

Abstract
Amylomaltase from Thermus aquaticus catalyzes intramolecular transglycosylation of α-1,4 glucans to produce cyclic α-1,4 glucans (cycloamyloses) with degrees of polymerization of 22 and higher. Although the amylomaltase mainly catalyzes the transglycosylation reaction, it also has weak hydrolytic activity, which results in a reduction in the yield of the cycloamyloses. In order to obtain amylomaltase with less hydrolytic activity, random mutagenesis was perfromed for the enzyme gene. Tyr54 (Y54) was identified as the amino acid involved in the hydrolytic activity of the enzyme. When Y54 was replaced with all other amino acids by site-directed mutagenesis, the hydrolytic activities of the mutated enzymes were drastically altered. The hydrolytic activities of the Y54G, Y54P, Y54T, and Y54W mutated enzymes were remarkably reduced compared with that of the wild-type enzyme, while those of the Y54F and Y54K mutated enzymes were similar to that of the wild-type enzyme. Introducing an amino acid replacement at Y54 also significantly affected the cyclization activity of the amylomaltase. The Y54A, Y54L, Y54R, and Y54S mutated enzymes exhibited cyclization activity that was approximately twofold higher than that of the wild-type enzyme. When the Y54G mutated enzyme was employed for cycloamylose production, the yield of cycloamyloses was more than 90%, and there was no decrease until the end of the reaction.
Cyclodextrins are well-known α-1,4 glucans with degrees of polymerization (DPs) ranging from 6 to 8, which are produced by the cyclization reaction catalyzed by cyclodextrin glycosyltransferase (CGTase) (EC 2.4.1.19) and have been used in various industries (2, 5, 15, 16). It was previously reported that larger cyclic α-1,4 glucans, cycloamyloses, with DPs ranging from 17 to several hundred, were produced by potato disproportionating enzyme (D-enzyme) (EC 2.4.1.25) (18). Gessler et al. (4) reported the structure of a cycloamylose with a DP of 26, which is not a doughnut shape like conventional cyclodextrins but has a single helical conformation with a central cavity. Cycloamyloses are highly soluble in water and also incorporate various molecules, such as an alcohol or a fatty acid, and form inclusion complexes (1, 7). Therefore, various applications of cycloamyloses can be expected in the pharmaceutical, cosmetic, food, toiletry, agricultural, and chemical industries. It was recently reported that cycloamylose functions as an artificial chaperone to refold denatured protein into an active form (9).
We have reported previously that cycloamylose can be synthesized by several enzymes, including CGTase (21), potato D-enzyme (18), yeast glycogen debranching enzyme (EC 3.2.1.33/EC 2.4.1.25) (26), and Thermus aquaticus amylomaltase (EC 2.4.1.25) (20). These enzymes, which belong to the 4-α-glucanotransferase group defined by Takaha and Smith (17), catalyze the glucan chain transfer expressed by the following equation: (α-1,4 glucan)m + (α-1,4 glucan)n ⇄ (α-1,4 glucan)m-x + (α-1,4 glucan)n+x.
This action is the intermolecular glucan transfer reaction and is often called the disproportionation reaction. Most 4-α-glucanotransferases also catalyze an intramolecular glucan transfer reaction (cyclization reaction) in a single linear glucan molecule to create a cyclic glucan product, as follows: (α-1,4 glucan)n ⇄ cyclic (α-1,4 glucan)x + (α-1,4 glucan)n-x.
Despite the similarities in the reactions catalyzed, the smallest DP of cycloamylose produced with 4-α-glucanotransferases depends on the enzymes used. The potato D-enzyme, yeast glycogen debranching enzyme, and T. aquaticus amylomaltase produce cycloamyloses with lowest DPs of 17 (17), 11 (26), and 22 (17), respectively. T. aquaticus amylomaltase is particularly useful for industrial production of cycloamyloses, since it exhibits high thermal stability and produces higher levels of cycloamylose (20). However, this enzyme has weak but significant hydrolytic activity along with its major transglycosylation activity. Therefore, the yield of cycloamylose at the end of the reaction is only about 50 to 60% of the maximum yield. In order to improve the properties of T. aquaticus amylomaltase for efficient production of cycloamyloses, we introduced random and saturation mutations into the gene encoding this enzyme.
MATERIALS AND METHODS
Chemicals.
