

Abstract
Microarray techniques hold great promise for identifying risk factors for schizophrenia (SZ) but have not yet generated widely reproducible results due to methodological differences between studies and the high risk of type I inferential errors. Here we established a protocol for conservative analysis and interpretation of gene expression data from the dorsolateral prefrontal cortex of SZ patients using statistical and bioinformatic methods that limit false positives. We also compared brain gene expression profiles with those from peripheral blood cells of a separate sample of SZ patients to identify disease-associated genes that generalize across tissues and populations and further substantiate the use of gene expression profiling of blood for detecting valid SZ biomarkers. Implementing this systematic approach, we: (i) discovered 177 putative SZ risk genes in brain, 28 of which map to linked chromosomal loci; (ii) delineated six biological processes and 12 molecular functions that may be particularly disrupted in the illness; (iii) identified 123 putative SZ biomarkers in blood, 6 of which (BTG1, GSK3A, HLA-DRB1, HNRPA3, SELENBP1, and SFRS1) had corresponding differential expression in brain; (iv) verified the differential expression of the strongest candidate SZ biomarker (SELENBP1) in blood; and (v) demonstrated neuronal and glial expression of SELENBP1 protein in brain. The continued application of this approach in other brain regions and populations should facilitate the discovery of highly reliable and reproducible candidate risk genes and biomarkers for SZ. The identification of valid peripheral biomarkers for SZ may ultimately facilitate early identification, intervention, and prevention efforts as well.
Keywords: microarray, ontology
Schizophrenia (SZ) has a substantial genetic basis (1), but its biological underpinnings remain largely unknown. Early attempts to profile the expression of specific neurochemicals in blood and postmortem brain detected several promising candidate risk factors for SZ (2, 3) that ultimately could not be substantiated (4, 5). Subsequent progress in mapping the human genome increased the viability of candidate gene association studies, which have since proliferated (6). Most candidate genes have been targeted based on their expression within systems widely implicated in the disorder (e.g., dopamine and glutamate neurotransmitter systems), and this approach is essential for clarifying the nature of dysfunction within these recognized candidate pathways; however, it may not be optimal for identifying additional novel risk factors outside of these systems.
The advent of microarrays that can survey the entire expressed human genome has made it possible to simultaneously investigate the roles of several thousand genes in a disorder. Relative to traditional candidate gene studies predicated on existing disease models, microarray analysis is a less-constrained strategy that could foster the discovery of novel risk genes that otherwise would not come under study. Because gene expression can reflect both genetic and environmental influences, it may be particularly useful for identifying risk factors for a complex disorder such as SZ, which is thought to have a multifactorial polygenic etiology in which many genes and environmental factors interact. However, the simultaneous consideration of thousands of dependent variables also increases the likelihood of false-positive results (7). In short, microarrays hold great promise for identifying etiologic factors for SZ but run the risk of being too liberal and failing to provide replicable results.
Several groups (8) have characterized gene expression profiles of SZ in postmortem tissue from the dorsolateral prefrontal cortex (DLPFC) of the brain, which has been consistently identified as dysfunctional in the illness (9). These studies have noted variable patterns of dysregulated gene expression in several domains, including G protein signaling, metabolism, mitochondrial function, myelination, and neuronal development. However, not all of these studies have reported significant alterations in each domain. Methodological differences, including ethnic and demographic disparities, alternative microarray platforms, and diverse methods of data analysis, as well as the high risk of false positives, have been cited as factors possibly contributing to this variability (7).
To overcome the limitations of prior microarray studies of SZ, we have adopted a rigorous and systematic approach to sequentially identifying, prioritizing, verifying, and validating potential etiologic factors in SZ. Importantly, our approach is also very conservative due to three critical design features, including: (i) the application of statistical and bioinformatic methods that substantially reduce type I error rates by using statistical significance criteria rather than fold-change values; (ii) the evaluation of potential confounds such as psychotropic medication use; and (iii) the comparison of gene expression profiles in two tissues (brain and blood) from two different samples. This approach has allowed us to identify numerous putative risk factors for SZ and further validate the use of gene expression profiling of blood for detecting SZ biomarkers, which we described in a pilot study earlier this year (10). The stringency of our methods bolsters the validity of the results and increases their likelihood of generalizing to other samples, which should prove essential for advancing our understanding of the biological basis of SZ. The identification of valid peripheral biomarkers for SZ may ultimately facilitate early identification, intervention, and prevention efforts as well.
Methods
Design. We first acquired data from cRNA microarrays surveying a vast portion of the expressed human genome in postmortem tissue from the DLPFC of SZ patients and nonpsychiatric control subjects. We analyzed the data with an innovative statistical tool that reduces the number of false positives relative to other methods and applied a bioinformatic algorithm to simplify the interpretation of the ontologies represented by the differentially expressed genes. In addition, following the comparative tissue approach adopted by Martin et al. (11) for studying breast cancer, we compared gene expression profiles in DLPFC with those derived from peripheral blood cells (PBCs) from a separate sample of SZ patients and nonpsychiatric control subjects. This comparison allowed for identification of those genes whose differential expression in SZ generalizes across tissues and populations and isolation of potential peripheral biomarkers for SZ. The differential expression of the strongest candidate SZ biomarker emerging from the microarray analyses (SELENBP1, which was significantly up-regulated in both brain and blood in SZ) was then verified in PBCs by quantitative RT-PCR. Finally, to demonstrate that SELENBP1 protein is expressed in brain and to preliminarily validate the differential expression of SELENBP1 between SZ patients and control subjects, we returned to the postmortem brain tissue to examine the expression of the protein product of this gene.
