

Abstract
Lymphocyte homeostasis is the result of a critical balance between cell proliferation and death. Disruption of this subtle equilibrium can lead to the onset of leukemia, an increase in the number of lymphocytes being potentially due to both of these parameters. The relative importance of cell proliferation vs. apoptosis during pathogenesis induced by the primate T cell lymphotropic viruses and bovine leukemia virus (BLV) has been difficult to assess because of conflicting data from a range of in vitro and ex vivo experimental systems. Here, we aim to resolve this issue by measuring the rates of cell proliferation and death in the BLV-ovine system, an animal model of human T lymphotropic virus (HTLV-1). We use a method based on the i.v. injection of 5-bromodeoxyuridine into BLV-infected sheep. We show that B lymphocytes in BLV+ asymptomatic sheep proliferate significantly faster than in uninfected controls (average proliferation rate: 0.020 per day vs. 0.011 per day). In contrast, the rates of cell death were not significantly different between aleukemic BLV-infected and control sheep (average death rate 0.089 per day vs. 0.094 per day, respectively). We conclude that the increase in the number of B cells during BLV-induced lymphocytosis results from higher proliferation rates but is not due to a significant decrease in apoptosis, in contrast to data from in vitro (ex vivo) experiments. The imbalance created by the net increase in proliferation in the absence of compensating cell death reveals a complex mechanism of feedback regulation controlling homeostasis in the blood compartment.
Complex oncoviruses, also called deltaretroviruses, comprise a series of pathogens infecting mammals such as humans, monkeys, and ruminants. These viruses infect either T or B lymphoid cells and sometimes cause hematological or neurological disorders (1, 2). The prototype virus, HTLV-1, propagates in CD4 or CD8 T cells and causes, in a minority of the infected hosts and after extended clinical latency periods of 30–50 years, a rapidly fatal leukemia called adult T cell leukemia/lymphoma (1, 2). Another member of this genus, bovine leukemia virus (BLV) infects ruminants (naturally cattle and experimentally sheep) and induces a B cell lymphoproliferative disease characterized by persistent lymphocytosis, leukemia, and/or tumors (lymphoma, lymphosarcoma; refs. 3 and 4). BLV and human T lymphotropic virus (HTLV-1) share many structural and functional homologies: both are exogenous to their host species (5, 6), have similar genomic organizations (7), integrate into dispersed sites within the host genome (8, 9), and are apparently silent in vivo, at least in the majority of detectable infected cells (10, 11).
Details of the virus–host interplay have been a very active area of research. A first milestone was the description of spontaneous incorporation of tritiated thymidine in peripheral blood mononuclear cells (PBMCs) cultivated in vitro (ex vivo) (12–14), indicating that lymphocytosis could result from increased rates of proliferation (13, 15). The concept of this model has been counterbalanced by the discovery of mechanisms related to apoptosis. Many viruses, including BLV and HTLV, have the capacity to interfere with apoptotic processes (16). For instance, in ex vivo cultures, BLV protects infected B lymphocytes from spontaneous programmed cell death (17–19).
Besides these observations on ex vivo-isolated lymphocytes, experiments based on the use of immortalized cell lines assigned a major role to the Tax protein during transformation. This viral transcriptional activator has the capacity to drive cells into cycle via multiple pathways: interaction with p53 (20), p16INK4a (21), cyclin-dependent kinases (22), cyclin D (23, 24), and E2F (25). Tax also modulates apoptosis but, unfortunately, opposite conclusions were drawn depending on the experimental conditions used. In T lymphocytes, Tax can act as an inducer of apoptosis (26) or, conversely, can protect from cell death (27).
A major weakness of these in vitro experiments is the nonphysiological assay conditions such as the choice of immortal cell lines or the levels of protein expression (Tax being toxic at high doses). Even ex vivo experiments depend on the type of culture medium, on the presence of activator molecules (cytokines, phorbol esters), and on the cell density. For example, cytotoxic response in infected lymphocytes requires close proximity of the cells in the cultures (10).
In an attempt to unravel the relative importance of cell proliferation vs. apoptosis during the process of leukemogenesis associated with infection by complex oncoviruses, we adopted a very direct strategy designed to measure these parameters in the BLV-infected sheep, an animal model of HTLV-1 in human.
Materials and Methods
Experimental Animals.
A total of 10 sheep infected with wild-type or mutant BLV viruses (as revealed by enzyme-linked immunosorbent assay; ref. 28) were studied (29–31). Three sheep (nos. 8, 105, and 293) were persistently infected with wild-type pBLVIX (29), whereas animals 104, 2658, 2667, and 2668 harbored provirus pBLVTax106 + 293, pLTR-NF1, pLTR-NF2, and pLTR-Ebox, respectively (31, 32). Importantly, all proviruses behaved as wild type during pathogenesis, despite the presence of some mutations. Sheep 8, 104, 105, and 293 were in the asymptomatic stage of the disease, whereas animals 2658, 2667, and 2668 were leukemic. Finally, three sheep (nos. 117, 1092, and 1097) were used as noninfected controls. The asymptomatic sheep and seronegative controls had similar total lymphocyte counts (ranging between 5,000 and 10,000 cells per mm3) with one exception (sheep 8, 18,988 cells per mm3).
