

Abstract
Rheumatoid arthritis (RA) is associated with certain MHC class II alleles and is characterized by a chronic autoimmune response in the joints. Using transgenic mice expressing human DR4 (DRB1*0401) and human CD4, but lacking endogenous MHC class II, we show that posttranslational glycosylation of type II collagen (CII) influences the level of T cell tolerance to this candidate cartilage-specific autoantigen. In such mice, the expression of human CII resulted in a tolerized murine T cell response to human CII. However, tolerance induction remained incomplete, preferentially deleting responses to the nonmodified CII 263–270 epitope, whereas T cell recognition of a glycosylated variant of this epitope was affected to a lesser degree. A similar dominance of T cell responses to CII-glycopeptides was recorded in a cohort of severely affected RA-patients (n = 14). Thus, RA T cells predominantly recognize the immunodominant CII peptide in its glycosylated form and may explain why previously it has been difficult to detect T cell responses to CII in RA patients.
Rheumatoid arthritis (RA) is an autoimmune disease that primarily affects peripheral joints with cartilage destruction and subsequent bone erosion. The role of T cells in RA is supported by the large number of activated CD4+ cells reported in the synovium of affected joints and by the association of RA to certain MHC (HLA) class II genes [e.g., DRB1*0401 (in DR4) and DRB1*0101 (in DR1)] that encode a specific peptide binding pocket, the so-called shared epitope.
Collagen type II (CII), the main constituent of hyaline cartilage, has been proposed as one possible autoantigen in RA because CII-specific antibodies are frequently found in RA patients and because an RA-like disease can be induced in certain mouse strains after immunization with CII. However, reports on T cell immunity to CII in RA patients as well as in healthy individuals are inconclusive, and the role of CII, and even T cells, in RA is still argued and remains to be proven.
Previous studies performed in DR4- and DR1-expressing mice have located the immunodominant T cell epitope to position 263–270 in CII by using synthetic peptides (1–3), but studies in RA patients have in general failed to identify DR4/DR1-restricted T cells specific for the same epitope (4, 5). One potential reason for this failure is that these studies did not directly address that CII can become posttranslationally modified. Within CII263–270, the lysines at positions 264 and 270 can be hydroxylated and further glycosylated with mono- or disaccharides, i.e., with a β-d-galactopyranosyl or an α-d-glucopyranosyl-(1-2)-β-d-galactopyranosyl residue. Such modifications have previously been shown to be of importance in the development of collagen-induced arthritis (CIA) in Aq-expressing mice (6, 7). Although the Aq-molecule is of a DQ isotype, its peptide-binding groove shows more similarity to the shared epitope variant of the DR4-molecule, because it binds almost the same CII epitope and presents the same amino acid side chains to T cells (3, 8–11). Furthermore, CIA is most commonly induced with heterologous CII, which is believed to induce a heteroreactive T cell response, followed by a B cell response, which in contrast to the T cell response is highly crossreactive to mouse CII (12, 13). Thus, earlier CIA experiments in DR4-expressing mice have not addressed T cell tolerance to self-CII. Therefore, to get a better understanding of autoimmunity to cartilage-derived proteins, we need to consider both posttranslational modifications and the aspect of T cell tolerance in these models. To achieve this result, we used a humanized mouse model expressing HLA-DRB1*0401/DRA1*0101, human CD4, and human CII (huCII) on a background deficient of murine class II expression (14, 15). In these mice, T cell responses to huCII peptides and the impact of posttranslational modification on the induction of CII-specific T cell tolerance was investigated.
Materials and Methods
Mice.
Crossing of different transgenic mice generated two groups of mice that were used for experiments: (i) DR4, expressing transgenic DRB*0401 and human CD4 but no murine class II molecule; and (ii) huCII/DR4, expressing DRB*0401, human CD4 and huCII but no murine class II molecule. To get these groups, huCII transgenic mice (15), mice expressing HLA-DR4 together with human CD4 (14), and mice lacking H-2 class II (16) were crossed as follows. First, HLA-DR4/huCD4 on a B10 background was introduced with H-2plus/− on a B6 background and backcrossed twice to B10 and subsequently intercrossed. HLA-DR4/huCD4, H-2−/− mice were then crossed with huCII mice on a C3H background. Offspring were backcrossed three generations to HLA-DR4/huCD4, H-2−/− on the B10 background. Finally, mice were intercrossed twice and selected for homozygosity in H-2−/−. Mice were bred and kept in the animal facility of Medical Inflammation Research (http://net.inflam.lu.se).
Antigens and in Vitro T Cell Assays.
