

Abstract
Cellular Fas-associated death domain-like interleukin-1-beta converting enzyme (FLICE) inhibitory proteins (cFLIPs) are endogenous caspase homologues that inhibit programmed cell death. We hypothesized that cFLIPs are differentially expressed in response to traumatic brain injury (TBI). cFLIP-alpha and cFLIP-delta mRNA were expressed in normal mouse brain—specifically cFLIP-delta (but not cFLIP-alpha) protein was robustly expressed. After controlled cortical impact (CCI), cFLIP-alpha expression increased initially then decreased to control levels at 12 h, increasing again at 24–72 h (P<0.05). cFLIP-delta expression was decreased in brain homogenates by 12 h after CCI, then increased again at 24 to 72 h (P<0.05). cFLIP-delta immunostaining was markedly reduced in injured cortex, but not hippocampus, at 3 to 72 h after CCI. In cortex, reduced cFLIP-delta staining was found in TUNEL-positive cells, but in hippocampus TUNEL-positive cells expressed cFLIP-delta immunoreactivity. cFLIP-delta was increased in a subset of reactive astrocytes in pericontusional cortex and hippocampus at 48 to 72 h. Low levels of both cFLIP isoforms were detected in human cortical tissue with no TBI, from four patients undergoing brain surgery for epilepsy and <24 h post mortem from three patients without CNS pathologic assessment. In cortical tissue surgically removed <18 h after severe TBI (n=3), cFLIP-alpha expression was increased relative to epilepsy controls (P<0.05) but not relative to post-mortem controls. The data suggest differential spatial and temporal regulation of cFLIP-alpha and cFLIP-delta expression that may influence the magnitude of cell death and further implicate programmed mechanisms of cell death after TBI.
Keywords: caspases, human, mice, neuronal cell death, programmed cell death, reactive astrocytes, trauma, traumatic brian injury
Introduction
Traumatic brain injury (TBI) induces delayed, progressive cell death that contributes to secondary injury and neurologic outcome. The tumor necrosis factor receptor (TNFR) superfamily of death receptors (e.g., Fas, TNFR1) are potent initiators of programmed cell death (Hengartner, 2000; Wajant et al, 2003; Wallach et al, 1999). After specific ligand binding, TNFRs recruit cytosolic adapter proteins such as Fas-associated protein with a death domain (FADD), which recruits initiator procaspases (e.g., 2, 8, and 10) to form a ‘death inducing signaling complex’ (DISC) (Hengartner, 2000). After cleavage and autoactivation at the DISC, initiator caspases cleave and activate effector (or ‘executioner’) caspases (e.g., caspases 3 and 7), which mediate cell death by proteolysis of critical cell constituents. Activated Fas can also initiate caspase-independent necrosis in some cell types including neurons, possibly via generation of oxygen free radicals (Kawahara et al, 1998; Vercammen et al, 1998; Matsumura et al, 2000; Raoul et al, 2002; Wajant et al, 2003).
There is mounting evidence that death receptor activation and DISC assembly are implicated in acute central nervous system (CNS) injury (Clark et al, 1999; Qiu et al, 2002; Zhang et al, 2003). Fas-FADD-procaspase-8 DISC assembles in mouse and human brain after TBI (Qiu et al, 2002), and tumor necrosis factor-alpha (TNFα) or Fas ligand suppression reduces cell death after closed head injury TBI (Shohami et al, 1996) or spinal cord transection (Demjen et al, 2004), respectively. Dual antagonism of both TNFα and Fas ligand reduces ischemic stroke in mice by up to 80% (Martin-Villalba et al, 2001). These and other studies suggest that death receptors may initiate cell death after acute CNS injury, and that endogenous mechanisms that inhibit death receptors and their downstream pathways might be neuroprotective (Lo et al, 2003; Nuttall et al, 2001; Robertson et al, 2000).
Cellular FLICE inhibitory proteins (cFLIPs) are endogenous caspase-8/10 homologues that lack active site amino-acid sequences and thus are catalytically inactive (Irmler et al, 1997; Krueger et al, 2001; Micheau, 2003; Rasper et al, 1998). Two splice variants, termed cFLIP-α (cFLIP-long, MW ~55 kD) and cFLIP-δ(cFLIP-short, MW 25 to 28 kD), are the most widely distributed and well studied isoforms (Irmler et al, 1997; Krueger et al, 2001; Micheau, 2003; Rasper et al, 1998). These proteins inhibit cell death by competing with procaspase-8 for binding to FADD and inhibiting assembly of a functional DISC (Rasper et al, 1998; Scaffidi et al, 1999). At least two other splice variants are predicted from mRNA transcripts, cFLIP-β and cFLIP-γ (Rasper et al, 1998; Tschopp et al, 1998). The functions of these two splice variants are currently unknown.
Numerous studies have suggested that cFLIPs mitigate cell death in experimental injury paradigms in vivo, and cFLIP-α and/or -δ protect against death receptor- or caspase-mediated cell death in vivo and in cultured cells, including endothelial cells, spinal motor neurons, and neuron-like PC-12 cells (Imanishi et al, 2000a; Raoul et al, 1999; Wu et al, 2004). However, evidence for a role for cFLIPs in CNS injury and development remains primarily circumstantial (Raoul et al, 1999; Engidawork et al, 2001; Zhao et al, 2003). Overexpression of cFLIP-α protects cultured PC12 cells from cell death mediated by Fas engagement, but not by serum withdrawal (Wu et al, 2004), suggesting that cFLIPs may selectively inhibit discrete cell death mechanisms after neuronal injury. If so, modulation of cFLIP expression could influence the extent of cell death after TBI. The aim of the current study was to characterize the expression of cFLIPs in brain, and to test the hypothesis that cFLIP is differentially expressed in contused brain as part of an endogenous response to TBI in mice and in humans.
Materials and methods
Controlled Cortical Impact
The controlled cortical impact (CCI) model was used as previously described with minor modifications (Qiu et al, 2002). The trauma protocol was approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee and complied with the NIH Guide for the Care and Use of Laboratory Animals. Male C57Bl/6 Mice 8–12 weeks of age (Charles River Laboratories, Wilmington, MA, USA) were anesthetized with 4% isoflurane (Anaquest, Memphis, TN, USA) in 70% N2O and 30% O2 using a Fluotec 3 vaporizer (Colonial Medical, Amherst, NH, USA) and positioned in a stereotaxic frame. Anesthesia was maintained using 2% to 3% isoflurane. Body temperature was monitored with a rectal probe and maintained at 35°C to 38°C with a heating pad. A 5-mm craniotomy was made using a portable drill, and a 5-mm trephine over the left parieto-temporal cortex and the bone flap was removed. Mice were then subjected to CCI using a pneumatic cylinder with a 3-mm flat-tip impounder, velocity 6 m/sec, depth of 0.6 mm, and 100 ms impact duration. The bone flap was replaced, and the scalp was sutured closed. The mice were allowed to recover in room air until able to ambulate (approximately 15 min), and returned to their cages.
Human Brain Samples
Human studies were approved by the Massachusetts General Hospital Institutional Review Board for Human Research Protection. TBI and epilepsy tissue samples were obtained from the R Adams Crowley Shock Trauma Center (Baltimore, MD, USA) and the University of Maryland (Baltimore, MD, USA), respectively. Control human brain samples were obtained from the Massachusetts General Hospital Department of Pathology. Contused brain tissue surgically removed within 18 h of injury from male patients (16 to 75 years of age) with severe TBI and refractory intracranial hypertension was immediately frozen in liquid nitrogen and stored at −80°C. Mechanisms of head injury included motor vehicle accidents, falls, or assault. All TBI patients received individualized critical care as dictated by the attending physicians. Brain tissue samples removed from adult patients with intractable epilepsy, or post-mortem brain samples taken within 24 h of death from adult patients who died of non-CNS causes, were also frozen in liquid nitrogen and stored at −80°C. Brain samples were thawed in homogenization buffer on ice in the presence of protease inhibitors and used for Western blot analyses (see below).
