

Abstract
By generating a population of Dictyostelium cells that are in the G1 phase of the cell cycle we have examined the influence of cell cycle status on cell fate specification, cell type proportioning and its regulation, and terminal differentiation. The lack of observable mitosis during the development of these cells and the quantification of their cellular DNA content suggests that they remain in G1 throughout development. Furthermore, chromosomal DNA synthesis was not detectable these cells, indicating that no synthesis phase had occurred, although substantial mitochondrial DNA synthesis did occur in prespore cells. The G1-phase cells underwent normal morphological development and sporulation but displayed an elevated prespore/prestalk ratio of 5.7 compared to the 3.0 (or 3:1) ratio normally observed in populations dominated by G2-phase cells. When migrating slugs produced by G1-phase cells were bisected, each half could reestablish the 5.7 (or 5.7:1) prespore/prestalk ratio. These results demonstrate that Dictyostelium cells can carry out the entire developmental cycle in the G1 phase of the cell cycle and that passage from G2 into G1 phase is not required for sporulation. Our results also suggest that the population asymmetry provided by the distribution of cells around the cell cycle at the time of starvation is not strictly required for cell type proportioning. Finally, when developed together with G2-phase cells, G1-phase cells preferentially become prespore cells and exclude G2-phase cells from the prespore-spore cell population, suggesting that G1-phase cells have an advantage over G2-phase cells in executing the spore cell differentiation pathway.
Growth-phase Dictyostelium cells must regulate their exit from the cell cycle when they encounter starvation conditions to initiate multicellular development (14). In a population of growing cells, most cells are in the G2 phase of the cell cycle since chromosome synthesis (S phase) and mitosis (M phase) are completed in ∼40 min out of an 8-h cell cycle (8, 41, 43). The relationship between cell fate determination and cell cycle regulation in Dictyostelium discoideum has been a topic of intense study over the past 30 years, and there is substantial evidence that the cell cycle status of growing cells at the time of starvation impinges on cell fate determination later in development. Cells that are in middle, or late, G2 phase at the time of starvation preferentially become prespore cells, whereas cells in the S, M, or early G2 phase preferentially become prestalk cells (3, 9, 10, 25, 27, 40, 45). Maeda and colleagues have proposed that cells exit the cell cycle at a particular point late in G2 called the “putative shift” (PS) point and that cells that take the longest time to reach the PS point after starvation will have a propensity to differentiate as prestalk cells (1, 21-23). Alternatively, it has been suggested that cells exiting the cell cycle early in G2 tend to differentiate as prestalk cells, whereas cells that exit the cell cycle late in G2 tend to differentiate as prespore cells (20). Various models have been proposed to explain how the cell cycle asymmetry produces the cell fate readout bias, such as through the differential sensitivity of cells to the chlorinated hexanophenone DIF-1 (36). It is significant that a mutation in the rtoA gene abrogates the cell fate bias imposed by the cell cycle with no apparent effect on cell type proportioning (42). This suggests that D. discoideum can use other parameters of population asymmetry to establish cell type proportions and produce a functioning multicellular organism.
In the studies cited above, models of cell cycle-based proportioning were constructed on the basis of experiments performed on cells synchronized by release from stationary phase or release from cold shock. Several issues confound interpretations of these studies. First, it is difficult to achieve perfect synchrony by these methods. There is also an implicit assumption in these studies that the cell cycle progresses with the growth-phase timing after release from the cell cycle block or after development is initiated by starvation. During growth, the G2 phase is a nearly featureless period of about 7 h. Only one molecular landmark has been described, and that is the accumulation of the mRNA encoding the small subunit of ribonucleotide reductase, RnrB (20). Even the M and S phases of the cell cycle are difficult to monitor in D. discoideum. Karyokinesis can be separated from cytokinesis by an hour or more in axenically grown populations, as evidenced by the presence of ∼20% binucleated cells, so the appearance of dividing cells is not a reliable marker for M phase or S phase (5). Thus, a cell that is assumed to be late in the G2 phase because it divided an hour or two later may actually be a binucleated cell with two G1 nuclei. DNA synthesis is not a perfect marker for S phase, since ∼30% of the cellular DNA is mitochondrial DNA (mtDNA), and mtDNA is not synthesized when chromosomes are replicated during development or after release from cold shock (reference 5 and unpublished observations). In part because of these uncertainties, we sought another method of synchronization to examine the effects of cell cycle status on cell fate determination.
We have recently shown that cell cycle timing is linked to cell differentiation during development (6). Upon starvation, all vegetative cells are arrested in G2 phase in the first 6 h of development. Between 12 and 20 h, prespore cells undergo mitosis and arrest in G1 prior to terminally encapsulating into spores, while prestalk cells appear to undergo mitosis after 22 h and become G1 stalk cells. The finding that prespore cells arrest in G1 after multicellularity is achieved well before terminal differentiation brings up several interesting questions. Does the cell fate imposed by the cell cycle status at the time of starvation influence later cell cycle events? Is G1 arrest required for commitment to the prespore fate or to encapsulate as a spore? Or, alternatively, is G1 arrest a consequence of becoming a prespore cell? What is the relationship, if any, between prespore cell differentiation and prespore-specific mtDNA replication? Answers to these questions will clarify our understanding of cell fate determination in D. discoideum.
It is clear that genes that become prespore enriched are expressed in some cells in the first few hours of development, well before prespore cell mitosis occurs and well before a distinct prespore tissue arises (11, 13, 38). Thus, cell cycle arrest in G1 is not required for the earliest steps in prespore differentiation. However, it has also been observed that certain mutants that are blocked in development prior to the multicellular phase do not undergo mitosis and block the production of prespore cells (6, 18, 19, 43). This indicates that mitosis is regulated during development but does not address whether it is required for cell differentiation or for sporulation.
It has been known for many years that D. discoideum amoebae that have recently hatched from germinating spores are capable of developing without an intervening growth phase (4, 29, 32). Since we now know that spores encapsulate in G1, these previous reports strongly suggest that G1 cells are fully capable of cell type proportioning and development without transiting the cell cycle. Our recent finding that heat-shocked spores germinating in growth medium stay in G1 phase for ∼20 h before resuming the cell cycle provides a means of testing this more completely (6). Here, we show that G1-phase cells, obtained from freshly germinated spores, develop fairly normally. Thus, the population asymmetry provided by the cell cycle at the time of starvation, and the timing of cell cycle exit, are not required developmental events. Interestingly, we find that the proportion of prespore cells is higher in a developing G1 population than in a G2 population and that G1-phase cells have an advantage over G2-phase cells in forming spores.
MATERIALS AND METHODS
Growth and development of D. discoideum.
All strains were derived from the D. discoideum strain AX4 (15). Marked strains were all derived from AX4 by standard molecular genetic manipulations (16, 24). Cells were grown on bacterial lawns and allowed to complete development in situ, and the spores were allowed to mature for 1 day. Spores were harvested into 0.1% NP-40 in 10 mM potassium phosphate buffer, pH 6.4. A 10-ml syringe fitted with an 18-gauge needle was used to disperse the spores and to disrupt nonspore cells. The spores were washed twice with 10 mM potassium phosphate buffer and were resuspended in buffer in glass tubes and incubated at 42°C for 30 min. The spores were diluted in HL-5 medium to 5 × 106 cells/ml and incubated in 150-mm petri dishes (30 ml/dish) at room temperature (6). At different times, cells were collected for development as previously described (34). Cells collected by centrifugation were washed once in PDF buffer (22.2 mM potassium phosphate [pH 6.4], 20 mM KCl, 1.0 mM CaCl2, 2.5 mM MgSO4), and 5 × 107 cells were deposited on each filter atop a cellulose pad saturated with PDF buffer (34).