HuCII was extracted from hip joints (obtained from replacement surgery) after pepsin digestion and purified as described (17). The following nonmodified and glycosylated CII peptides were synthesized as described (8, 18, 19): K264/270 (nonmodified CII261–275 with a lysine residue at position 264 and 270); Gal264 (CII261–278, glycosylated with a β-d-galactopyranose residue on l-hydroxylysine exclusively at position 264); Gal270 (CII261–278, glycosylated exclusively at position 270); and Gal264/270 (CII259–278, glycosylated at position 264 and 270). The following modified forms of the Gal264-glycopeptide, named deoxy-glycopeptides, were also synthesized (B.H., unpublished data): 2-deoxyGal (CII259–273, where the hydroxy-group at position 2 on the β-d-galactopyranosyl moiety has been eliminated); 3-deoxyGal (CII259–273, missing the hydroxy-group at position 3 on galactose); and 4-deoxyGal (CII259–273, missing the hydroxy-group at position 4 on galactose). As control for the deoxy-peptides, a shortened form of the Gal264-peptide (CII259–273) was used. Mice were immunized with huCII in complete Freund's adjuvant, and 10 days later cells from draining lymph nodes were stimulated in vitro for determination of antigen-specific proliferation and IFN-γ production as described (20). Establishment of T cell hybridoma clones and determination of antigen specificity was performed as described (8), with the exception that Gal264/270-peptide was used for immunization and for the first in vitro restimulation (20 μg/ml).
Analysis of Human T Cell Responses.
Fourteen patients fulfilling the RA-classification criteria of the American College of Rheumatology (21) were recruited for the study. The study protocol was approved by the review board of the Friedrich-Alexander-University Erlangen-Nuremberg, and informed consent was obtained from all individuals before entering the study. All patients had a severe course of the disease so that insufficient response to conventional therapy required an intensified treatment with TNFα-blocking antibodies [D2E7 (Knoll AG-BASF Pharma, Ludwigshafen, Germany) or infliximab (Essex Pharma, Munich, Germany]. The mean age of the patients (2 male and 12 female) was 63.6 ± 6.5 (SD) years, and the disease duration 10.9 ± 8.1 (SD) years. In 9 patients, information about the expression of HLA-DRB1*-alleles was available and is included in the result section. Peripheral blood mononucleated cells (PBMC) from the patients were separated by using Histoprep (BAG, Lich, Germany). For antigenic stimulation of 106 PBMC, 10 μg of CII-peptide and 1 μg anti-human CD28 (Becton Dickinson) were added per ml of culture medium. T cell receptor-specific responses were controlled in parallel by using culture conditions that either omitted any stimulation or only exposed the cells to the costimulatory anti-CD28 antibody overnight in the absence of antigen. T cell responsiveness to a common recall antigen was tested in parallel cultures of PBMC by using 10 μg/ml tetanus toxoid (Calbiochem-Nova Biochem) and anti-CD28 for stimulation. Monensin (2.5 mM; Sigma-Aldrich) was added to the overnight cultures, and the cells were incubated for additional 4 h before harvesting. Subsequently, the cells were washed twice in PBS and fixed in 4% paraformaldehyde/PBS solution for 7 min at 37°C, followed by a repeated washing procedure in PBS. A permeabilization step was performed for 10 min with 0.5% saponin/1% BSA/0.1% NaN3 in PBS; afterwards, the cells were washed twice with PBS/1% BSA. Cells were stained with 0.2 μg rat anti-human IL-2-PE (Becton Dickinson) and 3 μl CD4-FITC or CD3-FITC (Beckman Coulter) for 20 min at 4°C. Fluorescence intensities were determined by using a Coulter Epics XL-MCL flow cytometer and system-ii software. Large activated lymphocytes (blasts) were gated according to forward and side scatter as described previously (22, 23). Cells not treated with saponin were used to exclude background staining of anti-IL-2 antibody.
Results
Strong, but Incomplete, Negative Selection of the Autoreactive T Cell Population.
Previous studies have identified the immunodominant T cell epitope in bovine and human CII in DR4-transgenic mice, and they have also shown that these mice are susceptible to CIA (1–3). However, because bovine and human CII both differ from mouse CII within the identified epitope (at position 266, glutamic acid in heterologous CII, compared with aspartic acid in mouse CII), these reports did not include the aspect of T cell tolerance to self-CII. To generate an animal model of RA that would reflect the situation in humans more accurately, we crossed DR4/human CD4-transgenic and mouse MHC class II-deficient mice with huCII-transgenic mice to generate two lines of mice: DR4 mice (DR4/human CD4-transgenic and mouse MHC class II-deficient mice, expressing mouse CII in cartilage) and huCII/DR4 mice (DR4/human CD4-transgenic and mouse MHC class II-deficient mice, expressing huCII in cartilage). These mice were then immunized with huCII to examine the degree of T cell tolerance to endogenously expressed CII.
The heterologous immune response in DR4 mice to CII was biased toward the nonmodified peptide (K264/270), followed by a weaker response to peptides glycosylated with β-d-galactopyranosyl moieties on hydroxylysine residues at either position 264 (Gal264) or 270 (Gal270) (Fig. 1). The response to huCII protein was also weak but significantly above background level when measuring the production of IFN-γ. In sharp contrast, the response in huCII/DR4 mice was severely reduced against all CII antigens as compared with the response in DR4 mice, showing that strong tolerance to self-CII is present (P value less than 0.05 against all CII antigens except the Gal264/270-specific IFN-γ production). Most importantly, however, tolerance was not complete because huCII/DR4 mice were able to mount a significant response above background level against the Gal264-peptide, when the proliferation (P = 0.023) as well as IFN-γ production (P = 0.041) was measured (Fig. 1). In contrast, the response against the nonmodified K-peptide in huCII/DR4 mice was not significant above the background response (P > 0.05, Fig. 1).