Antibodies and Reagents
Anti-cFLIP antibodies used were anti-cFLIP #1159 (Alexis-Axxora Biochemicals, San Diego, CA, USA), and anti-cFLIP-δ (PX112; Cell Sciences, Norwood, MA, USA). Anti-cFLIP 1159 detected bands corresponding to cFLIP-α and cFLIP-δ, and anti-PX112 detected bands corresponding to cFLIP-δ in lysates of mouse heart tissue and Jurkat cells, known to express these cFLIP isoforms (Imanishi et al, 2000a, b; Rasper et al, 1998; Wang et al, 2002) (data not shown). Anti-cFLIP 1159 also recognized recombinant human cFLIP-α (at 55 to 60 kD), and cFLIP-δ at 28 kD in Jurkat cell lysates (a positive control for the delta splice variant). Mouse spleen (negative control) and heart (positive control) tissue homogenates were also used to confirm sensitivity and specificity of anti-cFLIP antibodies in Western blots. Beta-actin was detected using a monoclonal mouse anti-beta actin (Sigma, St Louis, MO, USA). Recombinant human cFLIP-α was obtained from R&D systems (Minneapolis, MN, USA). Human Jurkat cell lysates were from Transduction Laboratories (Lexington, KY, USA) and Upstate Biochemical (Lake Placid, NY, USA).
To increase sensitivity of immunohistochemical detection of cFLIPs, tyramide amplification kits were used according to the manufacturer’s instructions, with Alexa 594 or Alexa 488 as fluorophores (Molecular Probes, Eugene, OR, USA). Monoclonal antibodies to the neuronal marker NeuN (Chemicon, Temecula, CA, USA) and the astrocytic marker GFAP (Sigma, St Louis, MO, USA) were conjugated to Cy2 or Cy3 fluorophores, and were used in double labelling experiments. The endothelial marker was Rhodamine-conjugated Griffonia (Bandeiraea) Simplicifolia Lectin I (Vector Laboratories, Burlingame, CA, USA), used at 1:100 dilution.
3′-Rapid Amplification of cDNA Ends Reverse Transcriptase-Polymerase Chain Reaction
Total RNA was extracted from 300 mg of mouse brain using Trizol (Invitrogen, Paisley, UK) according to the manufacturer’s instructions. RNA was DNAse treated and reextracted with Trizol to remove contaminating genomic DNA. A 3′-rapid amplification of cDNA ends (3′-RACE) was performed on mouse cerebral cortex RNA using a mouse cFLIP-specific oligonucleotide ‘flip all’ (ATGAATACTCTCCAGGCTTCGCTCCCA) that was predicted to be present in all mouse splice variants based on the human cFLIP sequences (Irmler et al, 1997; Rasper et al, 1998). 3′-RACE was performed using a SMART RACE cDNA amplification kit (Clontech, Rockville, MD, USA). Reverse transcriptions were also performed in the absence of the enzyme or in the absence of template DNA, as controls for contaminating DNA. These control experiments resulted in no amplification products seen after PCR (data not shown). Amplified cDNAs (5 μL) were subjected to gel electrophoresis on a 1% (w/v) agarose gel. Amplification products were cloned into the TA vector pCR2.1 (Invitrogen) and subjected to sequence analysis.
Immunoblotting
Brain tissue dissected from contused cortex within the injured hemisphere or from the left hemisphere of uninjured control mice was homogenized on ice in a modified RIPA buffer (50 mmol/L Tris-HCl, pH 7.5, 50 mmol/L NaCl, 4 mmol/L urea, 0.5% sodium dodecyl sulfate, 0.5% NP-40, 0.5% Na-deoxycholate, 5 mmol/L phenylmethylsulfonylfluoride (PMSF), 1 mmol/L EDTA, 5 mmol/L EGTA, 10 mmol/L dithiothreitol, and protease inhibitor cocktail (1:100, Sigma). Homogenates were cleared by centrifugation at 20,800 g for 15 mins at 4°C. Protein content of the supernatant was assayed (Bio-Rad laboratories, Hercules, CA, USA) and aliquots of protein were boiled in denaturing sample buffer (62.5 mmol/L Tris pH 6.8, 2% SDS, 5 mmol/L EDTA, 10% glycerol, 0.25% 2-mercaptoethanol, 0.01% bromophenol blue). Heart, spleen, and Jurkat cell lysates were loaded at 40 μg total protein per lane, and brain samples were 20 μg per lane. Denatured protein was size fractionated on 10% SDS-polyacrylamide gels (Invitrogen, Carlsbad, CA, USA) and blotted onto Immobilon 0.45 μm PVDF membranes (Millipore, Bedford, MA, USA). Membranes were blocked for 1 h in 5% fat-free dried milk in Tris-buffered saline pH 7.4 containing 0.1% Tween 20 (TBST), then incubated overnight at 4°C in primary antibodies diluted (1:500) in TBST with 0.5% milk. Membranes were washed in TBST and 5% milk, then incubated for 1 h with the appropriate horseradish peroxidase-conjugated secondary antibody (1:20,000 in TBST, 5% milk) at room temperature. Proteins of interest were detected using the enhanced chemiluminescence Western blotting detection system kit (ECL Plus, Amersham, Buckinghamshire, UK) and Hyperfilm (Amersham, Oakville, Ontario, Canada). cFLIP expression was normalized to that of beta-actin, and was semiquantitated using densitometry performed with an M4 imaging system (Imaging Research, Inc., St Catherines, Ontario, Canada).
Immunohistochemistry and TUNEL
Coronal brain sections cut on a cryostat (10 to 15 μm) were placed on poly-l-lysine coated slides and stored at −80°C. Slides were fixed in 100% ethanol at room temparature for 10 mins then washed in phosphate-buffered saline (PBS) (pH 7.4) containing 0.1% Triton X-100 (PBST). Sections were boiled for 8 mins in antigen unmasking solution (Vector Laboratories, Burlingame, CA, USA), blocked for 1 h in PBST containing 5% normal goat serum (NGS), then incubated overnight with PX112 (1:100) at 4°C. Sections were washed in PBST and incubated with the appropriate secondary antibodies (all from Jackson Immunoresearch, or from Molecular Probes when tyramide amplification was used) for 60 mins. After washing in PBST, sections were incubated with anti-mouse NeuN-Cy2 conjugate (1:100; Chemicon Inc, Temecula, CA, USA) or anti-mouse GFAP-Cy3 (1:100; Sigma, St Louis, MO, USA) for 60 mins, and washed in PBST. For TUNEL staining, sections were incubated with terminal deoxynucleotidal transferase (TdT) buffer (30 mmol/L Tris, 140 mmol/L sodium cacodylate, 1 mmol/L cobalt chloride, pH 7.2) containing TdT (0.5 U/ml) and fluorescein-12-dUTP or biotin-16-dUTP (0.04 mmol/L) (all reagents from Boehringer Mannheim, Indianapolis, IN, USA) for 1 h at 37°C. The reaction was terminated by washing in PBS. Biotin-16-dUTP incorporated into tissue was reacted with streptavidin-Cy5 (1:1000, Jackson Immunoresearch) for 5 mins.
