

Abstract
A core region surrounded by a rim characterizes biological interfaces. We ascertain the importance of the core by showing the sequence entropies of the residues comprising the core to be smaller than those in the rim. Such a distinction is not seen in the 2-fold-related, nonphysiological interfaces formed in crystal lattices of monomeric proteins, thereby providing a procedure for characterizing the oligomeric state from crystal structures of protein molecules. This method is better than those that rely on the comparison of the sequence entropies in the interface and the rest of the protein surface, especially in cases where the surface harbors additional binding sites. To a good approximation there is a correlation between the accessible surface area lost because of complexation and ΔΔG values obtained through alanine-scanning mutagenesis (26-38 cal per Å2 of the surface buried) for residues located in the core, a relationship that is not discernable for rim residues. If, however, a residue participates in hydrogen bonding across the interface, the extent of stabilization is 52 cal/mol per 1 Å2 of the nonpolar surface area buried by the residue. As opposed to an amino acid classification used earlier, an environment-based grouping of residues yields a better discrimination in the sequence entropy between the core and the rim.
Keywords: protein-protein interaction, hot spots in the interface, residue conservation, crystal packing, quaternary structure prediction
Evolutionary conservation of amino acid residues may include active-site residues pertaining to the function of the molecule or tightly packed sites contributing to the stable core or indicative of the folding nucleus (1-4). For cellular function proteins need to interact with other molecules, and techniques such as the two-hybrid system and affinity purifications are being used to discover the physical association between proteins, leading to a fascinating view of cell-interaction maps (5, 6). Depending on the binary or even higher order of interaction, a large fraction of the protein surface is involved in molecular recognition and binding. Structurally conserved residues can also distinguish between binding sites and exposed protein surfaces (7-11). Indeed, the true oligomeric protein-protein contacts can be discriminated from nonspecific crystal contacts based on evolutionary consideration (12, 13).
Biophysical characterization of protein-binding interfaces has been achieved through alanine-scanning mutagenesis (14-16). A hot spot has been defined as a residue that when mutated to alanine leads to a significant drop in the binding constant (typically 10-fold or higher), as determined by the change in the free energy of binding (ΔΔG). A wealth of data on alanine scans is now accessible through the internet (17) for systematic analyses (18). There has also been development in computational methods for the prediction of the experimentally measured free-energy change by alanine substitution (19, 20). Although these methods incorporate free-energy function that includes terms that have been found to be important for protein stability, a simple physical model for characterizing protein-interaction hot spots is still lacking.
The role of hydrophobic residues in protein-protein recognition and the formation of multimeric protein assembly has long been recognized (21-23). A quantitative enumeration of the hydrophobic patches on the interface was achieved by dissecting it into core and rim regions, the former containing residues with some atoms fully buried in the interface, whereas the latter only contains atoms that retain partial accessibility (24, 25). The core possesses more hydrophobic residues and has a composition that is distinct from the rim or the rest of the protein surface. With the division into core and rim residues one can ask the question of whether as two groups these residues have different contributions to binding energy, and consequently have different evolutionary pressure for their conservation. The degree of conservation of each interface residue can be defined in terms of sequence entropy at that position in the polypeptide chain across all of the homologous proteins.
At least three types of contacts can be identified from the protein structures deposited in the Protein Data Bank (PDB) (26): (i) contacts between components of protein-protein complexes (24, 27); (ii) contacts between the subunits of homodimeric (and higher oligomeric) proteins (25, 28); and (iii) contacts between monomeric protein molecules in their crystal lattice (29, 30). Specific, biologically relevant interactions occur between the interfaces of the first two types, whereas those resulting from molecular packing in protein crystals are nonspecific.
Although the physiological interfaces generally have a core region surrounded by a partially exposed rim region, the interfaces formed in crystals are usually devoid of such features and are fragmented (30). If one still uses the same accessibility criterion to define the core and rim residues in crystal interfaces, even though they may not physically form the core and rim of the interface and then define sequence entropy for the two groups, the result would provide a benchmark against which the degree of conservation in the two regions of the physiological interfaces can be assessed.
Finally, the results of alanine-scanning mutagenesis can be mapped onto the location of the residue in the core and rim regions to correlate thermodynamic data with structural data obtained from crystallography. While calculating the sequence entropy, a classification of amino acids is used, which is found to provide a better contrast between core and rim residues than those used earlier.
Methods
Data Sets Used. The atomic coordinates of the protein chains were obtained from the PDB located at the Research Collaboratory for Structural Bioinformatics (26). The specific and physiological relevant interfaces are represented by 122 homodimers (25) and 70 heterocomplexes (24). The nonspecific interfaces are the 2-fold (crystallographic or noncrystallographic) symmetry-related contacts in crystals of 103 monomeric proteins, but having interface areas that lie in the realm of physiological interfaces (30).