Gene Expression in DLPFC. Samples. Gene expression data were obtained from cRNA microarrays of fresh-frozen postmortem DLPFC tissue samples (50 mg) from 19 SZ patients and 27 nonpsychiatric control subjects in the National Brain Databank (NBD) maintained by the Harvard Brain Tissue Resource Center. Patients and controls were closely matched on gender (68% vs. 70% male; P = 0.887) and mean age (57 vs. 56 yrs; P = 0.955), and DLPFC samples were very similar in laterality (58% vs. 52% right hemisphere; P = 0.875), mean pH (6.4 vs. 6.4; P = 0.981), and mean postmortem interval (21 vs. 20 h; P = 0.739). Ascertainment and diagnosis of these subjects according to Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) criteria (12), preparation of brain tissue, extraction, purification and hybridization of RNA, quantification of expression levels on cRNA microarrays, and quality-control procedures were all performed at the Harvard Brain Tissue Resource Center by standard methods, available in Supporting Text, which is published as supporting information on the PNAS web site.
Microarray data analysis. The gene expression data generated by these procedures were downloaded as cell intensity (CEL) files from the NBD web site and subjected to the statistical tool corgon (13), which is an academic software package developed in conjunction with two of the authors (J.C. and R.Š.) and freely available from them. corgon utilizes a novel statistical model that assumes multiplicative rather than additive noise and eliminates statistically significant outliers. corgon also assumes a uniform background level that is estimated from both mismatch and perfect-match probe intensities. Furthermore, corgon accounts for mRNA preparation, hybridization, normalization, and image analysis efficiencies.
There is no “gold standard” fold change in a gene that is known to be biologically relevant; thus, corgon identifies differentially expressed genes in conjunction with the Focus algorithm (14) based instead on their statistical significance beyond a threshold of P = 0.05. The P value of each gene is determined by two-tailed unadjusted permutation testing of 100,000 permutations of sample labels for each gene. For each permutation, the t statistic was calculated from log(expression) values, and the P value was estimated as the fraction of permutations for which the absolute value of the t statistic was greater than or equal to the absolute value of the unpermuted t statistic. [When applied to a series of arrays (15), corgon yields a type I error rate (4.4%) far superior to the rates of 29% and 15% attained by other widely accepted methods (Affymetrix microarray suite 5.0 and the method of Li and Wong (16), respectively]. Expression levels of all genes identified as differentially expressed by the corgon algorithm were then examined in relation to antipsychotic and other medication use to determine whether the elevated frequency of such exposures among patients would account for group differences in gene expression.
Following the method used by Iwamoto et al. (17), the effects of anticonvulsant, antidepressant, and anxiolytic medications were independently examined by comparing gene expression levels observed in treated and untreated groups with t tests for independent samples. Advancing beyond this scheme, antipsychotic medications were evaluated in a more quantitative manner by converting daily dosages to a common metric [maximum effective dose (18)] and examining correlations between this daily dose index and the expression level of each differentially expressed gene. Highly conservative family-wise corrections for multiple testing within each medication class were performed by using the Bonferroni correction.
Ontological profiling. To assist in the biological and molecular characterization of the differentially expressed genes, we classified these genes using the MicroArray Data Characterization and Profiling (MADCAP) algorithm (19), which was developed in conjunction with one of the authors (R.Š.). MADCAP compares the list of differentially expressed genes identified by corgon with the list of all genes on a microarray and determines which of the standardized gene ontology (GO) terms recognized by the GO Consortium (20) are more frequently represented by the corgon-selected genes than would be expected by chance based on the genes represented in the entire microarray. Unlike the gominer program (21), this method calculates conditional P values, which allows for the discovery of significant terms even if they are small in size. Additional details of the method are published as Supporting Text.
Gene Expression in PBCs. Samples. Peripheral whole-blood samples (10 ml) were obtained from a separate set of 30 SZ patients and 24 nonpsychiatric control subjects from Taiwan, as described (10). Patients and controls were of similar gender (40% vs. 58% male; P = 0.180) but differed in mean age (34 vs. 42 yrs; P = 0.014), warranting the examination of age as a potential covariate of gene expression in subsequent statistical analyses. All blood samples were collected into sterile violet-capped Vacutainer tubes (Becton Dickinson) containing K3 EDTA, temporarily stored at 4°C, and processed within 6 h of collection. Ascertainment and diagnosis of these subjects according to Diagnostic and Statistical Manual of Mental Disorders (DSM-IV, ref. 12) criteria; collection and preparation of blood samples; separation and lysis of PBCs; extraction, purification, and hybridization of RNA; quantification of expression levels on cRNA microarrays; and quality-control procedures were all performed by standard methods, which are described in greater detail elsewhere (10) and are published as Supporting Text.
Microarray data analysis. Gene expression data were analyzed by the corgon and Focus algorithms as outlined above for DLPFC samples, and the list of genes differentially expressed in the blood of these patients and controls was compared with the list of genes previously identified as differentially expressed in the DLPFC of SZ patients and controls in the NBD. Medication effects on gene expression levels were also examined as outlined above for DLPFC samples, and the effect of age (which differed between patients and controls) was evaluated by correlation as well.