After washing in PBS, sections were dehydrated in ascending ethanol series, immersed in xylene and cover-slipped with Permount (Fisher Scientific, Pittsburgh, PA, USA). Triple labelled sections were analyzed on a Leica DMRB/Bio-Rad MRC 1024 krypton–argon laser scanning confocal microscope. Double labelled sections were analyzed on a Nikon Eclipse T300 fluorescence microscope. Excitation/emission wavelengths were 488/506 nm for Cy2, 488/519 nm for Alexa 488, 543/585 nm for Cy3, 594/618 nm for Alexa 594, and 647/670 nm for Cy5, respectively. Negative controls included incubation with NGS instead of primary antibodies, or omission of biotin-16-dUTP or of secondary antibodies. Specificity controls also included varying the order of reaction with different antibodies and TUNEL.
Statistical Analyses
Western blot results are representative of 5 mice per time point. Immunohistochemical data are representative of 3 animals per time point. Bar charts show mean + one standard deviation. ANOVA and Dunnett’s test were used to analyze mouse densitometry measurements as only one measure per animal per time point was performed. Human densitometry data were analyzed by ANOVA followed by Tukey’s test. For all comparisons, P<0.05 was considered significant.
Results
All mice survived the trauma protocol and the experimental period. Recovery of consciousness was generally complete within 5 to 10 mins of discontinuing anesthesia. No mice became apneic at the level of injury used. Mice were able to ambulate and groom and feed themselves within 10 to 30 mins of awakening. No seizures or abnormal motor activity or behavior was noted.
Expression of cFLIP mRNA in Normal Mouse Brain
To identify cFLIP mRNA splice variants in mouse brain, a 3′RACE strategy was employed. We used a mouse gene-specific primer predicted to amplify all murine splice variants on the basis of homology with the human cFLIP mRNA species present in the EMBL database. After 35 cycles of amplification, two amplicons were obtained of 1.1 and 2.2 kb when reverse transcriptase was included in the first strand synthesis (Figure 1). A control reaction performed in the absence of reverse transcriptase was devoid of amplification products, as were amplifications where exogenous template was excluded or only one primer was included (Figure 1 and data not shown). The 1.1 and 2.2 kb species were cloned and subjected to sequence analysis and identified as mouse cFLIP-α and δ, respectively.
Figure 1.
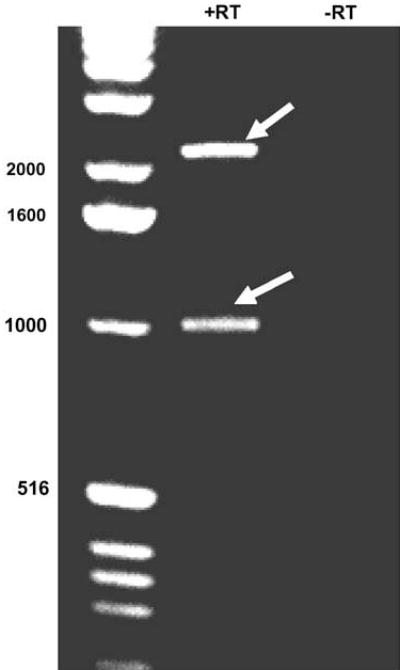
Detection of cFLIP mRNA in mouse brain. 3′-Rapid amplification of cDNA ends (RACE) RT-PCR was used to analyze cFLIP transcript expression in RNA extracted from uninjured mouse cortex. The 5′ primer was a mouse cFLIP-specific oligonucleotide (‘flip-all’), complementary to a predicted consensus sequence present in all mouse splice variants based on the human flip sequences. The 3′ primer was complementary to the polyA tail. First-strand cDNA preparations are shown, prepared in the presence ( + RT) and absence (−RT) of reverse transcriptase. Left lane contains 1 kb DNA ladder. After 35 cycles of amplification in the presence of reverse transcriptase, two amplicons were detected at ~2.2 and ~1.1 kb (arrows). Sequencing of the amplicons revealed that the former encodes mouse cFLIP-α and the latter mouse cFLIP-δ. mRNA corresponding to cFLIP-γ and cFLIP-β was not detected.
Expression of cFLIP Isoforms in Normal and Contused Mouse Brain
In cortical homogenates from normal adult mouse brain, cFLIP-α was barely detectable whereas cFLIP-δ was robustly detected on immunoblots (Figure 2A). After CCI, expression of cFLIP-α was increased in mouse cortical homogenates at 3 and 6 h, but decreased to control levels by 12 h, and increased again from 24 to 72 h after injury. In contrast, cFLIP-δ expression decreased from 3 to 12 h after CCI, then increased again at 24 to 72 h. Densitometric analysis of cFLIP expression showed that, compared with expression in control (naïve) mouse cortices, cFLIP-α was significantly increased at 3 and 6 h after CCI whereas cFLIP-δ expression was significantly decreased at 12 h after CCI (Figures 2B and 2C) (P<0.05 for all comparisons between CCI time points and control).
Figure 2.
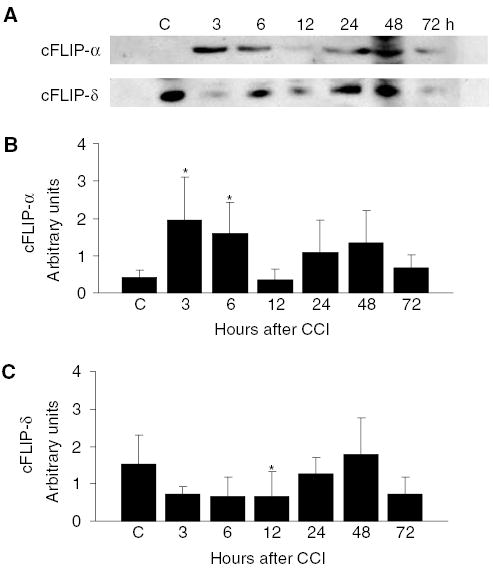
Expression of cFLIP isoforms in normal (uninjured) and contused mouse cortex after CCI. (A) Cortical lysates from uninjured mice (C) or from animals at 3, 6, 12, 24, 48, or 72 h after controlled cortical impact (CCI) were subjected to Western blot analysis using anti-cFLIP antibody 1159. Bands of 55 kD corresponding to cFLIP-α were barely detectable in naïve (C) mouse brain. Expression of cFLIP-α was detected by 3 h after CCI and remained detectable for up to 72 h. Expression of cFLIP-δ was decreased compared with control at 3 to 12 h after CCI, then increased at later times. (B) Densitometric analysis of cFLIP-α expression after CCI. Expression of cFLIP-α was normalized to that of beta-actin and densitometry performed on immunoblots (n = 5/time point). Compared with control, cFLIP-α expression was significantly increased at 3 and 6 h after CCI (*P<0.05 versus control). (C) Densitometric analysis of cFLIP-δ expression after CCI. Expression of cFLIP-δ was normalized to that of beta-actin and densitometry performed on immunoblots as in (B). Compared with control, cFLIP-δ expression was significantly decreased at 12 h after controlled cortical impact (*P<0.05 versus control).
Expression of cFLIP Isoforms in Human Brain
Figure 3 shows cFLIP expression in homogenates of human cortical brain samples. cFLIP-α was clearly detectable in contused brain surgically removed from 3 patients with traumatic brain injury. Faint bands at 28 kD, corresponding to the molecular weight of cFLIP-δ, were detectable in all the TBI and epilepsy lysates and in two of the three non-CNS cases. A band corresponding to cFLIP-α was faint or undetected in excised tissue from four patients with intractable epilepsy and three patients who died of non-CNS causes. Densitometric analysis was performed on these bands (Figure 3B), but the comparison between groups has low statistical power given the small sample numbers and heterogeneity of the samples. Despite the small numbers of patients, there was a significant increase in expression of cFLIP-α in TBI patients relative to epilepsy patients (Figure 3B), though the increase relative to post-mortem non-CNS cases was not statistically significant.