Sequence Alignments. The multiple sequence alignments for each protein studied were extracted from the HSSP (homology-derived secondary structure of proteins) database of sequence-structure alignments (31). The database provides for each PDB entry a list of protein sequences deemed structurally homologous to it on the basis of a homology-threshold curve.
Classification of Amino Acid Residues. The multiple sequence alignment files of the HSSP database contain estimates of sequence variability at each position along the sequence. However, by not making concessions for conservative mutations the scheme becomes too rigid. Unlike unconservative mutations, conservative ones preserve the essential nature of the side chain, and, hence, we may make some discounts for such mutations. Various classification schemes have been used in the literature. For example, following the suggestion of Mirny and Shakhnovich (1), Elcock and McCammon (12) divided the 20 aa into the following six groups: (i) Arg and Lys; (ii) Asp and Glu; (iii) His, Phe, Trp, and Tyr; (iv) Asn, Gln, Ser, and Thr; (v) Ala, Ile, Leu, Met, Val, and Cys; and (vi) Gly and Pro. Saha et al. (32) made a classification based on the similarity of the environment of each amino acid residue in protein structures, and we use a slightly modified version consisting of the following groups: (i) Ala, Val, Leu, Ile, Met, and Cys; (ii) Gly, Ser, and Thr; (iii) Asp and Glu; (iv) Asn and Gln; (v) Arg and Lys; (vi) Pro, Phe, Tyr, and Trp; and (vii) His.
Calculation of Sequence Entropy. We have used Shannon's information theoretic entropy to measure the variability at a particular position in a given protein sequence. Sequence entropy is given by the following expression:
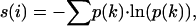 |
where p (k) is the probability that the ith position in the sequence is occupied by a residue of class k. A low value of sequence entropy, s(i), at position i in the multiple sequence alignment implies that the position has been subjected to relatively higher evolutionary conservation than another position in the same alignment having a higher sequence entropy value.
Calculation of Mean Sequence Entropies for the Core and Rim. The amino acid residues comprising the interface were segregated into core and rim based on the solvent accessibility of their constituent atoms in the bound state, the former type of residues having fully buried atoms, whereas the latter contain atoms that remain partially exposed to solvent (24). For calculating the mean sequence entropy of the core (or rim) region of the interface we considered the fractional contribution of each residue to the total interface area to correctly weigh the entropy values (12). The usefulness of this step lies in the fact that highly divergent or highly conserved positions should not be allowed to sharply increase or decrease the mean sequence entropies unless they form a significant part of the interface as evidenced by the fraction of interface area occupied by the corresponding residues. The expression used is given below.
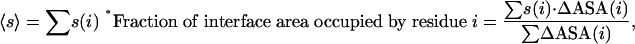 |
[1] |
where ΔASA(i) is the solvent accessible surface area of residue i that is buried on interface formation, and depending on the location of i in the core or rim, the summation is over that subset of interface residues; 〈s 〉 is the mean sequence entropy of the core (or rim) residues. Because of symmetry, the summation is over residues in one subunit for homodimers, whereas for the other interfaces both the chains in the assembly were considered. ASA values were computed with the program naccess (33), which implements the algorithm of Lee and Richards (34).
Although we used the above formula, we also calculated simple averages by using
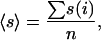 |
[2] |
where n is the number of core (or rim) residues, and found the parameter to be as discriminating as the one given by Eq. 1.
As the absolute value of mean sequence entropy (〈s 〉core or 〈s 〉rim) varies considerably between different proteins because of the variation in the number of homologs in a protein family and the evolutionary distance between their sequences, we used the ratio 〈s 〉core/ 〈s 〉rim to compare between proteins.
Results
Conservation of Core vs. Rim Residues in the Interfaces of Protein-Protein Complexes, Homodimers, and Crystal Contacts in Monomeric Proteins. Multiple sequence alignment for a few proteins in our database were not available in HSSP, and as such the mean sequence entropies could be calculated only for 121 homodimers, 69 heterocomplexes, and 102 crystal contacts. The average numbers of aligned sequences present in each HSSP file were 281, 1,380, and 444, respectively, for the three categories and the sequence identities ranged between 0.3 and 1.0. 〈s 〉core and 〈s 〉rim of each protein are plotted in Fig. 1, and a point above the diagonal indicates that 〈s 〉core is less than 〈s 〉rim, implying a core that is more conserved than the rim. Considering the nonorigin points, 73.6% (89/118) of the homodimers and 68.1% of the complexes are of this type; the average 〈s 〉core/ 〈s 〉rim ratio of <1.0 (Table 1) is indicative of the same fact. In contrast, only 50% of crystal contacts exhibit this behavior; i.e., points are divided equally above and below the diagonal. When using the Student's t test for paired samples, a P value of <0.01 implies that the observed difference between 〈s 〉core and 〈s 〉rim has a probability of <1% to occur by mere chance. P values for the homodimeric and complex datasets are both several orders of magnitude less than this value, implying that the observed difference between 〈s 〉core and 〈s 〉rim is highly significant. On the other hand, for the monomer data set the P value of 0.4 indicates that for crystal contacts there is no significant difference between the mean sequence entropies of the core and the rim.