Verification by RT-PCR. The level of mRNA expression of the strongest candidate biomarker gene (SELENBP1, which was significantly up-regulated in both DLPFC and PBCs in SZ) was quantified in PBCs by RT-PCR. Total blood RNA isolated by the TRIzol method was reversed-transcribed into single-stranded cDNA by using a High-Capacity cDNA Archive Kit (Applied Biosystems) in a 100-μl reaction. Each sample of cDNA (2 ng) was then mixed with SYBR green master mix (Qiagen, Valencia, CA) and primers in a 20-μl reaction. Forward and reverse primers were designed by using primerquest (Integrated DNA Technologies, Coralville, IA). PCR amplification was performed by using DNA Engine Opticon (MJ Research, Cambridge, MA). An automatically calculated melting point dissociation curve was examined to ensure specific PCR amplification and the lack of primer–dimer formation in each well. The comparative Ct equation (Applied Biosystems) was used to calculate relative fold changes between patient and control samples. Briefly, gene expression levels were represented as 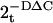 , where DΔCt = {[Δ t(single sample)]–[mean ΔCt(control samples)]}, ΔCt = {[Ct(target gene)]–[Ct(ACTB)]}, and ACTB is the housekeeping gene coding for β actin.
, where DΔCt = {[Δ t(single sample)]–[mean ΔCt(control samples)]}, ΔCt = {[Ct(target gene)]–[Ct(ACTB)]}, and ACTB is the housekeeping gene coding for β actin.
Protein Expression in DLPFC. Samples. We examined the expression of the protein product of the strongest candidate biomarker gene (SELENBP1) in DLPFC tissue from a randomly selected sample of four SZ patients and four control subjects in the Harvard Brain Tissue Resource Center from whom gene expression data were obtained.
Immunohistochemistry. Paraffin wax-embedded DLPFC brain tissue sections (10 μm) were treated with citrate buffer, microwaved for 10 min, and exposed for 24 h at 4°C to the mouse antiselenium-binding protein monoclonal antibody (1:250 dilution; MBL International, Woburn, MA). Antibody was detected by using the mouse monoclonal Vectastain ABC kit and the 3, 3′ diaminobenzidine substrate for peroxidase (Vector Laboratories). Sections were counterstained with hematoxylin, visualized on a Zeiss microscope, and analyzed by using image-pro plus software (Media Cybernetics, Silver Spring, MD).
Results
Gene Expression in DLPFC. Application of the corgon and Focus algorithms to the brain gene expression data from 19 SZ patients and 27 control subjects in the NBD identified 177 genes that were differentially expressed in the DLPFC of the two groups. Of these, 111 genes were up-regulated, and 66 were down-regulated in SZ. The Affymetrix probe number, accession number, gene symbol, gene product, and chromosomal locus of each differentially expressed gene, as well as its fold-change difference in expression between SZ patients and control subjects and the corresponding P value, are provided in Table 4, which is published as supporting information on the PNAS web site. Anticonvulsant-treated subjects showed significant down-regulation of six genes and up-regulation of one gene relative to untreated subjects, whereas anxiolytic treatment increased the expression of two genes and decreased the expression of two others. Antidepressant treatment influenced the expression of many more genes, with 10 showing significant up-regulation and 7 showing significant down-regulation with treatment. Daily dosage of antipsychotic medication had a significant positive impact on the expression of 13 genes but was not significantly related to down-regulation of any genes. The significance of all of these medication effects was abolished by Bonferroni correction for multiple testing.
The 177 differentially expressed genes were then profiled by MicroArray Data Characterization and Profiling, and 25 were found to be linked to one or more overrepresented ontologies. Thirteen of the 25 genes represented six Biological Process GO terms (Fig. 2A, which is published as supporting information on the PNAS web site). The most populated term within this ontology was Energy Pathways (P = 0.007), which described the involvement of six genes (Table 1). In addition, 12 Molecular Function GO terms were represented by 18 genes (Fig. 2B). The most populated term in this ontology was oxidoreductase activity (P = 0.031), which described the function of seven genes (Table 2). Three of these genes (NDUFA2, NDUFB5, and NDUFC1) were also classified as having NADH dehydrogenase activity. Genes representing terms in both the Biological Process and Molecular Function ontologies included ACOX1, COX7C, COX17, CNN3, NDUFA2, and NMT1, of which three genes (ACOX1, COX7C, and NDUFA2) had oxidoreductase activity within energy pathways. The remaining 152 differentially expressed genes were linked to GO terms that were not significantly overrepresented.
Table 1. Biological Process ontology terms overrepresented by genes differentially expressed in DLPFC in SZ.
| Ontology term | P | Gene symbol | Gene product | Gene fold change (P) in SZ |
|---|---|---|---|---|
| ATP-dependent proteolysis | 0.022 | CRBN | Cereblon | 1.08 (0.01660) |
| Circulation | 0.023 | LPL | Lipoprotein lipase | |
| RYR2 | Rvanodine receptor 2 (cardiac) | 1.14 (0.01030) | ||
| SRI | Sorcin | 1.20 (0.02480) | ||
| Energy pathways | 0.007 | ACOX1 | Acyl-Coenzyme A oxidase 1, palmitoyl | 1.20 (0.00480) |
| COX17 | COX17 homolog, cytochrome c oxidase assembly protein (yeast) | 1.10 (0.01780) | ||
| COX7C | Cytochrome c oxidase subunit VIIc | 1.07 (0.04030) | ||
| GLP1R | Glucagon-like peptide 1 receptor | -1.09 (0.03080) | ||
| NDUFA2 | NADH dehydrogenase (ubiquinone) 1 α subcomplex, 2, 8 kDa | 1.09 (0.02260) | ||
| SUCLG1 | Succinate-CoA ligase, GDP-forming, alpha subunit | 1.06 (0.02870) | ||
| Muscle contraction | 0.041 | CNN3 | Calponin 3, acidic | 1.37 (0.01660) |
| RYR2 | Ryanodine receptor 2 (cardiac) | 1.14 (0.01030) | ||
| SRI | Sorcin | 1.20 (0.02480) | ||
| SSPN | Sarcospan (Kras oncogene-associated gene) | 1.21 (0.04060) | ||
| Protein lipoylation | 0.004 | NMT1 | N-myristoyltransferase | 1.09 (0.00980) |
| Regulation of action potential | 0.013 | SRI | Sorcin | 1.20 (0.02480) |
Table 2. Molecular Function ontology terms overrepresented by genes differentially expressed in DLPFC in SZ.