Quantification of cells.
Two filters of developing cells were processed for each time point as described previously (6). For spores, two filters of fully developed cells were washed with 0.1% NP-40 in 10 mM potassium phosphate buffer, pH 6.4, and dispersed with syringe at least 10 times, and solution was added to equalize sample volumes. The cells in all samples were counted three times using a hemacytometer.
Flow cytometry.
Flow cytometry was carried out as described previously (6). To measure cellular DNA content, cells were fixed in 70% EtOH, digested with RNase, stained with propidium iodide, and analyzed by a Beckman-Coulter Epics XL-MCL apparatus, adjusting the parameters for the measurement of single, mononucleated cells by the use of the forward scatter plot as a guide. To study different cell populations in mixtures, one population was tagged with a reporter construct, ecmA/GFP, cotB/GFP, or actin15/GFP, as described previously (6). For comparison of cell type proportions of marked strains, we used the same culture to produce G1 cells and G2 cells to avoid potential issues of differential marker expression within the population.
DNA synthesis.
Bromo-deoxyuridine (BrdU) incorporation was used to label newly synthesized DNA during filter development, as described previously (6). Cells were grown in liquid medium (HL-5) and harvested by centrifugation, as described above. Pellets were washed at 22°C in PDF supplemented with 0.5 mM BrdU and resuspended in PDF with 0.5 mM BrdU at 1 × 108/ml. Cells (0.5 × 108) were spread on one filter with the underpad soaked with 1.5 ml 0.5 mM BrdU in PDF. After 30 h of development, cells from one filter were harvested and used to make high-molecular-weight DNA in a single 150-μl agarose block. Likewise, to monitor DNA synthesis during germination and recovery in nutrient medium and during development, germinated spores were recovered in HL-5 medium supplemented with 0.5 mM BrdU and developed on filters with 0.5 mM BrdU, as described above. The DNA synthesis was compared with the results obtained with samples from vegetative cells growing in HL-5 plus BrdU for 8 h (one cell doubling).
Agarose blocks were processed to produce high-molecular-weight DNA as described previously (17) except that cellulase and hemicellulase were used to digest the cell walls of spores. Pulsed-field gel electrophoresis and Southern hybridization were carried out as described previously (31). Detection of BrdU was carried out with anti-BrdU antibodies conjugated to peroxidase by use of an ECL detection kit (Amersham). Multiple exposures of each autoradiograph were used to assess the linearity of the signals, and quantification was carried out using standard image analysis software. Values determined for the BrdU signals were normalized either to the total chromosomal DNA content by use of the Southern hybridization signal from a DirsI probe or to the estimated amount of mtDNA probe. The mtDNA probe was a PCR product corresponding to a 3.6-kb fragment at 9,991 to 13,579 bp of the mtDNA sequence (26). The amplifying primers were as follows: Mito_5′ AGT TTA GAC ACT GCT GG and Mito_3′ CTA AAA CGC ACA CCT TCT C. By comparison with the signal obtained from growing cells, we estimate that we could detect the labeling of >2% of any cellular DNA species.
RESULTS
Germinated spores develop into fruiting bodies.
It has been known for decades that dictyostelid amoebae that emerge from dormant spores can develop to form fruiting bodies without an intervening period of cell growth (4, 29, 32). Since we had recently shown that D. discoideum spores encapsulate as G1-phase cells, we revisited this topic to examine the association of the cell cycle phase and cell fate patterning and the potential role of cell cycle regulation during development.
In an initial test of the developmental capacity of freshly emergent amoebae, we found that heat-shocked spores germinated and produced apparently normal fruiting bodies when plated directly on nitrocellulose filters (Fig. 1A). The spores germinated within 6 h after plating, and the emergent amoebae formed mature fruiting bodies in spite of significant cell loss that was presumably due to cell lysis. The quality of development correlated with the cell density at which the spores were plated. At 3 × 107 cells/cm2, or 108 cells/cm2, cells started to aggregate 4 h after germination and formed fruiting bodies by 18 h with significant numbers of spores (Table 1). To obtain a more accurate sporulation rate, spores were allowed to germinate while shaking in phosphate buffer and the surviving amoebae were harvested, counted, and put on filters to develop. The cells started to aggregate after 5 h and formed fingers by 10 h and fruiting bodies by 20 h. The sporulation rate was the same as when spores were allowed to germinate directly on filters, but the quality of the spores as judged by germination efficiency was relatively poor (Table 1).
FIG. 1.

Spores germinate and develop without growth phase. (A) Spores were heat shocked to stimulate germination, immediately plated on filters, and developed into mounds (12 h) and fruiting bodies (24 h). (B) Cellular DNA content was measured at various times of development by propidium iodide (PI) staining and flow cytometry of freshly germinated spores or vegetative cells. The cellular DNA content is summarized by the peak value (±standard errors of the means [SEM]) of mononucleated cells in the cytometry profile.
TABLE 1.
Spore production and viability
| Cell category and germination conditions | % Spores produceda (mean ± SEM) | % Germination rate (mean ± SEM) |
|---|---|---|
| Spores | ||
| Germinated and developed on filters (cell density in no. of cells/cm2) | ||
| 107 | 6.8 ± 0.6 | NDb |
| 3 × 107 | 23 ± 5 | ND |
| 108 | 26 ± 3 | ND |
| Germinated in bufferc for 12 h and developed on filters (cell density, 3 × 107/cm2) | 27 ± 4 | 29 ± 1 |
| Germinated in mediumd for the indicated duration (h) and developed on filters (cell density, 3 × 107/cm2) | ||
| 8 | 22 ± 3 | ND |
| 12 | 36 ± 3 | 65 ± 2 |
| 16 | 49 ± 3 | ND |
| 20 | 71 ± 3 | ND |
| Vegetative cells (cell density/cm2) | ||
| 1 × 107 nuclei | 100 ± 12 | 79 ± 4 |
| 1 × 107 (sporulation rate based on cells at 20 h of germination) | 62 ± 8 | 79 ± 4 |
Spore numbers were determined after 30 h of development. For comparison, the spores produced by vegetative cells were set at 100%.
ND, not done.
10 mM phosphate buffer, pH 6.1.
HL-5 medium.
Vegetative cells are mostly in the G2 phase of the cell cycle, and during development they undergo two cell divisions before culmination: the cytokinesis of multinuclear cells in the first 6 h and the mitosis of prespore cells from 12 h to 20 h (2, 6, 43). These cell divisions cause successive drops in the total cellular DNA content of a developing population of cells that can be visualized by flow cytometry of whole cells (6). Using this method we could assess whether the amoebae that had freshly emerged from spores were likely to have maintained a G1 state during development on filters. We found that the average cellular DNA content of the freshly emergent amoebae remained constant, suggesting that they maintained their G1 state during development (Fig. 1B).
Development of cells from germinating spores after recovery in growth medium.