Fig. 1.

Plot of mean sequence entropies 〈s 〉core and 〈s 〉rim of the core and rim regions in the interfaces in homodimers (a), protein-protein complexes (b), and crystal contacts of monomeric proteins (c). In a few cases (especially, when the protein family contains only a few members), 〈s 〉core and 〈s 〉rim are both zero. The numbers of points lying on the origin in the three plots are 3, 0, and 6, respectively. In b, ○ indicates antigen-antibody complexes.
Table 1. Comparison of 〈s〉core and 〈s〉rim values with statistics.
| Average*
|
No. of interfaces
|
||||||
|---|---|---|---|---|---|---|---|
| Interface type | Average no. of core/rim residues per interface | 〈s〉core | 〈s〉rim | 〈s〉core/〈s〉rim | Total | With 〈s〉core less than 〈s〉rim | Student's t test for paired samples, P values† |
| Homodimers | 32/21 | 0.63 (0.3) | 0.77 (0.4) | 0.87 (0.5) | 121 | 89 | 1.23E-13 |
| Complexes | 30/26 | 0.78 (0.4) | 0.88 (0.4) | 0.90 (0.3) | 69 | 47 | 1.56E-04 |
| Complexes excluding antigen—antibody | 32/26 | 0.65 (0.3) | 0.80 (0.3) | 0.82 (0.3) | 52 | 40 | 9.34E-07 |
| Crystal contacts in monomers | 13/19 | 0.98 (0.4) | 0.99 (0.4) | 1.14 (1.1) | 102 | 49 | 4.04E-01 |
SD is in parentheses
The P values show the significance levels for the one-tailed t test corresponding to the hypothesis that 〈s〉core is less than 〈s〉rim. If Eq. 2 is used to calculate the mean sequence entropy, the values for the last two columns for the four entries would be (95, 2.57E-20), (51, 2.21E-07), (47, 3.2E-11), and (49, 9.35E-02)
Although antigen-antibody complexes constitute a rich structural repertoire for analyzing the physicochemical features of specific interactions in biology (24), these may not be the right candidates for studying the evolutionary pressure on interface residues. Although the active sites of enzymes must be conserved for retaining their function, antibody sequences, on the other hand, must mutate and diversify for recognizing a wide arsenal of antigens. Indeed, Fig. 1b shows that the sequence entropies at both the core and rim are quite high for most of the 17 antibody-antigen complexes, and unlike the remaining ones these points occur predominantly below the diagonal line. If these points are excluded from the complex data set, there is a definite reduction of noise in the statistics, with 〈s 〉 values getting closer to those obtained for homodimers (Table 1).
The absolute values of mean entropies (core and rim) can vary within a certain range but the mean entropy for the core is less than the corresponding value for the rim in most cases when dealing with biological interfaces, which is not true when working with nonbiological ones. The distribution of 〈s 〉core/ 〈s 〉rim values is shifted toward the right for the crystal contacts, with the peak occurring at 1 (Fig. 2). In addition to the entropy ratio, the absolute value of 〈s 〉core and 〈s 〉rim is indicative of whether one is dealing with a homodimeric interface or a crystal interface; in Fig. 1a, 71% of points have both the entropy values <1.0, whereas in Fig. 1c the corresponding value is only 29%.
Fig. 2.

Histogram of the ratio of the mean sequence entropies, 〈s 〉core/ 〈s 〉rim, for the interfaces in homodimers, complexes, and crystal contacts.
The striking similarity of the division of a specific interface into core and rim regions and the separation based on the degree of conservation of interface residues can be seen in Fig. 3. The use of Eq. 2 for the calculation of the mean entropies (unweighted) provides essentially the same result; 78.5% (95/118) of the homodimers, 73.9% of the heterodimers, and 50% of the crystal contacts have 〈s 〉core/ 〈s 〉rim ratios <1.0.
Fig. 3.

Looking at the interface of one subunit of the homodimeric protein 2-phospho-d-glycerate hydrolase (PDB ID code 1ebh). (a) The interface is divided into core (in red) and rim (in blue) regions. (b) The sequence entropies of the interface residues are indicated, with the color code provided on top (red stands for the maximum conservation and blue, the minimum). The molecular surface representations have been made with grasp (43).