Synthetic amylose AS-30 and AS-110 (average molecular masses, 30 kDa and 110 kDa, respectively) were purchased from Nakano Vinegar Co. (Aichi, Japan). Maltotriose was purchased from Hayashibara Biochemical Laboratories Inc. (Okayama, Japan). Rhizopus sp. glucoamylase was purchased from Toyobo Co., Ltd. (Osaka, Japan), and porcine pancreas α-amylase was purchased from Sigma. A cycloamylose mixture with DPs ranging from 22 to about 50 was synthesized by using T. aquaticus amylomaltase. Unless otherwise specified, all chemicals were purchased from Wako Pure Chemical Industries Ltd. (Osaka, Japan).
Assays of enzyme activity.
The enzyme activities were determined by the following methods. The values for activities are the means of three independent assays.
Hydrolytic activity.
To determine hydrolytic activity, a cycloamylose mixture with DPs of 22 to 50 was used as a substrate. A 100-μl reaction mixture containing 0.5% (wt/vol) cycloamylose mixture, 50 mM sodium acetate buffer (pH 5.5), and diluted enzyme was incubated at 70°C for 60 min, and the subsequent total reducing power of glucan in the reaction mixture was determined by a modified Park-Johnson method (19). One unit of activity was defined as the amount of enzyme that produced 1 μmol of reducing sugar (as glucose) per min under these assay conditions.
Cyclization activity.
Amylose AS-30 (0.2 g) was dissolved in 10 ml of 90% (vol/vol) dimethyl sulfoxide. The enzyme solution (25 μl), which was diluted with 50 mM sodium acetate buffer (pH 5.5), was incubated at 70°C for 30 min with the AS-30 solution (25 μl). The reaction was terminated by addition of 1 ml of a chilled iodine solution. The iodine solution was made daily by mixing 0.1 ml of an iodine stock solution (0.26 g of I2 and 2.6 g of KI in 10 ml of water) and 0.1 ml of 1 N HCl and diluting the mixture to 26 ml. One unit of activity was defined as the amount of enzyme that decreased the A660 of the amylose-iodine complex by 10% per min. The activity determined in this way was considered cyclization activity, since it was thought that the decrease in A660 resulted from the decrease in the molecular weight of amylose due to the cyclization reaction.
Production and purification of the recombinant T. aquaticus amylomaltase and the mutated enzymes.
The recombinant amylomaltases were produced by the method described by Terada et al. (20), except that Escherichia coli strain BL21 cells were used as the host cells. The cells from a 40-ml culture were washed twice with 20 ml of 20 mM Tris-HCl buffer (pH 7.0). The suspended cells were broken by sonication at 4°C and centrifuged (10,000 × g, 30 min) to remove the cell debris. The crude extract was heated at 70°C for 30 min and centrifuged (10,000 × g, 30 min) to remove the aggregated protein. The supernatant was loaded onto a Phenyl-Toyopearl 650 M (Tosoh Co., Ltd., Tokyo, Japan) column equilibrated with the same buffer containing 0.5 M ammonium sulfate and washed with the same buffer containing 0.3 M ammonium sulfate. The enzyme was eluted with a linear 0.3 to 0 mM ammonium sulfate gradient in the same buffer. The active fraction was loaded onto a Source 15Q column (1 ml; Amersham Biosciences Corp., New Jersey), and the recombinant amylomaltase was eluted with a linear 0 to 100 mM NaCl gradient. Active fractions were dialyzed against 20 mM Tris-HCl buffer (pH 7.0)
Mutagenesis.
A Diversify PCR random mutagenesis kit (BD Biosciences Clontech, New Jersey), which is based on error-prone PCR, was used to introduce random mutations into the T. aquaticus amylomaltase gene. Error-prone PCR was carried out using plasmid pFQG8 (20) as the template DNA and oligonucleotide primers MQRM-F (5′-TTTCATATGGACCTTCCCCGCGCTTTCGGTCTGCTT-3′) and MQRM-R (5′-TTTGAATTCGGGCTGGTCCACCTAGAGCCGTTCCGT-3′), which were designed to create NdeI and EcoRI sites on both ends. The reaction conditions were same as those of the standard method except that the mixture contained an extra 0.04 mM dGTP. Few nucleotides per kbp DNA were substituted under these conditions. The amplified DNAs were digested with NdeI and EcoRI and then inserted into the NdeI-EcoRI site of pGEX-Nde, an expression vector described previously (20), and the plasmid library was then constructed. Site-directed mutagenesis was carried out using a Quick Change XL site-directed mutagenesis kit (Stratagene, La Jolla, Calif.). Plasmid pFQG8 was used as the template DNA, and the following two oligonucleotides were used as mutagenic primers: MQSM-F (5′-TTGGGCCCCACGGGCNNNGGCGACTCCCCCTACC-3′) and MQSM-R (5′-GGTAGGGGGAGTCGCCNNNGCCCGTGGGGCCCAAGG-3′). The introduction of a desired nucleotide change without second-site mutations was verified by DNA sequence analysis.