Germinating spores in buffer likely results in nutritionally compromised amoebae, and that may explain the low efficiency of sporulation that we observed. Thus, we sought conditions that would maintain emergent amoebae in a G1 state while allowing them to recover from spore dormancy to a nutritional status similar to that of vegetative cells. Spores that are heat shocked and allowed to germinate in nutrient medium enter S phase and begin to divide only after 18 to 20 h of shaken suspension (6). We replicated this experiment and confirmed the results in the present study (data not shown). We also found that cycB gene expression begins after between 16 and 20 h of recovery (Fig. 2A) . As expected for a mitotic cyclin gene like cycB, its expression correlates with the onset of mitosis in these germinating spores.
FIG. 2.
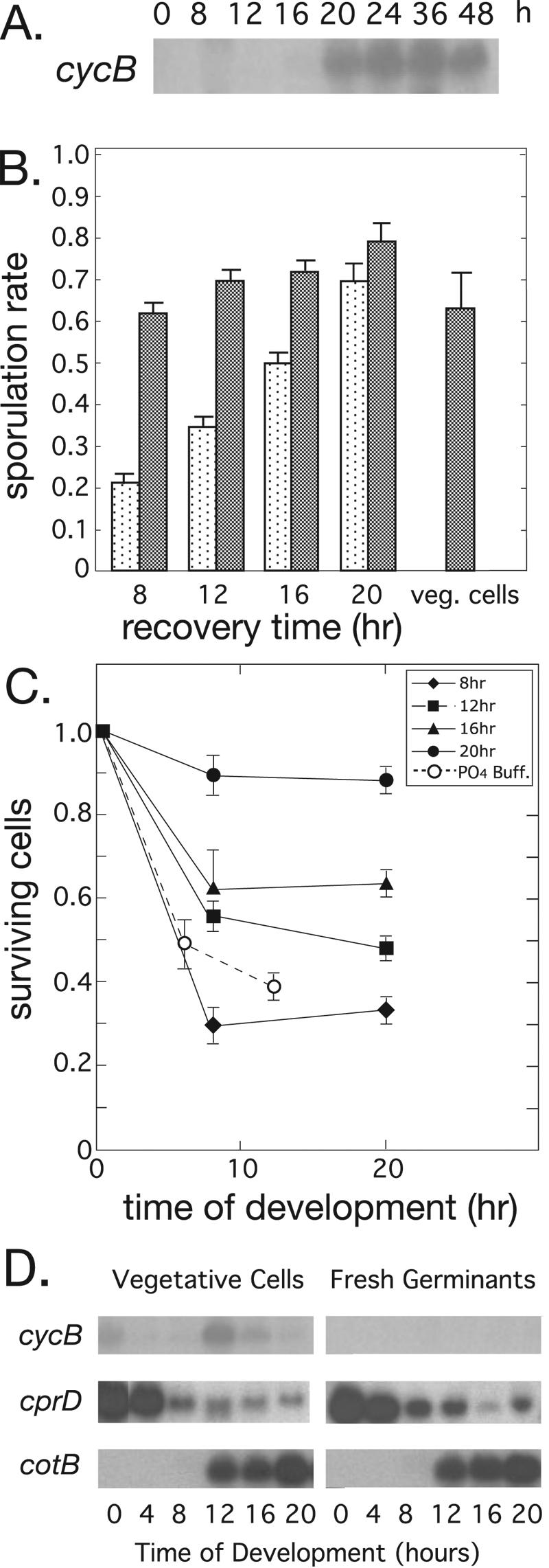
Development of emergent spores after germination and recovery in nutrient medium. Spores were heat shocked and suspended in nutrient medium to germinate and recover for various times, and the resulting amoebae were allowed to develop on filters. (A) Cyclin B gene (cycB) expression is shown during the recovery period by Northern blot hybridization. (B) The sporulation rates shown are 30-h spore totals divided by the initial number of cells plated (stippled bars) or divided by the number of cells present at 20 h of development (dark bars). Exponentially growing cells (veg. cells) were used as a comparison. The mean (±SEM) number of spores is shown for three determinations within a single experiment. (C) After the different recovery periods in nutrient medium, amoebae were plated on filters and counted (means ± SEM) at different times of development. Germination by heat shock and incubation in phosphate buffer for 12 h was used as a control. PO4 Buff., 10 mM potassium phosphate (pH 6.4). (D) Filter development of cells from spores that had been allowed to germinate and recover in nutrient medium for 16 h. Shown are the expression patterns determined by Northern blot hybridization for cycB; for cprD, the vegetative gene that encodes cysteine proteinase 4; and for cotB, which encodes a spore coat protein.
Using this procedure, we found that emergent amoebae could develop well, forming fruiting bodies in ∼24 h, independent of the time of recovery in growth medium. However, spore production increased significantly with increased recovery times (Fig. 2B). To determine whether cell viability could account for the increase in sporulation, we monitored cell number during development for cells allowed to recover for different times. There was significant cell loss in the first 8 h of development for every population tested (Fig. 2C). Since only the cells present at the time of culmination can form spores, the sporulation rates were normalized to the total number of cells present at 20 h of development. Surprisingly, the freshly emergent amoebae appeared to sporulate under all recovery conditions as efficiently as the control cells (Fig. 2B), although spore quality as judged by germination efficiency was a little lower (Table 1). This suggests that starved G1-phase cells have the same capacity to form spores as cells developing from a cycling G2 population. Consistent with the notion that these cells maintain a G1 state, we could detect no developmental expression of the cycB gene, while the regulatory patterns of the vegetative gene cprD and the prespore gene cotB were relatively normal (Fig. 2D). The expression of cycB normally rises just after starvation and again at the time of prespore cell division (18, 19).
No chromosome synthesis during spore germination, recovery, and development.
To ensure that the cells had remained in a G1 cell cycle state, we allowed spores to germinate, recover in medium for 16 h, and develop while we monitored the synthesis of cellular DNA by BrdU incorporation. We could detect no nuclear DNA synthesis after 16 h of recovery and 30 h of development, but significant mtDNA synthesis did occur (Fig. 3, Table 2). In the same experiment, G2 vegetative cells also displayed significant mtDNA synthesis but minimal synthesis of nuclear DNA, as previously reported (6, 31). The absence of detectable nuclear DNA synthesis during germination and recovery in medium is consistent with our previous results and confirms that the cells remain in a G1 state throughout the experiment (6). For simplicity, we refer to the freshly emergent amoebae that have recovered in growth medium for 16 h as “G1-phase cells” and to growing cells as “G2-phase cells.”
FIG. 3.