Isolation of PBMCs and Analysis of in Vitro (ex Vivo) Apoptosis.
PBMCs (4 × 106) were isolated on a Percoll gradient centrifugation (17) and cultivated at 37°C in a 5% CO2/air atmosphere in RPMI medium 1640 supplemented with 10% (vol/vol) FCS/2 mM l-glutamine/100 units of penicillin/100 μg of streptomycin per ml (Life Technologies, Rockville, MD) and, when indicated, in the presence of 200 nM phorbol 12-myristate 13-acetate (PMA) and 565 nM ionomycin (Sigma). After 18 h of culture, PBMCs were collected, labeled with 1H4 antibody recognizing surface immunoglobulin M (sIgM; ref. 33) and with a fluorescein isothiocyanate (FITC)-conjugate (Dako). The cells were stained with propidium iodide (PI; Sigma) and analyzed by flow cytometry, as described (17).
Analysis of 5-Bromo-2′-Deoxyuridine (BrdUrd) Incorporation in Vivo.
Sheep were injected intravenously with BrdUrd resuspended in physiologic serum (400 or 500 mg BrdUrd per injection; Sigma). To evaluate BrdUrd-incorporation, 1 ml of blood was collected, and red blood cells were lysed with 1× FACS lysing solution (Becton Dickinson) for 15 min at room temperature. Cells were washed twice with PBS containing 0.5% BSA (Sigma), incubated in the presence of biotinylated 1H4 monoclonal for 30 min at 4°C and, after two washes, labeled with streptavidin-phycoerythrin (Becton Dickinson). Then, cells were washed twice in PBS/0.5% BSA, treated with 1× FACS permeabilizing solution (Becton Dickinson), and incubated for 10 min at room temperature. Finally, leukocytes were stained with anti-BrdUrd FITC antibody in the presence of DNase (Becton Dickinson) for 30 min at room temperature and analyzed by flow cytometry.
To transiently induce viral expression, PBMCs were isolated by Percoll gradient centrifugation, cultivated for 18 h, washed twice in PBS/0.5% BSA, and fixed and permeabilized with the DAKO IntraStain Reagents A and B. Intracellular detection of the p24 viral protein was performed on fixed cells by sequential incubation with 4′G9 MAb and rat anti-mouse IgG1 phycoerythrin conjugate (Becton Dickinson) for 30 min at 4°C. After permeabilization with 1× FACS permeabilizing solution, BrdUrd incorporation was measured as described above.
Mathematical Modeling (Estimation of Proliferation and Death Rates).
BrdUrd labeling data can be very difficult to interpret correctly because of the complex kinetics of labeling and delabeling, including label dilution and postlabeling expansion (34). For this reason, we used a mathematical framework to help interpret the data (35–37).
The rates of proliferation and death within the B cell population were estimated by fitting the following model to the BrdUrd incorporation data obtained experimentally:
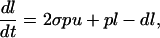 |
where u denotes the proportion of unlabeled B cells, and l denotes the proportion of labeled B cells, whereas p and d represent the proliferation and death rates, respectively. σ(t), the probability of labeling on division, is assumed to decline exponentially with time, reflecting the loss of unincorporated label after a single BrdUrd injection. It is to be expected that the estimated death rate is greater than the estimated proliferation rate because the death rate measured is the death rate of labeled cells only, whereas the proliferation rate measured is the average proliferation rate of all B cells. The formula was fitted to the data by nonlinear least squares regression with the program scop; SDs of the parameters were estimated by calculating the asymptotic covariance matrix. The formula was fitted to the data by nonlinear least squares regression by using the program SCOP; SDs of the parameters were estimated by calculating the asymptotic covariance matrix.
Results
Apoptosis in Short-Term Cultures of BLV-Infected Lymphocytes.