Figure 1.

Strong but incomplete tolerance to glycosylated CII in humanized mice. Recall in vitro response of lymph node cells from DR4 and huCII/DR4 mice immunized 10 days earlier with human CII (huCII). Cells were restimulated with huCII and the following CII-peptides: nonglycosylated (K264/270), glycosylated at hydroxylysine 264 (Gal264), glycosylated at hydroxylysine 270 (Gal270), or glycosylated at both hydroxylysine residues (Gal264/270) with β-d-galactopyranosyl residues. Ten animals (5 males and 5 females) of each mouse line were investigated. As a positive control, the recall response to mycobacteria antigen PPD (purified protein derivate, present in complete Freund's adjuvant) was measured. The response in huCII/DR4 mice was significantly reduced against all CII antigens (P < 0.05) as compared with DR4 mice, except for Gal264/270-specific IFN-γ production. *, Significant responses above background levels for huCII/DR4 mice. *, P < 0.05, Mann–Whitney U test; bars represent mean ± SEM. Antigen-specific proliferation and IFN-γ production: Δcpm and ΔU/ml respectively (response with antigen—response in the absence of antigen).
To further evaluate the shift in epitope selection between DR4 mice (with a dominant response to the K264/270-peptide) and huCII/DR4 mice (with a significant response against the Gal264-peptide alone), we made a pairwise comparison of the response to the K- and the Gal264-peptide (Fig. 2). The response to the K-peptide was stronger than to the Gal264-peptide in all DR4 mice at both concentrations tested (10 and 50 μg/ml, P = 0.0051). In contrast, a relatively stronger response against the Gal264-peptide than to the K-peptide was noted in 8 of 10 huCII/DR4 mice when cells were stimulated with the higher antigen concentration (P = 0.0218) and in all mice when stimulated with the lower antigen concentration (P = 0.0051). Hence, CII-specific T cells are strongly tolerized in DR4 mice expressing huCII in cartilage. However, glycopeptide-specific T cells appear to be less affected by tolerance than T cells specific for the nonmodified epitope.
Figure 2.

Comparative analysis of the individual response to the nonmodified and the galactosylated T cell epitope in DR4 and huCII/DR4 mice. Recall in vitro response of lymph node cells from DR4 and huCII/DR4 mice immunized 10 days earlier with human CII. Cells were restimulated with either 10 or 50 μg/ml of the K264/270- or the Gal264-peptide. A ratio above 1 indicates a stronger response against the K264/270-peptide whereas a ratio below 1 indicates a stronger response to the Gal264-peptide. The response of individual DR4 mice was stronger to the K264-peptide whereas the responses of huCII/DR mice were stronger to the Gal264-peptide (Wilcoxon signed rank test).
Different T Cell Fine Specificity of Auto- and Heteroreactive T Cell Clones.
To confirm our finding that DR4-restricted CII recognition includes T cells specific for glycosylated epitopes, hybridoma clones from Gal264/270-peptide-immunized DR4 and huCII/DR4 mice were established and characterized for comparison. Despite the weak in vitro response to glycosylated CII antigens seen earlier in DR4 and huCII/DR4 mice after CII immunization, glycopeptide-specific hybridomas were successfully established from both lines of transgenic mice (Table 1).
Table 1.
DR4-restricted T cell hybridoma responses to the CII259–273 epitope and human CII
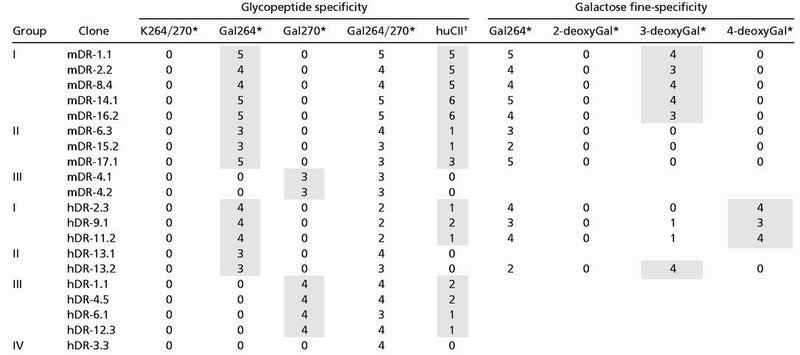 |
Semiquantitative scoring of T cell hybridoma response following stimulation with glycosylated or nonglycosylated collagen peptides (see Fig. 1). Shaded boxes highlight specific responses for the different groups of clones.