Proper Choice of Amino Acid Classes and the Achievement of a Better Discrimination Between the Core and the Rim Residues. Consideration of different properties has led to different classifications of amino acid residues, which are useful in the derivation of reduced sets of protein alphabets and the simplification of the theoretical simulation of the folding process (35). In their work on sequence conservation Mirny and Shakhnovich (1) and Elcock and McCammon (12) divided amino acids into six groups. We used a classification that has been derived on the basis of similarity of the residue environment in protein structures (32), the underlying assumption being that during evolution the residue contacts across the interface should have been optimized to match what is found within protein tertiary structures. We used both classifications to calculate 〈s 〉core and 〈s 〉rim, and the difference is plotted for the homodimeric interfaces (Fig. 4). Considering the points (85 in number) which lie in the positive quadrant, 73% have a greater value of 〈s 〉rim - 〈s 〉core (points below the line) with our classification than that obtained with the earlier classification, suggesting that the contrast in the conservation between core and rim residues is more with our classification.
Fig. 4.

Plot of (〈s 〉rim - 〈s 〉core), the difference between the mean sequence entropies of the rim and core regions (for the data set of homodimers), obtained by using our classification of amino acid residues and an earlier classification (1, 12).
Comparison of Core vs. Rim, Vis-à-Vis the Whole Interface vs. the Rest of the Protein Surface. A procedure that compares the core and rim regions of a given interface should be more powerful for predicting the biological relevance of an observed interface than all of the commonly used methods that invariably rely on comparing the interface with the rest of the protein surface. The latter criterion may give anomalous results in cases where the exposed surface contains binding sites for other molecules. Notwithstanding the physiological relevance of a particular interface if the remainder of the protein surface serves as binding site(s) for other molecules then parts of the surface will also be subjected to evolutionary conservation and the interface-to-surface entropy ratio will increase, diminishing the signal of the conserved residues emanating from the interface under study. In contrast, each interface is considered by itself in the present study, and the existence of other binding sites will not affect the results. To show the efficacy of our method, we compare our results with those obtained by using the criterion of the interface-to-noninterface entropy ratio for nine “known” dimeric proteins (12) (Table 2). Using our method we note a marked improvement in the conservation score of five proteins. In all of these cases, a considerable number of residues not part of the homodimerization interface are also under selective evolutionary pressures to maintain the integrity of functional interactions and resisting mutations, thereby resulting in a rather high interface-to-noninterface entropy ratio. The enzyme aldehyde ferredoxin oxidoreductase (PDB ID code 1aor) has binding sites for four cofactors, as well as its electron transfer partner, necessitating a large part of the surface to be well conserved, with the result that the interface-to-noninterface ratio is quite high (1.49). IL-8 (PDB ID code 1icw) is a cytokine, and its signaling activity depends on its receptor binding activity. Although interface-to-surface comparison results in a rather high value (1.15), our method gives a much smaller mean core-to-rim entropy ratio of 0.66. Thus, to discern a true homodimeric interface it is better to study the interface per se, with no consideration to the remaining surface.
Table 2. Analysis of “known” dimeric proteins with interfaces that are poorly conserved relative to the rest of the protein surface.
| Conservation parameter
|
|||
|---|---|---|---|
| PDB code | Protein name | EM 〈s〉interface/〈s〉noninterface | Ours 〈s〉core/〈s〉rim |
| 1aor | Aldehyde ferredoxin oxidoreductase | 1.49 | 1.07 |
| 1icw | IL-8 | 1.15 | 0.66 |
| 1czj | Octaheme cytochrome | 1.09 | 1.01 |
| 1pre | Proaerolysin toxin | 1.09 | 0.79 |
| 2tct | Tetracycline repressor | 1.07 | 1.07 |
| 1afw | 3-Ketoacetyl-CoA thiolase | 0.99 | 0.83 |
| 1smt | Smtb transcriptional repressor | 0.98 | 0.95 |
| 1alk | Alkaline phosphatase | 0.97 | 0.90 |
| 1a3c | Pyrimidine biosynthetic operon repressor | 0.90 | 0.60 |
Taken from table 4 of ref. 12, which also defines the parameter EM.