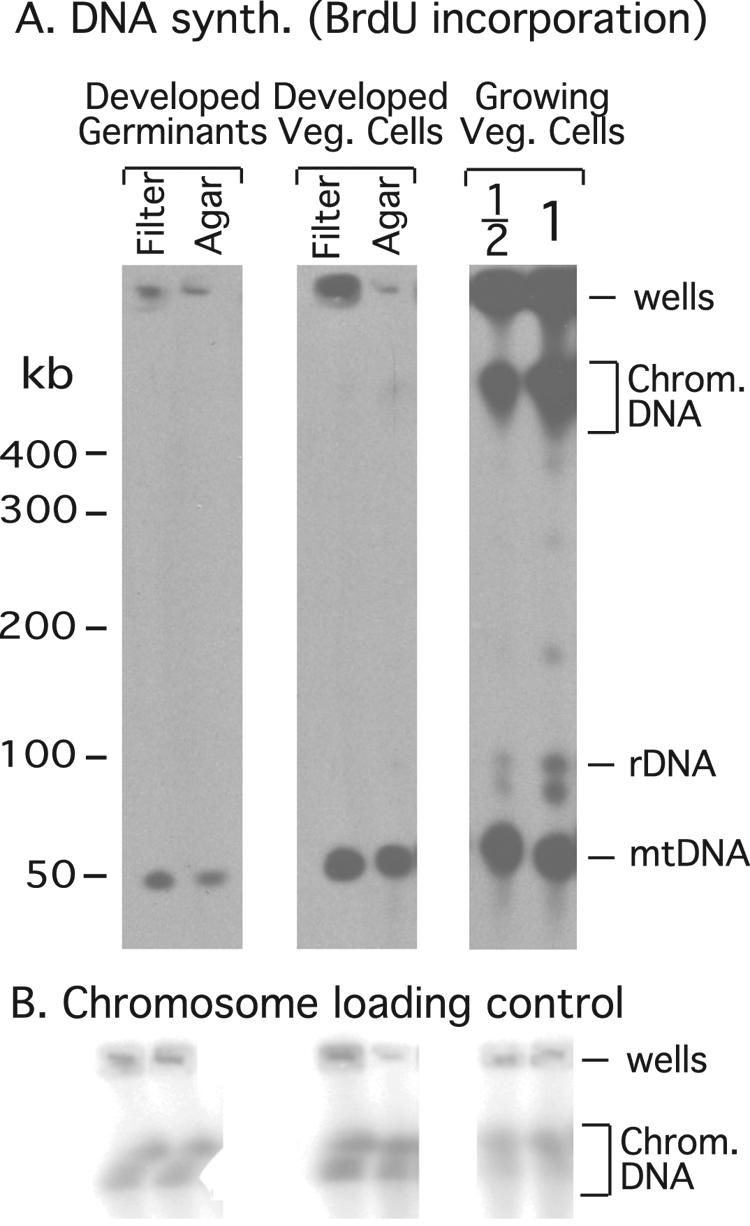
DNA synthesis during development. DNA synthesis was monitored by Southern transfer of cellular DNA after separation on standard pulsed-field gels, followed by BrdU immunostaining. (A) The amount of BrdU incorporation into chromosomal DNA (Chrom. DNA) and mtDNA is compared to the level of labeling observed in growing cells (Growing Veg. Cells) during one cell doubling. Germinants, spores that were allowed to germinate and recover for 16 h in nutrient medium and then allowed to develop on filters (or agar plates) for 36 h, all in the presence of BrdU. “Veg.” cells were allowed to develop on filters, or agar plates, in the presence of BrdU. “1/2” indicates that half of a standard sample block was loaded on the gel. rDNA, rDNA palindrome. (B) For normalization of the BrdU incorporation the amount of chromosomal DNA on the blot was determined by Southern blot hybridization with a chromosomal DirsI probe.
TABLE 2.
DNA synthesis during recovery from germination and during developmenta
| Sample | % Chromosomal DNA synthesis (mean ± SEM) | % mtDNA synthesis (mean ± SEM) |
|---|---|---|
| Growing cells in growth medium | 100 | 100 |
| Vegetative cells developed on filters | 2.3 ± 2.2 | 64 ± 9.1 |
| Vegetative cells developed on agar | 5.5 ± 5.2 | 64 ± 9.6 |
| Germinated spores developed on filters | <2.0 | 21 ± 4.7 |
| Germinated spores developed on agar | <2.0 | 13 ± 3.4 |
The DNA synthesis that occurs during development was quantified from the BrdU incorporation experiments whose results are shown in Fig. 3. The results are expressed as a percentage of the BrdU incorporation into vegetative cells that had undergone one cell doubling in growth medium. Spores were allowed to germinate, recover in growth medium, and develop, all in the presence of BrdU.
Developmental mtDNA synthesis in the absence of cell cycle progression.
During the development of G2-phase cells, mtDNA replication is observed mainly in prespore cells at about the time they undergo mitosis (6, 31). To determine whether the nuclear cell cycle impinges on mtDNA synthesis during development, we compared the BrdU incorporation into the mtDNA of G1- and G2-phase cells added at different stages of recovery and development (Fig. 4, Tables 2 and 3). Since we could not account for the variable retention of mtDNA in the wells of the pulsed-field gels, we estimated the amount of BrdU in the mtDNA band relative to the amount of mtDNA in that same band for each of the samples. This provides an estimate of the percentage of the mtDNA that was synthesized in each sample but provides no information on the total amount of mtDNA synthesized. We used the mtDNA synthesis observed in a G2 population during one cell doubling as a control. Assuming that all of the mtDNA is duplicated during every cell division and that mtDNA turnover is low during growth, the ratio of the BrdU incorporation signal to mtDNA hybridization signal in this control should represent the synthesis of ∼50% of the mtDNA.
FIG. 4.
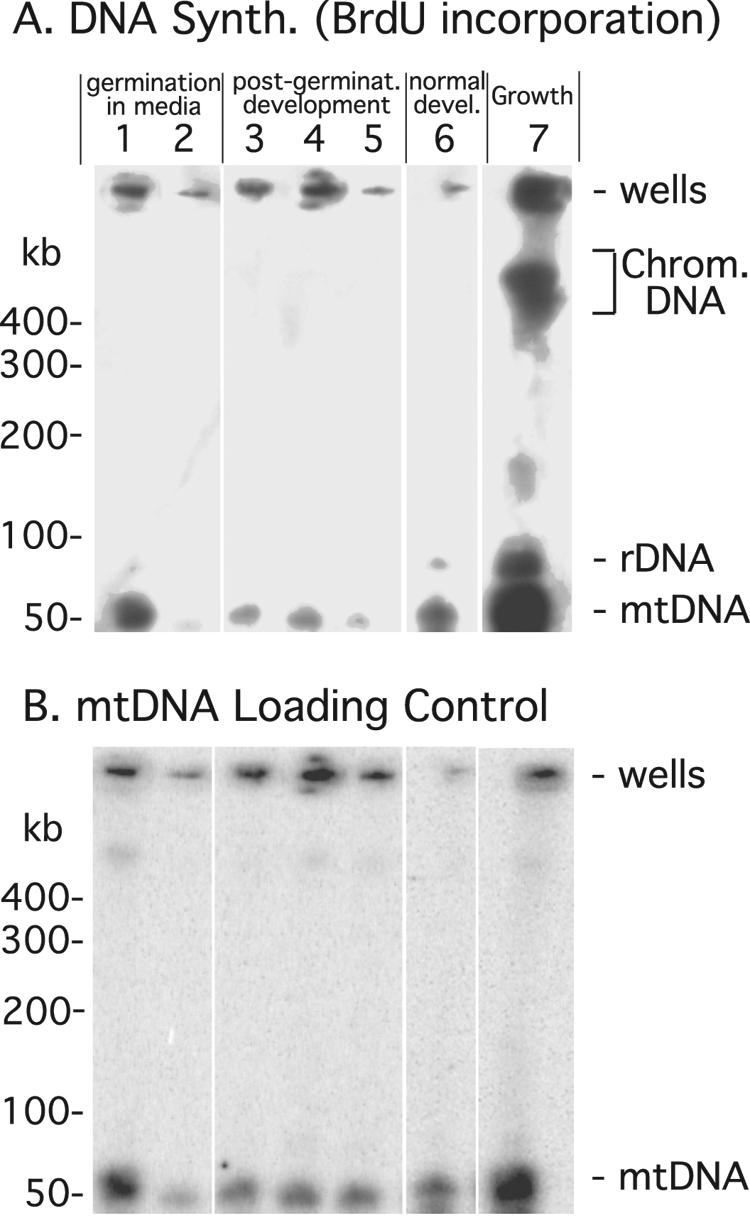
DNA synthesis in G1 cells during recovery and development. DNA synthesis was monitored by BrdU incorporation as described for Fig. 3. (A) BrdU incorporation in different samples. Lanes: 1, spores allowed to germinate and recover in growth medium for 16 h with BrdU; 2, cells allowed to recover in growth medium for 16 h with BrdU and then allowed to develop for 30 h without BrdU; 3, recovered cells with BrdU added upon initiation of development (0 to 30 h); 4, spores produced by cells treated as for sample 3; 5, recovered cells developing from 0 to 6 h of with BrdU and 6 to 30 h without BrdU; 6, vegetative cells developing with BrdU for 30 h; 7, vegetative cells grown with BrdU for 8 h (one cell doubling). rDNA, rDNA palindrome. (B) MtDNA detected by Southern blot hybridization.