Detection of mutated T. aquaticus amylomaltase with lower hydrolytic activity.
The genes of T. aquaticus amylomaltase that introduced random mutations were expressed in E. coli strain BL21. E. coli carrying the plasmid was grown at 37°C in 5 ml of LB medium containing 50 μg/ml ampicillin. After 18 h, 500 μl of lysis solution (5 mg/ml lysozyme, 1.0 U/ml DNase I, 200 mM magnesium chloride, 0.5% Triton X-100) was added to the culture medium directly, and a freeze-thaw treatment was carried out twice. The cell extract prepared in this way was heated at 70°C for 30 min and was centrifuged (10,000 × g, 20 min). The supernatant was used as an enzyme solution for an assay. In order to discover desired mutants, the following two assay methods were used. First, to detect the cyclization activity of amylomaltase, 50 μl of solution A (0.4% AS-110, 100 mM sodium acetate buffer [pH 5.5]) was incubated at 70°C with 50 μl of the enzyme solution for 18 h. The reaction was terminated by heating the preparation at 100°C for 10 min and then mixing it with 100 μl of an iodine solution (0.005% I2, 0.5% KI), and the absorbance at 660 nm was measured. Since amylases from the host cell were inactivated by heat treatment at 70°C for 30 min, degradation of amylose by the amylases was ruled out. Second, to test the hydrolytic activity, 50 μl of solution B (1.0% cycloamylose mixture, 100 mM sodium acetate buffer [pH 5.5]) was incubated at 70°C with the enzyme solution for 18 h, and the reaction was terminated by heating the preparation at 100°C for 10 min. After this, the increase in reducing sugars in the reaction mixture was determined using a modified Park-Johnson method. We selected mutated enzymes that had reduced hydrolytic activity but retained cyclization activity.
Synthesis of cycloamylose by T. aquaticus amylomaltase.
Amylose AS-110 (0.2 g) was dissolved in 10 ml of 90% (vol/vol) dimethyl sulfoxide. A 2.0-ml reaction mixture containing 40 U (cyclization activity) of the recombinant T. aquaticus amylomaltase, 0.2 ml of the AS-110 solution, and 50 mM sodium acetate buffer (pH 5.5) was incubated at 70°C, and the reaction was terminated by heating the reaction mixture at 100°C for 30 min. Then 40 μl of the reaction mixture was incubated with glucoamylase (1.8 U) with or without α-amylase (0.26 U) at 37°C for 3 h, and the reaction was terminated by heating the solution at 100°C for 10 min. After digestion, the amount of glucose released was determined by the glucose oxidase method (10). The amount of cycloamyloses was determined by determining the amount of glucoamylase-resistant glucan, which was calculated by subtracting the amount of glucose released by the glucoamylase and α-amylase digestions from the amount of glucose released by the glucoamylase digestion. The glucoamylase-treated glucan was purified by ethanol precipitation, redissolved in 50 μl of 0.15 N NaOH, and analyzed by high-performance anion-exchange chromatography (HPAEC).
HPAEC.
HPAEC was carried out using the DX-300 system (Dionex Corp., Sunnyvale, Calif.) with a pulsed electrochemical detector (model PED-II; Dionex) and a Carbopac PA-100 column (4 by 250 mm; Dionex). Twenty-five microliters of a glucan solution was injected and eluted with a sodium acetate gradient. The analysis conditions were the same as those described previously (20).
RESULTS
Random mutagenesis of T. aquaticus amylomaltase.
Random mutations were introduced into the structural gene of T. aquaticus amylomaltase by error-prone PCR, and a plasmid library was constructed in E. coli strain BL21. About 3,000 independent colonies were cultured one by one, and the cell extracts were prepared as described in Materials and Methods. Cell extracts prepared in this way were subjected to hydrolytic and cyclization activity assays to select clones with mutated amylomaltases that had less hydrolytic activity but retained cyclization activity. Three positive clones (MQ0634, MQ1986, and MQ2711) were isolated, and the plasmids were extracted. The nucleotide sequence of the gene coding for the amylomaltase of each clone was fully determined. Surprisingly, each clone had a single mutation that resulted in replacement of amino acid Tyr54 (Y54) with either Asp (MQ1986 and MQ2711) or Ser (MQ0634). These results suggested that Y54 of T. aquaticus amylomaltase affects the hydrolytic activity of this enzyme.