Contribution of the Side Chain of a Residue to ΔΔG. We wanted to see whether the higher degree of conservation of core residues matches with the experimental ΔΔG values obtained from alanine-scanning mutagenesis. For this process, we extracted information for 262 mutations in 13 protein-protein complexes (details in the legend to Fig. 5), as available from ASEdb, the Alanine Scanning Energetics database (17), and found their location in the core or the rim region of the interfaces. If we consider hot-spot residues as those that show a ΔΔG value of 2 kcal/mol or more upon mutation to alanine, there are 37 such hot spots in the core region and 11 in the rim. Averaging over the residues in the two regions gave a value of 1.4 (1.7) kcal/mol for the core and 0.8 (1.2) kcal/mol for the rim. Although the SDs are rather large, the values suggest that the core and the rim residues could contribute differently to the free energy of binding. As such we split the mutated residues into two groups, those belonging to the core or the rim, and examined the relationship of their ΔASA and ΔΔG values, where ΔASA represents the difference between the solvent ASA of the side chain in the complex and in the separated monomer. Instead of plotting the individual points, we combined all of the points in bins of size 20 Å2 in ΔASA and plotted their average ΔΔG values. ΔASA values provided in ASEdb include Cα atoms in the calculation, whereas we wanted to focus on the effect of only the side chain. We calculated ΔASA in two ways, by considering the contribution of the side chain (atoms Cβ onward) of the residue to the interface area (Fig. 5a) and that of the nonpolar carbon atoms (Cγ onward) (Fig. 5b). The logic underlying the second method is that the effect of the mutation of a long side chain to Ala should be caused by the atoms that lie beyond Cβ (for Ala), and by restricting to C atoms we are highlighting only the hydrophobic effect. The correlation coefficients for the points involving the core and rim residues for the two plots indicate large positive correlations only for the core residues. If one had directly used the ΔASA values from ASEdb in a similar plot, the correlation coefficient would have been inferior (0.77 for the core residues) to those obtained here. Linear regression analyses gave the quantitative estimate of the dependence between the two parameters, 26 cal/mol (in Fig. 5a) and 38 cal/mol (Fig. 5b) per1Å2 of the surface area buried.
Fig. 5.

Relationship between ΔASA and the change in free energy of binding (ΔΔG). (a and b) Along the x axis all of the values in a bin (of size 20 Å2) are pulled together and shown in the middle, while the y value corresponds to the mean of their ΔΔG values (the vertical bars representing the SDs). Values for the core and rim residues are marked in red and blue, respectively. The correlation coefficient (cc) between the variables and the equation for the least-squares line (passing through the origin) for the core residues are indicated. ΔASA values are calculated differently in the two plots. In a, ΔASA corresponds to the contribution of the side chain (Cβ onward) of a residue to the interface area, whereas in b only the contribution from hydrophobic atoms (Cγ onward) is considered. c is similar to b, except that the segregation into two categories is on the basis of the presence (red) or absence (black) of hydrogen bonding across the interface. Details (PDB ID code, mutated protein/partner protein, the number of mutations in interface/of the total available) of alanine-scanning mutagenesis data (17) used are: 1a4y, angiogenin/RNase inhibitor, 13/14; 1a4y, RNase inhibitor/angiogenin, 9/14; 1brs, barnase/barstar, 7/8; 1brs, barstar/barnase, 5/6; 1cbw, bovine pancreatic trypsin inhibitor (BPTI)/chymotrypsin, 7/9; 2ptc, BPTI/trypsin, 1/1; 1gc1, CD4/gp120, 17/49; 1dvf, D1.3/E5.2, 16/16; 1dvf, E5.2/D1.3, 9/9; 1vfb, D1.3/hen egg lysozyme (HEL), 14/17; 1vfb, HEL/D1.3, 12/12; 1dan, factor VII/tissue factor, 12/107; 1dan, tissue factor/factor VIIa, 33/78; 3hfm, HEL/HyHEL-10, 13/13; 3hfm, HyHEL-10/HEL, 3/3; 3hhr, human growth hormone (hGH)/hGH receptor (hGHbp),site1, 30/31; 3hhr, hGH/hGHbp,site?, 10/31; 3hhr, hGHbp,site1/hGH, 26/67; 1bxi, Im9/E9 DNase, 17/28; 1fc2, protein A/IgG1, 0/3; and 1ahw, tissue factor/Fab 5G9, 8/8.
We also examined whether a similar relationship exists for hydrogen-bonded residues. Of the 262 residues under study, 96 are hydrogen-bonded for a total of 117 interactions, which are of the following categories: 70 core-core, 32 core-rim, 9 rim-rim, and 6 involved interactions with a noninterface residue. Because of the paucity of data it was not feasible to split them into core and rim. Considering the individual points (plot not shown), the correlation coefficient is only 0.25, which changes to 0.43 and 0.19, if hydrogen-bonded and nonhydrogen-bonded residues are considered separately. ΔASA calculated by the second method given above (considering the hydrophobic atoms beyond Cβ) was used, as it gave better distinction between hydrogen-bonded and nonhydrogen-bonded residues, as compared with ΔASA calculated by the first method or that available in ASEdb. As done previously, if the points are grouped in bins (Fig. 5c), the correlation coefficient for the hydrogen-bonded set shows a remarkable improvement. The least-squares line indicates that for a hydrogen bond across the interface the free energy of binding would be 52 cal/mol per 1 Å2 of the nonpolar surface of the residue being buried. Assuming a 50-Å2 burial of the surface area for the residue, a hydrogen-bonding amounts to ≈2.5 kcal/mol.