| Ontology term | P | Gene symbol | Gene product | Gene fold change (P) in SZ |
|---|---|---|---|---|
| Beta-Catenin Binding | 0.041 | APC | Adenomatosis polyposis coli | 1.14 (0.02510) |
| Calpain activity | 0.009 | CAPN1 | Calpain 1, (mu/l) large subunit | -1.10 (0.02210) |
| CAPNS1 | Calpain, small subunit 1 | -1.10 (0.01740) | ||
| Copper ion transporter activity | 0.016 | COX17 | COX17 homolog, cytochrome c oxidase assembly protein (yeast) | 1.10 (0.01780) |
| Electron donor activity | 0.027 | ACOX1 | Acyl-Coenzyme A oxidase 1, palmitoyl | 1.20 (0.00480) |
| GABA receptor activity | 0.012 | GABRA2 | γ-aminobutyric acid (GABA) A receptor, α 2 | 1.22 (0.01050) |
| GABRB1 | γ-aminobutyric acid (GABA) A receptor, β 1 | 1.16 (0.04360) | ||
| Glycylpeptide N-tetradecanoyltransferase activity | 0.048 | NMT1 | N-myristoyltransferase | 1.09 (0.00980) |
| MHC protein binding | 0.048 | TAPBP | TAP-binding protein (tapasin) | -1.09 (0.02930) |
| NADH dehydrogenase activity | 0.019 | NDUFA2 | NADH dehydrogenase (ubiquinone) 1 α subcomplex, 2, 8 kDa | 1.09 (0.02260) |
| NDUFB5 | NADH dehydrogenase (ubiquinone) 1 β subcomplex, 5, 16 kDa | 1.09 (0.02910) | ||
| NDUFC1 | NADH dehydrogenase (ubiquinone) 1, subcomplex unknown, 1, 6 kDa | 1.07 (0.00900) | ||
| Oxidoreductase activity | 0.031 | ACOX1 | Acyl-Coenzyme A oxidase 1, palmitoyl | 1.20 (0.00480) |
| COX7C | Cytochrome c oxidase subunit VIIc | 1.07 (0.04030) | ||
| HSD17B12 | Hydroxysteroid (17-beta) dehydrogenase 12 | 1.11 (0.03900) | ||
| NDUFA2 | NADH dehydrogenase (ubiquinone) 1 α subcomplex, 2, 8 kDa | 1.09 (0.02260) | ||
| NDUFB5 | NADH dehydrogenase (ubiquinone) 1 β subcomplex, 5, 16 kDa | 1.09 (0.02910) | ||
| NDUFC1 | NADH dehydrogenase (ubiquinone) 1, subcomplex unknown, 1, 6 kDa | 1.07 (0.00900) | ||
| P4HA1 | Procollagen-proline, 2-oxoglutarate 4-dioxygenase, alpha polypeptide 1 | 1.12 (0.03640) | ||
| Peroxisome targeting signal receptor activity | 0.031 | PEX5 | Peroxisomal biogenesis factor 5 | -1.12 (0.00890) |
| Phosphoserine phosphatase activity | 0.009 | PSPHL | Phosphoserine phosphatase-like | -2.14 (0.04420) |
| Troponin C binding | 0.030 | CNN3 | Calponin 3, acidic | 1.37 (0.01660) |
Gene Expression in PBCs. Application of the corgon and Focus algorithms to the gene expression data from a separate sample of 30 SZ patients and 24 control subjects from Taiwan identified 123 genes that were differentially expressed in PBCs from the two groups. Of these, 67 genes were up-regulated and 56 down-regulated in SZ. The Affymetrix probe number, accession number, gene symbol, gene product, and chromosomal locus of each differentially expressed gene, as well as its fold-change difference in expression between SZ patients and control subjects and the corresponding P value, are provided in Table 5, which is published as supporting information on the PNAS web site. Eight genes increased and 10 genes decreased significantly in expression level with age. Anticonvulsant-treated subjects differed from untreated subjects in the expression of only one gene, which was up-regulated; however, antidepressant treatment was associated with significant up-regulation of 15 genes and significant down-regulation of two others. The effects of age and these classes of medication did not remain significant after correction for multiple testing. Remarkably, anxiolytic treatment was associated with significant up-regulation of 34 genes and down-regulation of another 40. Differential expression of 12 of these genes [including CSDA, EPB42, FBXO9, FKBP8, GSK3A HBA1 (two transcripts), HBA2, HBB (two transcripts), HLA-B, and UBB) remained significant after correcting for multiple testing. Daily dosage of antipsychotic medication was linearly related to the expression of only one gene (G0S2), which also remained statistically significant after corrections for multiple testing were applied.