Figure 3.
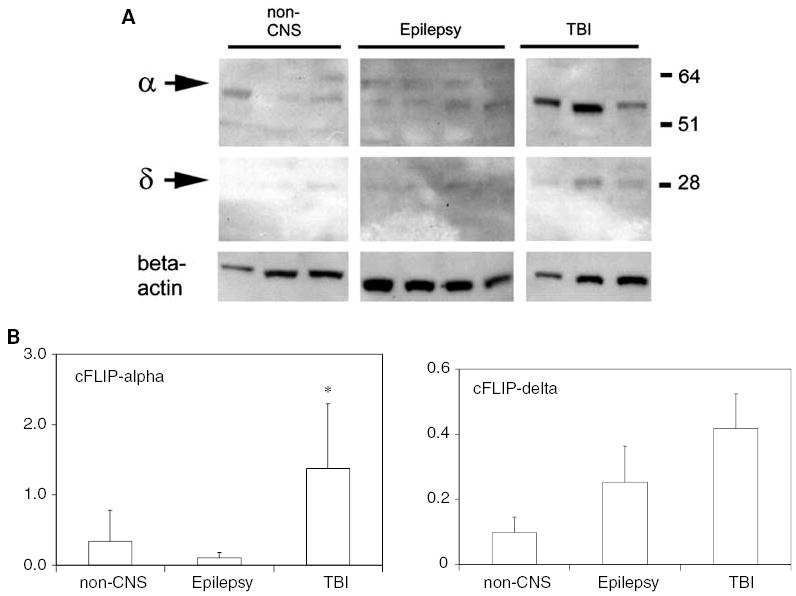
Western blot analysis of cFLIP species in human cortex. (A) Human cortical lysates were obtained from 3 post-mortem brains from patients dying of non-CNS pathologies, 4 patients with intractable epilepsy, and 3 patients with TBI. Lysates were subjected to Western blot analysis using anti-cFLIP antibody 1159. Beta-actin is shown as a marker for protein loading. In cortical samples from non-TBI cases, cFLIP-α was faintly detected. In the three TBI samples, cFLIP-α was abundant. (B) Abundance of cFLIP proteins was analyzed by semiquantitative densitometry. Bars show density relative to beta-actin, mean + 1 s.d. Expression of cFLIP-α was significantly increased in the TBI samples relative to the Epilepsy group (*P<0.05, ANOVA followed by Tukey’s test) but not relative to the non-CNS cases. Expression of cFLIP-δ was not significantly different between the three groups (P>0.05).
Immunohistochemical Localization of cFLIP-δ in Normal Mouse Brain and After Controlled Cortical Impact
Figure 4 shows immunohistochemical localization of cFLIP-δ in frozen sections of normal mouse brain. Immunoreactivity with PX112 (anti-cFLIP-delta) was detected in most, but not all, cells in cortical and subcortical brain regions (Figures 4A and 4B). cFLIP-δ colocalized with neurons (Figures 4C–4E) and non-neuronal cell types in cortical and hippocampal brain regions, and with endothelium of some medium- and large-sized blood vessels (Figures 4F–4H). cFLIP-δ was also weakly detected in astrocytes in cortex and hippocampus (Figure 6 and data not shown).
Figure 4.
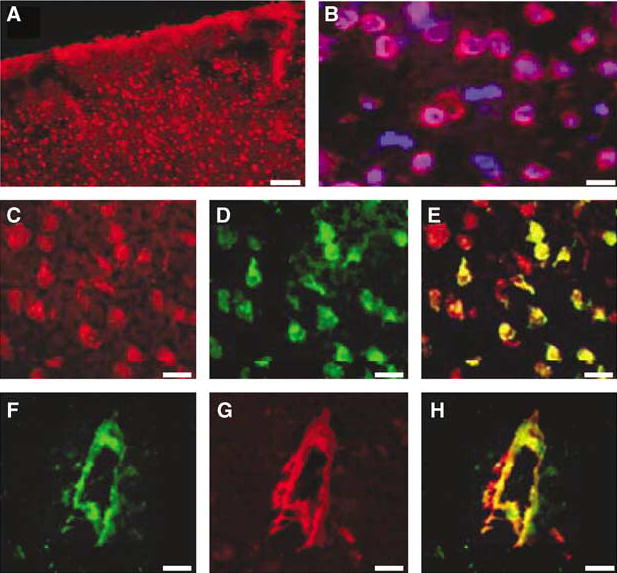
Immunohistochemical localization of cFLIP-δ in mouse brain. (A) cFLIP-δ immunoreactivity in normal mouse cortex (red: anti-cFLIP-δ antibody PX112). (B) High-power magnification of a region depicted in (A) counterstained with Hoechst nuclear stain (blue). Note that not all nucleated cells express cFLIP-δ. (C–E) cFLIP-δ is expressed in neurons. cFLIP-δ (red, panel C) and the neuronal marker NeuN (green, panel D) colocalized (overlay, yellow, panel E). (F–H) Expression of cFLIP-δ in vascular endothelium. cFLIP-δ (green, panel F) colocalized with a subpopulation of medium and large-sized blood vessels (red, panel G) in cortical and hippocampal regions (overlay, yellow, panel H). Scale bars: (A, 50 μm; B–H, 10 μm).
Figure 6.
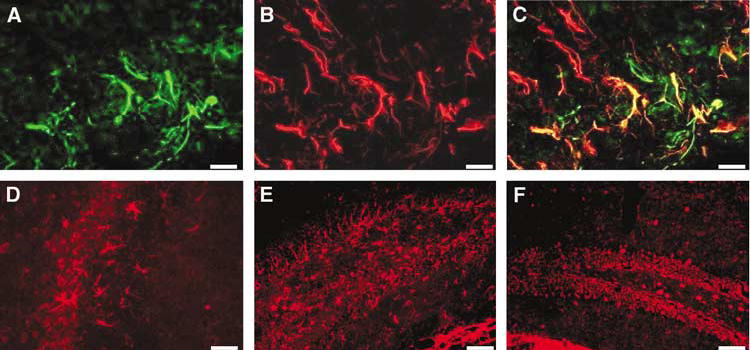
cFLIP-δ expression in reactive astrocytes in injured cortex and hippocampus after CCI. (A) PX112-labelled cells (green) morphologic features of reactive astrocytes stained positively with GFAP (red, panel B) in penumbral regions of the lesion at 7 after CCI (overlay, panel C). PX112-labelled cells (red) with morphologic features of astrocytes were robustly detected at 48 h CCI in injured CA3 (D) and dentate gyrus (E). cFLIP-δ was relatively weakly expressed in glial cells in contralateral (uninjured) dentate gyrus (F). Scale bars: A–D (40 μm); E, F (100 μm).
Figure 5 shows immunohistochemical localization of cFLIP-δ in cortex and hippocampus after CCI. At 1 h after CCI, cFLIP-δ expression was maintained or increased in neurons and other cell types in ipsilateral cortex and hippocampus (Figures 5A and 5F). However, from 3–72 h cellular cFLIP-δ immunoreactivity was markedly decreased in injured cortex (Figures 5C, 5E, 5G, 5I, 5K and 5M) compared with control, uninjured cortex (Figure 4A). The observed decrease in cortical cFLIP-δ immunoreactivity was not related to acute cell loss but to decreased cFLIP-δ in individual brain cells (Figures 5C and 5D). In contrast to expression in cortex, cFLIP-δ immunoreactivity was maintained in hippocampal brain regions at all time points examined (Figures 5F, 5H, 5J and L) relative to control hippocampus from uninjured mice (Figure 5B).
Figure 5.