Saturation mutagenesis of Y54.
Y54 was then replaced with all other amino acids by site-directed mutagenesis. Each mutated enzyme gene was expressed in E. coli BL21, and the enzyme was purified from the cell extract by heat treatment and hydrophobic chromatography, as described in Materials and Methods. All mutated enzymes prepared in this way exhibited a single band on sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels (data not shown) and were subjected for further analysis. The hydrolytic and cyclization activities of these mutated enzymes were measured and are summarized in Table 1. Surprisingly, the hydrolytic activity of the wild-type amylomaltase was the highest activity for the enzymes tested. Although the Y54F and Y54K mutated enzymes still exhibited levels of hydrolytic activity comparable to that of the wild-type enzyme, other mutated enzymes exhibited significantly less hydrolytic activity. In contrast to the hydrolytic activity, replacement of Y54 resulted in an increase in cyclization activity in most cases. The Y54R mutated enzyme exhibited the highest cyclization activity, which was double that of the wild-type enzyme, whereas the activity of the Y54W, Y54F, or Y54M mutated enzyme was slightly decreased. These results showed that amino acid replacement at Y54 not only has a significant effect that reduces the hydrolytic activity but also enhances the cyclization activity.
TABLE 1.
Hydrolytic and cyclization activities of wild-type and mutated enzymes
| Enzyme | Hydrolytic activity
|
Cyclization activity
|
Hydrolytic activity/cyclization activity ratio (108) | ||
|---|---|---|---|---|---|
| mU/mg | Relative valuea | U/mg | Relative valuea | ||
| Y54A | 19.5 | 0.39 | 2,140 | 1.92 | 9.11 |
| Y54C | 24.1 | 0.48 | 1,360 | 1.22 | 17.7 |
| Y54D | 19.2 | 0.38 | 1,980 | 1.77 | 9.72 |
| Y54E | 24.2 | 0.48 | 1,760 | 1.57 | 13.8 |
| Y54F | 48.4 | 0.96 | 1,020 | 0.91 | 47.5 |
| Y54G | 8.19 | 0.16 | 1,860 | 1.67 | 4.40 |
| Y54H | 38.5 | 0.76 | 1,830 | 1.64 | 21.0 |
| Y54I | 16.6 | 0.33 | 1,750 | 1.57 | 9.50 |
| Y54K | 49.6 | 0.99 | 1,970 | 1.76 | 25.2 |
| Y54L | 19.6 | 0.39 | 2,110 | 1.89 | 9.27 |
| Y54M | 19.4 | 0.39 | 900 | 0.81 | 21.6 |
| Y54N | 15.2 | 0.30 | 1,770 | 1.59 | 8.60 |
| Y54P | 2.71 | 0.05 | 530 | 0.48 | 5.11 |
| Y54Q | 35.1 | 0.70 | 1,870 | 1.68 | 18.8 |
| Y54R | 35.5 | 0.70 | 2,230 | 2.00 | 15.9 |
| Y54S | 19.2 | 0.38 | 2,080 | 1.86 | 9.25 |
| Y54T | 10.9 | 0.22 | 1,670 | 1.49 | 6.54 |
| Y54V | 14.4 | 0.29 | 1,780 | 1.60 | 8.11 |
| Y54W | 10.0 | 0.20 | 1,060 | 0.95 | 9.43 |
| Wild type | 50.4 | 1.00 | 1,120 | 1.00 | 45.0 |
Activity relative to the activity of the wild-type enzyme.
Synthesis of cycloamylose by mutated enzymes.
An enzyme suitable for cycloamylose production is expected to have high cyclization activity along with low hydrolytic activity. The ratio of hydrolytic activity to cyclization activity was highest for the Y54F mutated enzyme and second highest for the wild-type enzyme (Table 1), so these two enzymes were assumed to be unsuitable for cycloamylose production. The ratio for the Y54G mutated enzyme, on the other hand, was the lowest ratio and was less than one-tenth that of the wild-type enzyme (Table 1).