Discussion
Different Levels of Residue Conservation in the Core and the Rim of Protein Interfaces and the Discrimination of True Oligomeric Protein-Protein Contacts from Nonspecific Crystal Contacts. To understand the process of biological molecular recognition it is important to characterize the residues that form the interface between two protein chains when they associate and to delineate their relative importance in the binding process. The latter can be judged from the degree of conservation of residues in a series of homologous proteins. The biological interfaces considered here are of two types, the first type occurring in homodimers and the second forming when two independent molecules form a complex. In these cases, the interfaces can be dissected into a core region surrounded by a rim (24, 25). The question that we address is whether this spatial dissection is also reflected in the magnitude of the sequence entropies of the residues in the two regions. If the average sequence entropy in the core is less than for the rim (i.e., the residues in the former region are more conserved than those in the latter), we get a core-to-rim entropy ratio <1.0, which is found to be true in the majority of the cases in both data sets (Fig. 1 a and b). Fig. 3 shows the match between the degree of conservation and the core/rim definition in a specific example. Caffrey et al. (11), and recently, Bordner and Abagyan (36), have observed that the central interface residues are more conserved and thus more strongly predicted as belonging to the interface than the peripheral ones. Whereas these studies were restricted to obligate interfaces, we have also included protein-protein complexes. Additionally, a distance-based criterion used in ref. 36 means that the selection of central residues would depend on the shape and size of the interface and 25% of the interfaces do not have even six central residues. In contrast, our definition of the core is less restrictive and, in fact, it has more residues than the rim (Table 1).
The data set of monomeric proteins possessing crystal contacts across 2-fold axes of symmetry is used as a means of confirming whether our method can distinguish between biological and crystal contacts. In the case of the monomeric proteins, the surfaces come together in the crystal structure in an unpredictable manner. A monomeric protein may crystallize in many different crystal forms in which different regions of the surface are brought together to form the interfaces, which taken together essentially cover the whole protein surface (37). As such, the nonspecific crystal interfaces may or may not contain, wholly or partially, any binding site (for a substrate or a cofactor or another protein) that the molecule may have, and these functional epitopes, containing selectively conserved residues, may be anywhere (core or rim) of the interface. Hence, for the monomer data set a random distribution of the core-to-rim entropy ratio is expected, which is what is seen (Fig. 1c). This finding implies that the core-to-rim mean entropy ratio can be used as a criterion for distinguishing between physiologically relevant interfaces and those that are generated during the crystallization process (Table 1 and Fig. 2). The utility of the method lies in the fact that only the concerned interface needs to be analyzed with no need of additional information on the other regions of the protein surface, which increases the efficiency in situations where the protein harbors additional binding sites (Table 2).
Correlation Between Buried Area and Experimental ΔΔG. Alanine-scanning mutagenesis is frequently used as an experimental probe to determine the contribution of a residue to the binding affinity. Although there is little correlation between the buried surface area of the side chain and the free energy of binding (18), we wanted to see whether the situation improves on separating the data based on the location of the residues in the core or the rim, especially because the residues that are important for binding energetics tend to occur more near the center of the interface than at the edges. We selected from the ASEdb only those mutations that occurred at the interface of protein heterodimers and for whom the 3D structures were known. For each experimentally mutated residue that forms part of the protein-protein heterodimer interface, ASEdb gives us the corresponding ΔΔG value representing the change in free energy of binding upon mutation to alanine. Residues that contribute a large amount of binding energy are characterized by a large positive value of ΔΔG. Such residues have been labeled hot spots of binding energy. Of the 262 residues from the ASEdb considered here, 48 are hot spots, of which 77% are in the core. Among the 48 hot-spot residues 67% participate in hydrogen bonding (mostly through the side chain, 94%) across the interface, whereas only 30% of the 214 non-hot-spot residues are hydrogen-bonded. This finding indicates that the location in the core and simultaneous involvement in hydrogen bonding make a residue critical for binding.
Calculating ΔASA in two different ways, we find a very good correlation between ΔASA and ΔΔG (Fig. 5 a and b), with Fig. 5b giving a better correlation coefficient involving the core residues. Surprisingly, there is an anticorrelation for the rim residues. From the equations of the fitted lines, we get a value of 26-38 cal/mol per 1 Å2 of the buried area in the core region. Although the SDs associated with ΔΔG are large, indicating that there are factors other than the burial of the surface area that also contribute to ΔΔG, the burial of nonpolar area appears to be dominant. It is interesting to note that the effect of the burial of the surface during protein-protein association is quite similar to the hydrophobic contribution to the free energy of protein folding, which has been found to be ≈24 cal for each Å2 removed from contact with water (38, 39). Along the same line we wanted to find the relationship between the two parameters when the residues are involved in hydrogen bonding across the interface. Because of the paucity of the data we considered all mutations irrespective of their location in the core or the rim. The value obtained, 52 cal/mol per 1 Å2 of the buried area (Fig. 5c), indicates that hydrogen bonding may provide about twice the stabilization energy than the packing interactions of apolar residues. The higher value (≈2.5 kcal/mol per 50 Å2 of the buried surface) also conforms to the fact that the majority of the hot-spot residues participate in hydrogen bonds.