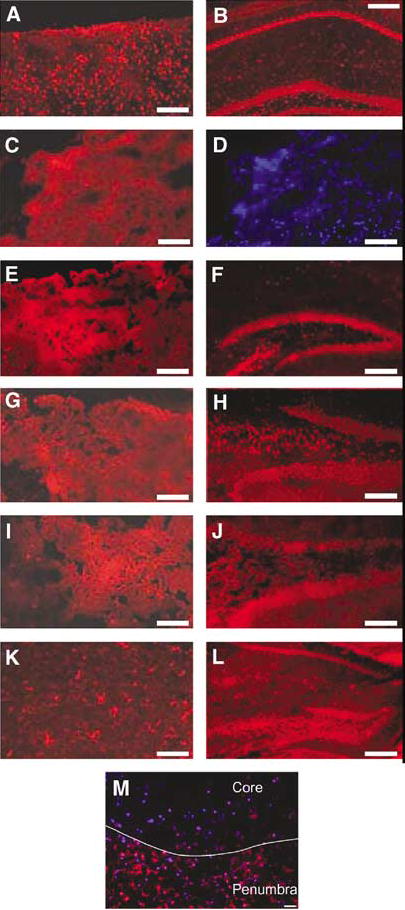
Differential expression of cFLIP-δ in mouse cortex and hippocampus after CCI. Cortical brain sections from within the contusion produced by CCI were reacted with anti-cFLIP-δ (red). Robust cellular expression of cFLIP was detected in injured cortex at 1 h (A) after CCI. Decreased cFLIP-δ expression in injured cortex was observed at 3 (C), 12 (E), 24 (G), 48 (I), and 72 h (K); however, cFLIP-δ was robustly expressed in cells with glial morphology in injured cortex at 72 h (K). In contrast, robust cFLIP-δ expression was detected in naïve hippocampus (B), and in injured hippocampus at 1 (F), 12 (H), 24 (J), and 72 h (L). Panel D shows Hoechst counterstain of the cortical brain region depicted in panel C. Note that decreased cFLIP-δ immunoreactivity is not associated with overt loss of nucleated cells. (M) cFLIP staining in penumbra and core areas of cerebral contusion at 12 h after CCI. Note decreased immunostaining in contusion core but robust staining in penumbra. Scale bars: 100 μm except B, F, L (200 μm) and M (40 μm).
At 24 h–7 days after CCI, cells with glial morphology labelled intensely with PX112 in cortical and subcortical brain regions surrounding the contusion (Figure 6). All PX112-labelled cells with astrocytic morphology colabelled with anti-GFAP, a marker for reactive astrocytes (Figures 6A–6C). By 48 to 72 h post-CCI, cFLIP-positive astrocytes could be seen within and surrounding the margins of contused cortex (e.g., Figure 5K), as well as within ipsilateral hippocampus (Figure 6D and 6E).
Association of cFLIP-δ Immunoreactivity with TUNEL
In injured cortex, discrete populations of TUNEL-positive and cFLIP-positive cells were observed at most time points examined (e.g., Figure 7A). However, TUNEL staining mainly occurred (from 3 to 48 h) in cells from injured cortex with decreased but still detectable cFLIP-δ expression (Figure 7B). In ipsilateral dentate gyrus and CA3, TUNEL staining at 3 to 48 h was observed in neurons and other cell types that maintained strong cFLIP-δ immunoreactivity (Figure 7B). Reactive astrocytes in the injured hemisphere that colocalized cFLIP-δ only occasionally were TUNEL-positive (Figure 7C).
Figure 7.
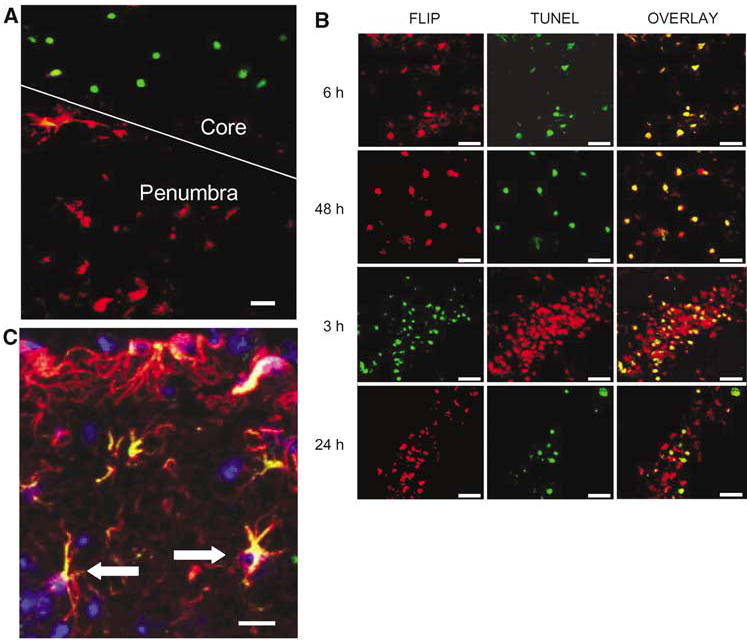
Relationship between cFLIP-δ immunoreactivity and TUNEL in cortex and hippocampus after CCI. (A) Region of injured cortex (core) and cortical penumbra taken from a mouse at 6 h after CCI. cFLIP-δ immunoreactivity (red) is robust in cells from the cortex penumbra whereas TUNEL staining (green) is robust in contused brain cells that lack detectable cFLIP-δ expression. (B) Top 2 rows: Colocalization of cFLIP-δ and TUNEL in injured cortical cells at 6 and 48 h after CCI. cFLIP-δ (red) was expressed in cells within contused cortex that were TUNEL-positive (green) at both time points (overlay). Bottom 2 rows: Colocalization of cFLIP-δ (red) and TUNEL (green) in ipsilateral hippocampal cells at 3 and 24 h after CCI. Cells robustly expressing cFLIP-δ also colocalized with TUNEL at both time points (overlay). (C) TUNEL in cFLIP-δ-positive astrocytes. Brain sections from the contusion at 48 h after CCI were reacted with anti-cFLIP-δ (green), anti-glial fibrillary acidic protein (red), and TUNEL (blue) and were photographed on a laser scanning confocal microscope. Overlap of red and green is shown as yellow. Some astrocytes expressing cFLIP-δ were TUNEL-positive (arrows). Scale bars: A, 40 μm; B, 40 μm; C, 20 μm.
Discussion
We studied the spatial and temporal distribution of cFLIP species in contused mouse and human brain, to test the hypothesis that cFLIP expression changes in contused brain as part of an endogenous response to TBI. The most striking finding of our study was that expression of both cFLIP-α and cFLIP-δ markedly decreased after CCI. Although cFLIP-α expression increased initially, expression of both cFLIP-α and cFLIP-δ decreased by 12 h after CCI, a time when cell death is robust. Cellular cFLIP-δ immunoreactivity was reduced in cortical neurons and other cell types as early as 3 h after injury and remained decreased for 72 h. The marked reduction in cellular cFLIP-δ expression was observed exclusively in injured cortex, and was not explained by overt cell loss, even at later times after injury (Figure 5 and data not shown). In contrast, cFLIP-δ expression was maintained in neurons in injured dentate gyrus, CA3, and CA1 at all time points studied. Thus, cerebral contusion induces a marked reduction in cFLIP-α and cFLIP-δ in brain regions that are vulnerable to cell death after TBI.