To compare the levels of apoptosis in vitro (ex vivo) and in vivo, we first investigated the extent of cell death occurring ex vivo. It has been demonstrated that infection by BLV is associated with a reduction in the number of B lymphocytes undergoing apoptosis ex vivo (17, 19). To this end, PBMCs from BLV-infected sheep (nos. 8, 105, and 293) and seronegative animals (sheep nos. 117, 1092, and 1097) were cultivated for 18 h, labeled with anti-sIgM 1H4 antibody, stained with PI, and analyzed by two-color flow cytometry to evaluate the proportion of B cells within the different phases of the cell cycle (Fig. 1A). Under these experimental conditions, apoptotic B lymphocytes staining in subG0/G1 represented 30% of the PBMC population of sheep 8 (Fig. 1A, Left). The majority of the lymphocytes were resting in G0/G1 (55%, Middle), and very few initiated the S phase of the cell cycle (5%, Right). The distribution of the cells throughout the cycle, i.e., mean percentages of B lymphocytes in subG1, G0/G1 and S/G2/M, was deduced from three independent experiments using three uninfected and three asymptomatic sheep (illustrated in Fig. 1A and summarized in 1B). Under spontaneous culture conditions (No chemical), the mean percentages of B cells undergoing apoptosis were significantly lower (two-tailed Student's t test, P = 0.017) in infected sheep (46.96 ± 3.38) compared with control sheep (65.51 ± 7.50). We conclude that, in ex vivo cultivation, B lymphocytes from infected sheep are less prone to undergo apoptosis, confirming and extending our previous observations (17). The reduction of the apoptotic B cell population is associated with a significant increase in G0/G1 resting cells (45.20 ± 4.50 vs. 26.02 ± 3.86; P = 0.005; Fig. 1B). There was no statistically significant difference appearing in the S/G2/M phase (Fig. 1C, No chemical).
Figure 1.

Apoptosis in short-term cultures of sheep PBMCs. (A) PBMCs were isolated from BLV-infected sheep (nos. 8, 105, and 293) and seronegative controls (nos. 117, 1092, and 1097). Cells were cultivated for 18 h in the absence (no chemical) or in the presence of PMA + Ionomycin and labeled with anti-IgM monoclonal antibody 1H4 and an FITC conjugate. After ethanol fixation, the cells were stained with PI and, after exclusion of the doublets, analyzed by two-color flow cytometry. Results from a representative experiment (10,000 events) are shown as dot plots (x axis, PI; y axis, B lymphocytes). Numbers within the plots represent the percentages of positively stained B cells within each region. (B) Mean percentages (± SD) of B lymphocytes at different stages of the cell cycle (subG1/apoptotic, G0/G1, S+G2/M) were calculated from three independent experiments by using cells isolated from infected animals (nos. 8, 105, and 293) and negative controls (nos. 117, 1092, and 1097). **, P < 0.01; *, P < 0.05; NS, not statistically significant, as determined by Student's t test. (C) Graphic representation of the mean values and SDs from B.
With the aim to increase cell viability and viral expression, two chemicals (PMA, a PKC activator, and ionomycin, a calcium ionophore) were added to the culture medium. Under these conditions, the majority of B cells were concentrated in the G0/G1 phase of the cell cycle, as illustrated in Fig. 1A (PMA + Ionomycin). Addition of PMA and ionomycin thus resulted, as expected, in a drastic decrease of the apoptotic levels: 20.13 and 18.59% vs. 46.96 and 65.51% for the cultures derived from infected and uninfected sheep, respectively (Fig. 1B). Importantly, the distributions of the B cells throughout the cycle (apoptotic, G0/G1, and S/G2/M) were similar among the asymptomatic and the control sheep (schematized in Fig. 1C, PMA + Ionomycin). In other words, the B lymphocytes from both categories of animals exhibit the same rates of apoptosis, and also, interestingly, similar levels of proliferation. Together, these data illustrating a reduction of cell death in ex vivo cultures of PBMCs isolated from BLV-infected sheep essentially confirm and extend our previous observations.
BrdUrd Incorporation into B Lymphocytes in Vivo.
To assess the biological relevance of these ex vivo studies, we next aimed to analyze cell proliferation and renewal in vivo. Our approach was based on the direct injection of BrdUrd into sheep. BrdUrd labels cells in the S-phase of the cycle after its incorporation into nascent DNA (38). Being labile in vivo (39), extracellular BrdUrd will only be incorporated into proliferating cells soon after injection. A single dose of BrdUrd was injected into three asymptomatic BLV-infected sheep (nos. 8, 104, and 293) and into three seronegative controls (animals 117, 1092, and 1097). To evaluate the kinetics of BrdUrd incorporation, an aliquot of blood from each animal was collected at regular intervals (1–3 days) after injection. After lysis of the red blood cells, the leukocytes were labeled with biotinylated 1H4 monoclonal antibody and streptavidin-phycoerythrin (PE) conjugate. The cells then were permeabilized, stained with anti-BrdUrd/FITC in the presence of DNase, and analyzed by two-color flow cytometry. Based on their size (FSC) and granularity (SSC), the lymphocyte and monocyte populations were selected to discard the granulocytes from the analysis. It appeared that more B+BrdUrd+ cells (arrows on Fig. 2A) are stained in samples from the three BLV-infected sheep (absolute cells count 187, 73, 139 among 10,000 events) compared with the controls (5, 4, 18). Because infected animals have more B cells, we calculated the proportion of BrdUrd+ cells within the sIgM+ cell population in the three infected sheep and three controls (Fig. 2B). A mathematical model was fitted to this data. The mathematical model includes (i) the rate of proliferation and death (apoptosis and necrosis) of the B lymphocytes; (ii) the probability of a cell becoming labeled, which declines exponentially with time reflecting the loss of unincorporated label after a single injection; and (iii) the dilution of the BrdUrd label upon division. The model assumes that the duration of the S phase was constant, and that the global B cell population remained stable throughout the experiment (as confirmed by the leukocyte counts, data not shown; see Materials and Methods for details). The fit of the theoretical formula to the data was good (Fig. 2B). The minimal proliferation and death rates (±SD) estimated from fitting the formula to the data are shown in Fig. 2C. The proliferation rates quantify the number of new cells that are produced by proliferation every day. For example, in BLV-infected sheep 8, the proliferation rate was 0.018 per day, corresponding to 1.8% of peripheral blood B cells produced by proliferation per day. Similarly the death rate quantifies the loss of B cells in infected and control animals (Fig. 2C). The experiment was repeated 2 months later in the same six sheep (experiment 2). The average proliferation rate (across both experiments) was 0.020 per day in infected and 0.011 per day in control sheep, considering similar B cell proportions (20%) in both categories of animals. These rates were statistically different as measured by the Wilcoxon–Mann–Whitney test. Surprisingly, and in apparent contradiction with the interpretation resulting from ex vivo experiments (Fig. 1), the rates of cell death were not statistically different between the two categories of sheep (0.089 per day vs. 0.094 per day; Fig. 2D). If the mathematical formula is altered to allow for an increase in cells in infected sheep, then it can be seen that there is a population growth rate of 7% per day at the start of the experiment.