Sensitivity of the T cell hybridomas was determined by the amount of peptide required for a CTLL response >1000 CPM above background; 0, no response; 1 = 50 μg/ml; 2 = 10 μg/ml; 3 = 2 μg/ml; 4 = 0.4 μg/ml; 5 = 0.08 μg/ml; 6 = 0.016 μg/ml.
Sensitivity of the T cell hybridomas to CII protein was determined as for the CII-peptides, but number indicates a 5 times higher concentration of antigen, i.e., 1 = 250 μg/ml; 2 = 50 μg/ml; 3 = 10 μg/ml; 4 = 2 μg/ml; 5 = 0.4 μg/ml; 6 = 0.08 μg/ml.
Specific recognition of the glycopeptide was evident because all clones responded to the Gal264/270-peptide but not to the nonglycosylated peptide. The majority of T cell clones from DR4 mice (named mDR for mouse CII and DR4) recognized the Gal264-peptide (8 of 10, group I and II), whereas only 2 clones, originally collected from the same subcloning, specifically responded to the Gal270-peptide (group III). Gal264-specific clones could further be divided in two groups where clones that displayed a higher sensitivity to the glycopeptide also had a strong response toward huCII (group I), whereas clones that responded weakly to the Gal264-peptide also responded weakly to huCII (group II). In line with this finding, the Gal270-specific clone(s) responded weakly to the Gal270-peptide and not at all to huCII.
Interestingly, T cell clones from huCII/DR4 mice (named hDR, for huCII and DR4) differed from mDR clones in their response to CII-peptides: (i) The biased recognition of the Gal264-peptide was not observed for hDR-clones (Table 1). (ii) Among the Gal264-specific clones (group I and II), the response was somewhat weaker, compared with the corresponding mDR clones. This difference in antigen sensitivity was even more evident in the response to intact huCII (Fig. 3 and Table 1). (iii) Gal270-specific hDR clones also responded to huCII (group III), although high antigen concentration was required (Table 1).
Figure 3.

An example of different responses to glycosylated CII between DR4 and huCII/DR4 mice. T cell hybridomas clones mDR-16.2 and hDR-9.1 were obtained from DR4 and huCII/DR4 mice, respectively, after immunization with the Gal264/270-peptide. Cells were stimulated with titrated amounts of huCII or CII-peptides (see Fig. 1) and investigated for production of IL-2.
To confirm that it was the posttranslational modification of CII that was specifically recognized by the obtained clones, Gal264-specific hybridomas were also tested against three differently modified glycopeptides, where one of the hydroxy-groups on the galactose moiety had been selectively removed at either position 2, 3, or 4, thereby generating three mono-deoxy glycopeptides. The two groups among the Gal264-specific mDR clones were confirmed in that they, apart from having different sensitivity to the Gal264-peptide, also recognized the sugar residue differently (Table 1). Similarly, the two groups of Gal264-specific hDR clones were also confirmed by the use of the deoxy-glycopeptides. Notably, differences in deoxy-glycopeptide responses between mDR and hDR clones indicated that T cells were differently selected if huCII was expressed. For example, all mDR clones were stringently dependent on the hydroxy-group at the C4-carbon of galactose, whereas the responses of 3 of 4 hDR clones did not depend on the C4-hydroxy-group.
Glycopeptide-Specific T Cells Dominate the CII Response in RA Patients.
Thus far, results from our humanized animal model showed that T cells specifically recognizing the glycostructure remain after tolerance induction and become dominant by the introduction of autologous huCII, whereas T cells with other fine specificities to the CII epitope seem to be functionally impaired or deleted to a greater extent. We argued that these findings could explain earlier reported difficulties in identifying CII specific T cell clones from joints or blood of RA patients, and therefore we tested a total number of 14 RA patients for T cell responses against the different glycosylated peptides. In the investigated cohort of RA patients, cytokine flow cytometry of in vitro-stimulated PBMC revealed specific IL-2 responses to the glycosylated CII-peptides. Using the increase in the percentage of IL-2-producing cells within the CD3 or CD4 positive T cell population above baseline of negative control as a parameter of specific T cell stimulation, 7 patients could be identified as CII-peptide responsive (Table 2). Interestingly, all CII-responding patients recognized the glycopeptides, but only 2 of these responded also to the nonglycosylated peptide. This result shows that T cell recognition was confined to the glycosylated CII-variants in 5 patients, representing more than 30% of the entire cohort. With one exception (patient 10) the baseline population of IL-2-producing T cells was low in all patients investigated (Table 2). However, also in patient 10, a specific rise in the percentage of IL-2-producing CD4+ T cells from 0.20 to 0.92% on stimulation with Gal264/270 was detectable. The individual glycospecific responses of patients were heterogenous. In some patients, the response was