TABLE 3.
mtDNA synthesis during germination and developmenta
| Expt | Cell category and treatment prior to harvesting (treatment duration) | mtDNA synthesis (% of mtDNA) |
|---|---|---|
| 1 | Vegetative cells grown in medium + BrdU for one cell doubling | 100 |
| 2 | Spores germinated and recovered in medium + BrdU (16 h) | 36 ± 9.8 |
| 3 | Spores germinated and recovered in medium + BrdU (16 h) and developed − BrdU (30 h) | 18 ± 10 |
| 4 | Vegetative cells grown in medium − BrdU (16 h) and developed + BrdU (30 h) | 70 ± 8.5 |
| 5 | Spores germinated and recovered in medium − BrdU (16 h) and developed + BrdU (30 h) | 26 ± 9.5 |
| 6 | Spores germinated and recovered in medium − BrdU (16 h) and developed + BrdU (30 h); spores only were harvested | 34 ± 7.0 |
| 7 | Vegetative cells grown in medium − BrdU (16 h) and developed − BrdU (0-6 h), then + BrdU (6-30 h) | 23 ± 6.0 |
| 8 | Spores germinated and recovered in medium − BrdU (16 h) and developed − BrdU (0-6 h), then + BrdU (6-30 h) | 13 ± 7.0 |
The mtDNA synthesis of developing cells was quantified from BrdU incorporation measured in experiments as described for Fig. 4. The BrdU incorporation observed in each band is shown relative to that observed during one cell cycle of G2 cells growing in liquid medium and normalized to the total amount of mtDNA in that band. Except for the experiment whose results are shown in row 6, all cells were harvested for processing.
During germination and recovery in growth medium, G1-phase cells synthesize about one-third of their mtDNA relative to G2-phase cells (Fig. 4, lane 1; Table 2; and Table 3, row 2). When cells labeled similarly were then allowed to develop without BrdU the percentage of labeled mtDNA dropped by about half, presumably due to mtDNA turnover and dilution by newly synthesized mtDNA during development (Fig. 4, compare lanes 1 to 3; Table 3, compare rows 2, 3, and 5). During development, G1-phase cells synthesize about one-third as much mtDNA as G2-phase cells (compare lanes 3 and 6 of Fig. 4; compare rows 4 and 5 of Table 3). About half of this synthesis occurs after 6 h of development (Table 3; compare rows 5 and 8). It appears that most of the mtDNA synthesis in G1-phase cells occurs in prespore cells, as observed previously, since the BrdU incorporation into purified spores is the expected ∼1.3-fold higher than that seen in the sample made from all cells (Table 3, rows 5 and 6) (6, 31). These results obtained with G1-phase cells indicate that developmental mtDNA synthesis is not strictly linked to the mitosis of prespore cells.
Regulation of cell type proportions in developing G1 cells.
Most developing organisms have robust cell type proportioning mechanisms and the ability to reestablish proper cell type proportions when their tissues are compromised by environmental or mechanical insults. During D. discoideum development, cell proportions are also dynamically maintained such that physically disrupted slugs can reestablish the proper patterning and ratio of cell types (28, 33). Pure populations of G1-phase cells produced prestalk and prespore cells with the proper location in slugs and fruiting bodies, as judged by the expression patterns of the prespore gene cotB and the prestalk gene ecmA (Fig. 5A). However, quantification of the cell types by flow cytometry revealed that G1-phase cells produce more prespore cells relative to a developing G2-phase population (Table 4). This proportioning difference is greater than the raw percentages suggest, since the proportions of developing G2-phase cells are produced by ∼60% of the cells differentiating into prespore cells followed by mitosis the of those prespore cells, whereas the G1-phase cells produced ∼85% prespore cells without cell division (6).
FIG. 5.

Regulation of cell type proportioning in migrating slugs. (A) Normal spatial patterning of the major cell types was observed in the slugs and fruiting bodies formed from G1-phase cells. Pairs of bright-field and fluorescence images indicate the presence of ecmA/GFP-positive prestalk cells located in the anterior of the slugs (on the right in all images) and in the stalk and upper and lower cups of the fruiting body (four left panels). The cotB/GFP-positive prespore cells and spores are patterned normally in slugs and fruiting bodies (four right panels). (B) The posterior half of a slug produced by cotB/GFP-marked G1-phase cells was dissected and removed from the agar, and the remaining cells were allowed to reform a slug and migrate for 12 h, demonstrating the ability of cells to reestablish proper proportions and patterning; fluorescent image (right panel) of the bright-field image (left panel). (C) The flow cytometry profiles of the anteriors of cotB/GFP-marked slugs produced from G1 cells that continued to migrate for 12 h and then disaggregated (solid line) or that were disaggregated immediately (dotted line) and subjected to flow cytometry. arb., arbitrary.
TABLE 4.
Regulation of the prespore cell fate in G1- and G2-phase cellsa
| Phase of Ax4[cotB/GFP] cells | % GFP-negative/GFP-positive slug cells | % GFP-negative/ GFP-positive cells from slug anteriors | % GFP-negative/ GFP-positive cells from slug anteriors after 12 h on agar |
|---|---|---|---|
| G2 | 25/75 | 44/56 | 23/77 |
| G1 | 15/85 | 30/70 | 16/84 |
To generate “G1” cells, spores were allowed to germinate and recover in growth medium for 16 h and were then washed and plated on agar. Slugs, or dissected slug anteriors, were disaggregated, and GFP-negative and -positive cell results were determined by flow cytometry. The results from a single determination for a representative experiment are shown.
We next tested the ability of developing G1-phase cells to regulate cell type proportions in response to a perturbation. We dissected slugs into two halves and monitored the prespore cells in the slug anteriors visually and by flow cytometry. Immediately after dissection, the anterior halves of G1- and G2-phase slugs had the expected percentage of prespore cells (Table 4). After the dissected anteriors were allowed to reform slugs and migrate for 12 h the G1-phase cells appeared to establish a normal prespore cell pattern (Fig. 5B). Interestingly, the G1-phase slug anteriors reestablished the proportion characteristic of developing G1-phase cells, as judged by flow cytometry (Table 4). An example of this experiment is shown in Fig. 5C. When prespore cells were examined by use of the cotB/GFP marker, about one-third of the prespore cells appear to have about 4-times-lower green fluorescent protein (GFP) expression than the rest (unpublished observations). It is this population of prespore cells that increases in the regulating G1-cell slug anteriors, apparently at the expense of the GFP-negative prestalk cell population.