Figure 2.

BrdUrd incorporation into B lymphocytes in vivo. (A) Three BLV-infected sheep (nos. 8, 104, and 293) and three controls (nos. 117, 1092, and 1097) were injected intravenously with 500 mg BrdUrd, and an aliquot of blood (1 ml) was collected 3 days later. After lysis of the red blood cells, B cells were labeled with biotinylated 1H4 monoclonal antibody and streptavidin-PE conjugate. Then, the cells were stained with anti-BrdUrd FITC antibody in the presence of DNase and analyzed by two-color flow cytometry (x axis, BrdUrd; y axis, B lymphocytes). Ten thousand cells (lymphocytes, monocytes, and granulocytes) were acquired, and PBMCs were selected by the FSC/SSC gating. The total numbers of B cells are indicated in the upper quadrants. (B) Kinetic analysis of BrdUrd+ B cells. Blood samples from six sheep (see A) were collected at different days after BrdUrd injection. The ratio (in %) of BrdUrd+ cells within the total B lymphocyte population was determined, and the data corresponding to the measured incorporation rates were fitted to a mathematical model, yielding a theoretical fit (see Materials and Methods). Figure shows the average data for the three infected and the three control sheep. (C) Minimal proliferation and death rates (± SD) estimated from fitting the source model to the data deduced from two independent experiments. (D) Summary of the proliferation and death rates (± SD) and estimation of the population growth.
B Cell Proliferation and Renewal in Leukemic Sheep.
To estimate the rates of B cell proliferation at later stages of the disease, we next analyzed a series of leukemic sheep. A single dose of 500 mg of BrdUrd was injected i.v. into three leukemic animals (nos. 2668, 2658, and 2667) exhibiting high leukocyte counts (36,000, 70,000, and 174,000 cells per μl, respectively; Fig. 3A). Because it was impossible to distinguish the different leukocyte populations from these leukemic sheep by using the FSC/SSC gating method, the whole population of white blood cells (i.e., granulocytes, lymphocytes, and monocytes) was analyzed, the B lymphocytes representing more than 80% of them. At regular intervals, the levels of BrdUrd incorporation were determined by two-color flow cytometry on the basis of 10,000 events, as described previously. The peak levels of BrdUrd incorporation in the leukemic sheep correlated with their total leukocyte count (Fig. 3B). The sheep with the lowest leukocyte count (sheep 2668), had a peak BrdUrd incorporation of 3.1%, comparable with the levels of incorporation in asymptomatic infected sheep. Sheep 2667 had an extremely high leukocyte count and died of leukemia 3 days after the injection of BrdUrd (Fig. 3A, †). In this animal, the proportion of B lymphocytes having incorporated BrdUrd reached 18%. Sheep 2658 was in between these two extremes, having an intermediate leukocyte count and an intermediate level of BrdUrd incorporation (9%). The minimal proliferation and death rates were estimated by fitting to BrdUrd labeling data and weighted leukocyte counts (fit shown in Fig. 3B, parameter estimates shown in 3C). This analysis suggested that tremendous cell proliferation occurred in the leukemic sheep (rates of 0.02 to 0.12 per day). The proliferation rates were correlated with the leukocyte count. We conclude that, in these three leukemic sheep, the proliferation rate increased with the progression of the disease.
Figure 3.

B cell proliferation and renewal in leukemic sheep. (A) Evolution of leukocyte counts (y axis in 103 cells per mm3) with time (x axis in months). Sheep 2667 died at day 3 of the study (†). Arrows indicate the estimated cell counts. (B) Percentages of B cells labeled with BrdUrd and the theoretical fit. (C) Minimal proliferation and death rates were estimated from fitting a theoretical model to the BrdUrd labeling and weighted extrapolated leukocyte count data.