biased to one of the glycosylation sites, whereas other patients had equal responses to both sites. In addition, in some patients, the response was restricted to one of the glycosylation sites, whereas the response to the peptide with glycosylation at both positions was weak or nonexistent. This latter response pattern was also seen in mice expressing both human DR4 and human CII (see Fig. 1). We also performed repetitive analysis of T cell responses to CII in 3 of the 14 patients. In the initial investigation, patient 3 responded exclusively to the Gal264-peptide because 0.24% of CD3+ cells produced IL-2 on stimulation with this peptide, compared with 0.07% in the absence of antigen. One week later, the same patient elicited an almost identical response (Fig. 4) where the two-color staining for CD4 and IL-2 show an increase in the double-positive T cell population on stimulation with the Gal264-peptide to 0.22% whereas exposure to the K264/270-peptide did not result in an increased IL-2 production. Similarly, patient 9 initially responded to the Gal264-peptide (Table 2), and this exclusive response was also noted when the patient was investigated 1 mo later (see footnotes in Table 2). However, in patient 10, the initially recorded response to the Gal264/270-peptide (Table 2) was undetectable 6 mo after the first assessment (see footnotes in Table 2). Thus, although CII-specific T cell responses may vary with time in some patients, possibly because of complex influences by treatment or spontaneous variations in the immune response of a characteristically chronic relapsing disease, the results obtained in a small cohort of RA patients with established diseases and severe course show the dominant targeting of glycosylated variants of the CII-peptide by the autoreactive T cell response. Finally, the available HLA-typing information on the patients reveals that all patients exhibiting T cell responses to the respective CII-peptide variants express at least one of the HLA-DRB1*-alleles containing the amino acid consensus motif QK(R)RAA in position 70–74. This motif constitutes the so-called “shared epitope” (24, 25), and is crucial for the binding of the CII-peptides to the respective class II molecules during antigen presentation (26).
Table 2.
Analysis of T cell recognition of CII-peptide 259–273 in RA patients
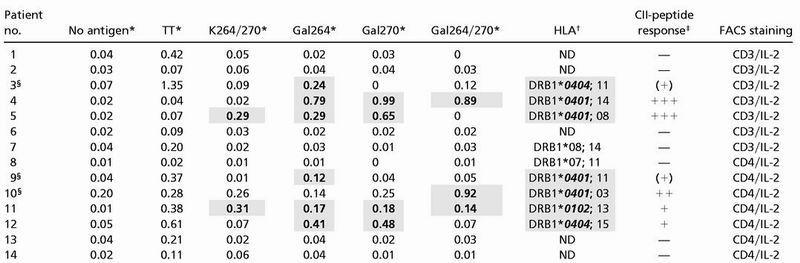 |
Percentage of IL-2-producing T cells after in vitro culture of PBMC without antigen (No antigen) or following stimulation with tetanus toxoid (TT1), or following stimulation with CII-peptides (see Fig. 1). IL-2 production was analyzed for the entire CD3+ T cell population in patients 1 to 7 or within the CD4+ subset in patients 8 to 14.
Percentage of double-positive T cells.
DRB1-alleles containing the “shared epitope” QKRAA or QRRAA in amino acid positions 70–74 are highlighted in shaded cells and bold italics; subtyping of DRB1* was performed only on 04 and 01 haplotypes. ND, Not determined.
Semiquantitative scoring of T cell response to any of the CII-peptides (% of IL-2-producing T cells): —, <0.1% or <3× (no antigen control); (+), ≥3× (no antigen control); +, ≥4× (no antigen control); ++, ≥5× (no antigen control); +++, ≥10× (no antigen control). Shaded cells and bold figures in the table highlight CII-peptide-specific responses fulfilling at least the (+) criterion.
Patients where repetitive analyses were performed. Patient 3 elicited a similar response exclusively to the Gal264-peptide in a second analysis performed 1 wk later (see Fig. 4). For patient 9, in a second study performed 1 mo later, 0.1% of CD3+ cells produced IL-2 upon stimulation with the Gal264-peptide, compared with 0.04% in the absence of antigen and 0.02% after stimulation with the K264/270- or Gal270-peptide. Patient 10 failed to respond to any CII-peptide when investigated 6 mo later.
Figure 4.

Specific recognition of CII-glycopeptides by human T cells. The two-color flow-cytometry of in vitro-stimulated T cells from an RA-patient shows fluorescence intensities for surface-binding of a FITC-labeled anti-CD4 antibody on the x axis whereas the y axis represents signal intensities for intracellular staining with a phycoerythrin-labeled anti-IL-2 antibody. PBMCs were cultured overnight without antigen, with tetanus toxoid, the K264/270-peptide, or the Gal264-peptide. The figures in the upper right quadrant of the different panels represent the percentage of double-positive cells.
Discussion
Posttranslationally glycosylated peptides from CII are presented by the RA-associated DR4 molecules as shown in human DR4-transgenic mice. In DR4-transgenic mice expressing huCII, and most likely also in humans, T cells recognizing the nonglycosylated 263–270 epitope are strongly tolerized, or even deleted, whereas T cells specific for the different glycosylated peptides persist. This is a potentially crucial finding because it provides an explanation not only for the role of the DR4 in the presentation of joint derived peptides, but also for the difficulties in providing evidence for T cells recognizing autologous CII.