In order to investigate the ability of the Y54 mutated enzymes to synthesize cycloamylose, each mutated enzyme was incubated with synthetic amylose, and the yield of cycloamyloses and the reducing power in the reaction mixture were measured at various times. The results for the Y54F, Y54G, Y54K, Y54W, and Y54P mutated enzymes are shown along with those for the wild-type enzyme in Fig. 1. In the case of the wild-type enzyme, the yield of cycloamylose increased rapidly to around 90% in 4 h and then decreased gradually to 60% during prolonged incubation. The reducing power increased significantly with time and reached 4% at the end of the reaction, which suggested that the presence of hydrolytic activity in the wild-type enzyme resulted in an increase in the reducing power and a concomitant decrease in the yield of cycloamylose. A similar profile was obtained with the Y54F mutated enzyme, which had levels of hydrolytic and cyclization activities comparable to those of the wild-type enzyme. The declines in the yield of cycloamylose and the increases in the reducing power were less in the mutated enzymes with lower hydrolytic activity (Y54K, Y54W, and Y54P). This phenomenon was obvious for the Y54G mutated enzyme, which had the highest cyclization activity and the second lowest hydrolytic activity; the yield was not decreased during the reaction and remained more than 90% until the end of the reaction, and the reducing power was not significantly increased. In order to investigate correlations with the activity ratio of hydrolysis to cyclization and the yield of cycloamyloses or in the ratio and the reducing power, the final yield of cycloamylose or the reducing power was plotted against the ratio of hydrolytic activity to cyclization activity. As shown in Fig. 2, there were good correlations between the yield of cycloamylose and the activity ratio (Fig. 2A) and between the reducing power and the activity ratio (Fig. 2B).
FIG. 1.

Time courses of cycloamylose production by the wild-type amylomaltase (A) and the Y54F (B), Y54G (C), Y54K (D), Y54P (E), and Y54W (F) mutated enzymes. Squares, cycloamylose (CA) yield; diamonds, reducing power in the reaction solution. The reaction conditions and the method used to determine the amount of cycloamyloses are described in Materials and Methods. The reducing power when all of the amylose is broken down to glucose is defined as 100%.
FIG. 2.
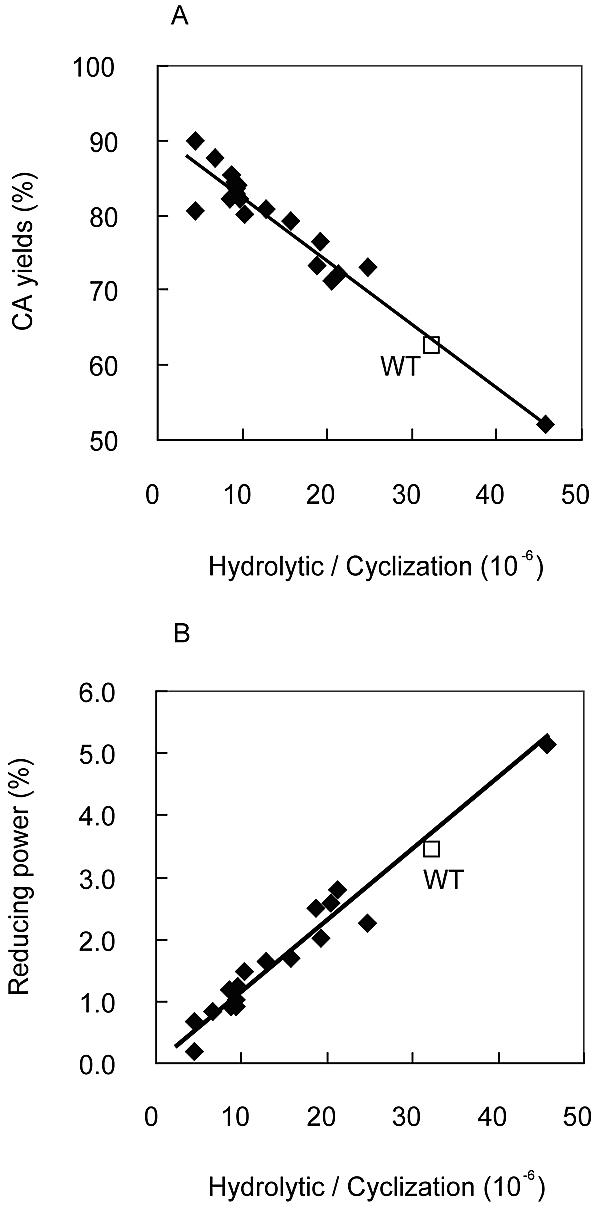
Correlation between the ratio of hydrolytic activity to cyclization activity and the yield of cycloamylose (A) or the reducing power in the reaction solution (B). Squares, wild-type enzyme (WT); diamonds, Y54G mutated enzyme. The correlation coefficients (R2) for the ratio and the yield of cycloamylose (CA) and for the ratio and the reducing power were 0.94 and 0.96, respectively.