Comparing the list of 123 genes differentially expressed in PBCs with that obtained from DLPFC identified six genes common to both (Table 3). BTG1, HRNPA3, and SFRS1 were significantly up-regulated in the DLPFC in SZ but significantly down-regulated in PBCs from the other sample of SZ patients; the reverse pattern of differential expression was observed for GSK3A, which was the only one of these six genes to show a significant relationship to psychotropic medication (i.e., anticonvulsant) use. In contrast, SELENBP1 was significantly up-regulated in both tissues from the two samples of SZ patients. HLA-DRB1 was significantly down-regulated in both DLPFC and PBCs in SZ; however, different probe sets (corresponding to different transcripts of the same gene) were associated with the illness in the two tissues.
Table 3. Six genes differentially expressed in both DLPFC and PBCs in SZ.
| Fold change (P) in SZ
|
||||||
|---|---|---|---|---|---|---|
| Affymetrix probe number | Accession number | Gene symbol | Gene product | Chromosomal locus | DLPFC | PBCs |
| 200920_s_at | AL535380 | BTG1 | B cell translocation gene 1, antiproliferative | 12q22 | 1.14 (0.04020) | -1.36 (0.00008) |
| 202210_x_at | NM_019884 | GSK3A | Glycogen synthase kinase 3 α | 19q13.2 | -1.09 (0.02490) | 1.59 (0.00044) |
| 209728_at | BC005312 | HLA-DRB1 | MHC class II, DR β 1 | 6p21.3 | -1.17 (0.04220) | NS* |
| 209312_x_at | U65585 | HLA-DRB1 | MHC, class II, DRB 1 | - | NS* | -1.27 (0.00007) |
| 215193_x_at | AJ297586 | HLA-DRB1 | MHC, class II, DRB 1 | - | NS* | -1.33 (0.00006) |
| 211929_at | AA527502 | HNRPA3 | Heterogeneous nuclear ribonucleoprotein A3 | 2q31.2 | 1.15 (0.01410) | -2.12 (0.00004) |
| 214433_s_at | NM_003944 | SELENBP1 | Selenium-binding protein 1 | 1q21-q22 | 1.16 (0.04510) | 1.95 (0.00093) |
| 211784_s_at | BC006181 | SFRS1 | Splicing factor, arginine/serine-rich 1 (splicing factor 2, alternate splicing factor) | 17q21.3-q22 | 1.12 (0.02460) | -1.71 (0.00005) |
NS, probe was not differentially expressed on the microarray used to profile the indicated tissue.
SELENBP1 was identified as the strongest candidate biomarker among all genes differentially expressed in SZ, because it was the only gene for which identical probe sets indicated significant differential expression in a similar direction in both brain and blood in SZ. This significant up-regulation was substantiated by RT-PCR in PBCs from a randomly selected subset of the same SZ patients (n = 21) and controls (n = 18) that were profiled by microarray analysis. A highly significant (P = 0.003) 2.2-fold increase in SELENBP1 was observed in PBCs of SZ patients by RT-PCR, which closely corresponded to the significant 2.0-fold up-regulation of the gene observed by microarray.
Protein Expression in DLPFC. Granular cytoplasmic staining of SELENBP1 protein was observed in a proportion of neurons and glia in DLPFC tissue from each of the four control subjects and four SZ patients. A representative example of the antibody-staining pattern observed in controls is shown in Fig. 1A, whereas a representative example of the antibody-staining pattern observed in patients is shown in Fig. 1B. Compared with control tissue, the intensity and ratio of glial/neuronal SELENBP1 antibody staining was noticeably increased in DLPFC tissue from at least three of the four SZ patients. The enhanced intraglial staining of SELENBP1 antibody in samples from SZ patients was most notable in a perinuclear rim of increased expression. No staining was observed in any cell when the primary antibody was omitted.
Fig. 1.

Expression of SELENBP1 protein in DLPFC. SELENBP1 protein expression in DLPFC was visualized through binding to the mouse antiselenium-binding protein monoclonal antibody. The pattern and intensity of antibody staining are shown at ×100 magnification for a representative control subject (A) and a representative SZ patient (B). Increased intraglial and decreased intraneuronal staining of SELENBP1 antibody were observed in DLPFC tissue from at least three of the four SZ patients. Colored arrows indicate antibody staining of cytoplasm in different cell types (black, glia; red, neurons).
Discussion
The pursuit of risk factors for SZ has been difficult, but systematic evaluation of widely implicated biological systems has identified several likely contributors to its pathogenesis, including central dopamine and glutamate pathways. Yet, SZ is an undeniably complex disorder likely to result from dysfunction in not only these but also numerous other biological substrates as well. This complexity, in conjunction with the restrictions on throughput of candidate gene and other single-marker approaches, has prompted a transition toward high-capacity technologies such as cRNA microarrays, which can simultaneously reflect genetically and environmentally mediated effects on the differential expression of potentially all human genes in an illness. However, as illustrated by the lack of uniformity among prior microarray studies of DLPFC in SZ, a more rigorous approach must be adopted if highly reliable and generalizable candidate genes are to be identified by this method.