Our findings support the speculation that cFLIPs protect against posttraumatic cortical cell death as long as their expression remains above a critical level. For example, TUNEL-positive cells were not detected in injured cortex at 1 h after CCI, a time when cellular cFLIP-δ expression was still robust (Figure 4), but TUNEL-positive cells were detected at later times when cFLIP-δ immunostaining was reduced. This inverse relationship between cFLIP-δ expression and cell death (assessed by TUNEL) in injured cortex persisted for up to 72 h after CCI. In contrast, brain cells at the margin of the contusion that strongly expressed cFLIP-δ remained TUNEL-negative. Loss of cFLIP immunoreactivity was inversely associated with caspase-3 cleavage and TUNEL in myocytes after cardiac ischemia–reperfusion injury (Rasper et al, 1998), and a cFLIP species detected by Western blot correlated inversely with prevalence of apoptotic cell death in a rat spinal cord compression model (Casha et al, 2001). Our data suggest that similar loss of cFLIP-δ expression could also contribute to posttraumatic cell death in injured cerebral cortex. The same could be true for cFLIP-α based on our immunoblot data; however, we were not able to examine the spatial profile of cFLIP-α expression because the antibodies used do not stain cFLIP-α by immunohistochemistry.
In contrast to cortical cell death, there was no apparent relationship between cFLIP-δ immunoreactivity and TUNEL in injured cells within ipsilateral dentate gyrus, CA3, or CA1. These data argue against a protective role for cFLIP-δ in injured hippocampus, as has been reported for bcl-2 and Akt in experimental TBI (Clark et al, 1997; Noshita et al, 2002). One explanation for the regional differences in cFLIP-δ expression observed in the current study could be that initiating mechanisms of posttraumatic cell death differ in cortex versus hippocampus. Preliminary support for this idea was reported in mice lacking TNFα and Fas receptor, which have immunohistochemically detectable cleaved caspase-3 in hippocampus, but not cortex, after CCI (Bermpohl et al, 2004). In cultured PC12 cells, cFLIP-α protects against death initiated by the extrinsic but not the intrinsic pathway (Wu et al, 2004). If extrinsic death mechanisms predominate in posttraumatic cortex but not hippocampus, a relationship between cFLIP-δ expression and cell death (assessed by TUNEL) would be expected in cortical but not hippocampal brain regions. Alternatively, robust expression of cFLIPs might be inadequate to prevent posttraumatic cell death in hippocampus or cFLIP expression might be an epiphenomenon of TBI. cFLIPs are worthy of further study because they provide additional evidence for programmed cell death mechanisms after TBI, and numerous prior reports implicate death receptor activation in cortical cell death after TBI (Beer et al, 2001; Keane et al, 2001; Knoblach et al, 2002; Qiu et al, 2002; Zhang et al, 2003). Furthermore, cFLIPs can inhibit caspase-mediated apoptosis, ‘programmed necrotic’ cell death (Vercammen et al, 1998; Chan et al, 2003; Wang et al, 2003; Vanden Berghe et al, 2004), and caspase-independent programmed cell death (Grambihler et al, 2003), all of which may contribute to TBI.
A marked increase in cFLIP-δ immunoreactivity was detected in a subset of reactive astrocytes surrounding the margins of contused cortex and within injured hippocampus at 48 to 72 h (Figures 5 and 6). Thus, astrocytes were the most likely source of cFLIP-δ upregulation detected by Western blot at later times after CCI. Increased expression of cFLIP-δ (or cFLIP-α) may contribute to resistance to cell death observed in astrocytes in vitro and in vivo (Goldberg and Choi, 1993; Petito et al, 1998). However, our data suggest that only incomplete protection might be afforded in the setting of TBI because occasionally astrocytes expressing cFLIP-δ were also TUNEL-positive (Figure 7). In addition to its anti-cell-death function, cFLIP-δ is a potent mitogen and stimulates cell division in T-lymphocytes (Siegmund et al, 2001; Lens et al, 2002). After traumatic CNS injury, astrocytes proliferate and migrate to the injury region where they protect tissue and preserve function, but astrocytes may also form a glial scar that inhibits axon regeneration (Silver and Miller, 2004; Faulkner et al, 2004). We speculate that cFLIP upregulation in a subset of astrocytes may influence their proliferation and/or differentiation in contused brain as well as secondarily injured regions such as hippocampus, where cFLIP-positive astrocytes were also robustly observed. If so, cFLIP-δ may also play a role in repair and regeneration of traumatically injured brain, in addition to a role in acute cell death.
cFLIP-δ and cFLIP-α are expressed in neurons and other cell types in human frontal cortex and hippocampus (Engidawork et al, 2001; Rasper et al, 1998; Zhao et al, 2003), and cFLIP-α is depleted from cortical and hippocampal neurons in Alzheimer patients (Engidawork et al, 2001; Zhao et al, 2003). Our data showing that cFLIP-α may increase in human brain after TBI (Figure 3) are consistent with the late (24 to 72 h, Figure 2) increase in cFLIP-α in the mouse CCI model. Direct comparison is difficult, however, because of the lack of precise information on time after injury for the clinical cases. cFLIP-δ expression was not increased in Western blots of brain homogenates from humans with TBI, compared with expression in subjects with seizure disorders or non-CNS pathologic study. Failure to adequately upregulate cFLIP-δ or cFLIP-α may favor DISC assembly after TBI, given the marked upregulation of procaspase-8-DISC that occurs after cortical contusion in humans (Qiu et al, 2002). Our data for cFLIP expression in human TBI are preliminary because the sample size is small and only a single time point examined per patient. Further analyses of cFLIP expression in contused human brain should clarify the effects of injury type/severity, and the possible influence of secondary insults, on regulation of cFLIP expression after human TBI.
Rapid upregulation of cFLIPs (and cFLIP mRNA) has been observed in cultured cells with agents that induce cFLIP expression, particularly through activation of nuclear factor kappa-B (NF-κB) and the phosphatidylinositol-3 kinase (PI-3K/Akt) pathways, both of which are induced early after TBI (Higuchi et al, 2003; Kreuz et al, 2001; Suzuki et al, 2003; Noshita et al, 2002; Sullivan et al, 1999). The marked depletion of cFLIP-δ early after CCI is somewhat surprising, but is consistent with a rapid cellular turnover of cFLIPs that results in an unusually short half-life (a few hours, Kreuz et al, 2001; Kim et al, 2002). Although the half-life of cFLIP-δ is similar to that of cFLIP-α, expression of cFLIP-α increased transiently after CCI while cFLIP-δ progressively decreased. Further work is needed to elucidate mechanisms responsible for the differential regulation of cFLIP isoforms after TBI.
We speculate that cFLIPs provide an endogenous protective response to acute brain injury, at least in injured cortex, and that loss of cFLIP expression could increase posttraumatic cell death. If cFLIPs play a significant role in the pathogenesis of TBI, endogenous upregulation or exogenous delivery of cFLIP species to traumatically injured brain might be novel therapeutic approaches to limit posttraumatic cell death, with the caveat that cFLIPs may also positively or negatively influence regeneration and repair mechanisms as well. Newly emerging techniques to overexpress or knockdown cFLIPs in brain, combined with delivery of cFLIP DNA, SiRNA, or protein to brain using viral vectors or protein transduction domains (Siegmund et al, 2002; Soutschek et al, 2004; Krautwald et al, 2004), should enable direct investigation of a possible role for cFLIPs in the pathogenesis of TBI and other acute brain injuries.
Acknowledgments
AHH thanks Drs CJ Guerin and HMTC Mulrooney for helpful comments.
Footnotes
This work was supported by KO8NS41969 (MJW), RO1NS37141 (MAM). Post Doc program of the German Academic Exchange Service (D/03/18557)(DB).