The compositions of the cycloamyloses produced by the wild-type and mutated enzymes were investigated next. Cycloamylose was recovered from the final reaction mixture as described in Materials and Methods and was analyzed by HPAEC. However, there were no differences in the lowest DPs and the distribution of DPs of cycloamyloses produced by the wild-type and mutated enzymes (data not shown).
DISCUSSION
An increasing number of 4-α-glucanotransferases are now known to catalyze the cyclization reaction of α-1,4 glucan to produce cycloamylose (3, 17). One of these enzymes, an amylomaltase from T. aquaticus, is extremely interesting, because this enzyme exhibited a high level of homology with potato D-enzyme (41% in the amino acid sequence) (20), but there are many differences between the enzymatic properties of these enzymes. Potato D-enzyme produced cycloamylose from a DP of 17, whereas T. aquaticus amylomaltase produced cycloamylose from a DP of 22. These two enzymes also differ in the smallest effective donor molecule and the smallest glucan chain unit that is transferred (maltotriose and maltosyl units, respectively, for potato D-enzyme and maltose and glucosyl units, respectively, for T. aquaticus amylomaltase) (17). Potato D-enzyme catalyzes the transglycosylation reaction almost exclusively and does not catalyze hydrolysis (17). On the other hand, T. aquaticus amylomaltase shows weak but significant hydrolytic activity together with its major transglycosylation activity, which causes the yield of cycloamylose to be reduced. When a practical enzyme for cycloamylose production is needed, T. aquaticus amylomaltase is extremely useful because this enzyme is highly thermostable compared to potato D-enzyme and produces cycloamylose with a DP that is 5 glucose units larger than the molecules produced by other enzymes. T. aquaticus amylomaltase, however, is not suitable for practical use because of its relatively high hydrolytic activity. Thus, in the current work we aimed primarily to obtain a T. aquaticus amylomaltase with less hydrolytic activity, but we also wanted to understand more about the function of this multifunctional enzyme. As described previously, T. aquaticus amylomaltase with dramatically decreased hydrolytic activity was successfully engineered by introducing a single amino acid replacement at Y54. The yield of cycloamylose with the Y54G mutated enzyme was not decreased with prolonged incubation, and it was about 30% higher than the yield with the wild-type enzyme.
The crystal structure of T. aquaticus amylomaltase has been determined in its native form (13) and together with acarbose molecules (14). The results indicated the presence of two acarbose binding sites in the enzyme; one site is in the active site groove at subsites −3 to 1, and the second site is around Y54, which is 14 Å from the catalytic site (14). It was strongly suggested that this enzyme interacts with substrate at this position as a secondary binding site. Przylas et al. proposed (14) that Y54 helps form a curved conformation of the glucan chain in this region and affects the composition and the smallest size of the cycloamylose produced. The current work obviously demonstrated that Y54 does not play such a role, because there was no significant effect on the composition of cycloamylose when Y54 was replaced by all other amino acids. An amino acid replacement at Y54, however, had a more fundamental effect on the reaction specificity of this enzyme, which enhanced the cyclization reaction and decreased the hydrolytic activity. Y54 is in subdomain B2 located between the second and third barrel strands (13). Subdomain B2 is present in amylomaltase and D-enzyme but absent in CGTase and α-amylase. Therefore, it is thought that this domain has a unique role in amylomaltase. From the results for the crystal structure of potato D-enzyme reported recently (6), it was concluded that potato D-enzyme also has subdomain B2. However, an amino acid corresponding to Y54 of T. aquaticus amylomaltase cannot be identified in potato D-enzyme, because the homology between the two enzymes is particularly low in this domain. Alignment of the available amylomaltase and D-enzyme sequences also indicated that Y54 is not conserved in this enzyme family.