A prespore cell fate bias in G1-phase cells.
Since G1-phase cells develop relatively normally as judged by gene expression patterns, their morphogenesis, and their sporulation rate, we attempted to detect more subtle effects of cell cycle status on cell fate determination and patterning by examining G1/G2 cell chimeras. We examined fate patterning in mixtures of G1-phase and G2-phase cells, where one or the other population was marked with a ubiquitously expressed act15/GFP reporter construct. At the slug stage, marked G1-phase cells were mainly observed in the prespore regions of slugs and almost all of these went on to form spores within sori (Fig. 6A and Table 5). The G1 cells appeared to be excluded from the stalk cell population. Conversely, the G2-phase cells were found almost exclusively in the anterior prestalk regions of slugs and they were underrepresented in the spore population (Fig. 6B and Table 5). In control experiments in which marked G1 cells were mixed with unmarked G1 cells and marked G2 cells were mixed with unmarked G2 cells, the marked cells showed no prestalk-prespore preference (data not shown).
FIG. 6.

Prespore cell fate bias in G1-phase cells. Wild-type cells (AX4) marked with a ubiquitously expressed GFP reporter gene (act15/GFP) were mixed at a 1:9 ratio with unlabeled cells and allowed to develop on agar into slugs and fruiting bodies. Fluorescent images (upper panels) and bright-field images (lower panels) of slugs (left panels) and a fruiting body (right panels) are shown in both A and B. (A) G1-phase, GFP-labeled cells mixed with unlabeled, G2-phase cells. (B) G2-phase, GFP-labeled cells mixed with G1-phase, unlabeled cells. The white circles indicate the slug anteriors.
TABLE 5.
Prespore cell fate bias of G1-phase cells in chimerasa
| Sample (marked cell:unmarked cell ratio, 1:9) | % GFP-positive slug cells (mean ± SEM) | % GFP-positive spores (mean ± SEM) |
|---|---|---|
| Ax4[act15/GFP] G1 cells + Ax4 G2 cells | 11 ± 2.4 | 10 ± 1.5 |
| Ax4[act15/GFP] G2 cells + Ax4 G1 cells | 12 ± 2.2 | 1.2 ± 0.6 |
G1-phase cells and G2-phase cells were prepared for development, mixed with G2 cells, and plated on agar. Ten percent of the GFP-marked cells were mixed with unmarked cells, and the mixture was plated on agar. Slugs or fruiting bodies (sori) were disaggregated, and GFP-positive cells (or spores) were determined by flow cytometry. The values are the means ± SEM of four determinations within a single experiment.
DISCUSSION
We have examined the role of the cell cycle in D. discoideum development using a new method to generate G1-phase cells. We exploited our previous findings that spores encapsulate in the G1 phase of the cell cycle and remain in G1 for up to 20 h after they germinate in nutrient medium (6). The developmental capacity of the cells improves during 16 h of incubation in nutrient medium, allowing them to recover much of their growth-phase levels of mtDNA, which presumably results in improved energy production. By preparing freshly hatched amoebae in this way, we were able to examine the development of G1-phase cells. We infer that the cells remain in G1, since we could detect no mitosis and no nuclear DNA synthesis in these cells while they developed. The lack of cyclin B expression also confirms that the developing G1-phase populations remain in G1. Cyclin B is normally observed at times of mitotic activity in developing G2-phase populations (19). Thus, spore germination is a relatively simple way to obtain a pure G1-phase cell population and this method may be useful for future studies.
G1-phase cells are capable of developing into fruiting bodies with spore production rates comparable to those of G2-phase cells. The G1-phase cells have a relatively normal body pattern at the slug stage, although they produce a higher proportion of prespore cells (see below). The developmental regulation characteristics of cprD and cotB are indistinguishable in G1 and G2 cells. The G1-phase cells are also capable of regulating cell type proportions in bisected slugs. These results imply that biases imposed by the cell cycle at the time of starvation are not strictly required for cell type proportioning. Indeed, the altered cell type proportions that we observe in G1-phase cells suggest that they are using an alternate mechanism. However, in the absence of initial cell cycle influences, G1-phase D. discoideum cells may still rely on factors thought to influence cell fate specification, such as population variations in the cytosolic Ca2+ concentration or pH (reviewed in references 3 and 12). Although the stalk-inducing hexanophenone DIF-1 is not strictly required for prestalk cell differentiation, it has been proposed that some aspects of cell fate bias imposed by the population heterogeneity at the time of starvation are mediated by differential sensitivity to DIF-1 (36). Any of these factors (Ca2+, pH, or DIF-1) may still differ sufficiently within a population of G1-phase cells to bias cell fate specification and affect the robust proportioning that we observe.
Significant mtDNA synthesis normally occurs in prespore cells with a timing that is roughly coincident with the mitosis of prespore cells midway through development (6, 31). Shaulsky and Loomis proposed that mtDNA synthesis is an important aspect of prespore cell differentiation (31). An extreme interpretation of their hypothesis is that mtDNA replication is required for prespore differentiation, on the basis of the idea that mitochondrial replication would produce more robust cells, allowing them to outcompete other cells for a position in the prespore population. For example, having more mitochondria might allow prespore cells to resist the potential uncoupling of oxidative phosphorylation by DIF-1. Alternatively, mtDNA synthesis may be a property of prespore cells, reflecting their need to supply daughter cells with mitochondria. We attempted to distinguish between these hypotheses by comparing the mtDNA synthesis data for developing G1- and G2-phase populations. We observed that developing G1-phase cells synthesize about one-third as much mtDNA as G2-phase cells. This synthesis cannot be explained by the need to supply daughter prespore cells with mitochondria, since G1-phase cells do not divide. Rather, these results suggest that mtDNA synthesis is an intrinsic property of prespore cell differentiation, but they leave open the issue of whether it is a required event.
Even though our results imply that cell fate bias that is imposed by the cell cycle cell is not required for development, the cell cycle may still play a role in the regulation of cell type proportions. In Drosophila development, for example, the plasticity of imaginal disk cells during trans-determination appears to depend on cells achieving a particular cell cycle state and on cell size (35). We examined this issue in two ways, by looking at cell fate regulation in bisected slugs and at cell fate tendencies of G1 and G2 cells in chimeras. Bisected slugs made from G1-phase cells appear to regulate proportions normally, so there does not appear to be a role for the cell cycle in that form of regulation. However, when G1 cells and G2 cells are mixed G1-phase cells preferentially become prespore cells and spores, while G2-phase cells are excluded from the prespore population and form mostly stalk cells. This result would seem to suggest that cell cycle status has a profound effect on cell fate determination. At a minimum it suggests that G1-phase cells have an advantage in executing the prespore differentiation pathway. Certainly, all prespore cells end up in G1 prior to encapsulation, and whether this state is achieved just after a cell differentiates as prespore or just prior to spore encapsulation, the G1 state follows the G2 state. Our previous results suggest that the G2-M-G1 transition occurs well after a cell becomes a prespore cell, since we could detect a substantial population of cotB/GFP-expressing prespore cells in the G2 phase (6). If G1 phase represents a distinct step in the commitment to sporulate, it could be that G1-phase cells are able to attain that state much earlier than their G2 competitors. In this simplistic scenario, sporulation could be viewed as a race to the G1 state and only in the contrived situation of an aggregate with a substantial population of G1 cells would the disadvantage of G2 cells become apparent. Evidence for the idea that G1 status represents a commitment to sporulate is scant. When prestalk cells are genetically ablated during slug migration by the expression of the ricin toxin under control of the ecmA promoter, prespore cells do not convert into prestalk cells (30). Since migrating slug cells are in G1, these experiments could be considered to support this idea. Further insight into this issue will require more information about the regulatory network that controls encapsulation and how G1 status might impinge on its execution.