The relevance of the findings made in mice expressing huCII and DR4 is evident because 30% of the investigated RA patients exhibited a predominant response to the glycosylated forms of the CII263–270 epitope. Thus, our data show that the physiological posttranslational modification of variable carbohydrate attachment converts the immunodominant naked self-peptide, which is tolerogenic, into several cryptic self-determinants that remain immunogenic.
In addition to reports showing that transgenic expression of DR4 and DR1 in mice permits development of CIA (2, 3, 27), there are several investigations describing T cell immunity to CII in either healthy individuals or RA patients or both (5, 28–36). However, less is known regarding the determinants recognized by CII-specific T cells in humans, and the interpretations of the reported findings are not all in agreement (4, 5, 34, 35). For example, one recent study failed to identify CII-specific T cells in RA patients when CII259–272 loaded DR4-tetramers were used (4), despite the earlier defined immunodominance of this peptide in DR4-transgenic mice (1).
By including the aspect of T cell tolerance to self-CII and recognition of glycosylated CII-epitopes in our humanized animal model of RA, we can now provide a possible explanation for the ambiguous results concerning CII recognition in RA patients. Our finding that T cells specific for glycosylated CII appeared less tolerized by endogenous expression of huCII than T cells specific for the nonmodified epitope is predictable from an earlier finding in Aq-expressing mice (15, 37). Mice with transgenic expression of either rat or human CII displayed strong T cell tolerance to the immunodominant CII epitope and were also partially protected from CIA when immunized with rat and human CII, respectively (15, 37). Moreover, mice transgenic for the Aq-restricted immunodominant T cell epitope CII256–270, present on heterologous CII, displayed relatively stronger tolerance against the nonglycosylated variant (20). Whether persistence of glycopeptide-specific T cells in huCII/DR4 mice depends on the possibility that the glycopeptides bind with lower affinity to DR4 than the nonmodified peptide, as was observed for Aq (10), was not addressed, but could be considered as a reasonable explanation. It should be emphasized, however, that the glycosylated side chain is more likely to be oriented toward the T cell receptor rather than the MHC, as we show here that glycopeptide-specific T cell responses are critically dependent on the galactose moiety. This assumption is further supported from earlier studies using the Aq-molecule (10) and is also indicated from other studies using glycopeptide-specific T cells (38). As an alternative or complementary explanation for a biased tolerance, it has been shown that relatively small differences in availability or levels of autoantigens in vivo have great impact on the size or status of the autoreactive T cell repertoire (39, 40). Thus, different expression levels of the posttranslational forms of the CII263–270 epitope in endogenous CII might modulate the repertoire selection of CII-specific T cells. It should be emphasized, however, that also glycopeptide-specific T cells were influenced by endogenous huCII, although to a lesser degree than nonglycosylated CII-specific T cells. The response to the individual glycopeptides was only slightly weaker in huCII/DR4 mice than in DR4 mice, but the reduction was more obvious when comparing the response to the intact CII protein, indicating anergized or low affinity T cells. In addition, with the use of deoxy-peptides, we found that T cells from huCII/DR4 mice recognized the glycopeptide differently, indicating also that huCII influences T cell repertoire selection.
The mice used in this report are complex, because they carry multiple genetic modifications on a mixed genetic background, and translation of our in vitro findings to the human system should be done with some caution. Nevertheless, despite the complexity of the experimental set-up, our data on T cell tolerance in huCII/DR4 mice are intact and support earlier findings (15, 20, 37, 41). Collectively, these data strongly suggest that endogenous CII is physiologically exposed to the immune system but does not lead to complete tolerance of a subset of CII-specific T cells.
In summary, the results in a humanized animal model of RA and the functional assays on cellular autoimmunity in RA patients provide convincing evidence for incomplete T cell tolerance to a set of closely related self-determinants that are physiologically modified by posttranslational glycosylation. It should be stressed, however, that our data do not prove that the remaining CII-specific T cells are causative or even involved in the pathogenesis of RA. Despite this fact, our findings are interesting because they may constitute the missing link between the CII-specific humoral response, observed in a substantial fraction of RA patients, and the difficulties in the identification of CII-specific T cells in RA patients. Furthermore, T cell recognition of glycosylated CII in RA may be useful as a disease progression marker and for classification of this heterogenous disease.
Acknowledgments
We thank Carlos Palestro for taking good care of the animals, as well as Alexandra and Caroline Treschow for critically reading the manuscript. The work was supported by grants from the King Gustaf V's 80-Year Foundation, Kock's Foundations, Österlund's foundation, the Swedish Association against Rheumatism, the Swedish Medical Research Council, the Swedish Foundation for Strategic Research, the Göran Gustafsson Foundation, the Deutsche Forschungsgemeinschaft (SFB 263, project C3), the Bundesministerium für Bildung und Forschung (MedNet Entzündlich rheumatische Erkrankungen, project C 2.1; BMBF, 01GI9948), the European Commission (Bio4-98-0479), and the Karen Elise Jensen and Novo Nordisk Foundations in Denmark.