Alterations in reaction specificity after amino acid substitution have been reported for CGTase (8, 11, 12, 22, 23, 24, 25). Nakamura et al. (11) introduced mutations into four aromatic residues, F183, Y195, F259, and F283, located in the center of the active cleft of CGTase from alkalophilic Bacillus sp. strain 1011 and reported that these mutations affected the hydrolytic activity and/or β-cyclodextrin-forming activity. Uitdehaag et al. (22) found that subsites −3 and −7 of CGTase from Bacillus circulans strain 251 were involved in product specificity. Leemhuis et al. (8) suggested that the hydrophobicity of F184 and F260 at subsite 2 limited the hydrolytic activity of CGTase from Thermoanaerobacterium thermosulfurigenes strain EM1. As described above, an aromatic amino acid residue located around the active site played an important role in determining the reaction specificity of CGTase and probably also other 4-α-glucanotransferases. Y54 of T. aquaticus amylomaltase, on the other hand, was 14 Å from the catalytic site, so it is unlikely that this residue was directly involved in determining the reaction specificity. We believe that binding of glucan to Y54 should cause a conformational change in this enzyme, which should alter the reaction specificity reported in this study.
Acknowledgments
This work was supported by a grant for the development of the next generation of bioreactor systems from the Society for Techno-Innovation of Agriculture, Forestry and Fisheries (STAFF), Tokyo, Japan, to Ezaki Glico Co., Ltd.
REFERENCES
- 1.Endo, T., M. Zheng, and W. Zimmermann. 2002. Enzymatic synthesis and analysis of large-ring cyclodextrins. Aust. J. Chem. 55:39-48. [Google Scholar]
- 2.Frömming, K.-H., and J. Szejtli. 1994. Cyclodextrins in pharmacy. Kluwer Academic Publishers, Dordrecht, The Netherlands.
- 3.Fujii, K., H. Takata, M. Yanase, Y. Terada, K. Ohdan, T. Takaha, S. Okada, and K. Kuriki. 2003. Bioengineering and application of novel glucose polymer. Biocatal. Biotransform. 21:167-172. [Google Scholar]
- 4.Gessler, K., I. Uson, T. Takaha, N. Krauss, S. M. Smith, S. Okada, G. M. Sheldrick, and W. Saenger. 1999. V-amylose at atomic resolution: X-ray structure of a cycloamylose with 26 glucoses. Proc. Natl. Acad. Sci. USA 96:4246-4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hedges, A. R. 1998. Industrial applications of cyclodextrins. Chem. Rev. 98:2035-2044. [DOI] [PubMed] [Google Scholar]
- 6.Imamura, K., T. Matsuura, M. Ye, T. Takaha, K. Fujii, M. Kusunoki, and Y. Nitta. 2005. Crystallization and preliminary X-ray crystallographic study of disproportionating enzyme from potato. Acta Crystallogr. 61:109-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kitamura, S., K. Nakatani, T. Takaha, and S. Okada. 1999. Complex formation of large-ring cyclodextrins with iodine in aqueous solution as revealed by isothermal titration calorimetry. Macromol. Rapid Commun. 20:612-615. [Google Scholar]
- 8.Leemhuis, H., B. W. Dijkstra, and L. Dijkhuizen. 2002. Mutations converting cyclodextrin glycosyltransferase from a transglycosylase into a starch hydrolase. FEBS Lett. 514:189-192. [DOI] [PubMed] [Google Scholar]
- 9.Machida, S., S. Ogawa, S. Xiaohua, T. Takaha, K. Fujii, and K. Hayashi. 2000. Cycloamylose as an efficient artificial chaperone for protein refolding. FEBS Lett. 486:131-135. [DOI] [PubMed] [Google Scholar]
- 10.Miwa, I., J. Okuda, K. Maeda, and G. Okuda. 1972. Mutarotase effect on colorimetric determination of blood glucose with β-d-glucose oxidase. Clin. Chim. Acta 37:538-540. [DOI] [PubMed] [Google Scholar]
- 11.Nakamura, A., K. Haga, and K. Yamane. 1994. Four aromatic resides in the active center of cyclodextrin glucanotransferase from alkalophilic Bacillus sp. 1011: effects of replacements on substrate binding and cyclization characteristics. Biochemistry 33:9929-9936. [DOI] [PubMed] [Google Scholar]
- 12.Penninga, D., B. Strokopytov, H. J. Rozeboom, C. L. Lawson, B. W. Dijkstra, J. Bergsma, and L. Dijkhuizen. 1995. Site-directed mutations in tyrosine 195 of cyclodextrin glycosyltransferase from Bacillus circulans strain 251 affect activity and product specificity. Biochemistry 34:3368-3376. [DOI] [PubMed] [Google Scholar]
- 13.Przylas, I., K. Tomoo, Y. Terada, T. Takaha, K. Fujii, W. Saenger, and N. Sträter. 2000. Crystal structure of amylomaltase from Thermus aquaticus, a glycosyltransferase catalyzing the production of large cyclic glucans. J. Mol. Biol. 296:873-886. [DOI] [PubMed] [Google Scholar]
- 14.Przylas, I., Y. Terada, K. Fujii, T. Takaha, W. Saenger, and N. Sträter. 2000. 2000. X-ray structure of acarbose bound to amylomaltase from Thermus aquaticus. Eur. J. Biochem. 267:6903-6913. [DOI] [PubMed] [Google Scholar]
- 15.Szejtli, J. 1998. Cyclodextrin technology, Kluwer Academic Publishers, Dordrecht, The Netherlands.