References
- Beer R, Franz G, Krajewski S, Pike BR, Hayes RL, Reed JC, Wang KK, Klimmer C, Schmutzhard E, Poewe W, Kampfl A. Temporal and spatial profile of caspase 8 expression and proteolysis after experimental traumatic brain injury. J Neurochem. 2001;78:862–73. doi: 10.1046/j.1471-4159.2001.00460.x. [DOI] [PubMed] [Google Scholar]
- Bermpohl D, You Z, Lai C, Moskowitz MA, Whalen MJ. Cell death and neurologic dysfunction after controlled cortical impact in immature mice deficient in TNF alpha and Fas genes. J Neurotrauma. 2004;21:1332. [Google Scholar]
- Casha S, Yu WR, Fehlings MG. Oligodendroglial apoptosis occurs along degenerating axons and is associated with FAS and p75 expression following spinal cord injury in the rat. Neuroscience. 2001;103:203–18. doi: 10.1016/s0306-4522(00)00538-8. [DOI] [PubMed] [Google Scholar]
- Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, Orenstein J, Moss B, Lenardo MJ. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem. 2003;278:51613–21. doi: 10.1074/jbc.M305633200. [DOI] [PubMed] [Google Scholar]
- Clark RS, Chen J, Watkins SC, Kochanek PM, Chen M, Stetler RA, Loeffert JE, Graham SH. Apoptosis-suppressor gene bcl-2 expression after traumatic brain injury in rats. J Neurosci. 1997;17:9172–82. doi: 10.1523/JNEUROSCI.17-23-09172.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RS, Kochanek PM, Chen M, Watkins SC, Marion DW, Chen J, Hamilton RL, Loeffert JE, Graham SH. Increases in Bcl-2 and cleavage of caspase-1 and caspase-3 in human brain after head injury. FASEB J. 1999;13:813–21. doi: 10.1096/fasebj.13.8.813. [DOI] [PubMed] [Google Scholar]
- Demjen D, Klussmann S, Kleber S, Zuliani C, Stieltjes B, Metzger C, Hirt UA, Walczak H, Falk W, Essig M, Edler L, Krammer PH, Martin-Villalba A. Neutralization of CD95 ligand promotes regeneration and functional recovery after spinal cord injury. Nat Med. 2004;10:389–95. doi: 10.1038/nm1007. [DOI] [PubMed] [Google Scholar]
- Engidawork E, Gulesserian T, Yoo BC, Cairns N, Lubec G. Alteration of caspases and apoptosis-related proteins in brains of patients with Alzheimer’s disease. Biochem Biophys Res Commun. 2001;281:84–93. doi: 10.1006/bbrc.2001.4306. [DOI] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–55. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci. 1993;13:3510–24. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grambihler A, Higuchi H, Bronk SF, Gores GJ. cFLIP-L inhibits p38 MAPK activation: an additional anti-apoptotic mechanism in bile acid-mediated apoptosis. J Biol Chem. 2003;278:26831–7. doi: 10.1074/jbc.M303229200. [DOI] [PubMed] [Google Scholar]
- Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–6. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- Higuchi H, Yoon JH, Grambihler A, Werneburg N, Bronk SF, Gores GJ. Bile acids stimulate cFLIP phosphorylation enhancing TRAIL-mediated apoptosis. J Biol Chem. 2003;278:454–61. doi: 10.1074/jbc.M209387200. [DOI] [PubMed] [Google Scholar]
- Imanishi T, McBride J, Ho Q, O’Brien KD, Schwartz SM, Han DK. Expression of cellular FLICE-inhibitory protein in human coronary arteries and in a rat vascular injury model. Am J Pathol. 2000a;156:125–37. doi: 10.1016/S0002-9440(10)64712-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imanishi T, Murry CE, Reinecke H, Hano T, Nishio I, Liles WC, Hofsta L, Kim K, O’Brien KD, Schwartz SM, Han DK. Cellular FLIP is expressed in cardiomyocytes and down-regulated in TUNEL-positive grafted cardiac tissues. Cardiovasc Res. 2000b;48:101–10. doi: 10.1016/s0008-6363(00)00154-1. [DOI] [PubMed] [Google Scholar]
- Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–5. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- Kawahara A, Ohsawa Y, Matsumura H, Uchiyama Y, Nagata S. Caspase-independent cell killing by Fas-associated protein with death domain. J Cell Biol. 1998;143:1353–60. doi: 10.1083/jcb.143.5.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane RW, Kraydieh S, Lotocki G, Alonso OF, Aldana P, Dietrich WD. Apoptotic and antiapoptotic mechanisms after traumatic brain injury. J Cereb Blood Flow Metab. 2001;21:1189–98. doi: 10.1097/00004647-200110000-00007. [DOI] [PubMed] [Google Scholar]
- Kim Y, Suh N, Sporn M, Reed JC. An inducible pathway for degradation of FLIP protein sensitizes tumor cells to TRAIL-induced apoptosis. J Biol Chem. 2002;277:22320–9. doi: 10.1074/jbc.M202458200. [DOI] [PubMed] [Google Scholar]
- Knoblach SM, Nikolaeva M, Huang X, Fan L, Krajewski S, Reed JC, Faden AI. Multiple caspases are activated after traumatic brain injury: evidence for involvement in functional outcome. J Neurotrauma. 2002;19:1155–70. doi: 10.1089/08977150260337967. [DOI] [PubMed] [Google Scholar]
- Krautwald S, Ziegler E, Tiede K, Pust R, Kunzendorf U. Transduction of the TAT-FLIP fusion protein results in transient resistance to Fas-induced apoptosis in vivo. J Biol Chem. 2004;279:44005–11. doi: 10.1074/jbc.M401327200. [DOI] [PubMed] [Google Scholar]
- Kreuz S, Siegmund D, Scheurich P, Wajant H. NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol Cell Biol. 2001;21:3964–73. doi: 10.1128/MCB.21.12.3964-3973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger A, Baumann S, Krammer PH, Kirchhoff S. FLICE-inhibitory proteins: regulators of death receptor-mediated apoptosis. Mol Cell Biol. 2001;21:8247–54. doi: 10.1128/MCB.21.24.8247-8254.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lens SM, Kataoka T, Fortner KA, Tinel A, Ferrero I, MacDonald RH, Hahne M, Beermann F, Attinger A, Orbea HA, Budd RC, Tschopp J. The caspase 8 inhibitor c-FLIP(L) modulates T-cell receptor-induced proliferation but not activation-induced cell death of lymphocytes. Mol Cell Biol. 2002;22:5419–33. doi: 10.1128/MCB.22.15.5419-5433.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- Martin-Villalba A, Hahne M, Kleber S, Vogel J, Falk W, Schenkel J, Krammer P. Therapeutic neutralization of CD95-ligand and TNF attenuates brain damage in stroke. Cell Death Differ. 2001;8:679–86. doi: 10.1038/sj.cdd.4400882. [DOI] [PubMed] [Google Scholar]
- Matsumura H, Shimizu Y, Ohsawa Y, Kawahara A, Uchiyama Y, Nagata S. Necrotic death pathway in Fas receptor signaling. J Cell Biol. 2000;151:1247–56. doi: 10.1083/jcb.151.6.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheau O. Cellular FLICE-inhibitory protein: an attractive therapeutic target? Expert Opin Ther Targets. 2003;7:559–73. doi: 10.1517/14728222.7.4.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noshita N, Lewen A, Sugawara T, Chan P. Akt phosphorylation and neuronal survival after traumatic brain injury in mice. Neurobiol Dis. 2002;9:294–304. doi: 10.1006/nbdi.2002.0482. [DOI] [PubMed] [Google Scholar]
