. R., M. E. Deato, C. Brignone, H. M. Chan, A. L. Kung, H. Tagami, Y. Nakatani, and D. M. Livingston. 2003. Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science 300:342-344. [DOI] [PubMed] [Google Scholar]
- 15.Hara, T., T. Kamura, K. Nakayama, K. Oshikawa, S. Hatakeyama, and K. I. Nakayama. 2001. Degradation of p27Kip1 at the G0-G1 transition mediated by a Skp2-independent ubiquitination pathway. J. Biol. Chem. 276:48937-48943. [DOI] [PubMed] [Google Scholar]
- 16.Hengst, L., and S. I. Reed. 1996. Translational control of p27Kip1 accumulation during the cell cycle. Science 271:1861-1864. [DOI] [PubMed] [Google Scholar]
- 17.Hiyama, H., M. Yokoi, C. Masutani, K. Sugasawa, T. Maekawa, K. Tanaka, J. H. Hoeijmakers, and F. Hanaoka. 1999. Interaction of hHR23 with S5a. The ubiquitin-like domain of hHR23 mediates interaction with S5a subunit of 26 S proteasome. J. Biol. Chem. 274:28019-28025. [DOI] [PubMed] [Google Scholar]
- 18.Ishida, N., T. Hara, T. Kamura, M. Yoshida, K. Nakayama, and K. I. Nakayama. 2002. Phosphorylation of p27Kip1 on serine 10 is required for its binding to CRM1 and nuclear export. J. Biol. Chem. 277:14355-14358. [DOI] [PubMed] [Google Scholar]
- 19.Kamura, T., T. Hara, M. Matsumoto, N. Ishida, F. Okumura, S. Hatakeyama, M. Yoshida, K. Nakayama, and K. I. Nakayama. 2004. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27Kip1 at G1 phase. Nat. Cell Biol. 6:1229-1235. [DOI] [PubMed] [Google Scholar]
- 20.Kamura, T., K. Maenaka, S. Kotoshiba, M. Matsumoto, D. Kohda, R. C. Conaway, J. W. Conaway, and K. I. Nakayama. 2004. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. 18:3055-3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaye, F. J., S. Modi, I. Ivanovska, E. V. Koonin, K. Thress, A. Kubo, S. Kornbluth, and M. D. Rose. 2000. A family of ubiquitin-like proteins binds the ATPase domain of Hsp70-like Stch. FEBS Lett. 467:348-355. [DOI] [PubMed] [Google Scholar]
- 22.Kim, I., K. Mi, and H. Rao. 2004. Multiple interactions of rad23 suggest a mechanism for ubiquitylated substrate delivery important in proteolysis. Mol. Biol. Cell 15:3357-3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kitagawa, M., S. Hatakeyama, M. Shirane, M. Matsumoto, N. Ishida, K. Hattori, I. Nakamichi, A. Kikuchi, K. I. Nakayama, and K. Nakayama. 1999. An F-box protein, FWD1, mediates ubiquitin-dependent proteolysis of β-catenin. EMBO J. 18:2401-2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kiyokawa, H., R. D. Kineman, K. O. Manova-Todorova, V. C. Soares, E. S. Hoffman, M. Ono, D. Khanam, A. C. Hayday, L. A. Frohman, and A. Koff. 1996. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27Kip1. Cell 85:721-732. [DOI] [PubMed] [Google Scholar]
- 25.Kleijnen, M. F., A. H. Shih, P. Zhou, S. Kumar, R. E. Soccio, N. L. Kedersha, G. Gill, and P. M. Howley. 2000. The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol. Cell 6:409-419. [DOI] [PubMed] [Google Scholar]
- 26.Kotoshiba, S., T. Kamura, T. Hara, N. Ishida, and K. I. Nakayama. 2005. Molecular dissection of the interaction between p27 and KPC, the ubiquitin ligase that regulates proteolysis of p27 in G1 phase. J. Biol. Chem. 280:17694-17700. [DOI] [PubMed] [Google Scholar]