Proliferation and Viral Expression.
With the aim of correlating cell turnover and viral expression, cells from three BLV-infected sheep (nos. 8, 2668, and 2658) and one control (no. 1097) were isolated 2 days after BrdUrd injection. PBMCs were transiently cultivated to trigger viral expression, labeled with anti-p24 and BrdUrd antibodies and analyzed by flow cytometry. Thirty-three, 7, and 1% of lymphocytes were p24+ in animals 8, 2668, and 2658, respectively (Fig. 4). Interestingly, the proportion of double-positive cells did not exceed the background level in any of the three infected sheep. We conclude that cells spontaneously expressing viral proteins ex vivo as revealed by the p24 major capsid protein did not incorporate BrdUrd in vivo. In other words, it seems that in vitro viral expression and proliferation in vivo are mutually exclusive.
Figure 4.

Proliferation and viral expression. (A) Two days after BrdUrd injection, PBMCs from noninfected (no. 1097), asymptomatic (no. 8), and lymphocytic (nos. 2668 and 2658) sheep were isolated and cultivated during 18 h. The cells then were fixed and incubated with anti-p24 antibody 4′G9, which recognizes the viral capsid protein, and with a PE-conjugated secondary antibody. Finally, cells were stained with anti-BrdUrd FITC with DNase and analyzed by flow cytometry. A representative experiment (out of three) is represented by dot plots (10,000 selected events). Numbers represent the percentages of positively stained cells in each quadrant. (B) Schematic representation of BrdUrd incorporation in vivo (hypothetical) and p24 labeling after short-term culture (ex vivo). Hatched areas represent the uninfected cell; black squares and full circles indicate BrdUrd and p24 markers, respectively. Crosses indicate that cells were undetectable by FACS.
Discussion
Viral dynamics of oncoviruses such as BLV and HTLV-1 is a very controversial area of research. On one side, cloning and sequencing data indicate that replication of the infected cell accounts for the expansion of the proviral pool that can be detected in the peripheral blood (40, 41). On the other hand, in vitro (ex vivo) assays lend support to an active replication of the virus associated with neo-infection of novel host cells and underline a very efficient immune response that depends on cytotoxic T lymphocytes (42). These two routes of viral spread are not necessarily contradictory and could co-exist. However, from ex vivo experiments, it is difficult to quantify the relative importance of clonal expansion vs. neo-infection. The more direct approach that we have used here casts some light onto a very active process of selection against infected cells. Indeed, dual flow cytometry (as illustrated in Fig. 4A) demonstrates an almost complete absence of p24+BrdUrd+ double-positive cells, revealing the mutually exclusive presence of both markers. In other words, among all infected cells proliferating in vivo as measured by BrdUrd uptake, none was found to express viral proteins. Because p24+ cells are spared from apoptosis ex vivo (17), p24+BrdUrd+ double positives were not lost during the culture but rather were eliminated in vivo (a model is schematized on Fig. 4B). If we postulate that viral expression and cell activation are closely linked, as largely illustrated by in vitro data for example, the promotion of the cycle by Tax and, conversely, the activation of viral expression by polyclonal lymphocyte activators (reviewed in refs. 11 and 43), the lack of p24+BrdUrd+ double-positive cells reveals a very efficient negative selection taking place in vivo. Another interpretation would be that only a subpopulation of cells harboring an integrated provirus is allowed to proliferate (i.e., incorporate BrdUrd) provided that no viral proteins are expressed. Of note, our flow cytometry analyses were performed with an anti-p24 antibody, and it remains possible that other viral proteins such as Tax are constitutively expressed at low levels in vivo, stimulating continuous proliferation. However, we tend to believe that this assumption is unlikely (but not impossible) because tax mRNA is barely detectable.
Another contribution of this report concerns the balance between cell proliferation and death. In particular, we have first underlined discrepancies that might occur between ex vivo (Fig. 1) and in vivo experiments (Fig. 2). Indeed, cell cycle analyses performed under basal and activated conditions did not reveal any increase in cell proliferation ex vivo. Although a higher proportion of B lymphocytes in the infected animals accounts for an absolute rise in proliferation, experimental and control B cells spontaneously cycle with similar relative efficiencies ex vivo. In contrast, in vivo, B lymphocytes from BLV-infected animals proliferated faster (average proliferation rate 0.020 per day vs. 0.011 per day for normal sheep), and this difference became even more pronounced at the late stage lymphocytosis immediately preceding the onset of overt leukemia (up to tenfold). This increase in proliferation could even be underestimated because bromodeoxyuridine is potentially cytotoxic after incorporation into the nascent DNA of the dividing cell (44). Although we cannot formally exclude this possibility, we think that this is unlikely because (i) BrdUrd cytotoxicity occurs mainly after continuous administration but not after a single injection; (ii) sheep were perfectly healthy and did not suffer from diarrhea, a reliable symptom of BrdUrd toxicity; and (iii) the results from serial independent experiments on the same animals were reproducible. Another possible shortcoming of our approach concerns the amount of bromodeoxyuridine used, which could limit the measured proliferation rates. This possibility could be ruled out because three consecutive injections each of 400 mg of BrdUrd, separated by 60 min, did not significantly modify the level of bromodeoxyuridine incorporation (data not shown).