Here we have established a protocol for conservative analysis and interpretation of gene expression microarray data by using the corgon and Focus algorithms to limit type I errors and MicroArray Data Characterization and Profiling to identify the ontologies represented by differentially expressed genes. In its present application to gene expression data from DLPFC in SZ, this approach identified 177 genes, 6 biological processes, and 12 molecular functions that should be identified as high-priority targets for further candidate gene association analyses and hypothesis-driven functional studies. Twenty-eight of these genes also code to chromosomal loci strongly implicated in SZ by linkage analysis (22) (Table 4) and are thus particularly attractive candidates. These include four genes (ACOX1, NDUFA2, SUCLG1, and TAPBP) that were also linked to significantly overrepresented GO terms, and SFRS1 and HLA-DRB1, which were differentially expressed in both DLPFC and PBCs in SZ. These genes provide reference points for the commencement of future fine-mapping and positional cloning of putative SZ risk loci.
Quite surprisingly, neither this study nor most of the prior microarray studies of SZ identified significant differences between SZ patients and controls in the expression of “traditional” candidate genes, such as those coding for dopamine and glutamate receptors, transporters, or catalytic enzymes. Conversely, the GO terms overrepresented in DLPFC in the present study were generally related to neurotransmitter systems not typically implicated in SZ (e.g., GABA receptor activity), neuronal processes not specific to a given neurotransmitter system (e.g., regulation of action potential), or biological processes not specific to the nervous system (e.g., energy pathways). More precisely, genes related to energy metabolism were predominant among those differentially expressed in DLPFC in SZ. These results are quite consistent with the report of Prabakaran et al. (23), who found significant alterations in SZ of several genes involved in the mitochondrial electron transport chain, NADH dehydrogenase complex, or mitochondrial membrane. Iwamoto et al. (17) also found global down-regulation of 76 mitochondrial genes in the DLPFC in SZ. Our results substantiate these observations, because at least four of the genes implicated in our study are involved in electron transfer, and three of these are also part of the NADH dehydrogenase complex located in the inner mitochondrial membrane. These data suggest that dysfunction within particular pathways or processes, but perhaps not necessarily in specific genes, might be important in the etiology of SZ.
Whereas each of the 177 genes differentially expressed in DLPFC of SZ patients is a promising candidate risk gene, the 123 genes differentially expressed in PBCs from such patients can be considered putative biomarkers for the illness. Two other studies examining differences in blood-based gene expression in SZ have appeared since our initial report (10). The study of Zvara et al. (24) used an arbitrary fold-change criterion (>2.0-fold) rather than P values to identify differentially expressed genes, the limitations of which we have described above. Thus, although the two genes identified in that study as differentially expressed in PBCs in SZ (DRD2 and Kir2.3) were not among the 123 genes we identified, it cannot be determined whether this is due to nonreplication or the different criteria used for identifying differential expression. Supporting the latter possibility (and underscoring the heightened sensitivity of our current statistical methods), we note that many of the most reliable candidate biomarker genes identified in the present report were not among the most highly differentially expressed genes we previously identified based solely on fold-change criteria (10). In the other existing study of PBC gene expression in SZ reported by Middleton et al. (25), ≈300 genes were identified as differentially expressed between patients and their unaffected siblings, but the authors reported the identity of only the 40 genes with the largest fold-change difference in expression between groups. Of those 40 genes, S100A12 (which codes for S100 calcium-binding protein A12 [calgranulin C]) was significantly up-regulated 1.67-fold in PBCs from SZ patients, similar to the significant 1.50-fold up-regulation observed in our sample. Thus, although no prior report is directly comparable to the present study (either in analytic methods or sample composition), there is already some evidence for replication of results.
Six of the putative biomarker genes we identified in PBCs were also differentially expressed in the brain in SZ. Among these were BTG1, which regulates cell proliferation, and three genes (GSK3A, HNRPA3, and SFRS1) that regulate RNA splicing or transcription. These results endorse the view that, in the search for SZ risk genes and biologically meaningful disease markers, inherited mutations regulating gene expression should be evaluated as thoroughly as those that induce structural or functional changes in proteins.
Although conceivable, no specific homeostatic mechanism is known whereby up-regulation of a gene in one tissue (e.g., DLPFC) is directly related to down-regulation of that gene in another (e.g., PBCs); therefore, the genes noted above should be considered secondary candidate SZ biomarkers to SELENBP1 and HLA-DRB1, which showed altered expression in the same direction in both DLPFC and PBCs (up- and down-regulation, respectively). Adding to this rationale, HLA-DRB1 maps to the MHC region on chromosome 6p21.3, which is a prime candidate locus for SZ (22), and SELENBP1 maps to chromosome 1q21-22, a locus that has been strongly linked to SZ in some but not most genome-wide linkage studies (26).
The utility of SELENBP1 as a potential peripheral biomarker was substantiated by RT-PCR verification of its up-regulation in PBCs. The altered levels of SELENBP1 transcripts measured in DLPFC and PBCs also translated into observable consequences at the functional level, because preliminary analyses suggested that expression of SELENBP1 protein was denser in glia and less dense in neurons of the DLPFC in SZ; however, further stereological quantification of these changes are required and are presently being undertaken in our laboratory. Little is known of the functions of SELENBP1 beyond its clear role in binding the antioxidant selenium. Epidemiological evidence inversely relating selenium intake to the prevalence of colorectal and other cancers (27) is more compelling in light of an established reduction in the rate of these cancers among SZ patients (28). Links between selenium deficiency and glutamate-induced excitotoxicity (29) are also provocative, because the increased expression of SELENBP1 could cause increased sequestration of selenium (or perhaps be in response to already low levels of selenium, ref. 30), which may thus promote neurodegeneration in SZ.