It is instructive to view the results from other studies from the perspective of this work. Chakravarty and Varadarajan (40) found that the change in free energy of association on mutation had a stronger correlation with the depth of the residue than with ΔASA. This finding is in accordance with our results, which indicate that the core residues, which would necessarily have a greater depth as compared with those in the rim, contribute more to binding. Likewise, the observation by Halperin et al. (41) that the hot spots are organized in regions of high packing density is essentially a manifestation of their predominant occurrence in the core region, which would have a greater number of contacts across the interface. By correlating structural changes on mutation with binding free energies in an antigen-antibody complex, Li et al. (42) observed that the hydrophobic interaction at a central site was 46 cal/mol per 1 Å2, twice that at the periphery. This value is quite close to 38 cal/mol per 1 Å2 obtained from Fig. 5b. According to the O-ring hypothesis, hot-spot residues are surrounded by less important peripheral residues that serve to occlude bulk solvent. Instead of individual hot-spot residues, our model visualizes a core in which the contribution of a residue to the free energy of binding depends on the hydrophobic area buried, 26-38 cal/mol per 1 Å2 (Fig. 5 a and b) or 52 cal/mol per 1 Å2 if there is a hydrogen bond formed across the interface (Fig. 5c).
Summing up, we show that for biological interfaces, the core is more conserved than the rim region, and for interfaces that are vestiges of crystallization procedures, the results are random. As the core residues are easily identifiable from a crystal structure, the calculation of sequence entropy provides an elegant way of ascertaining whether a particular interface observed in the crystal corresponds to a biologically relevant assembly, without taking recourse to the features in the rest of the protein surface. Moreover, the surface area buried by a core residue provides an estimate of its contribution toward the free energy of binding.
Acknowledgments
This work was supported by the Department of Biotechnology, Ministry of Science and Technology, New Delhi.
Author contributions: P.C. designed research; M.G. performed research; M.G. and P.C. analyzed data; and M.G. and P.C. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: PDB, Protein Data Bank; ASA, accessible surface area; ΔASA, the change in ASA on complexation; ASEdb, Alanine Scanning Energetics database.
References
- 1.Mirny, L. A. & Shakhnovich, E. I. (1999) J. Mol. Biol. 291, 177-196. [DOI] [PubMed] [Google Scholar]
- 2.Ptitsyn, O. B. & Ting, K.-L. H. (1999) J. Mol. Biol. 291, 671-682. [DOI] [PubMed] [Google Scholar]
- 3.Panchenko, A. R., Kondrashov, F. & Bryant, S. (2004) Protein Sci. 13, 884-892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liao, H., Yeh, W., Chiang, D., Jernigan, R. L. & Lustig, B. (2005) Protein Eng. Design Selection 18, 59-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salwinski, L. & Eisenberg, D. (2003) Curr. Opin. Struct. Biol. 13, 377-382. [DOI] [PubMed] [Google Scholar]
- 6.Aloy, P. & Russell, R. B. (2004) Nat. Biotechnol. 22, 1317-1321. [DOI] [PubMed] [Google Scholar]
- 7.Lichtarge, O., Bourne, H. R. & Cohen, F. E. (1996) J. Mol. Biol. 257, 342-358. [DOI] [PubMed] [Google Scholar]
- 8.Valdar, S. J. & Thornton, J. M. (2001) Proteins Struct. Funct. Genet. 42, 108-124. [PubMed] [Google Scholar]
- 9.Lichtarge, O. & Sowa, M. E. (2002) Curr. Opin. Struct. Biol. 12, 21-27. [DOI] [PubMed] [Google Scholar]