Our results showing that G1 cells overproduce prespore cells in pure populations (and are biased to produce prespore cells in chimeras) appear to contradict all previous studies on the cell cycle and cell fate bias (39). Other than the uninformative rationalization that these are novel experiments, we can offer only one explanation. All previous experiment utilized axenic cells that have a high percentage of binucleated cells, like the Ax4 cell line used here that has ∼20% binucleated cells (43, 44). Since tetranucleated cells are present in only about 1% of the population, a substantial portion of cells undergoing cell division at any one time are actually G1-phase binucleated cells undergoing cytokinesis. Thus, many cells that are defined as being in late G2 phase because they divide 1 to 2 h later are actually G1-phase cells. Matters could be even more skewed if all cells passed through a binucleated stage before mitosis, because in this situation nearly all “late G2 cells” would actually be in G1. If true, such an offset in cell cycle timing would actually place the Maeda's PS point at G1 of the cell cycle, bringing our results in line with previous work.
We are still left with this question: why did D. discoideum evolve the capacity to develop from G1-phase cells? In the absence of any specific requirement for G2 phase or mitosis in regulating development, the relatively normal development of G1-phase cells might by viewed as a curious happenstance of laboratory manipulation. Alternatively, the development of G1-phase cells may represent a survival mechanism that could serve to correct mass premature germination. Spores can be triggered to germinate by heat, moisture, and nutritional cues, which cannot be perfect reporters of new food sources in the environment (7). The ability of G1 cells to develop gives the species a kind of salvage pathway that could rescue a potential future population after they had acted on a false germination signal. There may be other situations that result in the accumulation of vegetative cells in G1, and the ability of these populations to carry out development and generate spores would promote the long-term survival of the species.
Acknowledgments
We thank Gad Shaulsky and Bill Loomis for helpful discussions during the course of this work and Dorothy Lewis and Jeff Scott from the Flow Cytometry laboratory in the Immunology Department at Baylor College of Medicine for expert advice and assistance.
This work was supported by U.S. Public Heath Service grant GM52359 from the National Institutes of Health.
REFERENCES
- 1.Araki, T., T. Abe, J. G. Williams, and Y. Maeda. 1997. Symmetry breaking in Dictyostelium morphogenesis: evidence that a combination of cell cycle stage and positional information dictates cell fate. Dev. Biol. 192:645-648. [DOI] [PubMed] [Google Scholar]
- 2.Atryzek, V. 1976. Alteration in timing of cell differentiation resulting from cell interactions during development of the cellular slime mold, Dictyostelium discoideum. Dev. Biol. 50:489-501. [DOI] [PubMed] [Google Scholar]
- 3.Azhar, M., P. K. Kennady, G. Pande, M. Espiritu, W. Holloman, D. Brazill, R. H. Gomer, and V. Nanjundiah. 2001. Cell cycle phase, cellular Ca2+ and development in Dictyostelium discoideum. Int. J. Dev. Biol. 45:405-414. [PubMed] [Google Scholar]
- 4.Bonner, J. T., B. D. Joyner, A. A. Moore, H. B. Suthers, and J. A. Swanson. 1985. Successive asexual life cycles of starved amoebae in the cellular slime mold Dictyostelium mucoroides var. stoloniferum. J. Cell Sci. 77:19-26. [DOI] [PubMed] [Google Scholar]
- 5.Chen, G. 2003. The growth-development transition and tissue-specific cell cycle control in Dictyostelium discoideum. Ph.D. dissertation. Baylor College of Medicine, Houston, Tex.
- 6.Chen, G., G. Shaulsky, and A. Kuspa. 2004. Tissue-specific G1-phase cell-cycle arrest prior to terminal differentiation in Dictyostelium. Development 131:2619-2630. [DOI] [PubMed] [Google Scholar]
- 7.Cotter, D. A., D. C. Mahadeo, D. N. Cervi, Y. Kishi, K. Gale, T. Sands, and M. Sameshima. 2000. Environmental regulation of pathways controlling sporulation, dormancy and germination utilizes bacterial-like signaling complexes in Dictyostelium discoideum. Protist 151:111-126. [DOI] [PubMed] [Google Scholar]
- 8.Durston, A. J., C. J. Weijer, J. F. Jongkind, A. Verkerk, A. Timmermans, and W. Te Kulve. 1984. A flow fluorimetric analysis of the cell cycle during growth and differentiation in Dictyostelium discoideum. Wilhelm Roux Arch. Dev. Biol. 194:18-24. [DOI] [PubMed] [Google Scholar]
- 9.Gomer, R. H., and R. R. Ammann. 1996. A cell-cycle phase-associated cell-type choice mechanism monitors the cell cycle rather than using an independent timer. Dev. Biol. 174:82-91. [DOI] [PubMed] [Google Scholar]
- 10.Gomer, R. H., and R. A. Firtel. 1987. Cell-autonomous determination of cell-type choice in Dictyostelium development by cell-cycle phase. Science 237:758-762. [DOI] [PubMed] [Google Scholar]
- 11.Good, J. R., M. Cabral, S. Sharma, J. Yang, N. Van Driessche, C. A. Shaw, G. Shaulsky, and A. Kuspa. 2003. TagA, a putative serine protease/ABC transporter of Dictyostelium that is required for cell fate determination at the onset of development. Development 130:2953-2965. [DOI] [PubMed] [Google Scholar]
- 12.Gross, J. D., J. Bradbury, R. R. Kay, and M. J. Peacey. 1983. Intracellular pH and the control of cell differentiation in Dictyostelium discoideum. Nature 303:244-245. [DOI] [PubMed] [Google Scholar]
- 13.Iranfar, N., D. Fuller, R. Sasik, T. Hwa, M. Laub, and W. F. Loomis. 2001. Expression patterns of cell-type-specific genes in Dictyostelium. Mol. Biol. Cell 12:2590-2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kessin, R. H. 2001. Dictyostelium—evolution, cell biology, and the development of multicellularity, p. xiv and p. 294. Cambridge University Press, Cambridge, United Kingdom.