We observed that after an initial peak, the proportion of BrdUrd-labeled cells decreased with time (Fig. 2B and 3B). This decline could result from dilution of the BrdUrd label resulting from proliferation as well as from cell death. Fitting the theoretical formula (see Materials and Methods) to the data shows that the decrease in label is due to the fact that the death rate of cells exceeded their proliferation rate. Most importantly, the death rates were not significantly different between infected and control sheep (Fig. 2D), indicating that the apoptotic process was not modulated by BLV. This conclusion holds true if the B lymphocyte population is considered as homogeneous, and if all putative subpopulations of cells replicate at similar rates. Of note, phenotypic characterization by means of CD5 or CD11b markers, two molecules suggested to be associated with BLV infection (33, 45), did not reveal any preferential proliferation of a given B cell subtype (data not shown). However, long-term analysis of BrdUrd incorporation demonstrated that a significant proportion of B lymphocytes might remain positive even at 44 days after labeling (Fig. 2B and data not shown), indicating that a minority of cells are protected from apoptosis and survive in a resting stage for extended periods of time. These lymphocytes might account for the persistence of the virus and explain the long-term maintenance of given cell clones within the infected host. Furthermore, continuous accumulation of these cells, possibly memory lymphocytes, could account for the expansion of the proviral pool. Our in vivo analysis demonstrates that BLV infection is mainly associated with an increase in cell proliferation but not with a reduction in apoptosis. This process contrasts with the mechanism recently described for another retrovirus, SIV, whose infection in rhesus macaques provokes a substantial increase of both cell death and proliferation (37). Conversely, chronic lymphocytic leukemia, a B cell neoplasm resembling persistent lymphocytosis in BLV-infected cattle, is primarily characterized by a problem with apoptosis rather than abnormal proliferation (46, 47).
Finally, and most interestingly, the dynamical analysis revealed an unexpected result related to the still unclear mechanism of lymphocyte homeostasis. Indeed, the net increase in the number of cells produced in infected animals compared with the controls was estimated at 7% in the first day (Fig. 2D), corresponding to a very fast doubling of the population. Thus, the expected increase in cell numbers seems to be far above what is actually observed. Therefore, other processes, such as a limited but significant reduction of B lymphocyte release from the tissues are required to maintain the cell population in the peripheral blood at steady-state levels. Inhibition of cell release from the tissues is a mechanism of feedback regulation controlling homeostasis that has been elegantly characterized in mice (48). Homeostatic control of cell production from the lymph nodes, the bone marrow, or the Peyer's patches could thus be a major determinant of leukemogenesis induced by complex oncoviruses.
Acknowledgments
We thank K. Walravens (Centre d'Étude et de Recherches Vétérinaires et Agrochimiques, Uccle, Belgium) and J. J. Letesson (Facultés Universitaires Notre-Dame de la Paix de Namur, Belgium) for kindly providing the 1H4 antibody. We thank J. M. Londes, M. Nuttinck, T. Peremans, and G. Vandendaele for excellent technical help, and C. R. M. Bangham and E. Hanon for helpful discussions. For financial support, we thank the Fortis Bank Assurance, the Belgian Federation against Cancer, the Fonds National de la Recherche Scientifique, the Loterie Nationale, the Service de Programmation pour la Politique Scientifique, the Action de Recherche Concertée du Ministère de la Communauté Française, and the Wellcome Trust. R.K. and L.W. are research directors of the Fonds National de la Recherche Scientifique; C.D. is a fellow of the Action de Recherche Concertée du Ministère de la Communauté Française.