- 10.Ma, B., Elkayam, T., Wolfson, H. & Nussinov, R. (2003) Proc. Natl. Acad. Sci. USA 100, 5772-5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caffrey, D. R., Somaroo, S., Hughes, J. D., Mintseris, J. & Huang, E. S. (2004) Protein Sci. 13, 190-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elcock, A. H. & McCammon, J. A. (2001) Proc. Natl. Acad. Sci. USA 98, 2990-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valdar, S. J. & Thornton, J. M. (2001) J. Mol. Biol. 313, 399-416. [DOI] [PubMed] [Google Scholar]
- 14.Clackson, T. & Wells, J. A. (1995) Science 267, 383-386. [DOI] [PubMed] [Google Scholar]
- 15.DeLano, W. L., Ultsch, M. H., de Vos, A. M. & Wells, J. A. (2000) Science 287, 1279-1283. [DOI] [PubMed] [Google Scholar]
- 16.DeLano, W. L. (2002) Curr. Opin. Struct. Biol. 12, 14-20. [DOI] [PubMed] [Google Scholar]
- 17.Thorn, K. S. & Bogan, A. A. (2001) Bioinformatics 17, 284-285. [DOI] [PubMed] [Google Scholar]
- 18.Bogan, A. A. & Thorn, K. S. (1998) J. Mol. Biol. 280, 1-9. [DOI] [PubMed] [Google Scholar]
- 19.Kortemme, T. & Baker, D. (2002) Proc. Natl. Acad. Sci. USA 99, 14116-14121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guerois, R., Nielsen, J. E. & Serrano, L. (2002) J. Mol. Biol. 320, 369-387. [DOI] [PubMed] [Google Scholar]
- 21.Young, L., Jernigan, R. L. & Covell, D. G. (1994) Protein Sci. 3, 717-729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lijnzaad, P. & Argos, P. (1997) Proteins Struct. Funct. Genet. 28, 333-343. [PubMed] [Google Scholar]
- 23.Tsai, C. J., Lin, S. L., Wolfson, H. J. & Nussinov, R. (1997) Protein Sci. 6, 53-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chakrabarti, P. & Janin, J. (2002) Proteins Struct. Funct. Genet. 47, 334-343. [DOI] [PubMed] [Google Scholar]
- 25.Bahadur, R. P., Chakrabarti, P., Rodier, F. & Janin, J. (2003) Proteins Struct. Funct. Genet. 53, 708-719. [DOI] [PubMed] [Google Scholar]
- 26.Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N. & Bourne, P. E. (2000) Nucleic Acids Res. 28, 235-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lo Conte, L., Chothia, C. & Janin, J. (1999) J. Mol. Biol. 285, 2177-2198. [DOI] [PubMed] [Google Scholar]
- 28.Ponstingl, H., Henrick, K. & Thornton, J. M. (2000) Proteins Struct. Funct. Genet. 41, 47-57. [DOI] [PubMed] [Google Scholar]
- 29.Dasgupta, S., Iyer, G. H., Bryant, S. H., Lawrence, C. E. & Bell, J. A. (1997) Proteins Struct. Funct. Genet. 28, 494-514. [DOI] [PubMed] [Google Scholar]
- 30.Bahadur, R. P., Chakrabarti, P., Rodier, F. & Janin, J. (2004) J. Mol. Biol. 336, 943-955. [DOI] [PubMed] [Google Scholar]
- 31.Sander, C. & Schneider, R. (1991) Proteins Struct. Funct. Genet. 9, 56-68. [DOI] [PubMed] [Google Scholar]
- 32.Saha, R. P., Bahadur, R. P. & Chakrabarti, P. (2005) J. Proteome Res., in press. [DOI] [PubMed]
- 33.Hubbard, S. J. (1992) naccess, A Program for Calculating Accessibilities (Dept. of Biochemistry and Molecular Biology, Univ. College of London, London).
- 34.Lee, B. & Richards, F. M. (1971) J. Mol. Biol. 55, 379-400. [DOI] [PubMed] [Google Scholar]
- 35.Fan, K. & Wang, W. (2003) J. Mol. Biol. 328, 921-926. [DOI] [PubMed] [Google Scholar]
- 36.Bordner, A. J. & Abagyan, R. (2005) Proteins Struct. Funct. Bioinf. 60, 353-366. [DOI] [PubMed] [Google Scholar]
- 37.Crosio, M. P., Janin, J. & Jullien, M. (1992) J. Mol. Biol. 228, 243-251. [DOI] [PubMed] [Google Scholar]
- 38.Chothia, C. (1974) Nature 248, 338-339. [DOI] [PubMed] [Google Scholar]
- 39.Eisenhaber, F. (1996) Protein Sci. 5, 1676-1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chakravarty, S. & Varadarajan, R. (1999) Structure (London) 7, 723-732. [DOI] [PubMed] [Google Scholar]
- 41.Halperin, I., Wolfson, H. & Nussinov, R. (2004) Structure (London) 12, 1027-1038. [DOI] [PubMed] [Google Scholar]
- 42.Li, Y., Huang, Y., Swaminathan, C. P., Smith-Gill, S. J. & Mariuzza, R. A. (2005) Structure (London) 13, 297-307. [DOI] [PubMed] [Google Scholar]
- 43.Nicholls, A., Sharp, K. & Honig, B. (1991) Proteins Struct. Funct. Genet. 11, 281-296. [DOI] [PubMed] [Google Scholar]