- 27.Kumar, S., A. L. Talis, and P. M. Howley. 1999. Identification of HHR23A as a substrate for E6-associated protein-mediated ubiquitination. J. Biol. Chem. 274:18785-18792. [DOI] [PubMed] [Google Scholar]
- 28.Loda, M., B. Cukor, S. W. Tam, P. Lavin, M. Fiorentino, G. F. Draetta, J. M. Jessup, and M. Pagano. 1997. Increased proteasome-dependent degradation of the cyclin-dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nat. Med. 3:231-234. [DOI] [PubMed] [Google Scholar]
- 29.Marti-Renom, M. A., A. C. Stuart, A. Fiser, R. Sanchez, F. Melo, and A. Sali. 2000. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29:291-325. [DOI] [PubMed] [Google Scholar]
- 30.Maruyama, S., S. Hatakeyama, K. Nakayama, N. Ishida, K. Kawakami, and K. I. Nakayama. 2001. Characterization of a mouse gene (Fbxw6) that encodes a homologue of Caenorhabditis elegans SEL-10. Genomics 78:214-222. [DOI] [PubMed] [Google Scholar]
- 31.Masutani, C., M. Araki, K. Sugasawa, P. J. van der Spek, A. Yamada, A. Uchida, T. Maekawa, D. Bootsma, J. H. Hoeijmakers, and F. Hanaoka. 1997. Identification and characterization of XPC-binding domain of hHR23B. Mol. Cell. Biol. 17:6915-6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Medema, R. H., G. J. Kops, J. L. Bos, and B. M. Burgering. 2000. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 404:782-787. [DOI] [PubMed] [Google Scholar]
- 33.Millard, S. S., A. Vidal, M. Markus, and A. Koff. 2000. A U-rich element in the 5′ untranslated region is necessary for the translation of p27 mRNA. Mol. Cell. Biol. 20:5947-5959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miura, M., S. Hatakeyama, K. Hattori, K. Nakayama, and K. I. Nakayama. 1999. Structure and expression of the gene encoding mouse F-box protein, Fwd2. Genomics 62:50-58. [DOI] [PubMed] [Google Scholar]
- 35.Mori, M., K. Mimori, T. Shiraishi, S. Tanaka, H. Ueo, K. Sugimachi, and T. Akiyoshi. 1997. p27 expression and gastric carcinoma. Nat. Med. 3:593. [DOI] [PubMed] [Google Scholar]
- 36.Murray, A. W. 2004. Recycling the cell cycle: cyclins revisited. Cell 116:221-234. [DOI] [PubMed] [Google Scholar]
- 37.Nakayama, K., N. Ishida, M. Shirane, A. Inomata, T. Inoue, N. Shishido, I. Horii, D. Y. Loh, and K. I. Nakayama. 1996. Mice lacking p27Kip1 display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell 85:707-720. [DOI] [PubMed] [Google Scholar]
- 38.Nakayama, K., H. Nagahama, Y. A. Minamishima, M. Matsumoto, I. Nakamichi, K. Kitagawa, M. Shirane, R. Tsunematsu, T. Tsukiyama, N. Ishida, M. Kitagawa, K. I. Nakayama, and S. Hatakeyama. 2000. Targeted disruption of Skp2 results in accumulation of cyclin E and p27Kip1, polyploidy and centrosome overduplication. EMBO J. 19:2069-2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakayama, K., H. Nagahama, Y. A. Minamishima, S. Miyake, N. Ishida, S. Hatakeyama, M. Kitagawa, S. Iemura, T. Natsume, and K. I. Nakayama. 2004. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev. Cell 6:661-672. [DOI] [PubMed] [Google Scholar]
- 40.Nakayama, K., and K. I. Nakayama. 1998. Cip/Kip cyclin-dependent kinase inhibitors: brakes of the cell cycle engine during development. Bioessays 20:1020-1029. [DOI] [PubMed] [Google Scholar]