- 15.Knecht, D. A., S. M. Cohen, W. F. Loomis, and H. F. Lodish. 1986. Developmental regulation of Dictyostelium discoideum actin gene fusions carried on low-copy and high-copy transformation vectors. Mol. Cell. Biol. 6:3973-3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuspa, A., T. Dingermann, and W. Nellen. 1995. Analysis of gene function in Dictyostelium. Experientia 51:1116-1123. [DOI] [PubMed] [Google Scholar]
- 17.Kuspa, A., D. Maghakian, P. Bergesch, and W. F. Loomis. 1992. Physical mapping of genes to specific chromosomes in Dictyostelium discoideum. Genomics 13:49-61. [DOI] [PubMed] [Google Scholar]
- 18.Luo, Q., C. Michaelis, and G. Weeks. 1994. Overexpression of a truncated cyclin B gene arrests Dictyostelium cell division during mitosis. J. Cell Sci. 107:3105-3114. [DOI] [PubMed] [Google Scholar]
- 19.Luo, Q., C. Michaelis, and G. Weeks. 1995. Cyclin B and Cdc2 expression and Cd2 kinase activity during Dictyostelium differentiation. DNA Cell Biol. 14:901-908. [DOI] [PubMed] [Google Scholar]
- 20.MacWilliams, H., P. Gaudet, H. Deichsel, C. Bonfils, and A. Tsang. 2001. Biphasic expression of rnrB in Dictyostelium discoideum suggests a direct relationship between cell cycle control and cell differentiation. Differentiation 67:12-24. [DOI] [PubMed] [Google Scholar]
- 21.Maeda, Y. 1993. Pattern formation in a cell-cycle dependent manner during the development of Dictyostelium discoideum. Dev. Growth Differ. 35:609-616. [DOI] [PubMed] [Google Scholar]
- 22.Maeda, Y. 1997. Cellular and molecular mechanisms of the transition from growth to differentiation in Dictyostelium cells, p. 207-218. In Y. Maeda, K. Inouye, and I. Takeuchi (ed.), Dictyostelium—a model system for cell and developmental biology. Universal Academy Press, Tokyo, Japan.
- 23.Maeda, Y., T. Ohmori, T. Abe, F. Abe, and A. Amagai. 1989. Transition of starving Dictyostelium cells to differentiation phase at a particular position of the cell cycle. Differentiation 41:169-175. [DOI] [PubMed] [Google Scholar]
- 24.Mann, S. K. O., P. N. Devreotes, S. Eliott, K. Jermyn, A. Kuspa, M. Fechheimer, R. Furukawa, C. A. Parent, J. Segall, G. Shaulsky, P. H. Vardy, J. Williams, K. L. Williams, and R. A. Firtel. 1994. Cell biological, molecular genetic, and biochemical methods to examine Dictyostelium, p. 412-451. In J. E. Celis (ed.), Cell biology—a laboratory handbook. Academic Press, San Diego, Calif.
- 25.McDonald, S. A., and A. J. Durston. 1984. The cell cycle and sorting behaviour in Dictyostelium discoideum. J. Cell Sci. 66:195-204. [DOI] [PubMed] [Google Scholar]
- 26.Ogawa, S., R. Yoshino, K. Angata, M. Iwamoto, M. Pi, K. Kuroe, K. Matsuo, T. Morio, H. Urushihara, K. Yanagisawa, and Y. Tanaka. 2000. The mitochondrial DNA of Dictyostelium discoideum: complete sequence, gene content and genome organization. Mol. Gen. Genet. 263:514-519. [DOI] [PubMed] [Google Scholar]
- 27.Ohmori, R., and Y. Maeda. 1987. The developmental fate of Dictyostelium discoideum cells depends greatly on the cell-cycle position at the onset of starvation. Cell Differ. 22:11-18. [DOI] [PubMed] [Google Scholar]
- 28.Raper, K. B. 1940. Pseudoplasmodium formation and organization in Dictyostelium discoideum. J. Elisha Mitchell Sci. Soc. 56:241-282. [Google Scholar]
- 29.Russell, G. K., and J. T. Bonner. 1960. A note on spore germination in the cellular slime mold Dictyostelium mucoroides. Bull. Torrey Bot. Club. 87:187-191. [Google Scholar]
- 30.Shaulsky, G., and W. F. Loomis. 1993. Cell type regulation in response to expression of ricin-A in Dictyostelium. Dev. Biol. 160:85-98. [DOI] [PubMed] [Google Scholar]
- 31.Shaulsky, G., and W. F. Loomis. 1995. Mitochondrial DNA replication but no nuclear DNA replication during development of Dictyostelium. Proc. Natl. Acad. Sci. USA 92:5660-5663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Snyder, H. M., III, and C. Ceccarini. 1966. Interspecific spore inhibition in the cellular slime moulds. Nature 209:1152. [Google Scholar]
- 33.Sternfeld, J., and C. N. David. 1981. Cell sorting during pattern formation in Dictyostelium. Differentiation 20:10-21. [Google Scholar]
- 34.Sussman, M. 1987. Cultivation and synchronous morphogenesis of Dictyostelium under controlled experimental conditions, p. 9-29. In J. A. Spudich (ed.), Methods in cell biology. Academic Press, Orlando, Fla. [DOI] [PubMed]
- 35.Sustar, A., and G. Schubiger. 2005. A transient cell cycle shift in Drosophila imaginal disc cells precedes multipotency. Cell 120:383-393. [DOI] [PubMed] [Google Scholar]
- 36.Thompson, C. R., and R. R. Kay. 2000. Cell-fate choice in Dictyostelium: intrinsic biases modulate sensitivity to DIF signaling. Dev. Biol. 227:56-64. [DOI] [PubMed] [Google Scholar]
- 37.Thompson, C. R. L., and R. R. Kay. 2000. The role of DIF-1 signaling in Dictyostelium development. Mol. Cell 6:1509-1514. [DOI] [PubMed] [Google Scholar]
- 38.VanDriessche, N., C. Shaw, M. Katoh, T. Morio, R. Sucgang, M. Ibarra, H. Kuwayama, T. Saito, H. Urushihara, M. Maeda, I. Takeuchi, H. Ochiai, W. Eaton, J. Tollett, J. Halter, A. Kuspa, Y. Tanaka, and G. Shaulsky. 2002. A transcriptional profile of multicellular development in Dictyostelium discoideum. Development 129:1543-1552. [DOI] [PubMed] [Google Scholar]
- 39.Weeks, G., and C. J. Weijer. 1994. The Dictyostelium cell cycle and its relationship to differentiation. FEMS Microbiol. Lett. 124:123-130. [DOI] [PubMed] [Google Scholar]
- 40.Weijer, C. J., G. Duschl, and C. N. David. 1984. Dependence of cell-type proportioning and sorting on cell cycle phase in Dictyostelium discoideum. J. Cell Sci. 70:133-145. [DOI] [PubMed] [Google Scholar]
- 41.Weijer, C. J., G. Duschl, and C. N. David. 1984. A revision of the Dictyostelium discoideum cell cycle. J. Cell Sci. 70:111-131. [DOI] [PubMed] [Google Scholar]
- 42.Wood, S. A., R. R. Ammann, D. A. Brock, L. Li, T. Spann, and R. H. Gomer. 1996. RtoA links initial cell type choice to the cell cycle in Dictyostelium. Development 122:3677-3685. [DOI] [PubMed] [Google Scholar]
- 43.Zada-Hames, I. M., and J. M. Ashworth. 1978. The cell cycle and its relationship to development in Dictyostelium discoideum. Dev. Biol. 63:307-320. [DOI] [PubMed] [Google Scholar]
- 44.Zada-Hames, I. M., and J. M. Ashworth. 1978. The cell cycle during the vegetative stage of Dictyostelium discoideum and its response to temperature change. J. Cell Sci. 32:1-20. [DOI] [PubMed] [Google Scholar]
- 45.Zimmerman, W., and C. J. Weijer. 1993. Analysis of cell cycle progression during the development of Dictyostelium and its relationship to differentiation. Dev. Biol. 160:178-185. [DOI] [PubMed] [Google Scholar]