Abbreviations
- BLV
bovine leukemia virus
- HTLV-1
human T lymphotropic virus
- PBMC
peripheral blood mononuclear cell
- PMA
phorbol 12-myristate 13-acetate
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Gallo R C. Nat Med. 1995;1:753–759. doi: 10.1038/nm0895-753. [DOI] [PubMed] [Google Scholar]
- 2.Yoshida M. Gann. 1983;74:777–789. [PubMed] [Google Scholar]
- 3.Ferrer J F, Marshak R R, Abt D A, Kenyon S J. Ann Rech Vet. 1978;9:851–857. [PubMed] [Google Scholar]
- 4.Burny A, Cleuter Y, Kettmann R, Mammerickx M, Marbaix G, Portetelle D, Van den Broecke A, Willems L, Thomas R. Adv Vet Sci Comp Med. 1988;32:149–170. doi: 10.1016/b978-0-12-039232-2.50010-4. [DOI] [PubMed] [Google Scholar]
- 5.Kettmann R, Portetelle D, Mammerickx M, Cleuter Y, Dekegel D, Galoux M, Ghysdael J, Burny A, Chantrenne H. Proc Natl Acad Sci USA. 1976;73:1014–1018. doi: 10.1073/pnas.73.4.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Longo D L, Gelmann E P, Cossman J, Young R A, Gallo R C, O'Brien S J, Matis L A. Nature (London) 1984;310:505–506. doi: 10.1038/310505a0. [DOI] [PubMed] [Google Scholar]
- 7.Seiki M, Hattori S, Yoshida M. Proc Natl Acad Sci USA. 1982;79:6899–6902. doi: 10.1073/pnas.79.22.6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kettmann R, Cleuter Y, Mammerickx M, Meunier-Rotival M, Bernardi G, Burny A, Chantrenne H. Proc Natl Acad Sci USA. 1980;77:2577–2581. doi: 10.1073/pnas.77.5.2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seiki M, Eddy R, Shows T B, Yoshida M. Nature (London) 1984;309:640–642. doi: 10.1038/309640a0. [DOI] [PubMed] [Google Scholar]
- 10.Hanon E, Hall S, Taylor G P, Saito M, Davis R, Tanaka Y, Usuku K, Osame M, Weber J N, Bangham C R. Blood. 2000;95:1386–1392. [PubMed] [Google Scholar]
- 11.Willems L, Burny A, Collete D, Dangoisse O, Dequiedt F, Gatot J S, Kerkhofs P, Lefebvre L, Merezak C, Peremans T, et al. AIDS Res Hum Retroviruses. 2000;16:1787–1795. doi: 10.1089/08892220050193326. [DOI] [PubMed] [Google Scholar]
- 12.Muscoplat C C, Alhaji I, Johnson D W, Pomeroy K A, Olson J M, Larson V L, Stevens J B, Sorensen D K. Am J Vet Res. 1974;35:1053–1055. [PubMed] [Google Scholar]
- 13.Itoyama Y, Minato S, Kira J, Goto I, Sato H, Okochi K, Yamamoto N. Neurology. 1988;38:1302–1307. doi: 10.1212/wnl.38.8.1302. [DOI] [PubMed] [Google Scholar]
- 14.Prince H E, Lee H, Jensen E R, Swanson P, Weber D, Fitzpatrick L, Doyle M, Kleinman S. Blood. 1991;78:169–174. [PubMed] [Google Scholar]
- 15.Mann D L, Martin P, Hamlin-Green G, Nalewaik R, Blattner W. Clin Immunol Immunopathol. 1994;72:312–320. doi: 10.1006/clin.1994.1147. [DOI] [PubMed] [Google Scholar]
- 16.Teodoro J G, Branton P E. J Virol. 1997;71:1739–1746. doi: 10.1128/jvi.71.3.1739-1746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dequiedt F, Hanon E, Kerkhofs P, Pastoret P P, Portetelle D, Burny A, Kettmann R, Willems L. J Virol. 1997;71:630–639. doi: 10.1128/jvi.71.1.630-639.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dequiedt F, Cantor G H, Hamilton V T, Pritchard S M, Davis W C, Kerkhofs P, Burny A, Kettmann R, Willems L. J Virol. 1999;73:1127–1137. doi: 10.1128/jvi.73.2.1127-1137.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwartz-Cornil I, Chevallier N, Belloc C, Le Rhun D, Laine V, Berthelemy M, Mateo A, Levy D. J Gen Virol. 1997;78, Pt. 1:153–162. doi: 10.1099/0022-1317-78-1-153. [DOI] [PubMed] [Google Scholar]
- 20.Pise-Masison C A, Choi K S, Radonovich M, Dittmer J, Kim S J, Brady J N. J Virol. 1998;72:1165–1170. doi: 10.1128/jvi.72.2.1165-1170.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suzuki T, Kitao S, Matsushime H, Yoshida M. EMBO J. 1996;15:1607–1614. [PMC free article] [PubMed] [Google Scholar]
- 22.Schmitt I, Rosin O, Rohwer P, Gossen M, Grassmann R. J Virol. 1998;72:633–640. doi: 10.1128/jvi.72.1.633-640.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neuveut C, Low K G, Maldarelli F, Schmitt I, Majone F, Grassmann R, Jeang K T. Mol Cell Biol. 1998;18:3620–3632. doi: 10.1128/mcb.18.6.3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lemoine F J, Marriott S J. J Biol Chem. 2001;276:31851–31857. doi: 10.1074/jbc.M105195200. [DOI] [PubMed] [Google Scholar]