Abbreviations
- CII
type II collagen
- CIA
collagen-induced arthritis
- huCII
human type II collagen
- PBMC
peripheral blood mononucleated cells
- RA
rheumatoid arthritis
- mDR
T cell clones from mouse CII and DR4
- hDR
T cell clones from huCII/DR4
Footnotes
See commentary on page 9611.
References
- 1.Fugger L, Rothbard J B, Sonderstrup McDevitt G. Eur J Immunol. 1996;26:928–933. doi: 10.1002/eji.1830260431. [DOI] [PubMed] [Google Scholar]
- 2.Rosloniec E F, Brand D D, Myers L K, Esaki Y, Whittington K B, Zaller D M, Woods A, Stuart J M, Kang A H. J Immunol. 1998;160:2573–2578. [PubMed] [Google Scholar]
- 3.Andersson E C, Hansen B E, Jacobsen H, Madsen L S, Andersen C B, Engberg J, Rothbard J B, McDevitt G S, Malmström V, Holmdahl R, et al. Proc Natl Acad Sci USA. 1998;95:7574–7579. doi: 10.1073/pnas.95.13.7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kotzin B L, Falta M T, Crawford F, Rosloniec E F, Bill J, Marrack P, Kappler J. Proc Natl Acad Sci USA. 2000;97:291–296. doi: 10.1073/pnas.97.1.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim H Y, Kim W U, Cho M L, Lee S K, Youn J, Kim S I, Yoo W H, Park J H, Min J K, Lee S H, et al. Arthritis Rheum. 1999;42:2085–2093. doi: 10.1002/1529-0131(199910)42:10<2085::AID-ANR8>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 6.Michaëlsson E, Malmström V, Reis S, Engström Å, Burkhardt H, Holmdahl R. J Exp Med. 1994;180:745–749. doi: 10.1084/jem.180.2.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corthay A, Bäcklund J, Broddefalk J, Michaëlsson E, Goldschmidt T J, Kihlberg J, Holmdahl R. Eur J Immunol. 1998;28:2580–2590. doi: 10.1002/(SICI)1521-4141(199808)28:08<2580::AID-IMMU2580>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 8.Michaëlsson E, Andersson M, Engstrom Å, Holmdahl R. Eur J Immunol. 1992;22:1819–1825. doi: 10.1002/eji.1830220722. [DOI] [PubMed] [Google Scholar]
- 9.Brunsberg U, Gustafsson K, Jansson L, Michaëlsson E, Ahrlund Richter L, Pettersson S, Mattsson R, Holmdahl R. Eur J Immunol. 1994;24:1698–1702. doi: 10.1002/eji.1830240736. [DOI] [PubMed] [Google Scholar]
- 10.Kjellen P, Brunsberg U, Broddefalk J, Hansen B, Vestberg M, Ivarsson I, Engstrom Å, Svejgaard A, Kihlberg J, Fugger L, Holmdahl R. Eur J Immunol. 1998;28:755–767. doi: 10.1002/(SICI)1521-4141(199802)28:02<755::AID-IMMU755>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 11.Dessen A, Lawrence C M, Cupo S, Zaller D M, Wiley D C. Immunity. 1997;7:473–481. doi: 10.1016/s1074-7613(00)80369-6. [DOI] [PubMed] [Google Scholar]
- 12.Holmdahl R, Andersson M, Goldschmidt T J, Gustafsson K, Jansson L, Mo J A. Immunol Rev. 1990;118:193–232. doi: 10.1111/j.1600-065x.1990.tb00817.x. [DOI] [PubMed] [Google Scholar]
- 13.Holmdahl R, Vingsbo C, Mo J A, Michaëlsson E, Malmström V, Jansson L, Brunsberg U. Immunol Rev. 1995;144:109–135. doi: 10.1111/j.1600-065x.1995.tb00067.x. [DOI] [PubMed] [Google Scholar]
- 14.Fugger L, Michie S A, Rulifson I, Lock C B, McDevitt G S. Proc Natl Acad Sci USA. 1994;91:6151–6155. doi: 10.1073/pnas.91.13.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malmström V, Ho K K, Lun J, Tam P P, Cheah K S, Holmdahl R. Scand J Immunol. 1997;45:670–677. doi: 10.1046/j.1365-3083.1997.d01-446.x. [DOI] [PubMed] [Google Scholar]
- 16.Cosgrove D, Gray D, Dierich A, Kaufman J, Lemeur M, Benoist C, Mathis D. Cell. 1991;66:1051–1066. doi: 10.1016/0092-8674(91)90448-8. [DOI] [PubMed] [Google Scholar]
- 17.Andersson M, Holmdahl R. Eur J Immunol. 1990;20:1061–1066. doi: 10.1002/eji.1830200517. [DOI] [PubMed] [Google Scholar]
- 18.Broddefalk J, Bäcklund J, Almqvist F, Johansson M, Holmdahl R, Kihlberg J. J Am Chem Soc. 1998;120:7676–7683. [Google Scholar]
- 19.Holm B, Broddefalk J, Flodell S, Wellner E, Kihlberg J. Tetrahedron. 2000;56:1579–1586. [Google Scholar]