- 41.Nakayama, K. I., and K. Nakayama. 2005. Regulation of the cell cycle by SCF-type ubiquitin ligases. Semin. Cell Dev. Biol. 16:323-333. [DOI] [PubMed] [Google Scholar]
- 42.Ng, J. M., W. Vermeulen, G. T. van der Horst, S. Bergink, K. Sugasawa, H. Vrieling, and J. H. Hoeijmakers. 2003. A novel regulation mechanism of DNA repair by damage-induced and RAD23-dependent stabilization of xeroderma pigmentosum group C protein. Genes Dev. 17:1630-1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ortolan, T. G., P. Tongaonkar, D. Lambertson, L. Chen, C. Schauber, and K. Madura. 2000. The DNA repair protein Rad23 is a negative regulator of multi-ubiquitin chain assembly. Nat. Cell Biol. 2:601-608. [DOI] [PubMed] [Google Scholar]
- 44.Pagano, M., S. W. Tam, A. M. Theodoras, P. Beer-Romero, G. Del Sal, V. Chau, P. R. Yew, G. F. Draetta, and M. Rolfe. 1995. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science 269:682-685. [DOI] [PubMed] [Google Scholar]
- 45.Polyak, K., M. H. Lee, H. Erdjument-Bromage, A. Koff, J. M. Roberts, P. Tempst, and J. Massague. 1994. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 78:59-66. [DOI] [PubMed] [Google Scholar]
- 46.Porter, P. L., K. E. Malone, P. J. Heagerty, G. M. Alexander, L. A. Gatti, E. J. Firpo, J. R. Daling, and J. M. Roberts. 1997. Expression of cell-cycle regulators p27Kip1 and cyclin E, alone and in combination, correlate with survival in young breast cancer patients. Nat. Med. 3:222-225. [DOI] [PubMed] [Google Scholar]
- 47.Raasi, S., I. Orlov, K. G. Fleming, and C. M. Pickart. 2004. Binding of polyubiquitin chains to ubiquitin-associated (UBA) domains of HHR23A. J. Mol. Biol. 341:1367-1379. [DOI] [PubMed] [Google Scholar]
- 48.Rao, H., and A. Sastry. 2002. Recognition of specific ubiquitin conjugates is important for the proteolytic functions of the ubiquitin-associated domain proteins Dsk2 and Rad23. J. Biol. Chem. 277:11691-11695. [DOI] [PubMed] [Google Scholar]
- 49.Richly, H., M. Rape, S. Braun, S. Rumpf, C. Hoege, and S. Jentsch. 2005. A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell 120:73-84. [DOI] [PubMed] [Google Scholar]
- 50.Riley, B. E., Y. Xu, H. Y. Zoghbi, and H. T. Orr. 2004. The effects of the polyglutamine repeat protein ataxin-1 on the UBL-UBA protein A1Up. J. Biol. Chem. 279:42290-42301. [DOI] [PubMed] [Google Scholar]
- 51.Rodier, G., A. Montagnoli, L. Di Marcotullio, P. Coulombe, G. F. Draetta, M. Pagano, and S. Meloche. 2001. p27 cytoplasmic localization is regulated by phosphorylation on Ser10 and is not a prerequisite for its proteolysis. EMBO J. 20:6672-6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saeki, Y., T. Sone, A. Toh-e, and H. Yokosawa. 2002. Identification of ubiquitin-like protein-binding subunits of the 26S proteasome. Biochem. Biophys. Res. Commun. 296:813-819. [DOI] [PubMed] [Google Scholar]
- 53.Schauber, C., L. Chen, P. Tongaonkar, I. Vega, D. Lambertson, W. Potts, and K. Madura. 1998. Rad23 links DNA repair to the ubiquitin/proteasome pathway. Nature 391:715-718. [DOI] [PubMed] [Google Scholar]
- 54.Seeger, M., R. Hartmann-Petersen, C. R. Wilkinson, M. Wallace, I. Samejima, M. S. Taylor, and C. Gordon. 2003. Interaction of the anaphase-promoting complex/cyclosome and proteasome protein complexes with multiubiquitin chain-binding proteins. J. Biol. Chem. 278:16791-16796. [DOI] [PubMed] [Google Scholar]