- 16.Szejtli, J. 1998. Introduction and general overview of cyclodextrin chemistry. Chem. Rev. 98:1743-1753. [DOI] [PubMed] [Google Scholar]
- 17.Takaha, T., and S. M. Smith. 1999. The function of 4-α-glucanotransferases and their use for the production of cyclic glucans. Biotechnol. Genet. Eng. Rev. 16:257-280. [DOI] [PubMed] [Google Scholar]
- 18.Takaha, T., M. Yanase, H. Takata, S. Okada, and S. M. Smith. 1996. Potato D-enzyme catalyzes the cyclization reaction of amylose to produce cycloamylose, a novel cyclic glucan. J. Biol. Chem. 271:2902-2908. [DOI] [PubMed] [Google Scholar]
- 19.Takeda, Y., H.-P. Guan, and J. Preiss. 1993. Branching of amylose by the branching isoenzymes of maize endosperm. Carbohydr. Res. 240:1600-1606. [Google Scholar]
- 20.Terada, Y., K. Fujii, T. Takaha, and S. Okada. 1999. Thermus aquaticus ATCC 33923 amylomaltase gene cloning and expression and enzyme characterization: production of cycloamylose. Appl. Environ. Microbiol. 65:910-915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Terada, Y., M. Yanase, H. Takata, T. Takaha, and S. Okada. 1997. Cyclodextrins are not the major cyclic a-1,4-glucans produced by the initial action of cyclodextrin glucanotransferase on amylose. J. Biol. Chem. 272:15729-15733. [DOI] [PubMed] [Google Scholar]
- 22.Uitdehaag, J. C. M., B. A., van der Veen, L. Dijkhuizen, and B. W. Dijkstra. 2002. Catalytic mechanism and product specificity of cyclodextrin glycosyltransferase, a prototypical transglycosylase from the α-amylase family. Enzyme Microb. Technol. 30:295-304. [Google Scholar]
- 23.van der Veen, B. A., J. C. M. Uitdehaag, B. W. Dijkstra, and L. Dijkhuizen. 2000. The role of arginine 47 in the cyclization and coupling reactions of cyclodextrin glycosyltransferase from Bacillus circulans strain 251: implications for product inhibition and product specificity. Eur. J. Biochem. 267:3432-3441. [DOI] [PubMed] [Google Scholar]
- 24.van der Veen, B. A., J. C. M. Uitdehaag, B. W. Dijkstra, and L. Dijkhuizen. 2000. Engineering of cyclodextrin glycosyltransferase reaction and product specificity. Biochim. Biophys. Acta 1543:336-360. [DOI] [PubMed] [Google Scholar]
- 25.van der Veen, B. A., J. C. M. Uitdehaag, D. Penninga, G. J. van Alebeek, L. M. Smit, B. W. Dijkstra, and L. Dijkhuizen. 2000. Rational design of cyclodextrin glycosyltransferase from Bacillus circulans strain 251 to increase alpha-cyclodextrin production. J. Mol. Biol. 296:1027-1038. [DOI] [PubMed] [Google Scholar]
- 26.Yanase, M., H. Takata, T. Takaha, T. Kuriki, S. M. Smith, and S. Okada. 2002. Cyclization reaction catalyzed by glycogen debranching enzyme (EC 2.4.1.25/EC 3.2.1.33) and its potential for cycloamylose production. Appl. Environ. Microbiol. 68:4233-4239. [DOI] [PMC free article] [PubMed] [Google Scholar]