Although our approach has several strengths, such as careful control of type I errors, evaluation of potential confounding effects of psychotropic medications, ontological profiling of differentially expressed genes, and comparison of gene expression in two tissues and samples, this work also has important limitations. Foremost among these, studies of postmortem brain tissue are naturalistic, and as such, we could not directly control for the effects of many subject factors (e.g., diet, exercise, substance use, psychotropic medication use, etc.) that could influence gene expression and that also might differ among subject groups, thus inducing either false-negative or false-positive results. We have attempted to control for some of these factors through appropriate matching of patients and controls and through statistical modeling when possible. Statistical evaluation of psychotropic medication effects is perhaps the most critical in this study, because this factor is the most likely to systematically differ between patients and controls. Ideally, first-episode or unmedicated patients should be studied to further eliminate the possibility of medication effects on gene expression. The inclusion of unaffected (and thus unmedicated) first-degree biological relatives of SZ patients could also be effective in this regard and could also help rule out the effects of illness-associated factors such as chronicity or degeneration after onset.
Another potential limitation is that the various tissues examined in this study were obtained from two very different populations, with DLPFC samples coming from subjects in the NBD identified mostly as “white” or alternatively as “unknown” ethnicity and PBC samples coming from subjects from Taiwan. This design feature may have caused us to miss some additional genes whose differential expression in DLPFC and PBCs might overlap if assessed from the same ethnic or geographic population; however, this may also strengthen the design, in that our results may be overly (or appropriately) conservative. Studying two very different samples also provides a form of replication that is rare in microarray studies and may facilitate the generalization of these putative biomarkers to other ethnically diverse samples. Finally, it is important to reiterate that SZ is an etiologically complex and heterogeneous disorder that invariably thwarts classification schemes relying on a single dimension to differentiate affected and unaffected persons. Thus, although we have initially analyzed SELENBP1 most thoroughly, the other putative risk genes identified here should also be pursued, verified, validated, and incorporated into causal models of the illness.
Conclusion
Through the implementation of a systematic approach toward the analysis of gene expression microarray data, we: (i) discovered 177 putative SZ risk genes in DLPFC, 28 of which map to chromosomal loci linked to the disorder; (ii) delineated 6 biological processes and 12 molecular functions that may be particularly disrupted in the illness; (iii) identified 123 putative SZ biomarkers in PBCs, 6 of which had corresponding differential expression in DLPFC; (iv) verified the up-regulation of the strongest candidate SZ biomarker (SELENBP1) in PBCs; and (v) demonstrated an altered pattern of expression of SELENBP1 protein in DLPFC in SZ. The continued application of this approach in other brain regions (e.g., both implicated and nonimplicated structures, to identify ubiquitous and region-specific changes) and populations (e.g., bipolar disorder patients, to establish disease-specific changes) should facilitate the discovery of highly reliable and reproducible candidate risk genes and biomarkers for SZ. Further work to replicate and validate SZ biomarkers in peripheral blood may ultimately provide the means to identify individuals at risk for SZ before disease onset, which may in turn allow these individuals to be targeted for early intervention and prevention efforts.
Supplementary Material
Acknowledgments
We thank Arthur B. Pardee, Adam Dempsey, and Nadine Nossova for helpful comments on the manuscript; Vural Ozdemir for recommendations regarding antipsychotic medication dose equivalences; and the Harvard Brain Tissue Resource Center for providing gene expression microarray data and tissue samples from the NBD. This work was supported in part by the University of California at San Diego Center for AIDS Research Genomics Core and National Institutes of Health Grants P30MH062512 (Igor Grant), R01AG018386, R01AG022381, R01AG022982 (to W.S.K.); and R01DA012846, R01DA018662, R01MH065562, and R01MH071912 (to M.T.T.). J.C. is the Holder of a Canada Research Chair in Genomics.
Author contributions: S.J.G., I.P.E., W.S.K., C.-C.L., and M.T.T. designed research; S.J.G., I.P.E., J.C., R.š., N.K., M.H., C.-C.L., and M.T.T. performed research; J.C. and R.š. contributed new reagents/analytic tools; S.J.G., I.P.E., J.C., and R.š. analyzed data; and S.J.G., I.P.E., W.S.K., R.š., N.K., M.H., and M.T.T. wrote the paper.
Abbreviations: DLPFC, dorsolateral prefrontal cortex; GO, gene ontology; NBD, National Brain Databank; PBC, peripheral blood cell; SZ, schizophrenia.