- Nuttall ME, Lee D, McLaughlin B, Erhardt JA. Selective inhibitors of apoptotic caspases: implications for novel therapeutic strategies. Drug Discov Today. 2001;6:85–91. doi: 10.1016/s1359-6446(00)01601-9. [DOI] [PubMed] [Google Scholar]
- Petito CK, Olarte JP, Roberts B, Nowak Jr TS, Pulsinelli WA. Selective glial vulnerability following transient global ischemia in rat brain. J Neuropathol Exp Neurol. 1998;57:231–8. doi: 10.1097/00005072-199803000-00004. [DOI] [PubMed] [Google Scholar]
- Qiu J, Whalen MJ, Lowenstein P, Fiskum G, Fahy B, Darwish R, Aarabi B, Yuan J, Moskowitz MA. Upregulation of the Fas receptor death-inducing signaling complex after traumatic brain injury in mice and humans. J Neurosci. 2002;22:3504–11. doi: 10.1523/JNEUROSCI.22-09-03504.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoul C, Estevez AG, Nishimune H, Cleveland DW, deLapeyriere O, Henderson CE, Haase G, Pettmann B. Motoneuron death triggered by a specific pathway downstream of Fas. Potentiation by ALS-linked SOD1 mutations. Neuron. 2002;35:1067–83. doi: 10.1016/s0896-6273(02)00905-4. [DOI] [PubMed] [Google Scholar]
- Raoul C, Henderson CE, Pettmann B. Programmed cell death of embryonic motoneurons triggered through the Fas death receptor. J Cell Biol. 1999;147:1049–62. doi: 10.1083/jcb.147.5.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasper DM, Vaillancourt JP, Hadano S, Houtzager VM, Seiden I, Keen SL, Tawa P, Xanthoudakis S, Nasir J, Martindale D, Koop BF, Peterson EP, Thornberry NA, Huang J, MacPherson DP, Black SC, Hornung F, Lenardo MJ, Hayden MR, Roy S, Nicholson DW. Cell death attenuation by ‘Usurpin’, a mammalian DED-caspase homologue that precludes caspase-8 recruitment and activation by the CD-95 (Fas, APO-1) receptor complex. Cell Death Differ. 1998;5:271–88. doi: 10.1038/sj.cdd.4400370. [DOI] [PubMed] [Google Scholar]
- Robertson GS, Crocker SJ, Nicholson DW, Schulz JB. Neuroprotection by the inhibition of apoptosis. Brain Pathol. 2000;10:283–92. doi: 10.1111/j.1750-3639.2000.tb00262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi C, Schmitz I, Krammer PH, Peter ME. The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem. 1999;274:1541–8. doi: 10.1074/jbc.274.3.1541. [DOI] [PubMed] [Google Scholar]
- Shohami E, Bass R, Wallach D, Yamin A, Gallily R. Inhibition of tumor necrosis factor alpha (TNFalpha) activity in rat brain is associated with cerebroprotection after closed head injury. J Cereb Blood Flow Metab. 1996;16:378–84. doi: 10.1097/00004647-199605000-00004. [DOI] [PubMed] [Google Scholar]
- Siegmund D, Hadwiger P, Pfizenmaier K, Vornlocher HP, Wajant H. Selective inhibition of FLICE-like inhibitory protein expression with small interfering RNA oligonucleotides is sufficient to sensitize tumor cells for TRAIL-induced apoptosis. Mol Med. 2002;8:725–32. [PMC free article] [PubMed] [Google Scholar]
- Siegmund D, Mauri D, Peters N, Juo P, Thome M, Reichwein M, Blenis J, Scheurich P, Tschopp J, Wajant H. Fas-associated death domain protein (FADD) and caspase-8 mediate up-regulation of c-Fos by Fas ligand and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) via a FLICE inhibitory protein (FLIP)-regulated pathway. J Biol Chem. 2001;276:32585–90. doi: 10.1074/jbc.M100444200. [DOI] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–56. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M, Elbashir S, Geick A, Hadwiger P, Harborth J, John M, Kesavan V, Lavine G, Pandey RK, Racie T, Rajeev KG, Rohl I, Toudjarska I, Wang G, Wuschko S, Bumcrot D, Koteliansky V, Limmer S, Manoharan M, Vornlocher HP. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432:173–8. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- Sullivan P, Bruce-Keller A, Rabchevsky A, Christakos S, Clair D, Mattson MP, Scheff S. Exacerbation of damage and altered NF-kappaB activation in mice lacking tumor necrosis factor receptors after traumatic brain injury. J Neuroscience. 1999;19:6248–56. doi: 10.1523/JNEUROSCI.19-15-06248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Kusakai G, Kishimoto A, Lu J, Ogura T, Esumi H. ARK5 suppresses the cell death induced by nutrient starvation and death receptors via inhibition of caspase 8 activation, but not by chemotherapeutic agents or UV irradiation. Oncogene. 2003;22:6177–82. doi: 10.1038/sj.onc.1206899. [DOI] [PubMed] [Google Scholar]
- Tschopp J, Irmler M, Thome M. Inhibition of fas death signals by FLIPs. Curr Opin Immunol. 1998;10:552–8. doi: 10.1016/s0952-7915(98)80223-9. [DOI] [PubMed] [Google Scholar]
- Vanden Berghe T, van Loo G, Saelens X, van Gurp M, Brouckaert G, Kalai M, DeClercq W, Vandenabeele P. Differential signaling to apoptotic and necrotic cell death by Fas- associated death domain protein FADD. J Biol Chem. 2004;279:7925–33. doi: 10.1074/jbc.M307807200. [DOI] [PubMed] [Google Scholar]
- Vercammen D, Brouckaert G, Denecker G, Van de Craen M, Declercq W, Fiers W, Vandenabeele P. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med. 1998;188:919–30. doi: 10.1084/jem.188.5.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajant H, Pfizenmaier K, Scheurich P. Non-apoptotic Fas signaling. Cytokine Growth Factor Rev. 2003;14:53–66. doi: 10.1016/s1359-6101(02)00072-2. [DOI] [PubMed] [Google Scholar]
- Wallach D, Varfolomeev EE, Malinin NL, Goltsev YV, Kovalenko AV, Boldin MP. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu Rev Immunol. 1999;17:331–67. doi: 10.1146/annurev.immunol.17.1.331. [DOI] [PubMed] [Google Scholar]
- Wang W, Prince CZ, Mou Y, Pollman MJ. Notch3 signaling in vascular smooth muscle cells induces c-FLIP expression via ERK/MAPK activation. Resistance to Fas ligand-induced apoptosis. J Biol Chem. 2002;277:21723–9. doi: 10.1074/jbc.M202224200. [DOI] [PubMed] [Google Scholar]
- Wang X, Ryter SW, Dai C, Tang ZL, Watkins SC, Yin XM, Song R, Choi AM. Necrotic cell death in response to oxidant stress involves the activation of the apoptogenic caspase-8/bid pathway. J Biol Chem. 2003;278:29184–29191. doi: 10.1074/jbc.M301624200. [DOI] [PubMed] [Google Scholar]
- Wu S, Zhou L, Rose M, Xiao X, Graham SH. c-FLIP-L recombinant adeno-associated virus vector infection prevents Fas-mediated but not nerve growth factor withdrawal-mediated cell death in PC12 cells. Brain Res Mol Brain Res. 2004;122:79–87. doi: 10.1016/j.molbrainres.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Zhang X, Graham SH, Kochanek PM, Marion DW, Nathaniel PD, Watkins SC, Clark RS. Caspase-8 expression and proteolysis in human brain after severe head injury. FASEB J. 2003;17:1367–9. doi: 10.1096/fj.02-1067fje. [DOI] [PubMed] [Google Scholar]
- Zhao M, Cribbs DH, Anderson AJ, Cummings BJ, Su JH, Wasserman AJ, Cotman CW. The induction of the TNF alpha death domain signaling pathway in Alzheimer’s disease brain. Neurochem Res. 2003;28:307–18. doi: 10.1023/a:1022337519035. [DOI] [PubMed] [Google Scholar]