- 20.Malmström V, Bäcklund J, Jansson L, Kihlberg J, Holmdahl R. Arthritis Res. 2000;2:315–326. doi: 10.1186/ar106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arnett F C, Edworthy S M, Bloch D A, McShane D J, Fries J F, Cooper N S, Healey L A, Kaplan S R, Liang M H, Luthra H S, et al. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 22.Assenmacher M, Schmitz J, Radbruch A. Eur J Immunol. 1994;24:1097–1101. doi: 10.1002/eji.1830240513. [DOI] [PubMed] [Google Scholar]
- 23.Assenmacher M, Lohning M, Scheffold A, Manz R A, Schmitz J, Radbruch A. Eur J Immunol. 1998;28:1534–1543. doi: 10.1002/(SICI)1521-4141(199805)28:05<1534::AID-IMMU1534>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 24.Stastny P. N Engl J Med. 1978;298:869–871. doi: 10.1056/NEJM197804202981602. [DOI] [PubMed] [Google Scholar]
- 25.Gregersen P K, Silver J, Winchester R J. Arthritis Rheum. 1987;30:1205–1213. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- 26.Diab B Y, Lambert N C, L'Faqihi F E, Loubet-Lescoulie P, de Preval C, Coppin H. Immunogenetics. 1999;49:36–44. doi: 10.1007/s002510050461. [DOI] [PubMed] [Google Scholar]
- 27.Rosloniec E F, Brand D D, Myers L K, Whittington K B, Gumanovskaya M, Zaller D M, Woods A, Altmann D M, Stuart J M, Kang A H. J Exp Med. 1997;185:1113–1122. doi: 10.1084/jem.185.6.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trentham D E, Dynesius R A, Rocklin R E, David J R. N Engl J Med. 1978;299:327–332. doi: 10.1056/NEJM197808172990703. [DOI] [PubMed] [Google Scholar]
- 29.Stuart J M, Postlethwaite A E, Townes A S, Kang A H. Am J Med. 1980;69:13–18. doi: 10.1016/0002-9343(80)90494-5. [DOI] [PubMed] [Google Scholar]
- 30.Solinger A M, Bhatnagar R, Stobo J D. Proc Natl Acad Sci USA. 1981;78:3877–3881. doi: 10.1073/pnas.78.6.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Londei M, Savill C M, Verhoef A, Brennan F, Leech Z A, Duance V, Maini R N, Feldmann M. Proc Natl Acad Sci USA. 1989;86:636–640. doi: 10.1073/pnas.86.2.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ofosu-Appiah W A, Warrington R J, Wilkins J A. Clin Immunol Immunopathol. 1989;50:264–271. doi: 10.1016/0090-1229(89)90134-7. [DOI] [PubMed] [Google Scholar]
- 33.Lacour M, Rudolphi U, Schlesier M, Peter H H. Eur J Immunol. 1990;20:931–934. doi: 10.1002/eji.1830200432. [DOI] [PubMed] [Google Scholar]
- 34.Yan T, Burkhardt H, Ritter T, Broker B, Mann K H, Bertling W M, von der Mark K, Emmrich F. Eur J Immunol. 1992;22:51–56. doi: 10.1002/eji.1830220109. [DOI] [PubMed] [Google Scholar]
- 35.Snowden N, Reynolds I, Morgan K, Holt L. Arthritis Rheum. 1997;40:1210–1218. doi: 10.1002/1529-0131(199707)40:7<1210::AID-ART4>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 36.Berg L, Ronnelid J, Sanjeevi C B, Lampa J, Klareskog L. Arthritis Res. 2000;2:75–84. doi: 10.1186/ar71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malmström V, Michaëlsson E, Burkhardt H, Mattsson R, Vuorio E, Holmdahl R. Proc Natl Acad Sci USA. 1996;93:4480–4485. doi: 10.1073/pnas.93.9.4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rudd P M, Elliott T, Cresswell P, Wilson I A, Dwek R A. Science. 2001;291:2370–2376. doi: 10.1126/science.291.5512.2370. [DOI] [PubMed] [Google Scholar]
- 39.Cibotti R, Kanellopoulos J M, Cabaniols J P, Halle-Panenko O, Kosmatopoulos K, Sercarz E, Kourilsky P. Proc Natl Acad Sci USA. 1992;89:416–420. doi: 10.1073/pnas.89.1.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akkaraju S, Ho W Y, Leong D, Canaan K, Davis M M, Goodnow C C. Immunity. 1997;7:255–271. doi: 10.1016/s1074-7613(00)80528-2. [DOI] [PubMed] [Google Scholar]
- 41.Malmström V, Kjellen P, Holmdahl R. J Autoimmun. 1998;11:213–221. doi: 10.1006/jaut.1998.0198. [DOI] [PubMed] [Google Scholar]