References
- 1.Faraone, S. V., Seidman, L. J., Kremen, W. S., Toomey, R., Pepple, J. R. & Tsuang, M. T. (2000) Biol. Psychiatry 48, 120–126. [DOI] [PubMed] [Google Scholar]
- 2.Breakefield, X. O. & Edelstein, S. B. (1980) Schizophr. Bull. 6, 282–288. [DOI] [PubMed] [Google Scholar]
- 3.Rossor, M. (1984) J. Psychiatr. Res. 18, 457–465. [DOI] [PubMed] [Google Scholar]
- 4.Fleissner, A., Seifert, R., Schneider, K., Eckert, W. & Fuisting, B. (1987) Eur. Arch. Psychiatry Neurol. Sci. 237, 8–15. [DOI] [PubMed] [Google Scholar]
- 5.Gasque, P., Dean, Y. D., McGreal, E. P., VanBeek, J. & Morgan, B. P. (2000) Immunopharmacology 49, 171–186. [DOI] [PubMed] [Google Scholar]
- 6.Abusaad, I., Mackay, D., Zhao, J., Stanford, P., Collier, D. A. & Everall, I. P. (1999) J. Comp. Neurol. 408, 560–566. [DOI] [PubMed] [Google Scholar]
- 7.Pounds, S. & Cheng, C. (2004) Bioinformatics 20, 1737–1745. [DOI] [PubMed] [Google Scholar]
- 8.Harrison, P. J. & Weinberger, D. R. (2005) Mol. Psychiatry 10, 40–68. [DOI] [PubMed] [Google Scholar]
- 9.Hill, K., Mann, L., Laws, K. R., Stephenson, C. M., Nimmo-Smith, I. & McKenna, P. J. (2004) Acta Psychiatr. Scand.. 110, 243–256. [DOI] [PubMed] [Google Scholar]
- 10.Tsuang, M. T., Nossova, N., Yager, T., Tsuang, M. M., Guo, S. C., Shyu, K. G., Glatt, S. J. & Liew, C. C. (2005) Am. J. Med. Genet. 133, 1–5. [DOI] [PubMed] [Google Scholar]
- 11.Martin, K. J., Graner, E., Li, Y., Price, L. M., Kritzman, B. M., Fournier, M. V., Rhei, E. & Pardee, A. B. (2001) Proc. Natl. Acad. Sci. USA 98, 2646–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.American Psychiatric Association (1994) Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) (Am. Psychiatric Assoc., Washington, DC).
- 13.Sasik, R., Calvo, E. & Corbeil, J. (2002) Bioinformatics 18, 1633–1640. [DOI] [PubMed] [Google Scholar]
- 14.Cole, S. W., Galic, Z. & Zack, J. A. (2003) Bioinformatics 19, 1808–1816. [DOI] [PubMed] [Google Scholar]
- 15.Sasik, R., Woelk, C. H. & Corbeil, J. (2004) J. Mol. Endocrinol. 33, 1–9. [DOI] [PubMed] [Google Scholar]
- 16.Li, C. & Wong, W. H. (2001) Proc. Natl. Acad. Sci. USA 98, 31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iwamoto, K., Kakiuchi, C., Bundo, M., Ikeda, K. & Kato, T. (2004) Mol. Psychiatry 9, 406–416. [DOI] [PubMed] [Google Scholar]
- 18.Davis, J. M. & Chen, N. (2004) J. Clin. Psychopharmacol. 24, 192–208. [DOI] [PubMed] [Google Scholar]
- 19.Lozach, J., Sasik, R., Ogawa, S., Glass, C. K. (2003) in European Conference on Computational Biology (Institut National de la Recherche Agronomique, Paris), p. GE24.
- 20.Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., Davis, A. P., Dolinski, K., Dwight, S. S., Eppig, J. T., et al. (2000) Nat. Genet. 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeeberg, B. R., Feng, W., Wang, G., Wang, M. D., Fojo, A. T., Sunshine, M., Narasimhan, S., Kane, D. W., Reinhold, W. C., Lababidi, S., et al. (2003) Genome Biol. 4, R28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lewis, C. M., Levinson, D. F., Wise, L. H., DeLisi, L. E., Straub, R. E., Hovatta, I., Williams, N. M., Schwab, S. G., Pulver, A. E., Faraone, S. V., et al. (2003) Am. J. Hum. Genet. 73, 34–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prabakaran, S., Swatton, J. E., Ryan, M. M., Huffaker, S. J., Huang, J. T., Griffin, J. L., Wayland, M., Freeman, T., Dudbridge, F., Lilley, K. S., et al. (2004) Mol. Psychiatry 9, 684–697. [DOI] [PubMed] [Google Scholar]
- 24.Zvara, A., Szekeres, G., Janka, Z., Kelemen, J. Z., Cimmer, C., Santha, M. & Puskas, L. G. (2005) Disease Markers 21, 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Middleton, F. A., Pato, C. N., Gentile, K. L., McGann, L., Brown, A. M., Trauzzi, M., Diab, H., Morley, C. P., Medeiros, H., Macedo, A., et al. (2005) Am. J. Med. Genet. 136, 12–25. [DOI] [PubMed] [Google Scholar]
- 26.Brzustowicz, L. M., Hodgkinson, K. A., Chow, E. W. C., Honer, W. G. & Basett, A. S. (2000) Science 288, 678–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jacobs, E. T., Jiang, R., Alberts, D. S., Greenberg, E. R., Gunter, E. W., Karagas, M. R., Lanza, E., Ratnasinghe, L., Reid, M. E., Schatzkin, A., et al. (2004) J. Natl. Cancer Inst. 96, 1669–1675. [DOI] [PubMed] [Google Scholar]
- 28.Kotler, M., Barak, P., Cohen, H., Averbuch, I. E., Grinshpoon, A., Gritsenko, I., Nemanov, L. & Ebstein, R. P. (1999) Am. J. Med. Genet. 88, 628–633. [PubMed] [Google Scholar]
- 29.Savaskan, N. E., Brauer, A. U., Kuhbacher, M., Eyupoglu, I. Y., Kyriakopoulos, A., Ninnemann, O., Behne, D. & Nitsch, R. (2003) FASEB J. 17, 112–114. [DOI] [PubMed] [Google Scholar]
- 30.Vaddadi, K. S., Soosai, E. & Vaddadi, G. (2003) Br. J. Clin. Pharmacol. 55, 307–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.