- 25.Ohtani K, Iwanaga R, Arai M, Huang Y, Matsumura Y, Nakamura M. J Biol Chem. 2000;275:11154–11163. doi: 10.1074/jbc.275.15.11154. [DOI] [PubMed] [Google Scholar]
- 26.Chlichlia K, Moldenhauer G, Daniel P T, Busslinger M, Gazzolo L, Schirrmacher V, Khazaie K. Oncogene. 1995;10:269–277. [PubMed] [Google Scholar]
- 27.Copeland K F, Haaksma A G, Goudsmit J, Krammer P H, Heeney J L. AIDS Res Hum Retroviruses. 1994;10:1259–1268. doi: 10.1089/aid.1994.10.1259. [DOI] [PubMed] [Google Scholar]
- 28.Portetelle D, Mammerickx M, Burny A. J Virol Methods. 1989;23:211–222. doi: 10.1016/0166-0934(89)90135-3. [DOI] [PubMed] [Google Scholar]
- 29.Willems L, Thienpont E, Kerkhofs P, Burny A, Mammerickx M, Kettmann R. J Virol. 1993;67:1086–1089. doi: 10.1128/jvi.67.2.1086-1089.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Twizere J C, Kerkhofs P, Burny A, Portetelle D, Kettmann R, Willems L. J Virol. 2000;74:9895–9902. doi: 10.1128/jvi.74.21.9895-9902.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Merezak C, Pierreux C, Adam E, Lemaigre F, Rousseau G G, Calomme C, Van Lint C, Christophe D, Kerkhofs P, Burny A, et al. J Virol. 2001;75:6977–6988. doi: 10.1128/JVI.75.15.6977-6988.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Willems L, Grimonpont C, Kerkhofs P, Capiau C, Gheysen D, Conrath K, Roussef R, Mamoun R, Portetelle D, Burny A, et al. Oncogene. 1998;16:2165–2176. doi: 10.1038/sj.onc.1201765. [DOI] [PubMed] [Google Scholar]
- 33.Letesson J J, Van den Broecke A, Marbaix-Cleuter Y, Delcommenne M, Mager A, Mammerickx M, Burny A, Depelchin A. Vet Immunol Immunopathol. 1991;27:207–213. doi: 10.1016/0165-2427(91)90102-i. [DOI] [PubMed] [Google Scholar]
- 34.Hellerstein M K. Immunol Today. 1999;20:438–441. doi: 10.1016/s0167-5699(99)01529-7. [DOI] [PubMed] [Google Scholar]
- 35.Rosenzweig M, DeMaria M A, Harper D M, Friedrich S, Jain R K, Johnson R P. Proc Natl Acad Sci USA. 1998;95:6388–6393. doi: 10.1073/pnas.95.11.6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bonhoeffer S, Mohri H, Ho D, Perelson A S. J Immunol. 2000;164:5049–5054. doi: 10.4049/jimmunol.164.10.5049. [DOI] [PubMed] [Google Scholar]
- 37.Mohri H, Bonhoeffer S, Monard S, Perelson A S, Ho D D. Science. 1998;279:1223–1227. doi: 10.1126/science.279.5354.1223. [DOI] [PubMed] [Google Scholar]
- 38.Gratzner H G. Science. 1982;218:474–475. doi: 10.1126/science.7123245. [DOI] [PubMed] [Google Scholar]
- 39.Kriss J P, Revesz L. Cancer Res. 1962;22:254–265. [PubMed] [Google Scholar]
- 40.Mortreux F, Leclercq I, Gabet A S, Leroy A, Westhof E, Gessain A, Wain-Hobson S, Wattel E. J Natl Cancer Inst. 2001;93:367–377. doi: 10.1093/jnci/93.5.367. [DOI] [PubMed] [Google Scholar]
- 41.Wattel E, Cavrois M, Gessain A, Wain-Hobson S. J Acquired Immune Defic Syndr Hum Retrovirol. 1996;13, Suppl. 1:S92–S99. doi: 10.1097/00042560-199600001-00016. [DOI] [PubMed] [Google Scholar]
- 42.Bangham C R. J Clin Pathol. 2000;53:581–586. doi: 10.1136/jcp.53.8.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoshida M. Annu Rev Immunol. 2001;19:475–496. doi: 10.1146/annurev.immunol.19.1.475. [DOI] [PubMed] [Google Scholar]
- 44.Reome J B, Johnston D S, Helmich B K, Morgan T M, Dutton-Swain N, Dutton R W. J Immunol. 2000;165:4226–4230. doi: 10.4049/jimmunol.165.8.4226. [DOI] [PubMed] [Google Scholar]
- 45.Chevallier N, Berthelemy M, Le Rhun D, Laine V, Levy D, Schwartz-Cornil I. J Virol. 1998;72:4413–4420. doi: 10.1128/jvi.72.5.4413-4420.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reed J C. Semin Oncol. 1998;25:11–18. [PubMed] [Google Scholar]
- 47.Voutsadakis I A. Acta Oncol. 2000;39:151–156. doi: 10.1080/028418600430707. [DOI] [PubMed] [Google Scholar]
- 48.Agenes F, Rosado M M, Freitas A A. Curr Top Microbiol Immunol. 2000;252:68–75. [PubMed] [Google Scholar]