- 55.Sheaff, R. J., M. Groudine, M. Gordon, J. M. Roberts, and B. E. Clurman. 1997. Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev. 11:1464-1478.9192873 [Google Scholar]
- 56.Sherr, C. J., and J. M. Roberts. 1999. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 13:1501-1512. [DOI] [PubMed] [Google Scholar]
- 57.Shi, J., T. L. Blundell, and K. Mizuguchi. 2001. FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol. 310:243-257. [DOI] [PubMed] [Google Scholar]
- 58.Shirane, M., Y. Harumiya, N. Ishida, A. Hirai, C. Miyamoto, S. Hatakeyama, K. I. Nakayama, and M. Kitagawa. 1999. Down-regulation of p27Kip1 by two mechanisms, ubiquitin-mediated degradation and proteolytic processing. J. Biol. Chem. 274:13886-13893. [DOI] [PubMed] [Google Scholar]
- 59.Sutterluty, H., E. Chatelain, A. Marti, C. Wirbelauer, M. Senften, U. Muller, and W. Krek. 1999. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat. Cell Biol. 1:207-214. [DOI] [PubMed] [Google Scholar]
- 60.Tomoda, K., Y. Kubota, and J. Kato. 1999. Degradation of the cyclin-dependent-kinase inhibitor p27Kip1 is instigated by Jab1. Nature 398:160-165. [DOI] [PubMed] [Google Scholar]
- 61.Toyoshima, H., and T. Hunter. 1994. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell 78:67-74. [DOI] [PubMed] [Google Scholar]
- 62.Tsvetkov, L. M., K. H. Yeh, S. J. Lee, H. Sun, and H. Zhang. 1999. p27Kip1 ubiquitination and degradation is regulated by the SCFSkp2 complex through phosphorylated Thr187 in p27. Curr. Biol. 9:661-664. [DOI] [PubMed] [Google Scholar]
- 63.Verma, R., R. Oania, J. Graumann, and R. J. Deshaies. 2004. Multiubiquitin chain receptors define a layer of substrate selectivity in the ubiquitin-proteasome system. Cell 118:99-110. [DOI] [PubMed] [Google Scholar]
- 64.Vlach, J., S. Hennecke, and B. Amati. 1997. Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27. EMBO J. 16:5334-5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Walters, K. J., M. F. Kleijnen, A. M. Goh, G. Wagner, and P. M. Howley. 2002. Structural studies of the interaction between ubiquitin family proteins and proteasome subunit S5a. Biochemistry 41:1767-1777. [DOI] [PubMed] [Google Scholar]
- 66.Wilkinson, C. R., M. Seeger, R. Hartmann-Petersen, M. Stone, M. Wallace, C. Semple, and C. Gordon. 2001. Proteins containing the UBA domain are able to bind to multi-ubiquitin chains. Nat. Cell Biol. 3:939-943. [DOI] [PubMed] [Google Scholar]
- 67.Yamasaki, L., and M. Pagano. 2004. Cell cycle, proteolysis and cancer. Curr. Opin. Cell Biol. 16:623-628. [DOI] [PubMed] [Google Scholar]
- 68.Yoshinaga, S., M. Kohjima, K. Ogura, M. Yokochi, R. Takeya, T. Ito, H. Sumimoto, and F. Inagaki. 2003. The PB1 domain and the PC motif-containing region are structurally similar protein binding modules. EMBO J. 22:4888-4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhu, Q., G. Wani, M. A. Wani, and A. A. Wani. 2001. Human homologue of yeast Rad23 protein A interacts with p300/cyclic AMP-responsive element binding (CREB)-binding protein to down-regulate transcriptional activity of p53. Cancer Res. 61:64-70. [PubMed] [Google Scholar]